
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElastic MXene conductive layers and electrolyte engineering enable robust potassium storage†
Xinyue
Xu‡
a,
Qingqing
Jiang‡
 *a,
Chenyu
Yang‡
b,
Jinxi
Ruan
b,
Weifang
Zhao
a,
Houyu
Wang
b,
Xinxin
Lu
b,
Zhe
Li
a,
Yuanzhen
Chen
c,
Chaofeng
Zhang
*b,
Juncheng
Hu
*a and
Tengfei
Zhou
*b
*a,
Chenyu
Yang‡
b,
Jinxi
Ruan
b,
Weifang
Zhao
a,
Houyu
Wang
b,
Xinxin
Lu
b,
Zhe
Li
a,
Yuanzhen
Chen
c,
Chaofeng
Zhang
*b,
Juncheng
Hu
*a and
Tengfei
Zhou
*b
aKey Laboratory of Catalysis and Energy Materials Chemistry of Ministry of Education, South-Central Minzu University, Wuhan 430074, China. E-mail: qqjiang@mail.scuec.edu.cn; jchu@mail.scuec.edu.cn
bInstitutes of Physical Science and Information Technology, Key Laboratory of Structure and Functional Regulation of Hybrid Material (Ministry of Education), Anhui University, Hefei 230601, China. E-mail: cfz@ahu.edu.cn; tengfeiz@ahu.edu.cn
cState Key Laboratory for Mechanical Behavior of Materials, School of Materials Science and Engineering, Xi'an Jiaotong University, Xi'an, 710049, China
First published on 19th January 2024
Abstract
The precisely engineered structures of materials greatly influence the manifestation of their properties. For example, in the process of alkali metal ion storage, a carefully designed structure capable of accommodating inserted and extracted ions will improve the stability of material cycling. The present study explores the uniform distribution of self-grown carbon nanotubes to provide structural support for the conductive and elastic MXene layers of Ti3C2Tx-Co@NCNTs. Furthermore, a compatible electrolyte system has been optimized by analyzing the solvation structure and carefully regulating the component in the solid electrolyte interphase (SEI) layer. Mechanistic studies demonstrate that the decomposition predominantly controlled by FSI− leads to the formation of a robust inorganic SEI layer enriched with KF, thus effectively inhibiting irreversible side reactions and major structural deterioration. Confirming our expectations, Ti3C2Tx-Co@NCNTs exhibits an impressive reversible capacity of 260 mA h g−1, even after 2000 cycles at 500 mA g−1 in 1 M KFSI (DME), surpassing most MXene-based anodes reported for PIBs. Additionally, density functional theory (DFT) calculations verify the superior electronic conductivity and lower K+ diffusion energy barriers of the novel superstructure of Ti3C2Tx-Co@NCNTs, thereby affirming the improved electrochemical kinetics. This study presents systematic evaluation methodologies for future research on MXene-based anodes in PIBs.
Introduction
Potassium-ion batteries (PIBs) are increasingly regarded as a prospective substitute for lithium-ion batteries (LIBs) for large-scale energy storage systems owing to plentiful potassium resources, lower redox potential, and smaller solvation radius.1 Nonetheless, the large size of potassium ions and the resulting high repulsive forces lead to structural damage and slow kinetics during repeated intercalation/deintercalation processes, significantly restricting their applicability.2 Consequently, there is a requirement to design high-performance electrode materials with sturdy structures capable of accommodating K+ ions, thereby enhancing rate capability and cycle life.MXenes, which are transition metal carbides/nitrides, possess inherently prime conductivity, mechanical flexibility, and adjustable layer spacing, all of which are extremely desirable for PIBs.3 However, both theoretical and experimental studies confirm that pristine MXenes exhibit low potassium storage capabilities and experience rapid capacity decay owing to susceptible self-aggregation caused by van der Waals interactions or hydrogen bonds between adjacent exfoliated layers.4 The irreversibility of restacking or spontaneous collapse greatly restricts the optimum utilization of MXene-based electrode materials. Therefore, the design of heterostructures is essential to effectively preventing the aggregation or self-restacking of MXene nanosheets in electrochemical energy storage applications.5 Due to their high electrical conductivity and rapid potassium transfer properties, Ti3C2Tx nanosheets are better suited for constructing an electrode framework that ensures fast electron and ion transfer, rather than serving as active materials.6
Carbon nanotubes (CNTs) with high electrical conductivity have been used as the embedding spacer to prevent the restacking of MXene sheets.7 Unfortunately, the ex situ mixing growth method often suffers from disordered dispersion of CNTs and high contact resistance. It is widely acknowledged that vertically aligned arrays of CNTs show great promise as electrical contacts.8 Consequently, various methods have been explored for the in situ growth of highly integrated CNTs between Ti3C2Tx layers, including chemical vapor deposition, hydrothermal reaction, microwave irradiation, and high-temperature pyrolysis treatment.9 Moreover, CNTs can be further enhanced through heteroatom doping, such as the introduction of transition metal active sites or incorporating n sites, which alters the charge density of carbon atoms, provides additional active sites, and improves overall conductivity.10 Drawing inspiration from this, we present a novel “step-by-step” assembly strategy for in situ growth of Co, N co-doped CNTs (Co@NCNTs) on the surface of MXenes. In this approach, Co@NCNTs could serve as spacers to prevent the accumulation of MXene nanosheets as well as function as intraparticle and interparticle charge collectors, thereby facilitating enhanced ion/electron transport channels.
Electrolytes, including potassium salt and solvent, are the bridge that connects the cathode and anode.11 The electrochemical performance of batteries such as reversible capacity, lifetime, rate capability, operational voltages, and safety is closely related to the choice of electrolyte system.12 The electrolyte's decomposition and solvation effects substantially impact the construction of the SEI film and the kinetics of K+ diffusion.13 A robust and flexible SEI is vital for mitigating capacity degradation and minimizing irreversible side reactions.14 However, there is a scarcity of reliable reports that accurately correlate electrolyte salts and solvents with the electrochemical performance of MXene-based electrodes in practical intercalation batteries (PIBs).
In this study, highly conductive elastic layers of Ti3C2Tx MXenes were integrated with uniformly dispersed self-grown Co, N co-doped carbon nanotubes (Co@NCNTs) through high-temperature pyrolysis of zeolitic imidazolate frameworks-67/Ti3C2Tx. This high-temperature pyrolysis of Ti3C2Tx/ZIF-67 resulted in the formation of Ti3C2Tx-Co@NCNTs. The performance of Ti3C2Tx-Co@NCNTs electrodes was systematically investigated using different electrolyte systems containing two potassium salts (KFSI and KPF6) in various solvents (DME and EC/DEC). The Ti3C2Tx-Co@NCNTs composite exhibited remarkable advantages, including: (1) highly conductive nanocomposites with multiple ion-diffusion pathways, facilitating electron/K+ transport and leading to outstanding rate capability, (2) the inherent ability of Ti3C2Tx to accommodate strain resulting from volume changes due to its adaptive interlayer spacing, (3) strong interaction between the vertically aligned Co@NCNTs arrays and the Ti3C2Tx matrix, contributing to robust structural stability and efficient electron transfer, and (4) the influence of electrolyte chemistry in regulating the construction of a robust SEI layer, enabling extended cycle life and enhanced rate performance. As anticipated, the Ti3C2Tx-Co@NCNTs electrode demonstrated a capital reversible capacity of 260 mA h g−1, even at a demanding high current density of 500 mA g−1, after 2000 cycles in 1 M KFSI (DME) electrolyte.
Results and discussion
Material characterization
Fig. 1a illustrates the “step by step” process for the preparation of Ti3C2Tx-Co@NCNTs hybrids. First, the Al atomic layers of the Ti3AlC2 MAX phase were selectively etched with hydrofluoric acid (HF) to obtain Ti3C2Tx with multilayered morphology. The negative charge surface termination groups, such as OH−, O2−, and F− on the Ti3C2Tx nanosheet facilitated the absorption of Co2+ ions through electrostatic interaction. Then, 2-methylimidazole molecules were coordinated with the surface-anchored Co2+ ions to construct Ti3C2Tx/ZIF-67 hybrids. Afterwards, the as-prepared Ti3C2Tx/ZIF-67 nanocomposites were pyrolyzed at 700 °C under an Ar gas atmosphere with the addition of dicyandiamide (DCDA). The 2-methylimidazole ligands were converted into N-doped carbon species, and Co2+ was reduced to a metallic Co phase during the high-temperature pyrolysis process. At high temperature, the Co species catalyze the transformation of N-doped carbon species into N-doped graphitic carbon nanotubes (NCNTs), leading to the successful fabrication of Ti3C2Tx-Co@NCNTs hybrids. Additionally, the presence of DCDA induces the formation of NCNTs at relatively low temperature by accelerating the graphitic structure.7b | ||
| Fig. 1 (a) Scheme for the synthesis route of Ti3C2Tx-Co@NCNTs. SEM images of (b) Ti3C2Tx, (c) the exfoliated Ti3C2Tx, (d) Ti3C2Tx/ZIF-67, and (e) Ti3C2Tx-Co@NCNTs, TEM images of (f) Ti3C2Tx, (g) Ti3C2Tx, (h) Ti3C2Tx/ZIF-67, and (i)Ti3C2Tx-Co@NCNTs and (j) elemental mapping of Ti3C2Tx-Co@NCNTs. | ||
The Ti3C2Tx nanosheets obtained from etching Ti3C2Tx with micrometer dimensions exhibit a loosely structured, accordion-like layer configuration (Fig. 1b, S1a and b†). Subjecting this accordion-like structure to ultrasonic treatment results in the exfoliation of the Ti3C2Tx nanosheets, which possess a smooth surface and an ultrathin thickness of 8–10 layers, with a lateral size of several micrometers (Fig. 1c and S1c†). Additionally, the interlayer spacing of the exfoliated Ti3C2Tx in the c-direction is approximately 13.6 Å, whereas the interlayer spacing of the Ti3C2Tx with micrometer dimensions is about 10.1 Å (Fig. 1f and g). The presence of surface termination groups facilitates the easy adsorption of Co2+ ions on both sides of Ti3C2Tx MXene through electrostatic interactions. Subsequently, the 2-methylimidazole molecules coordinate with the adsorbed Co2+ ions, resulting in the uniform growth and anchoring of ZIF-67 on both sides of Ti3C2Tx (Fig. 1d and h). The distribution of ZIF-67 nanoparticles with varying Co2+ concentrations is shown in Fig. S1d–l.† Following high-temperature pyrolysis, the Co2+ ions are reduced to a metallic Co phase, while the N-containing organic ligands transform into N-doped carbon nanotubes (Fig. 1e, i and S2†). Notably, the Co@NCNTs are evenly dispersed within the conductive layers of Ti3C2Tx, while the Co metal nanoparticles are confined within the NCNTs. Fig. 1i illustrates a lattice distance of 0.25 nm, which corresponds to the (0010) layer of Ti3C2Tx, suggesting that the Ti3C2Tx phase is preserved after high-temperature reduction treatment.15 Furthermore, two distinct visible lattice spacings are observed, with 0.21 nm corresponding to the (222) layer of the Co metal phase and 0.34 nm corresponding to the (002) plane of graphitic carbon.16 The presence of metal Co nanoparticles enhances conductivity and facilitates rapid electron transportation. Elemental mapping technology (EDX) confirms the even distribution of C, N, Co, and Ti elements in the Ti3C2Tx-Co@NCNTs hybrids (Fig. 1j). In contrast, the pyrolysis of the Ti3C2Tx/ZIF-67 precursor at high temperatures without the addition of DCDA leads to the formation of Ti3C2Tx-Co@NC hybrids without the production of NCNTs (Fig. S3†).
The X-ray diffraction technique was used to analyze the phase structure of Ti3AlC2, Ti3C2Tx, Ti3C2Tx/ZIF-67, and Ti3C2Tx-Co@NCNTs (Fig. 2a and S4†). The disappearance of the characteristic (104) peak at 39° of Ti3AlC2 confirms the removal of the Al atomic layers.17 The typical (002) peak of Ti3C2Tx broadens and shifts from 9.6° to 6.5° after ultrasonic treatment, indicating an increase in interlayer spacing, which is consistent with HRTEM images (Fig. 1f and g).18 In the Ti3C2Tx/ZIF-67 hybrids, distinct peaks corresponding to ZIF-67 can be observed. The peaks at 7.3° and 12.8° are sharp and intense, indicating the presence of highly crystalline ZIF-67 particles.19 After high-temperature pyrolysis, the characteristic (002) peak of Ti3C2Tx at ∼7° still appears in the XRD patterns, confirming the presence of the Ti3C2Tx phase in the hybrids. The broad peak at approximately 26.5° corresponds to the (002) plane of graphitized carbon. Additionally, the prominent peaks at 44.1° can be assigned to the metallic Co phase.20
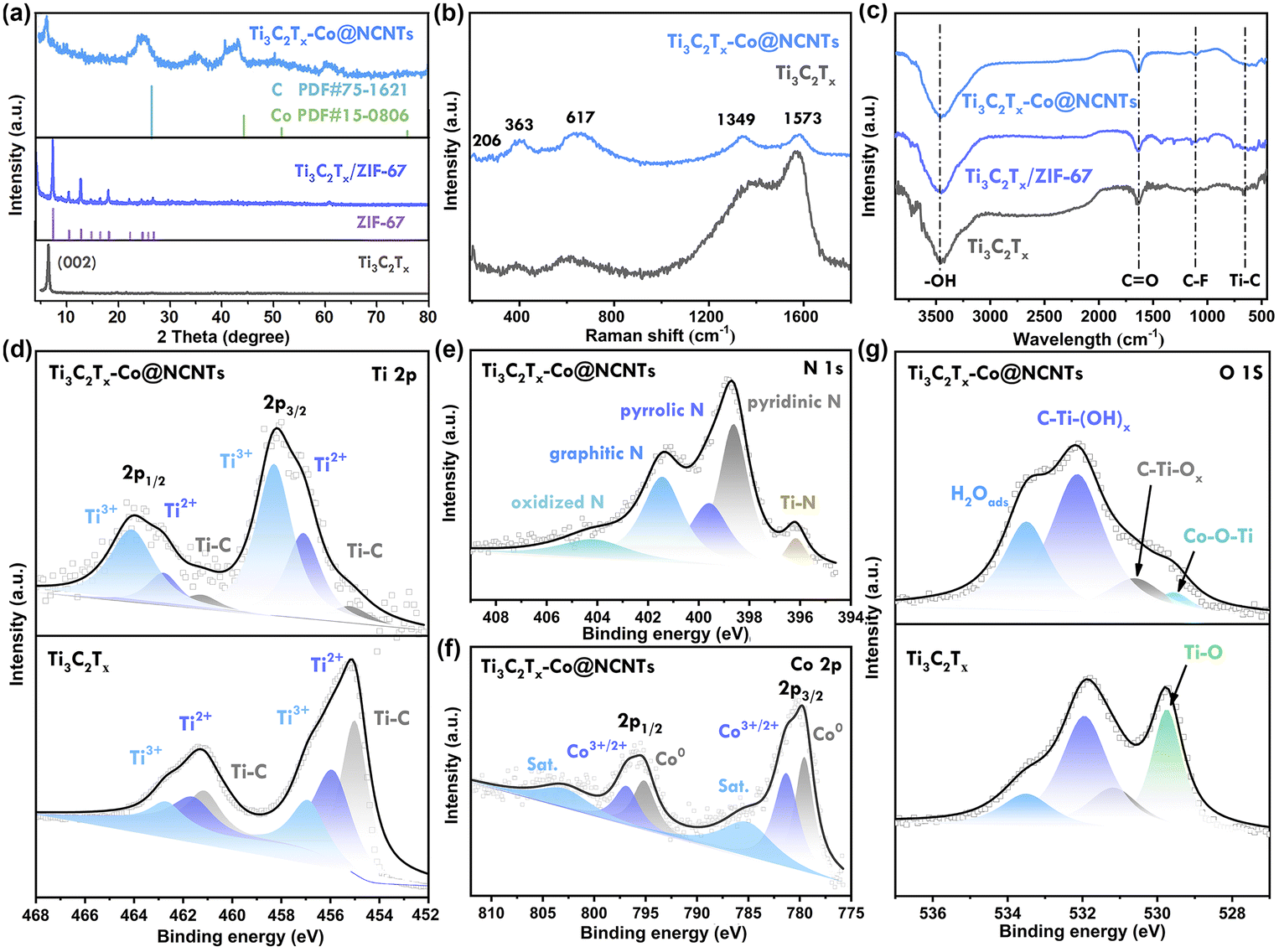 | ||
| Fig. 2 (a) XRD patterns of Ti3C2Tx, Ti3C2Tx/ZIF-67, and Ti3C2Tx-Co@NCNTs, (b) Raman spectrum of Ti3C2Tx, and Ti3C2Tx-Co@NCNTs, (c) FT-IR spectrum of Ti3C2Tx, Ti3C2Tx/ZIF-67 and Ti3C2Tx-Co@NCNTs, and high-resolution XPS spectra of (d) Ti 2p, (e) N 1s, (f) Co 2p, and (g) O 1s. | ||
The Raman spectra of Ti3C2Tx and Ti3C2Tx-Co@NCNTs show multiple similar features within the 200–800 cm−1 range (Fig. 2b), providing further evidence of the presence of the Ti3C2Tx phase in the hybrids. The peak at approximately 206 cm−1 corresponds to the A1g vibration mode (out-of-plane) of C–Ti–X (X = –F, –OH) in Ti3C2Tx MXenes. Additionally, the peaks at 363 cm−1 and 617 cm−1 correspond to the Eg group vibrations, which include in-plane (shear) modes of C–Ti–X (X = –F, –OH).5a The Raman peaks of Ti3C2Tx-Co@NCNTs show a noticeable shift to the right compared to pristine Ti3C2Tx, indicating an increase in interlayer spacing following the growth of NCNTs.21 The value of ID/IG (the disordered carbon-induced band at 1355 cm−1 and the graphite carbon band at 1590 cm−1) is 0.92, indicating a significant number of defect sites in the nitrogen-doped carbon nanotubes. These defect sites can act as active sites for K+ storage.22
The chemical bonds at the surface were further investigated using Fourier transform infrared (FT-IR) spectroscopy (Fig. 2c). In the Ti3C2Tx curve, the peaks at 3432, 1648, 1110, and 652 cm−1 correspond to the –OH, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–F, and Ti–C stretching modes, respectively. In the Ti3C2Tx/ZIF-67 hybrid, the peaks at 683 cm−1 and 743 cm−1 are characteristic of the out-of-plane bending of the imidazole ring. The peaks corresponding to the in-plane bending of the imidazole ring are in the range of 900–1500 cm−1. The characteristic peaks at 1629 cm−1 and 3429 cm−1 in Ti3C2Tx-Co@NCNTs are assigned to the CO and –OH bonds, respectively.23
O, C–F, and Ti–C stretching modes, respectively. In the Ti3C2Tx/ZIF-67 hybrid, the peaks at 683 cm−1 and 743 cm−1 are characteristic of the out-of-plane bending of the imidazole ring. The peaks corresponding to the in-plane bending of the imidazole ring are in the range of 900–1500 cm−1. The characteristic peaks at 1629 cm−1 and 3429 cm−1 in Ti3C2Tx-Co@NCNTs are assigned to the CO and –OH bonds, respectively.23
The X-ray photoelectron spectroscopy (XPS) surveys of Ti3C2Tx and Ti3C2Tx-Co@NCNTs are presented in Fig. S5.† In the spectra of Ti3C2Tx-Co@NCNTs, the peak corresponding to the F element disappears after undergoing high-temperature strong reduction processes (Fig. S6†). The presence of surface –F groups with high concentrations hinders the excellent electrical conductivity properties of Ti3C2Tx MXenes;24 thus the high-temperature reduction process promotes the restoration of the MXenes' intrinsic electrical conductivity. In the Ti 2p spectrum, the three dominant peaks at 461.3 eV, 462.7 eV, and 464.2 eV are associated with Ti–C, Ti2+, and Ti3+ in the 2p1/2 state, while the peaks at 454.9 eV, 457.1 eV, and 458.3 eV correspond to Ti–C, Ti2+, and Ti3+ in the 2p3/2 state, respectively (Fig. 2d).25 The intensity of these peaks in the spectra of Ti3C2Tx-Co@NCNTs follows an opposite trend compared to Ti3C2Tx, indicating that the surface of Ti3C2Tx is covered by oxygen functional groups (–OH and –O–) following the high-temperature reduction treatment.26 In the O 1s XPS region of Ti3C2Tx-Co@NCNTs, four peaks can be fitted at 529.4, 530.7, 532.1, and 533.5 eV, respectively (Fig. 2g). Interestingly, for Ti3C2Tx-Co@NCNTs, the peak at 529.6 eV can be attributed to Ti–O–Co bonds, confirming the strong interaction between Co metal nanoparticles and Ti3C2Tx MXenes.27a For pure Ti3C2Tx, the peak at 529.6 eV can be attributed to Ti–O bonds owing to the surface oxidation.27b The peak at 530.6 eV corresponds to C–Ti–Ox bonds, while the peak at 532.1 eV is assigned to C–Ti–(OH)x.28 The N 1s spectrum can be fitted into five peaks at 396.1 eV (Ti–N), 398.5 eV (pyridinic N), 399.2 eV (pyrrolic N), 401.2 eV (graphitic N), and 404.5 eV (oxidized N), respectively (Fig. 2e). Notably, pyrrolic-N and pyridinic-N contribute to the formation of defects and active sites, which can enhance potassium ion storage performance. Regarding the Co 2p spectrum, the peaks at approximately 780.5 eV, 786.4 eV, 795.9 eV, and 802.5 eV are attributed to metal Co, Co–O/Co–C–N, Co–N bonds, and the satellite peak of Co, respectively (Fig. 2f), indicating the presence of the Co metallic phase.4a,29
Electrochemical performance for PIBs
The electrochemical performance of pure Ti3C2Tx, Ti3C2Tx-Co@NCNTs, and Ti3C2Tx-Co@NC was compared as anode materials in PIBs using CR2032 coin-type cells with K foil as the counter electrode. The purpose of the comparison was to demonstrate the unique microstructure of Ti3C2Tx-Co@NCNTs. Fig. 3a illustrates the cycling performance of the three samples at 100 mA g−1 in 1 M KFSI (DME) electrolyte. After 80 cycles, Ti3C2Tx-Co@NCNTs exhibited a high reversible specific capacity of 360 mA h g−1, surpassing that of neat Ti3C2Tx (110 mA h g−1) and Ti3C2Tx-Co@NC (117 mA h g−1). | ||
| Fig. 3 (a) Cycling performance at 100 mA g−1 and (b) rate performance from 100 to 5000 mA g−1 of the as-prepared Ti3C2Tx, Ti3C2Tx-Co@NC and Ti3C2Tx-Co@NCNTs electrode in 1 M KFSI (DME). (c) Cycling performance at 100 mA g−1 and (d) rate performance of Ti3C2Tx-Co@NCNTs in the electrolyte of 1 M KFSI (DME), 0.8 M KPF6 (EC/DEC) and 1 M KFSI (EC/DEC). (e) Cycling and (f) rate performance of Ti3C2Tx-Co@NCNTs in KFSI (DME) with different concentrations. (g) Long-term cycling stability at 500 mA g−1. (h) Comparison of the present work and recently reported literature. (i) Schematic illustration of KPB‖Ti3C2Tx-Co@NCNTs full cell configuration. (j) The GDV curves for the cathode, anode and full-battery, and (k) the cycling performance of the full cell at 100 mA g−1 in 3 M KFSI (DME) and 0.8 M KPF6 (EC/DEC). The hollow spheres in the figures represent the charge capacity and the coulombic efficiency. | ||
The rate performance of the prepared electrodes in 1 M KFSI (DME) was evaluated and is shown in Fig. 3b. Notably, a high specific capacity of 220 mA h g−1 was achieved even at a current density of 5.0 A g−1. Furthermore, the specific discharge capacity immediately recovered to 460 mA h g−1 once the current density was reduced to 0.1 A g−1, suggesting that Ti3C2Tx-Co@NCNTs exhibited strong tolerance for rapid K+ uptake/release in 1 M KFSI (DME) electrolyte. The superior rate performance of the Ti3C2Tx-Co@NCNTs electrode was further supported by the charge/discharge curves at different current densities (Fig. S7†).
In contrast, Ti3C2Tx-Co@NC exhibited reversible specific capacities of 183, 123, 99, 80, 62, and 40 mA h g−1 at various current densities, while Ti3C2Tx showed reversible specific capacities of 125, 97, 63, 46, 35, and 24 mA h g−1. The unique architecture of Ti3C2Tx-Co@NCNTs, with Co@NCNT arrays vertically grafted between Ti3C2Tx MXene nanosheets, served as a conductive network to enable high electronic conductivity and multiple K+/electrolyte transfer pathways. This distinctive feature allowed for a short electron/K+ transfer path through the conductive bridge arrays, resulting in superior rate performance.
Electrolyte systems consisting of two potassium salts (KFSI and KPF6) in two different solvents (DME and EC/DEC) were systematically studied for Ti3C2Tx-Co@NCNTs electrodes (Fig. 3c). The initial discharge capacity in 1 M KFSI (DME) was 1051 mA h g−1 with an initial coulombic efficiency (CE) of 37%. After 80 cycles, Ti3C2Tx-Co@NCNTs achieved a high reversible specific capacity of 360 mA h g−1. In 0.8 M KPF6 (EC/DEC), the discharge capacity was 371 mA h g−1 after 80 cycles. However, in 0.8 M KPF6 (EC/DEC), it exhibited poor coulombic efficiency within the first 80 cycles. In 1 M KFSI (EC/DEC), the specific discharge capacity was 128 mA h g−1 after 80 cycles. The specific capacities in EC/DEC solvent were much lower than those in DME solvent. The CE value was nearly 100% in 1 M KFSI (EC/DEC) during the cycling tests.
The rate performance of Ti3C2Tx-Co@NCNTs in different electrolyte systems is illustrated in Fig. 3d. The 1 M KFSI (DME) electrolyte system demonstrated the most outstanding rate performance. However, in 0.8 M KPF6 (EC/DEC), it displayed poor coulombic efficiency initially. It delivered reversible capacities of 302, 242![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 194, 173, 147 and 121 mA h g−1 at 100, 200, 500, 1000, 2000 and 5000 mA g−1, respectively. The discharge capacity quickly recovered to 252 mA h g−1 once the current density was reduced to 100 mA g−1. The Ti3C2Tx-Co@NCNTs anode in 1 M KFSI (EC/DEC) displayed poor but stable reversible capacities at different current densities. Fig. S8–S10† in the manuscript display the electrochemical performance of Ti3C2Tx, Ti3C2Tx/ZIF-67, and Ti3C2Tx-Co@NCNTs in different electrolyte systems.
194, 173, 147 and 121 mA h g−1 at 100, 200, 500, 1000, 2000 and 5000 mA g−1, respectively. The discharge capacity quickly recovered to 252 mA h g−1 once the current density was reduced to 100 mA g−1. The Ti3C2Tx-Co@NCNTs anode in 1 M KFSI (EC/DEC) displayed poor but stable reversible capacities at different current densities. Fig. S8–S10† in the manuscript display the electrochemical performance of Ti3C2Tx, Ti3C2Tx/ZIF-67, and Ti3C2Tx-Co@NCNTs in different electrolyte systems.
Fig. 3e and f illustrate the electrochemical performance of Ti3C2Tx-Co@NCNTs in KFSI (DME) electrolyte with different concentrations. It can be observed that there is no significant change in cycling capacity at 100 mA g−1 as the electrolyte concentration increases. However, a higher concentration of the electrolyte leads to increased viscosity and lower ionic conductivity, resulting in poor rate performance. The reversible specific capacities at different current densities (1000, 2000, and 5000 mA g−1) in 2 M KFSI (DME) and 3 M KFSI (DME) are as follows: 197, 175, and 110 and 169, 145, and 73 mA h g−1, respectively. In comparison, the reversible specific capacities in 1 M KFSI (DME) are higher, with values of 268, 245, and 220 mA h g−1 at 1000, 2000, and 5000 mA g−1, respectively.
Moreover, the long-term cyclability of Ti3C2Tx-Co@NCNTs was assessed in different electrolyte systems (Fig. 3g). In 1 M KFSI (DME) electrolyte, Ti3C2Tx-Co@CNTs exhibited a stable reversible capacity of approximately 260 mA h g−1 at 500 mA g−1 for up to 2000 cycles. The average capacity fading was only 0.0053 per cycle, demonstrating the superiority of the Ti3C2Tx-Co@NCNTs anode. In 0.8 M KPF6 (EC/DEC) electrolyte, the long-term cycling capacity retention was 90%, retaining a reversible specific capacity of 180 mA h g−1 at 500 mA g−1 after 2000 cycles. In 3 M KFSI (DME), the reversible capacity maintains 190 mA h g−1 at 500 mA g−1 after 1000 cycles, which is lower than that in 1 M KFSI (DME). Comparatively, the potassium storage performance of the Ti3C2Tx-Co@NCNTs electrode was found to be preferable in comparison to most previously reported MXene-based materials (Fig. 3h).4a,5a,30 Optimization of the electrolyte system revealed that the Ti3C2Tx-Co@NCNTs anode exhibited superior cycling stability and rate performance when paired with a 1 M KFSI (DME) electrolyte.
The unique architecture of the Ti3C2Tx-Co@NCNTs electrode demonstrates superior potassium-storage performance in half-battery setups, encouraging further investigation of its practicability in full-batteries. To construct a full-battery setup, potassium Prussian blue (KPB) was selected as the cathode and the pre-treated Ti3C2Tx-Co@NCNTs was used as the anode. Fig. 3j and S11† depict the typical galvanostatic charge/discharge (GCD) curves for K0.72Fe[Fe(CN)6] at a current density of 0.5 A g−1. The voltage window for the assembled full cell was tested between 0.9 V and 4.0 V. At a concentration of 1 M KFSI (DME), the full cell demonstrates peak charge/discharge capacity, achieving coulombic efficiency (CE) values of 92%; however, it maintains this performance for only 62 cycles. With 3 M KFSI (DME), the cell's charge/discharge capacity stabilizes around 100 mA h g−1 following 200 cycles, with near-complete CE values of approximately 100%. At 0.8 M KPF6 (EC DEC), the cell exhibits a charge/discharge capacity below 100 mA h g−1 and a relatively low CE of 80%. Utilizing 3 M KFSI (EC DEC), the full cell maintains a specific capacity of 190 mA h g−1 over 128 cycles (as shown in Fig. 3k and S12†). Consequently, a high-concentration KFSI electrolyte system appears more suitable for the KPB‖Ti3C2Tx-Co@NCNTs full cell.
Electrochemical reaction mechanism
The electrochemical reaction behaviors of Ti3C2Tx-Co@CNTs anodes in different electrolyte systems were investigated using cyclic voltammetry (CV) tests at a scanning rate of 0.1 mV s−1 (Fig. 4a). In 1 M KFSI (DME) electrolyte, cathodic peaks appeared at 0.9–1.5 V in the first cycle, but disappeared in subsequent cycles, possibly due to the reductive decomposition of the electrolyte system. However, when EC and DEC were used as solvents, specifically in 0.8 M KPF6 (EC/DEC) and 1 M KFSI (EC/DEC) systems, cathodic peaks were observed at 1.57 V and 0.68 V, respectively, indicating the decomposition of EC/DEC. The redox peaks for the DME electrolyte were at lower voltages compared to the EC/DEC electrolyte, indicating different decomposition behaviors of the electrolytes. These peaks corresponded to the first voltage platform in the charge–discharge curve (Fig. S13†). Subsequent cycles showed overlapping curves, indicating high reversibility. Furthermore, the curve shapes of the subsequent cycles in the three electrolytes were similar, except for the first cycle. During the cathodic scanning, the insertion of K+ occurred within the voltage range of 0.68–0.01 V, while the anodic scanning exhibited a peak ranging from 0.4 V to 0.6 V, which indicated the process of K+ extraction from Ti3C2Tx-Co@NCNTs.10a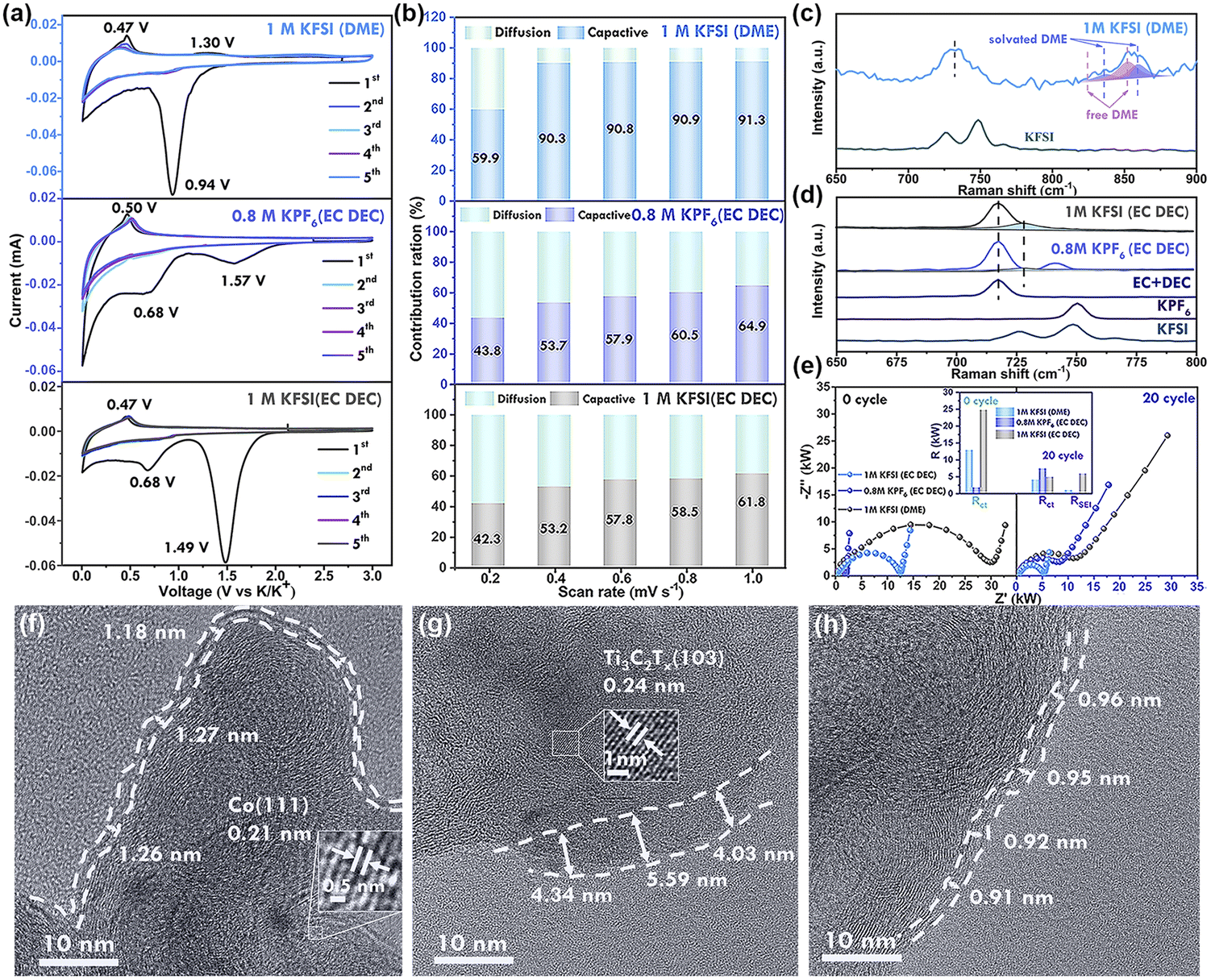 | ||
| Fig. 4 (a) Cyclic voltammetry curves of Ti3C2Tx at 0.1 mV s−1 in different electrolyte systems. (b) Capacitive charge storage performances of Ti3C2Tx at different scan rates in 1 M KFSI (DME), 0.8 M KPF6 (EC/DEC) and 1 M KFSI (EC/DEC). (c) Raman spectra of KFSI-DME electrolytes. (d) Raman spectra of electrolyte systems (KFSI and KPF6 in EC/DEC). (e) EIS data in different electrolytes and modeling results of RSEI and Rct for 0 cycles and 20 cycles in different electrolyte systems. SEI layer on the Ti3C2Tx-Co@NCNTs electrode in (f) 1 M KFSI(DME), (g) 0.8 M KPF6 (EC DEC) and (h) 1 M KFSI (EC DEC). | ||
In order to gain a comprehensive understanding of the K+ storage kinetics in the fabricated electrode, the ratio of redox pseudo-capacitance is calculated through cyclic voltammetry (CV) measurements at scanning rates ranging from 0.2 to 1.0 mV s−1 (Fig. S14†). Moreover, the kinetic behavior can be inferred by examining the current–voltage (i–v) relationship described by eqn (1) and (2).
| i = avb | (1) |
| logi = blogv + loga | (2) |
The value of parameter b can be determined through analysis of the cyclic voltammetry (CV) curves. A value of 0.5 for b signifies a diffusion-controlled process for potassium storage, while a value of 1 indicates a surface-controlled process or pseudocapacitive storage. In the electrolyte systems of 1 M KFSI (DME), 0.8 M KPF6 (EC/DEC), and 1 M KFSI (EC/DEC), the Ti3C2Tx-Co@NCNTs electrode exhibits b values of 0.80, 0.70, and 0.72, respectively (Fig. S14 d–f†). These results suggest that the stable storage of potassium ions in the Ti3C2Tx-Co@NCNTs electrode is influenced by both surface-controlled capacitive storage and diffusion-controlled processes.
| i = k1v + k2v1/2 | (3) |
In the context of our study, the contribution ratios of the pseudo-capacitive effect increase gradually as the scanning rate is enhanced for the Ti3C2Tx-Co@NCNTs electrode in different electrolyte systems (Fig. 4b). Specifically, in the 1 M KFSI (DME) electrolyte, the calculated contribution ratios are 59.9%, 90.3%, 90.8%, 90.9%, and 91.3% for varying scan rates, highlighting the dominance of the pseudocapacitive effect in this electrolyte system. A substantial presence of pseudo-capacitance significantly accelerates reaction kinetics, consequently leading to improved rate performance (as observed in Fig. 3d). The relatively large ionic radius of potassium hinders diffusion kinetics within the crystals, consequently emphasizing the effectiveness of surface-controlled capacitive storage in achieving superior electrochemical performance, particularly at high current densities.31
Galvanostatic intermittent titration technique (GITT) tests were conducted to investigate the kinetic characteristics of the Ti3C2Tx-Co@NCNTs electrode prepared (Fig. S15†). The GITT profiles obtained demonstrate a consistent decline in overpotential, indicating a reliable K+ insertion process during each relaxation step. The diffusion coefficient of K+ (DK) can be calculated using eqn (4).20
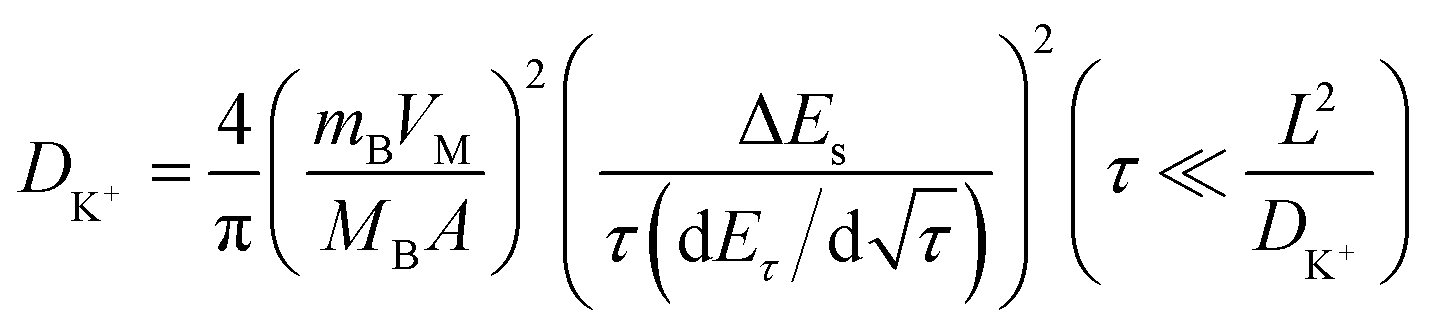 | (4) |
In the 1 M KFSI (DME) electrolyte, the diffusion coefficient ranges from 4 × 10−10 cm2 S−1 to 1 × 10−8 cm2 S−1 during discharge and from 2 × 10−8 cm2 S−1 to 9 × 10−8 cm2 S−1 during charge. In the EC/DEC solvent with a 0.8 M KPF6 (EC DEC) electrolyte, the diffusion coefficient ranges from 2 × 10−7 cm2 S−1 to 8 × 10−9 cm2 S−1 during discharge and from 1 × 10−7 cm2 S−1 to 1 × 10−8 cm2 S−1 during charge. Alternatively, in the 1 M KFSI (EC DEC) electrolyte, the diffusion coefficient ranges from 1 × 10−9 cm2 S−1 to 3 × 10−10 cm2 S−1 during discharge and from 1 × 10−9 cm2 S−1 to 9 × 10−9 cm2 S−1 during charge (Fig. S15†).
The DK coefficients calculated for the Ti3C2Tx-Co@NCNTs electrode demonstrate faster kinetic behavior compared to most other MXene-based hybrids, falling within the range of 10−7–10−10 cm2 s−1. However, it is evident that the DK values decrease over longer diffusion pathways. This can be attributed to the repulsive force exerted by the previously adsorbed K+ ions at the surface sites, which hinders further insertion. Consequently, the excellent K-ion storage capacity of Ti3C2Tx-Co@NCNTs is not fully utilized throughout the entire charge/discharge process. Furthermore, it is important to note that the calculated DK value is influenced by the type of electrolyte. Specifically, for the potassium salt KFSI, the diffusion coefficient in DME (dimethyl ether) is larger than that in the EC/DEC solvent. This difference can be attributed to the size of solvated K+ ions and the viscosity of the solvent. Ester-based electrolytes typically have higher viscosity and lower diffusivity due to the strong polarization and high dielectric constant of EC molecules. Moreover, the size of K+-DME ions is smaller than that of K+-EC/DEC, leading to faster K+ diffusion in DME compared to that in EC/DEC. Additionally, it has been demonstrated that the diffusion coefficient is influenced by the choice of potassium salts.32
To clarify the potassium storage and diffusion features, as well as the interaction between NCNTs and Ti3C2Tx, further density functional theory (DFT) calculations were conducted. Fig. 5a–c illustrate the adsorption energy of K+ on the surfaces of NCNTs, Ti3C2Tx, and Ti3C2Tx-Co@NCNTs.
 | ||
| Fig. 5 Theoretical simulations of potassium adsorption/diffusion on the heterointerface between NCNTs and Ti3C2Tx. Potassium adsorption on the surface of (a) NCNTs, (b) Ti3C2Tx, and (c) Ti3C2Tx-Co@NCNTs, (d) potassium diffusion paths on the Ti3C2Tx-Co@NCNTs, (e) the corresponding diffusion barrier energy, and (f) the electron density differences. | ||
The adsorption sites on carbon nanotubes include the upper end of the carbon atom, the C–C bridge, and the vacancy point, with adsorption energies of −0.773 eV, −0.881 eV, and −1.141 eV, respectively. The results indicate that K+ is more likely to diffuse at the vacancy point. On the surface of Ti3C2Tx, there are three adsorption sites: the upper end of the carbon atom, the upper end of the Ti atom, and the vacancy point, with adsorption energies of −0.631 eV, −0.349 eV, and −0.326 eV, respectively. The findings suggest that K+ is more likely to diffuse at the upper end of the carbon atom of Ti3C2Tx. In the case of Ti3C2Tx-Co@NCNTs, two scenarios can be considered: the carbon skeleton region and the nitrogen-doped carbon skeleton region. When K+ diffuses in the carbon skeleton region, there are three adsorption sites: the upper end of the carbon atom, the C–C bridge, and the vacancy point, with adsorption energies of −0.395 eV, −0.819 eV, and −0.944 eV, respectively. When K+ diffuses in the nitrogen-doped carbon skeleton region, there are three adsorption positions: the upper end of the nitrogen atom, the vacancy point, and the N–N bridge, with adsorption energies of −0.940 eV, −0.972 eV, and −0.931 eV, respectively. The vacancy point in the nitrogen-doped carbon skeleton region allows for easier K+ diffusion. Fig. 5d illustrates the K+ migration paths in the Ti3C2Tx-Co@NCNTs electrode, showing that the diffusion energy barrier for K+ is higher on the surface of carbon nanotubes and lower on Ti3C2Tx-Co@NCNTs compared to the surface of carbon nanotubes. The Ti3C2Tx MXenes, serving as the conductive matrix, not only enhance overall conductivity but also facilitate the diffusion and migration of K+-ions (Fig. 5e). Fig. 5f provides the charge density analysis of the Ti3C2Tx, NCNTs, and Ti3C2Tx-Co@NCNTs heterostructures. The yellow cloud represents the electron-rich region, while the blue cloud represents the electron-deficient region. The differences observed demonstrate substantial charge depletions near the surface-adsorbed potassium-ion due to significant charge transfer from potassium atoms to the Ti3C2Tx-Co@NCNTs heterostructures. Notably, the inclusion of N and Co elements significantly enhances the charge transfer, confirming the superiority of the hybrid structure. Thus, both the experimental results and theoretical calculations confirm that the design of such a heterostructure can expedite charge/K+ transport in the battery.30c
The electrolyte degradation (SEI formation) mechanism
The interaction between electrolyte salts and solvents in the electrolyte was investigated using Raman spectroscopy. A peak at 718 cm−1 in the EC + DEC solvents corresponds to the CO bending vibration mode. Upon the addition of potassium salt, a new peak emerges at 729 cm−1, which can be attributed to K+-solvated EC (Fig. 4d). The proportion and intensity of the peak at 729 cm−1 are higher in the KFSI (EC DEC) electrolyte system compared to the KPF6 (EC DEC) electrolyte, indicating stronger solvation with KFSI.33 This strong solvation effect reduces the number of free solvent molecules in the electrolyte system, mitigating parasitic reactions between solvent molecules and K+ metal, leading to higher CE values. In the case of KFSI, the peaks at 725 and 750 cm−1 correspond to the SO stretching vibration mode in FSI−.34 These peaks merge into one peak at a higher wavenumber compared to solid KFSI, indicating fewer free FSI− anions and a stronger interaction between solvated K+ and FSI−. Upon the addition of KFSI to DME solvent, new peaks at 835–860 cm−1 appear for K+-solvated DME. These peaks can be distinguished between K+-solvated DME and free DME molecules. The ratio and intensity of the peaks corresponding to K+-solvated molecules in KFSI-based electrolytes are larger than those in KPF6-based electrolytes, demonstrating a stronger solvation effect of KFSI.35
High-resolution transmission electron microscopy (HRTEM) was conducted to investigate the thickness of the solid electrolyte interphase (SEI) film in different electrolytes. The SEI film displayed an average thickness of approximately 1.3 nm in 1 M KFSI (DME), 4–6 nm in 0.8 M KPF6 (EC DEC), and about 0.9 nm in 1 M KFSI (EC DEC) (Fig. 4f–h, S16, S17, and S18†). In the case of 1 M KFSI (DME) electrolyte, the SEI layer uniformly covered the particles of the electrolyte material, maintaining its integrity. The thicker SEI film derived from the 0.8 M KPF6 (EC DEC) electrolyte resulted in low initial coulombic efficiency (CE) values. Moreover, the increased thickness of the SEI layer effectively countered volume expansion during the charge/discharge process, thereby hindering anode deactivation and severe capacity decay, consequently leading to stable long-term cycling performance.36
EIS measurements were conducted to evaluate the influence of electrolytes on the internal resistance and formation of the solid electrolyte interphase (SEI) on the Ti3C2Tx-Co@NCNTs anode (Fig. 4e). The resistances, including charge-transfer resistance (Rct), SEI resistance (RSEI), and internal resistance (Rs), were determined using the equivalent circuit model depicted in Fig. S19.† (ref. 37) Notably, the key distinctions among the different electrolytes lie in RSEI and Rct, while Rs only accounts for a minor portion of the overall resistance (R = Rs + RSEI + Rct). In a 0.8 M KPF6 (EC/DEC) electrolyte, the Rct value significantly increased from 2 kΩ to 8 kΩ, indicating that the formation of the SEI layer hinders the diffusion of K+. Conversely, the Rct values in the KFSI-based electrolyte decreased after the SEI films were formed. This suggests that the SEI film induced by the decomposition of KFSI effectively enhances the charge transport kinetics, facilitating the diffusion of potassium ions through the SEI layer to the electrode material.38 Additionally, the Rct value in the DME electrolyte system was lower compared to that in the EC DEC solvent, indicating faster K+ transfer in DME solvent, consistent with the GITT test (Fig. S15†). The highest RSEI value was observed in the 1 M KFSI (EC/DEC) electrolyte, resulting in the lowest battery capacity.
The solid electrolyte interphase (SEI) film plays a crucial role in determining the coulombic efficiency (CE) value and long-term stability of potassium-ion batteries. Therefore, the components of the SEI film were further analyzed using the ex situ X-ray photoelectron spectroscopy (XPS) technique (Fig. 6 and S20†). In the C 1s spectra, all four samples exhibited peaks corresponding to C–C, C–O, K2CO3, and RCOOK. The peaks of K2CO3 and RCOOK are attributed to the decomposition of carbonate species, while the C–O peak is generated by the decomposition of ether-based solvents. The decomposition of carbonate species leads to the formation of C–O and CO species on the surface layer in EC DEC solvent.39 In the O 1s spectra, the peak at 530.5 eV can be attributed to KxPOyFz or SOxF, resulting from the decomposition of K+ salt (KPF6 and KFSI). The peaks at 533.4 eV and 534.2 eV are assigned to RO-COOK or R–OK, derived from the decomposition of carbonate-based and ether-based solvents, respectively. The F 1s spectra reveal that the SEI components in the KPF6-based electrolyte system consist of KxPFy, KxPOyFz, and K–F.40
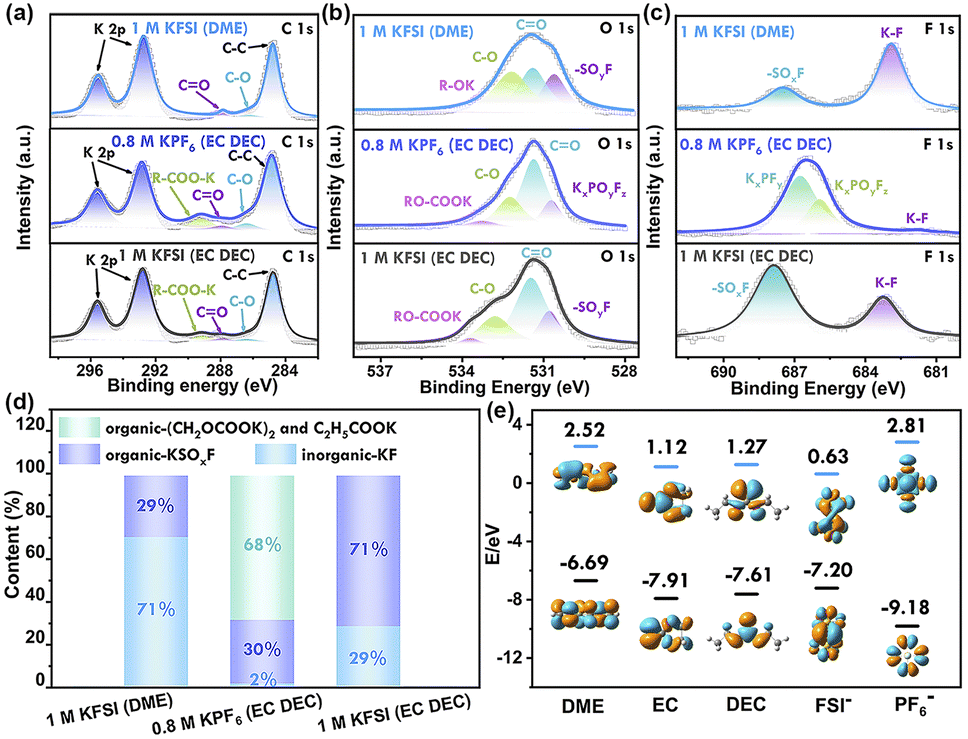 | ||
| Fig. 6 XPS fitting results for the SEI layer in different electrolytes. (a) C 1s, (b) O 1s, and (c) F 1s, (d) the molecular energy levels of the HOMO and the LUMO of the solvent molecules and potassium salts: FSI− and DME in the DME electrolyte; FSI− in the EC/DEC electrolyte and KPF6 in the EC/DEC electrolyte, and (e) SEI species ratio. | ||
The species distribution of solid-electrolyte interphase (SEI) layers was quantified by calculating the integral area of each peak and the proportion of each component. In a 1 M potassium fluorosilicate (KFSI) solution in dimethoxyethane (DME), the SEI film consisted of 71% inorganic salt component (KF) and 29% organic species. The inorganic salt component KF, known for its low electronic conductivity and high surface energy, facilitated the embedding and removal of K+ ions and exhibited higher charge–discharge capacity compared to ester-based electrolytes. The SEI layer rich in potassium fluoride (KF) exhibited smaller charge transfer (Rct) and SEI (RSEI) resistance, as well as a larger potassium-ion diffusion coefficient, thus promoting faster reaction dynamics, as confirmed by electrochemical impedance spectroscopy (EIS). Additionally, this dense inorganic layer limited electrolyte permeability and suppressed further electrolyte decomposition, resulting in enhanced cell performance.41 In a 0.8 M potassium hexafluorophosphate (KPF6) solution in ethylene carbonate (EC) and diethyl carbonate (DEC), the SEI layer primarily consisted of organic components (98%). The presence of organic components led to SEI instability and a constant process of destruction and rebuilding, resulting in low coulombic efficiency. Conversely, the SEI layer in a 1 M KFSI solution in EC/DEC consisted of both inorganic components (29% KF from the decomposition of KFSI) and organic components ((CH2OCOOK)2 and (C2H5COOK) from the decomposition of EC/DEC), leading to high coulombic efficiency nearing 100%.
The disparity in SEI layers can be elucidated by examining the LUMO energy levels of potassium salts and solvents. DFT calculations illustrate that KPF6 and KPF6-solvent complexes have higher LUMO energies compared to KFSI and KFSI-solvent complexes, respectively.42 This suggests that the reduction of the KFSI species is easier compared to that of KPF6 (Fig. 6e). Hence, the mechanism of SEI layer formation varies in different electrolytes. In the KFSI electrolyte system, the SEI layer is mainly formed through reduction of the potassium salt, whereas in the KPF6 electrolyte system, the SEI film is predominantly a result of solvent reduction. Considering the solvent effect, KFSI has a lower LUMO energy level compared to DME, but is similar to that of EC and DEC. Consequently, when KFSI is combined with DME, electrolyte degradation primarily stems from KFSI. Conversely, in the KFSI (EC/DEC) electrolyte, there is simultaneous reduction of both EC/DEC solvent and KFSI. This explanation is consistent with the ex situ XPS analysis. In the KFSI (DME) electrolyte, the SEI layer is predominantly composed of KF (resulting from FSI− decomposition), whereas in the KPF6 (EC/DEC) electrolyte, the SEI layer consists primarily of organic compounds. In contrast, the SEI layer of the KFSI (EC/DEC) electrolyte had random distribution of organic and inorganic components, which led to permeability and ongoing electrolyte decomposition, resulting in poor cyclability.12 This study concludes that the formation of a KF-rich solid electrolyte interface layer on the surface of the Ti3C2Tx-Co@NCNTs electrode is advantageous. This can be achieved through the regulation of a KFSI-DME electrolyte system, which promotes the formation of resilient and KF-rich SEI layers.
Conclusions
In summary, this study aimed to promote the electrochemical performance of potassium-ion storage through the construction of a hierarchical structure and the regulation of the electrolyte system. To prevent MXene aggregation and improve the conductivity of the architectures, Co@NCNTs derived from ZIF-67 were uniformly dispersed on the surface of two-dimensional layered Ti3C2Tx. Additionally, the relationship between electrolyte salts, solvents, and the electrochemical performance of MXene-based electrodes in PIBs was investigated. The combination of KFSI salt and DME solvent facilitated sufficient K+-ion solvation and the formation of a high-KF SEI. This, in turn, effectively prevented SEI corrosion, greatly enhancing cycling stability and rate capability. This work provides insights into the multifunctionality of electrolytes in PIBs and proposes systematic evaluation methodologies for future electrolyte engineering.Data availability
All the data supporting this article have been included in the main text and the ESI.†Author contributions
X. X., Q. J., and C. Y. designed the experiments, participated in analysing the data, and drafted the manuscript; J. R., H. W., X. L., Z. L. and W. Z. discussed the characterization data; Y. C., C. Z., J. H., and T. Z. conceived the project and provided guidance and supervision throughout the work. All authors have approved the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21806187, 22272207, and 52302205), Fundamental Research Funds for the Central Universities of South-Central Minzu University (CZY23015, CZZ23008), the Anhui Provincial Natural Science Foundation for Outstanding Young Scholar (2208085Y05), the Anhui Provincial Scientific Reuter Foundation for Returned Scholars (2022LCX030), and the Excellent Research and Innovation Team Project of Anhui Province (2022AH010001).Notes and references
- (a) M. Shen, H. Ding, L. Fan, A. M. Rao, J. Zhou and B. Lu, Adv. Funct. Mater., 2023, 33, 2213362 CrossRef CAS; (b) X. Sun, Z. Li, Z. Liu, X. Lv, K. Shi, R. Chen, F. Wu and L. Li, Adv. Funct. Mater., 2023, 33, 2300125 CrossRef CAS; (c) S. Imtiaz, N. Kapuria, I. S. Amiinu, A. Sankaran, S. Singh, H. Geaney, T. Kennedy and K. M. Ryan, Adv. Funct. Mater., 2022, 33, 2209566 CrossRef; (d) J. Chen, D. An, S. Wang, H. Wang, Y. Wang, Q. Zhu, D. Yu, M. Tang, L. Guo and H. Wang, Angew. Chem., Int. Ed., 2023, 62, e202307122 CrossRef CAS PubMed; (e) M. Tang, S. Dong, J. Wang, L. Cheng, Q. Zhu, Y. Li, X. Yang, L. Guo and H. Wang, Nat. Commun., 2023, 14, 6004 CrossRef PubMed.
- (a) Y. Chen, B. Xi, M. Huang, L. Shi, S. Huang, N. Guo, D. Li, Z. Ju and S. Xiong, Adv. Mater., 2022, 34, 2108621 CrossRef CAS PubMed; (b) L. Wu, H. Fu, S. Li, J. Zhu, J. Zhou, A. M. Rao, L. Cha, K. Guo, S. Wen and B. Lu, Nat. Commun., 2023, 14, 644 CrossRef CAS PubMed.
- Y. Wu, Y. Sun, J. Zheng, J. Rong, H. Li and L. Niu, Chem. Eng. J., 2021, 404, 126565 CrossRef CAS.
- (a) X. Xu, Y. Zhang, H. Sun, J. Zhou, Z. Liu, Z. Qiu, D. Wang, C. Yang, Q. Zeng, Z. Peng and S. Guo, Adv. Mater., 2021, 33, 2100272 CrossRef CAS PubMed; (b) D. Sha, C. Lu, W. He, J. Ding, H. Zhang, Z. Bao, X. Cao, J. Fan, Y. Dou, L. Pan and Z. Sun, ACS Nano, 2022, 16, 2711–2720 CrossRef CAS PubMed.
- (a) B. Cao, H. Liu, P. Zhang, N. Sun, B. Zheng, Y. Li, H. Du and B. Xu, Adv. Funct. Mater., 2021, 31, 2102126 CrossRef CAS; (b) C. Liu, Y. Bai, W. Li, F. Yang, G. Zhang and H. Pang, Angew. Chem., Int. Ed., 2022, 61, e202116282 CrossRef CAS PubMed; (c) S. H. Yang, Y. J. Lee, H. Kang, S.-K. Park and Y. C. Kang, Nano-Micro Lett., 2021, 14, 17 CrossRef PubMed.
- X. Cao, Y. You, D. Sha, H. Xia, H. Wang, J. Zhang, R. Hu, Y. Wei, Z. Bao, Y. Xu, L. Pan, C. Lu, W. He, M. Zhou and Z. Sun, Adv. Funct. Mater., 2023, 33, 2303275 CrossRef CAS.
- (a) L. P. Yu, X. H. Zhou, L. Lu, L. Xu and F. J. Wang, ChemSusChem, 2021, 14, 5079–5111 CrossRef CAS PubMed; (b) P. Su, H. Xiao, J. Zhao, Z. Shao, C. Li and Q. Yang, Chem. Sci., 2013, 4, 2941 RSC.
- K. Yang, M. Luo, D. Zhang, C. Liu, Z. Li, L. Wang, W. Chen and X. Zhou, Chem. Eng. J., 2022, 427, 132002 CrossRef CAS.
- (a) X. Wang, S. Wang, J. Qin, X. Xie, R. Yang and M. Cao, Inorg. Chem., 2019, 58, 16524–16536 CrossRef CAS PubMed; (b) H. Li, R. Chen, M. Ali, H. Lee and M. J. Ko, Adv. Funct. Mater., 2020, 30, 2002739 CrossRef CAS; (c) Y. Yue, Y. Wang, X. Xu, C. Wang, Z. Yao and D. Liu, Ceram. Int., 2022, 48, 6338–6346 CrossRef CAS; (d) W. Wang, F. Xiong, S. Zhu, J. Chen, J. Xie and Q. An, Escience, 2022, 2, 278–294 CrossRef.
- (a) W. Zhao, X. Xu, L. Wang, Y. Liu, T. Zhou, S. Zhang, J. Hu and Q. Jiang, J. Energy Chem., 2022, 75, 55–65 CrossRef CAS; (b) D. Xu, Q. Cheng, P. Saha, Y. Hu, L. Chen, H. Jiang and C. Li, Adv. Funct. Mater., 2023, 33, 2211661 CrossRef CAS.
- M. Zhou, P. Bai, X. Ji, J. Yang, C. Wang and Y. Xu, Adv. Mater., 2021, 33, 2003741 CrossRef CAS PubMed.
- Q. Li, Z. Cao, W. Wahyudi, G. Liu, G.-T. Park, L. Cavallo, T. D. Anthopoulos, L. Wang, Y.-K. Sun, H. N. Alshareef and J. Ming, ACS Energy Lett., 2020, 6, 69–78 CrossRef.
- M. Gu, L. Fan, J. Zhou, A. M. Rao and B. Lu, ACS Nano, 2021, 15, 9167–9175 CrossRef CAS PubMed.
- G. Ma, Y. Wang, J. Song, K. Song, N. Wang, J. Yang and Y. Qian, Carbon Energy, 2022, 4, 332–345 CrossRef CAS.
- X. Liu, Y. Liu, S. Dong, X. Zhang, Y. Wei, L. Lv and S. He, ACS Appl. Nano Mater., 2023, 6, 9579–9587 CrossRef CAS.
- Z. Liang, N. Kong, C. Yang, W. Zhang, H. Zheng, H. Lin and R. Cao, Angew. Chem., Int. Ed., 2021, 60, 12759–12764 CrossRef CAS PubMed.
- Y. Zhu, Z. Wang, R. Zhao, Y. Zhou, L. Feng, S. Gai and P. Yang, ACS Nano, 2022, 16, 3105–3118 CrossRef CAS PubMed.
- L. Liu, M. Orbay, S. Luo, S. Duluard, H. Shao, J. Harmel, P. Rozier, P. L. Taberna and P. Simon, ACS Nano, 2022, 16, 111–118 CrossRef CAS PubMed.
- Y. Zhang, J. Wu, S. Zhang, N. Shang, X. Zhao, S. M. Alshehri, T. Ahamad, Y. Yamauchi, X. Xu and Y. Bando, Nano Energy, 2022, 97, 107146 CrossRef CAS.
- L. Wang, Q. Jiang, K. Yang, Y. Sun, T. Zhou, Z. Huang, H.-J. Yang and J. Hu, J. Colloid Interface Sci., 2021, 601, 60–69 CrossRef CAS PubMed.
- A. Sarycheva, M. Shanmugasundaram, A. Krayev and Y. Gogotsi, ACS Nano, 2022, 16, 6858–6865 CrossRef CAS PubMed.
- J. S. Lee, R. Saroha and J. S. Cho, Nano-Micro Lett., 2022, 14, 113 CrossRef CAS PubMed.
- M. Xu, L. Liang, J. Qi, T. Wu, D. Zhou and Z. Xiao, Small, 2021, 17, 2007446 CrossRef CAS PubMed.
- Y. An, Y. Tian, Q. Man, H. Shen, C. Liu, S. Xiong and J. Feng, Nano Lett., 2023, 23, 5217 CrossRef CAS PubMed.
- S. Kim, H. Shin, J. Lee, C. Park, Y. Ahn, H.-J. Cho, S. Yuk, J. Kim, D. Lee and I.-D. Kim, ACS Nano, 2023, 17, 19387–19397 CrossRef CAS PubMed.
- C. E. Park, G. H. Jeong, J. Theerthagiri, H. Lee and M. Y. Choi, ACS Nano, 2023, 17, 7539–75479 CrossRef CAS PubMed.
- (a) L. Yao, Q. Gu and X. Yu, ACS Nano, 2021, 15, 3228–3240 CrossRef CAS PubMed; (b) J. Cao, Z. Sun, J. Li, Y. Zhu, Z. Yuan, Y. Zhang, D. Li, L. Wang and W. Han, ACS Nano, 2021, 15, 3423–3433 CrossRef CAS PubMed.
- H. Huang, J. Cui, G. Liu, R. Bi and L. Zhang, ACS Nano, 2019, 13, 3448–3456 CrossRef CAS PubMed.
- P. Liu, S. Gao, G. Zhang, Y. Huang, W. You and R. Che, Adv. Funct. Mater., 2021, 31, 2102812 CrossRef CAS.
- (a) J. Cao, L. Wang, D. Li, Z. Yuan, H. Xu, J. Li, R. Chen, V. Shulga, G. Shen and W. Han, Adv. Mater., 2021, 33, 2101535 CrossRef CAS PubMed; (b) R. Zhang, Y. Tian, T. Otitoju, Z. Feng, Y. Wang and T. Sun, Small, 2023, 19, 2302148 CrossRef CAS PubMed; (c) J. Cao, Z. Sun, J. Li, Y. Zhu, Z. Yuan, Y. Zhang, D. Li, L. Wang and W. Han, ACS Nano, 2021, 15, 3423–3433 CrossRef CAS PubMed; (d) H. Liu, Y. He, H. Zhang, S. Wang, K. Cao, Y. Jiang, X. Liu and Q.-S. Jing, J. Colloid Interface Sci., 2022, 606, 167–176 CrossRef CAS PubMed.
- (a) X. Chen, S. Xia, T. Tan, Y. Zhu, L. Li, Q. Zhu and W. Zhang, Inorg. Chem. Front., 2023, 10, 4414–4424 RSC; (b) T. Lei, M. Gu, H. Fu, J. Wang, L. Wang, J. Zhou, H. Liu and B. Lu, Chem. Sci., 2023, 14, 2528 RSC; (c) P. Wang, X. Dai, P. Xu, S. Hu, X. Xiong, K. Zou, S. Guo, J. Sun, C. Zhang, Y. Liu, T. Zhou and Y. Chen, Escience, 2023, 3(1), 100088 CrossRef; (d) X. Wu, Z. Li, J. Liu, W. Luo, J. Gaumet and L. Mai, Rare Met., 2022, 41, 3446–3455 CrossRef CAS.
- L. Wang, J. Yang, J. Li, T. Chen, S. Chen, Z. Wu, J. Qiu, B. Wang, P. Gao, X. Niu and H. Li, J. Power Sources, 2019, 409, 24 CrossRef CAS.
- H. Wang, D. Yu, X. Wang, Z. Niu, M. Chen, L. Cheng, W. Zhou and L. Guo, Angew. Chem., Int. Ed., 2019, 58, 16451 CrossRef CAS PubMed.
- L. Li, S. Zhao, Z. Hu, S.-L. Chou and J. Chen, Chem. Sci., 2021, 12, 2345–2356 RSC.
- H. J. Liang, Z. Y. Gu, X. X. Zhao, J. Z. Guo, J. L. Yang, W. H. Li, B. Li, Z. M. Liu, W. L. Li and X. L. Wu, Angew. Chem., Int. Ed., 2021, 60, 26837–26846 CrossRef CAS PubMed.
- Y. Liu, C. Gao, L. Dai, Q. Deng, L. Wang, J. Luo, S. Liu and N. Hu, Small, 2020, 16, 2004096 CrossRef CAS PubMed.
- Z. Wu, J. Zou, S. Shabanian, K. Golovin and J. Liu, Chem. Eng. J., 2022, 427, 130972 CrossRef CAS.
- B. Li, J. Zhao, Z. Zhang, C. Zhao, P. Sun, P. Bai, J. Yang, Z. Zhou and Y. Xu, Adv. Funct. Mater., 2019, 29, 1807137 CrossRef.
- Y. Xu, T. Ding, D. Sun, X. Ji and X. Zhou, Adv. Funct. Mater., 2023, 33, 2211290 CrossRef CAS.
- (a) L. Zhou, Z. Cao, J. Zhang, H. Cheng, G. Liu, G. T. Park, L. Cavallo, L. Wang, H. N. Alshareef, Y. K. Sun and J. Ming, Adv. Mater., 2021, 33, 2005993 CrossRef CAS PubMed; (b) J. Zhao, C. Li, G. Chen, F. Ji, Y. Shen, J. Peng and W. Wang, Rare Met., 2022, 41, 2259–2267 CrossRef CAS.
- X. Du and B. Zhang, ACS Nano, 2021, 15, 16851–16860 CrossRef CAS PubMed.
- J. Li, Y. Hu, H. Xie, J. Peng, L. Fan, J. Zhou and B. Lu, Angew. Chem., Int. Ed., 2022, 61, e202208291 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc06079a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |