
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photochemical H2 activation by an Zn–Fe heterometallic: a mechanistic investigation†
Marina
Perez-Jimenez
 and
Mark R.
Crimmin
*
and
Mark R.
Crimmin
*
Department of Chemistry, Molecular Sciences Research Hub, Imperial College London, 82 Wood Lane, White City, London, W12 0Z, UK. E-mail: m.crimmin@imperial.ac.uk
First published on 14th December 2023
Abstract
Addition of H2 to a Zn–Fe complex was observed to occur under photochemical conditions (390 or 428 nm LED) and leads to the formation of a heterometallic dihydride complex. The reaction does not occur under thermal conditions and DFT calculations suggest this is an endergonic, light driven process. Through a combined experimental and computational approach, the plausible mechanisms for H2 activation were investigated. Inhibition experiments, double-label cross-over experiments, radical trapping experiments, EPR spectroscopy and DFT calculations were used to gain insight into this system. The combined data are consistent with two plausible mechanisms, the first involving ligand dissociation followed by oxidative addition of H2 at the Fe centre, the second involving homolytic fragmentation of the Zn–Fe heterometallic and formation of radical intermediates.
Introduction
In the past few years there has been growing interest in using heterometallic complexes for H2 activation. Transition metal complexes supported by Lewis-acidic B,1–3 Al,4 Zn,5,6 Mg,7,8 and Sn9,10 ligands have all been documented to react with H2 under thermal conditions. Despite the rapid growth of this area, examples of photolytic H2 splitting using these types of heterometallic complexes are rare. To the best of our knowledge, photolytic reactivity is limited to two examples involving the addition of H2 to Ru–Zn and Ru–Al heterometallic complexes.11,12These findings contrast the rich chemistry of single-site transition metal complexes, where examples of photochemical H2 activation are more common. Under photolytic conditions, 18-electron transition metal carbonyl complexes are prone to ligand dissociation to generate coordinatively unsaturated intermediates that can react with H2. For example, irradiation of mixtures [M(CO)6] and H2 at low temperature leads to the formation of the corresponding dihydrogen complexes [M(CO)5(η2-H2)] (M = Cr, W).13 In contrast, photochemical reaction of [Ru(CO)3(PPh3)2] with H2 leads to oxidative addition to form [Ru(CO)2(PPh3)2(H)2].14
Bimetallic transition metal carbonyl complexes such as [Ru(η5-C5H5)(CO)2]2 also react with H2 under photochemical conditions.15 In these cases, photolysis can promote either dissociation of CO or homolysis of the metal–metal bond forming 17-electron species.16–18 As a result, a diverse set of mechanistic pathways involving both neutral and radical intermediates are possible for H2 splitting.19,20
In this paper, we report the reaction of a heterometallic Zn–Fe carbonyl complex with H2 under photochemical conditions; this is an uphill, light-driven reaction (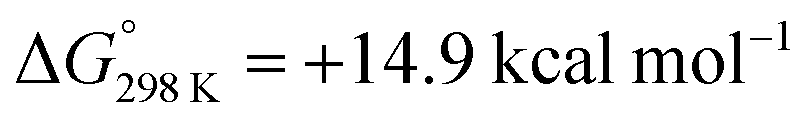 ; ΔH° = +17.5 kcal mol−1). Using a combined experimental (inhibition, cross-over, radical trapping, EPR spectroscopy) and computational (DFT) approach, we present a detailed investigation into the plausible pathways of H2 activation at the heterometallic complex. We conclude that the most likely pathways involve (i) a closed-shell mechanism involving CO dissociation and subsequent oxidative addition at Fe, and (ii) an open-shell pathway involving homolysis of the Zn–Fe bond to form radical intermediates capable of reacting with H2. Our findings provide rare insight into a light-driven process that operates at a heterometallic complex,21–24 and add to the growing examples that invoke the formation of radical intermediates.
; ΔH° = +17.5 kcal mol−1). Using a combined experimental (inhibition, cross-over, radical trapping, EPR spectroscopy) and computational (DFT) approach, we present a detailed investigation into the plausible pathways of H2 activation at the heterometallic complex. We conclude that the most likely pathways involve (i) a closed-shell mechanism involving CO dissociation and subsequent oxidative addition at Fe, and (ii) an open-shell pathway involving homolysis of the Zn–Fe bond to form radical intermediates capable of reacting with H2. Our findings provide rare insight into a light-driven process that operates at a heterometallic complex,21–24 and add to the growing examples that invoke the formation of radical intermediates.
Results and discussion
Photochemical H2 activation
Irradiation of [Zn(H){CH(CMeNAr)2}] (1a, Ar = 2,6-iPr2C6H3) and 0.5 equiv. of [Fe(η5-C5H5)(CO)2]2 for 6 hours with either a 390 or 428 nm LED lamp (40 W) leads to the formation of a mixture of heterometallic species [(η5-C5H5)(CO)Fe–Zn{CH(CMeNAr)2}] (2a) and [(η5-C5H5)(CO)Fe(H)2Zn{CH(CMeNAr)2}] (3a) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (Scheme 1a). The mixture of 2a and 3a in benzene-d6 showed no changes over time at 25 °C, or upon heating to 100 °C. Both 2a and 3a are diamagnetic and can be assigned formal oxidation states of Fe(0) and Fe(II) respectively. Complex 2a is a Zn–Fe adduct which has been previously reported by Mankad and co-workers,25 while complex 3a features two bridging hydride groups linking the Zn and Fe centres. Formation in a 1:1 ratio conserves the reaction stoichiometry. Generation of 3a could be considered to derive from: (i) 1a reacting with [Fe(η5-C5H5)(CO)2]2 to form 2a and [Fe(η5-C5H5)(H)(CO)2]26–28 as a transient intermediate which is then trapped by a second equiv. of 1a to form 3a or (ii) addition of in situ generated H2 across the Zn–Fe bond of 2a under photochemical conditions. To test this second potential mechanism involving photochemical H2 splitting, 1a and [Fe(η5-C5H5)(CO)2]2 were reacted with dihydrogen (1 bar) under LED light. A change in the ratio of the two products was observed reaching a 0.2:0.8 (2a:3a) mixture in 6 hours (Scheme 1a).
:1 ratio (Scheme 1a). The mixture of 2a and 3a in benzene-d6 showed no changes over time at 25 °C, or upon heating to 100 °C. Both 2a and 3a are diamagnetic and can be assigned formal oxidation states of Fe(0) and Fe(II) respectively. Complex 2a is a Zn–Fe adduct which has been previously reported by Mankad and co-workers,25 while complex 3a features two bridging hydride groups linking the Zn and Fe centres. Formation in a 1:1 ratio conserves the reaction stoichiometry. Generation of 3a could be considered to derive from: (i) 1a reacting with [Fe(η5-C5H5)(CO)2]2 to form 2a and [Fe(η5-C5H5)(H)(CO)2]26–28 as a transient intermediate which is then trapped by a second equiv. of 1a to form 3a or (ii) addition of in situ generated H2 across the Zn–Fe bond of 2a under photochemical conditions. To test this second potential mechanism involving photochemical H2 splitting, 1a and [Fe(η5-C5H5)(CO)2]2 were reacted with dihydrogen (1 bar) under LED light. A change in the ratio of the two products was observed reaching a 0.2:0.8 (2a:3a) mixture in 6 hours (Scheme 1a).
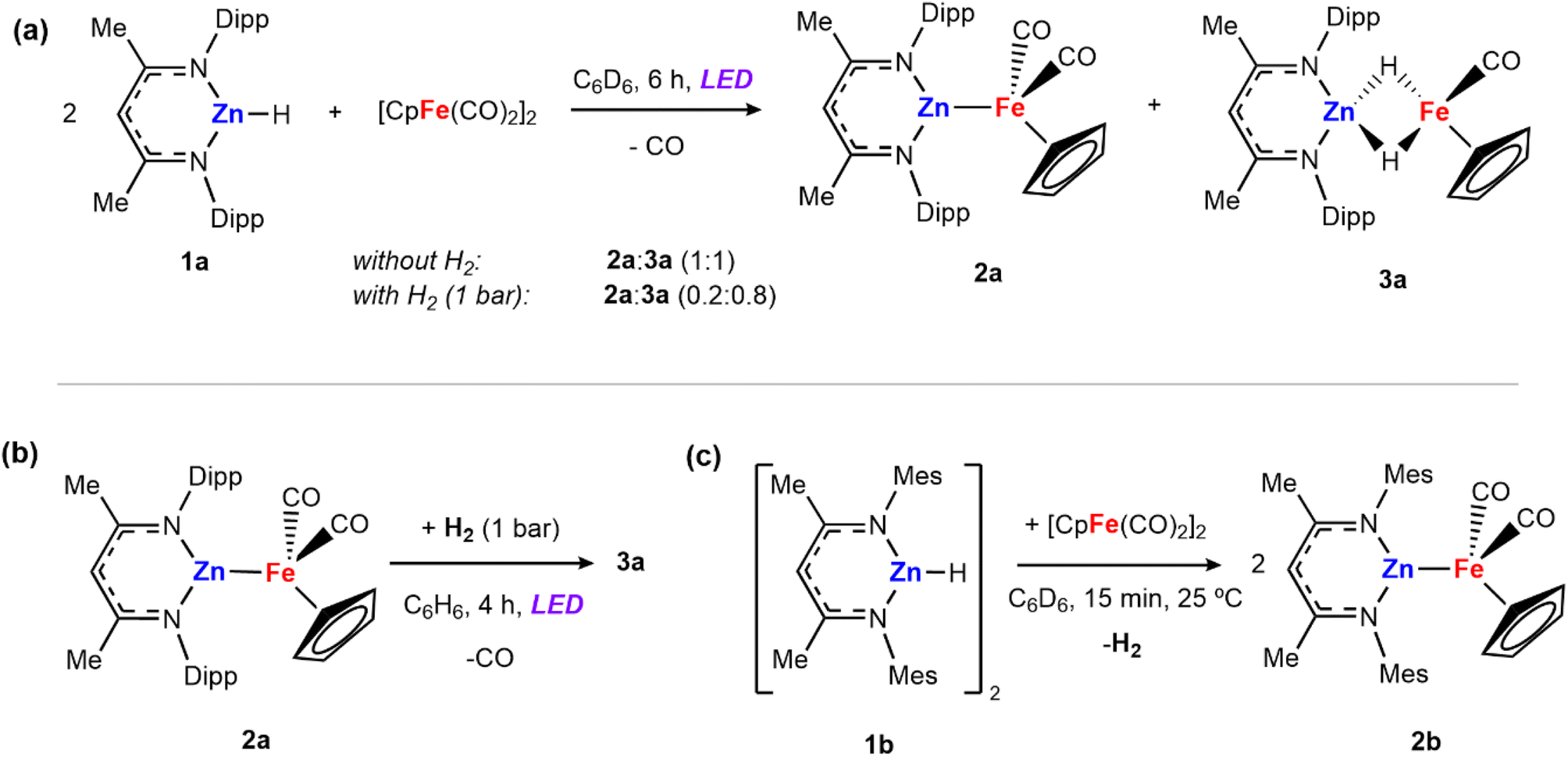 | ||
| Scheme 1 (a) Reactions of 1a with [Fe(η5-C5H5)(CO)2]2 to form a mixture of compounds 2a and 3a; (b) reaction of 2a with dihydrogen under photochemical conditions; (c) reaction of 1b with [Fe(η5-C5H5)(CO)2]2 to form 2b and release H2. | ||
Independently prepared samples of 2a also react with H2 under photochemical conditions (blue LED lamp, 40 W, 390 nm) to form 3a in 55% yield by 1H NMR spectroscopy after 4 h (Scheme 1b). No reaction occurred under thermal conditions (24 h at 80 °C). While this experiment cannot rule out the direct formation of 3a from 1a and [Fe(η5-C5H5)(CO)2]2 it suggests that 2a is capable of activating H2 under photolysis. The reaction was monitored by 1H NMR spectroscopy to quantify the formation of 3a, as well as minor products 1a and [Fe(η5-C5H5)(CO)2]2 which are formed alongside the product. When employing either 390 or 428 nm LED Kessil lamps, longer exposures times led to decreases in the yield of 3a.
UV-vis spectra were recorded for 2a and the dihydride product 3a. Both species present similar absorption bands with a maximum absorption peak around 350 nm. The light absorption of both starting material and product at the same wavelength might explain the challenge of achieving higher yields and the instability of 3a during photolysis. From all the conditions tested, the best results obtained were: 1 bar H2, benzene solvent, 4 hours irradiation using a 390 LED lamp (3a, 45% isolated yield or 55% NMR yield). Experiments using D2 showed 92% deuterium incorporation in 3a, demonstrating that the hydride groups come from dihydrogen, rather than from other possible sources i.e. reaction with the solvent.
The propensity for H2 activation in this system proved sensitive to the precise sterics of the ligand environment on zinc and reaction of [Zn(H){CH(CMeNMes)}] (1b, Mes = 2,4,6-Me3C6H2), bearing smaller aryl flanking groups, with [Fe(η5-C5H5)(CO)2]2 led to the immediate formation of ([(η5-C5H5)(CO)Fe–Zn{CH(CMeNMes)2}] (2b) with elimination of dihydrogen at 25 °C (Scheme 1c). A control reaction was conducted in which 2b was exposed to H2 in the presence of light. However, no hydride-containing products were detected at short reaction times (1 bar H2, 1–6 hours) or long exposure times (24 hours) and under high H2 pressures (4 bar).
Characterisation and bonding analysis
Complex 3a was characterised by X-ray crystallography, along with multinuclear NMR and IR spectroscopy. The 1H NMR spectrum shows characteristic signals in the hydride region at δ = −15.98 ppm (2H). The IR also exhibited bands at 1780 and 1810 cm−1 for the bridging hydrides and at 1948 cm−1 for the CO group. The structure was confirmed after obtaining crystals suitable for X-ray diffraction studies from the reaction mixture of 1a and [Fe(η5-C5H5)(CO)2]2 (Fig. 1a). Two molecules co-crystallized in the same unit cell, one corresponding to the Zn–Fe adduct (2a) and the other to the dihydride complex (3a). The Zn–Fe distance of 2.384(1) Å in complex 2a is elongated to 2.404(1) Å in complex 3a due to the presence of two bridging hydrides (Fig. 1a). The hydrogen atoms were located in the Fourier difference map during X-ray experiments and their positions confirmed by DFT calculations (Table S3†).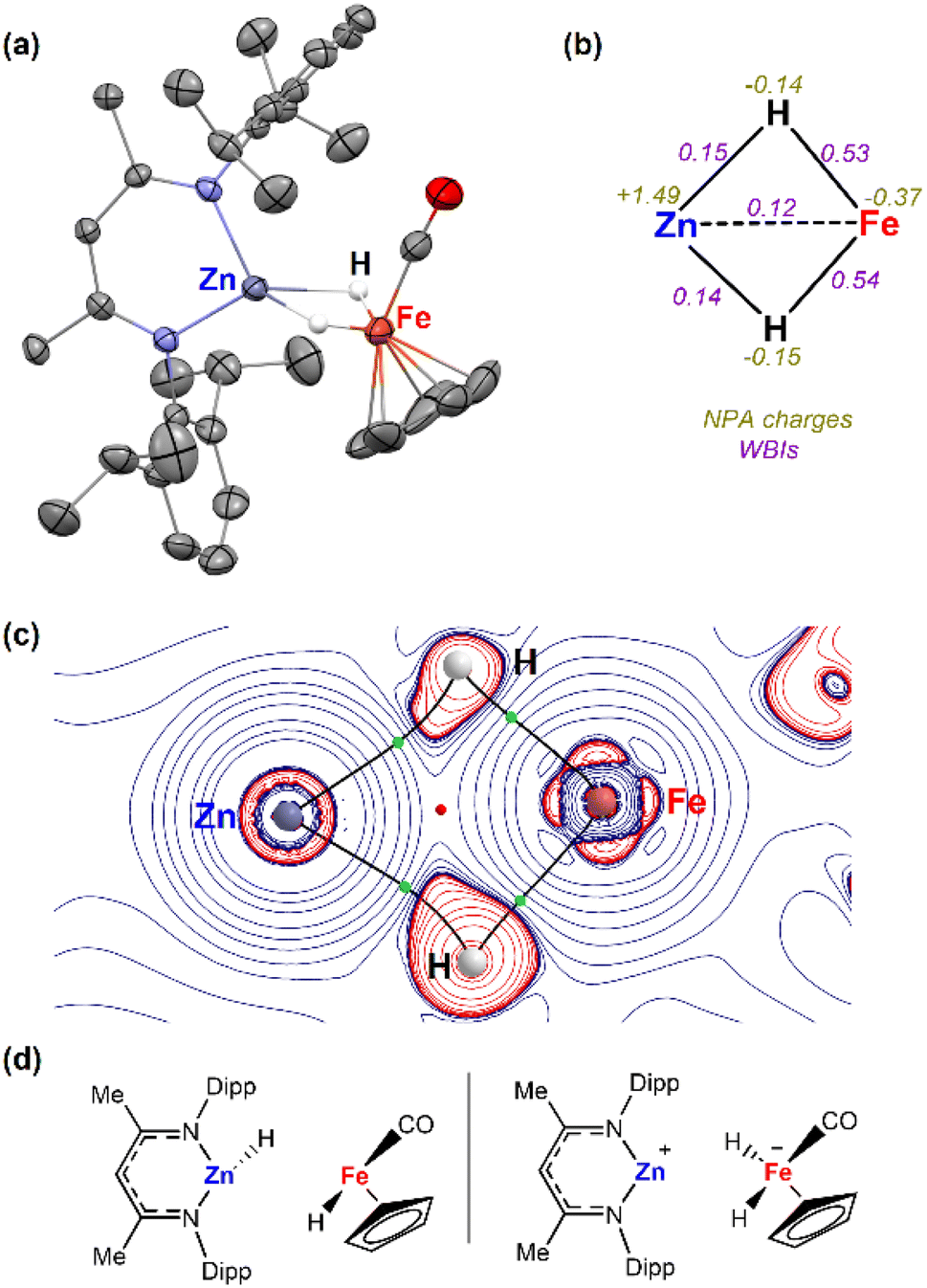 | ||
| Fig. 1 (a) Single-crystal X-ray structures of complex 3a (50% probability ellipsoids) hydrogen atoms were removed from the ORTEP structure for clarity except the bridging hydrides; (b) NBO, (c) QTAIM molecular graphs of 3a. Showing key bond critical points (BCPs, green spheres) and ring critical points (RCPs, red spheres), and (d) fragmentations analysed by ETS-NOCV. | ||
Natural Bonding Orbital (NBO) calculations of complex 3a provided information on the bonding of the heterometallic motif (Fig. 1b). Based on the analysis the hydrides are best considered as bridging ligands which participate in 3-centre 2-electron bonding with Zn and Fe. There is limited support for a significant Zn⋯Fe interaction. Wiberg Bond Indices (WBI) for the Fe–H bonds are higher than those of the corresponding Zn–H bonds. In comparison the Zn⋯Fe WBI (0.12) of 3a is lower than those between the metals and hydrides and lower compared to the Zn⋯Fe WBI in 2a (0.31). NPA charges are negative on the Fe and H atoms but electropositive on Zn (Fig. 2b). The calculations suggest that the hydrides form the most significant covalent interaction with Fe rather than Zn. QTAIM analysis were also undertaken on 3a showing four bond critical points (bcps) between each of the H atoms and Fe or Zn atoms but not between the metals themselves (Fig. 1c). A ring critical point (rcp) was found in the middle of the four membered ring. ETS-NOCV calculations were also performed on 3a. The molecule was separated into two neutral metal hydride fragments, [Fe]–H and [Zn]–H. A total interaction energy ΔEORB = −80.4 kcal mol−1 was obtained, with the two principal contributions corresponding to donation from the [Zn]–H bond towards the [Fe] centre (Δρ1 = −41.1 kcal mol−1) and donation from the [Fe]–H bond to the [Zn] centre (Δρ2 = −25.8 kcal mol−1). A second option was also considered (Fig. 1d), dividing the molecule into [Zn]+ and [Fe]H2− fragments. In this case, a similar total interaction energy of ΔEORB = −93.5 kcal mol−1 was obtained, with the main contribution (Δρ1 = −43.6 kcal mol−1) corresponding to the donation from the two [Fe]–H bonds to the Zn atom.
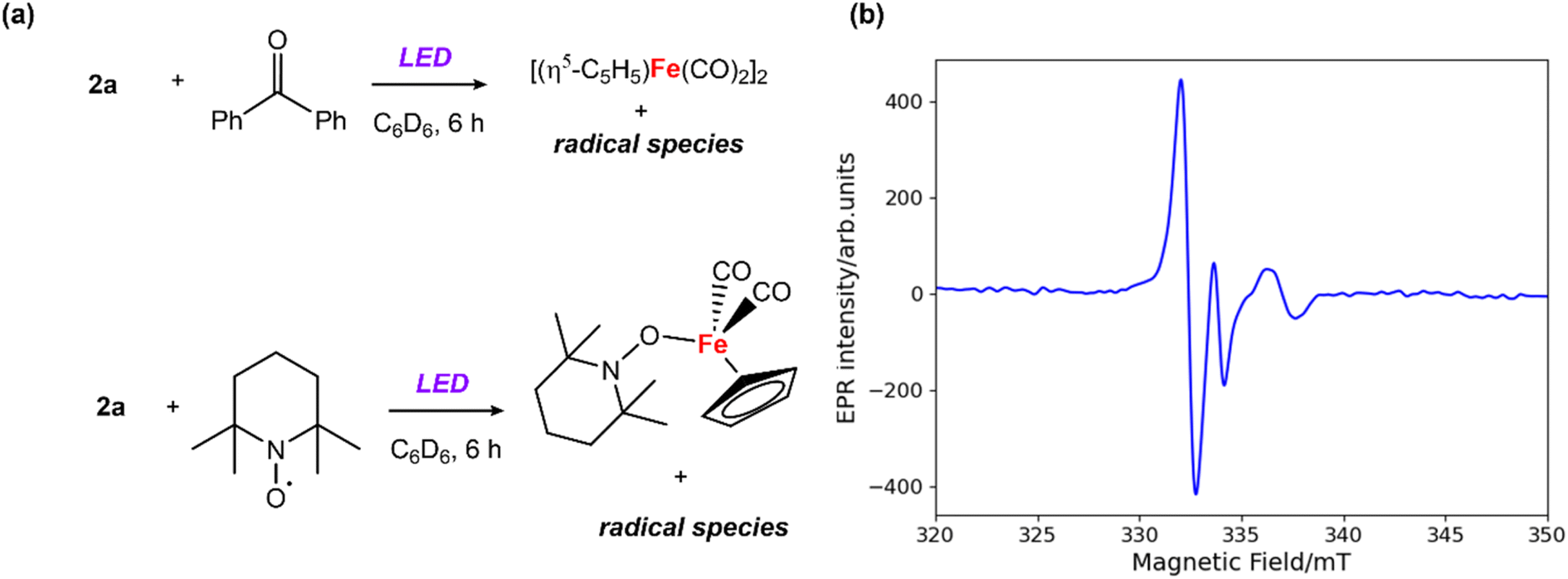 | ||
| Fig. 2 (a) Reactions between 2a and benzophenone/TEMPO; (b) CW X-band EPR spectrum recorded in benzene solution at ambient temperature; microwave frequency, 9.44667 GHz; field modulation amplitude, 0.25 mT. | ||
Plausible mechanism(s) of H2 activation
Although H2 activation with heterometallic complexes consisting of both transition and main group metals is gaining increasing attention, to date most examples involve thermal conditions. The discovery of a new photolytic reaction in this area, raises questions as to how the mechanism proceeds under photochemical conditions. The overall reaction of 2a + H2 to form 3a is calculated to be endergonic with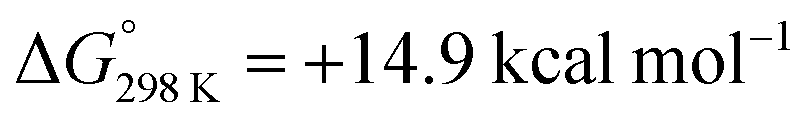 and ΔH° = +17.5 kcal mol−1, consistent with a light-driven process. 2a contains an Fe site with a formal 18-electron count that does not to react in the absence of light. Irradiation is expected to result in this complex accessing an excited state through changes in geometry, ligand dissociation, or fragmentation of the metal–metal bond. Several plausible pathways for photochemical H2 activation were considered (Scheme 2). These included: (Mechanism A) ligand dissociation from 2a to generate a 16-electron coordinatively unsaturated intermediate that reacts with H2 by either direct addition to Fe or across the Fe–Zn bond; (Mechanism B) heterolysis of the metal–metal bond to generate zwitterionic fragments that react with H2 by a closed-shell pathway; (Mechanism C) homolysis of the metal–metal bond to form a radical pair that reacts with H2 by an open-shell pathway. Experiments and calculations were undertaken to try and draw conclusions on the most likely possibilities.
and ΔH° = +17.5 kcal mol−1, consistent with a light-driven process. 2a contains an Fe site with a formal 18-electron count that does not to react in the absence of light. Irradiation is expected to result in this complex accessing an excited state through changes in geometry, ligand dissociation, or fragmentation of the metal–metal bond. Several plausible pathways for photochemical H2 activation were considered (Scheme 2). These included: (Mechanism A) ligand dissociation from 2a to generate a 16-electron coordinatively unsaturated intermediate that reacts with H2 by either direct addition to Fe or across the Fe–Zn bond; (Mechanism B) heterolysis of the metal–metal bond to generate zwitterionic fragments that react with H2 by a closed-shell pathway; (Mechanism C) homolysis of the metal–metal bond to form a radical pair that reacts with H2 by an open-shell pathway. Experiments and calculations were undertaken to try and draw conclusions on the most likely possibilities.
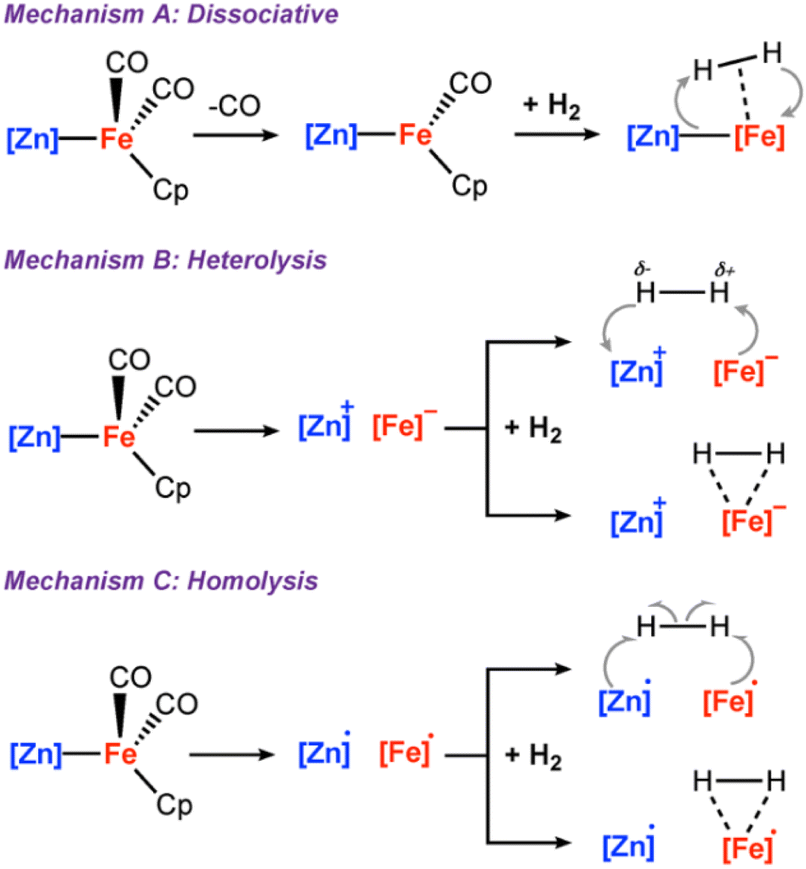 | ||
| Scheme 2 (A) ligand dissociation followed by H2 addition; (B) metal–metal heterolysis; (C) metal–metal homolysis. | ||
Mechanism A: during the reaction with H2, CO must dissociate from 2a to ultimately form the product 3a. Dissociation could potentially occur prior to, or after, the H2 splitting step. The order of these events was probed through inhibition and isotope labelling experiments. The addition of 1:1 mixture of CO:H2 to a solution of 2a resulted in a complete inhibition of H2 activation, even under the exposure of LED light over 24 hours. Reaction of 2a with 13CO demonstrated that ligand exchange was possible at 25 °C after 20 h in the absence of light. This reaction was quicker under photochemical conditions and complete in 2 h on irradiation with 390 nm LEDs. These experiments support the facile and reversible dissociation of CO from 2a under photochemical conditions and based on these findings it is likely this occurs before H2 activation step. Dissociation of CO from 2a would generate a coordinatively unsaturated 16-electron intermediate that has the potential to directly react with H2 by an oxidative addition step.29
Mechanism B and C: the alternative mechanisms for H2 activation with 2a both involve fragmentation of the heterometallic into monomeric units. To probe this possibility, we conducted double-label cross-over experiments. Complex 2c was prepared by salt metathesis reaction of Na[Fe(η5-C5H4CH3)(CO)2]2 with [ZnCl{CH(CMeN-2,6-Et2C6H3)2}] and exposed to H2 to generate the corresponding dihydride species 3c. Then, a mixture of 2a and 2c were dissolved in benzene-d6 and the mixture was exposed to 1 bar of dihydrogen and the 390 nm LED lamp. After 4 hours, an equimolar ratio of the four hydride species 3a, 3c, 3d and 3e was detected by 1H NMR experiments (Scheme 3a).
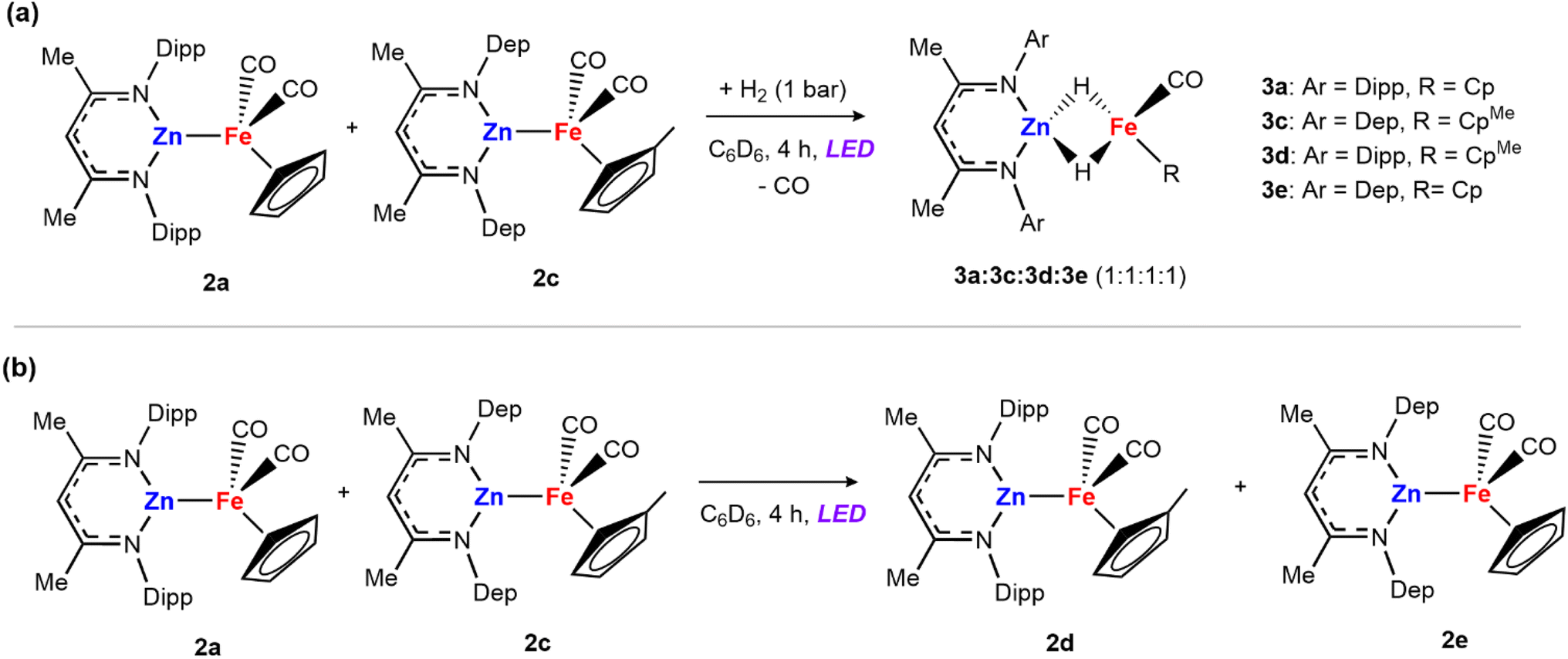 | ||
| Scheme 3 Cross-over experiments: (a) mixture of 2a and 2c in the presence of dihydrogen; (b) reaction between 2a and 2c in the absence of H2. | ||
The formation of cross-over products is consistent with fragmentation of the heterometallic under the reaction conditions. However, the independent mixture of 3a and 3c, also generated the mixed hydrides 3d and 3e, under the same photochemical conditions, meaning that the fragmentation of the dihydride could also account for the products observed. To further support that fragmentation comes from the initial adducts via metal–metal bond cleavage, a solution of 2a and 2c was treated under the same reaction conditions in the absence of the H2 atmosphere (Scheme 3b). After 4 hours under LED light the mixed iron product [Fe2(η5-C5H4CH3)(η5-C5H5)(CO)4] was detected, alongside 2d and 2e. Adduct 2e was prepared independently via the salt metathesis route and spectroscopic data match those of the cross-over experiment. These cross-over experiments suggest that breaking of the metal–metal bond can occur under photochemical conditions.
To further understand whether metal–metal bond cleavage could occur by heterolytic or homolytic pathways, a series of DFT calculations were undertaken. The energies for the homolytic and heterolytic cleavage of the metal–metal bond were calculated for 2a (Scheme 4). These steps were evaluated for structures with CO associated and CO dissociated. While barriers to cleavage are universally high, the energies for the homolytic dissociation were significantly lower (<25 kcal mol−1) compared to the heterolysis. The dissociation energies were calculated without including semiempirical dispersion corrections, as invoking these resulted in deviations from experimental results and over-estimated binding energies.30,31 Furthermore, similar barriers for homolysis of Al–Fe bonds have been calculated by Mankad and coworkers for a closely related system.32
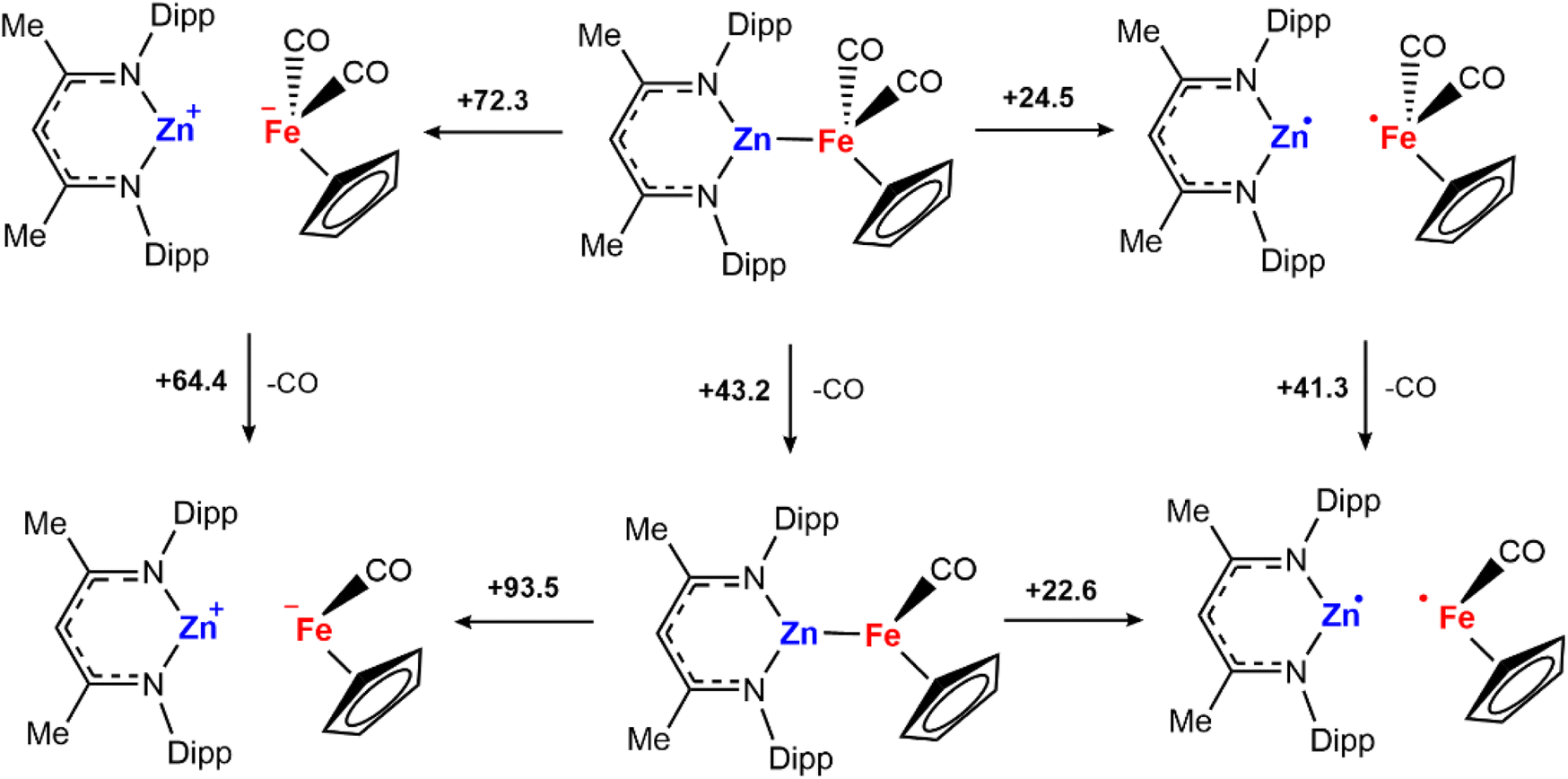 | ||
| Scheme 4 CO and metal–metal bond dissociation Gibbs free energies at 298 K in kcal mol−1. G09: BP86/def2TZVP (C, H, N, O)/SDDAll (Fe, Zn)//BP86/6-31g**(C, H)/6-311+g*(N, O)/SDDAll (Fe, Zn). Solvent corrections (benzene, epsilon = 2.2706) which were modelled using the polarized continuum model (PCM). | ||
The calculations suggest that the results of cross-over experiments are most likely due to homolysis rather than heterolysis of the metal–metal bond. To further probe this hypothesis, reactions with H2 were conducted with a series of different solvents. Heterolysis results in charge separation and as such, it might be expected that more polar solvents would facilitate this pathway increasing yields at shorter reaction times. The reaction of H2 with 2a in different solvents (benzene, ε = 2.3; THF, ε = 7.6; fluorobenzene, ε = 5.4) during the same reaction times (6 h) led to similar NMR yields for formation of 3a.33 Given these results, we consider that the homolytic metal–metal bond cleavage to form radical intermediates is more probable to occur compared to the heterolytic version.
EPR experiments were performed to probe the potential formation of radical intermediates. Addition of 1 equiv. of benzophenone to a solution of 2a in benzene and 6 hours exposure to LED light led to a distinctive signal in the X-band EPR spectrum (giso = 2.031) at 25 °C (Fig. 2b). A similar EPR signal was observed from the reaction the analogous Zn–Zn bonded complex [Zn2{CH(CMeN-2,4,6-Me3C6H2)2}2] with benzophenone under the same photochemical conditions (Fig. S17†). The short lifetime of the radical species, presumed to be a benzophenone radical anion coordinated to zinc, prevented its isolation and characterisation. While attempts to crystallise this species from the reaction mixture resulted in isolation of the starting material, a closely related magnesium analogue has been prepared by Jones and coworkers.34 EPR studies on the Mg-benzophenone radical reported by Jones34 (giso = 2.004) and the Al-benzophenone radical by Mankad32 (giso = 2.006) present similar g values to our measurements. During these experiments formation of [Fe(η5-C5H5)(CO)2]2 was observed by 1H NMR spectroscopy. In contrast, the addition of 1 equiv. of 2,2,6,6-tetramethylpyridine 1-oxyl (TEMPO) to 2a under the same conditions led to formation of [Fe(η5-C5H5)(CO)2(TEMPO)] (detected by 1H NMR experiments),35 and inhibited the formation of [Fe(η5-C5H5)(CO)2]2 (Fig. 2a).36 The EPR measurements and radical trapping experiments support homolysis of the Zn–Fe bond under photolytic conditions, with clear evidence for the formation and trapping of Fe-based radicals.
In combination, the cross-over experiments, calculations, solvent effects, and EPR data suggest that while heterolytic fragmentation of 2a is unlikely under the reaction conditions, homolytic cleavage leading to a radical pathway remains a distinct mechanistic possibility for H2 activation. Following homolysis of the metal–metal bond, the addition of H2 could potentially occur through either a single-site mechanism involving addition at the Fe fragment to generate a Fe-dihydride radical, which recombines with the zinc radical or a radical pair mechanism in which both metal radicals react with H2 in a concerted manner (Scheme 2C). The thermodynamics for these options were calculated by DFT, both are exergonic and feasible.
Conclusions
In summary, we report a rare example of photochemical H2 activation with a Zn–Fe heterometallic complex. Through a combined experimental, spectroscopic, and computation approach we have interrogated plausible mechanisms for bond breaking of H2. We concluded that there is no unequivocal evidence that supports a single mechanism for the reaction. The combination of experimental and computational data suggests two mechanisms are possible and may operate simultaneously. One involves photochemical dissociation of CO from the Zn–Fe complex to form a coordinatively saturated intermediate that can react with H2via oxidative addition and the other involves homolytic cleavage of the Zn–Fe bond to form a pair of radicals that react directly with H2 through a radical pathway.Data availability
Data are available within the ESI.† Crystallographic data can be obtained viahttps://www.ccdc.cam.ac.uk/data_request/cif, or by emailing E-mail: data_request@ccdc.cam.ac.uk.Author contributions
MPJ conducted all the experimental and computational work. All authors were involved in preparing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Spanish Ministry of Universities/University of Sevilla for a Margarita Salas fellowship (MPJ). The PEPR facility at Imperial College London and Alberto Collauto are thanked for guidance in collection and interpretation of EPR data. We thank Pete Haycock and Andrew J. P. White for their help with NMR and XRD experiments.Notes and references
- H. Fong, M.-E. Moret, Y. Lee and J. C. Peters, Organometallics, 2013, 32, 3053 CrossRef CAS PubMed.
- B. R. Barnett, C. E. Moore, A. L. Rheingold and J. S. Figueroa, J. Am. Chem. Soc., 2014, 136, 10262 CrossRef CAS PubMed.
- G. Zeng and S. Sakaki, Inorg. Chem., 2013, 52, 2844 CrossRef CAS PubMed.
- M. Devillard, R. Declercq, E. Nicolas, A. W. Ehlers, J. Backs, N. Saffon-Merceron, G. Bouhadir, J. C. Slootweg, W. Uhl and D. Bourissou, J. Am. Chem. Soc., 2016, 138, 4917 CrossRef CAS PubMed.
- I. M. Riddlestone, N. A. Rajabi, J. P. Lowe, M. F. Mahon, S. A. Macgregor and M. K. Whittlesey, J. Am. Chem. Soc., 2016, 138, 11081 CrossRef CAS PubMed.
- F. M. Miloserdov, N. A. Rajabi, J. P. Lowe, M. F. Mahon, S. A. Macgregor and M. K. Whittlesey, J. Am. Chem. Soc., 2020, 142, 6340 CrossRef CAS PubMed.
- M. Garçon, A. Phanopoulos, A. J. P. White and M. R. Crimmin, Angew. Chem., Int. Ed., 2023, 135, e202213001 CrossRef.
- Y. Cai, S. Jiang, T. Rajeshkumar, L. Maron and X. Xu, J. Am. Chem. Soc., 2022, 144, 16647 CrossRef CAS PubMed.
- P. M. Keil, A. Soyemi, K. Weisser, T. Szilvási, C. Limberg and T. J. Hadlington, Angew. Chem., Int. Ed., 2023, 62, e202218141 CrossRef CAS PubMed.
- M. Widemann, K. Eichele, H. Schubert, C. P. Sindlinger, S. Klenner, R. Pöttgen and L. Wesemann, Angew. Chem., Int. Ed., 2021, 60, 5882 CrossRef CAS PubMed.
- F. M. Miloserdov, C. J. Isaac, M. L. Beck, A. L. Burnage, J. C. B. Farmer, S. A. Macgregor, M. F. Mahon and M. K. Whittlesey, Inorg. Chem., 2020, 59, 15606 CrossRef CAS PubMed.
- C. J. Isaac, C. I. Wilson, A. L. Burnage, F. M. Miloserdov, M. F. Mahon, S. A. Macgregor and M. K. Whittlesey, Inorg. Chem., 2022, 61, 20690 CrossRef CAS PubMed.
- S. L. Matthews and D. M. Heinekey, J. Am. Chem. Soc., 2006, 128, 2615 CrossRef CAS PubMed.
- J. P. Dunne, D. Blazinaa, S. Aiken, H. A. Carteret, S. B. Duckett, J. A. Jones, R. Poli and A. C. Whitwood, Dalton Trans., 2004, 21, 3616 RSC.
- T. E. Bitterwolf, J. C. Linehan and J. E. Shade, Organometallics, 2000, 19, 4915 CrossRef CAS.
- R. J. Sullivan and T. L. Brown, J. Am. Chem. Soc., 1991, 113, 9155 CrossRef CAS.
- S. Zhang and T. L Brown, Organometallics, 1992, 11, 2122 CrossRef CAS.
- B. D. Moore, M. B. Simpson, M. Poliakoff and J. J. Turner, J. Chem. Soc., Chem. Commun., 1984, 972 RSC.
- T. J. Meyer and J. V. Caspar, Chem. Rev., 1985, 85, 187 CrossRef CAS.
- A. S. Weller and J. S. McIndoe, Eur. J. Inorg. Chem., 2007, 28, 4411 CrossRef.
- T. E. Bitterwolf, Coord. Chem. Rev., 2001, 211, 235 CrossRef CAS.
- S. R. Parmelee, T. J. Mazzacano, Y. Zhu, N. P. Mankad and J. A. Keith, ACS Catal., 2015, 5, 3689 CrossRef CAS.
- J. T. Davis, E. E. Martinez, K. J. Clark, D.-H. Kwon, M. R. Talley, D. J. Michaelis, D. H. Ess and M. C. Asplund, Organometallics, 2021, 40, 1859 CrossRef CAS.
- S. Sinhababu and N. P. Mankad, Organometallics, 2022, 41, 1917 CrossRef CAS.
- T. J. Mazzacano and N. P. Mankad, J. Am. Chem. Soc., 2013, 135, 17258 CrossRef CAS PubMed.
- T. A. Shackleton, S. C. Mackie, S. B. Fergusson, L. J. Johnston and M. C. Baird, Organometallics, 1990, 9, 2248 CrossRef CAS.
- D. P. Estes, A. K. Vannucci, A. R. Hall, D. L. Lichtenberger and J. R. Norton, Organometallics, 2011, 30, 3444 CrossRef CAS.
- M. R. Radzhabov and N. P. Mankad, Organometallics, 2023, 42, 2111 CrossRef CAS.
- R. A. Fischer, E. Herdtweck and T. Priermeier, Inorg. Chem., 1994, 33, 934 CrossRef CAS.
- T. Weymuth, E. P. A. Couzijn, P. Chen and M. Reiher, J. Chem. Theory Comput., 2014, 10, 3092 CrossRef CAS PubMed.
- T. Husch, L. Freitag and M. Reiher, J. Chem. Theory Comput., 2018, 14, 2456 CrossRef CAS PubMed.
- S. Sinhababu, M. R. Radzhabov, J. Telser and N. P. Mankad, J. Am. Chem. Soc., 2022, 144, 3210 CrossRef CAS PubMed.
- Only slightly lower yields, <8%, were observed for the THF and fluorobenzene reactions compared to the benzene one.
- D. M. Murphy, L. E. McDyre, E. Carter, A. Stasch and C. Jones, Magn. Reson. Chem., 2011, 49, 159 CrossRef CAS PubMed.
- J. P. Lomont, S. C. Nguyen and C. B. Harris, J. Am. Chem. Soc., 2013, 135, 11266 CrossRef CAS PubMed.
- The 1H NMR monitoring of the TEMPO trapping experiment showed other minor BDI-containing species (<10 % NMR yield) and formation of [Zn]–TEMPO cannot be dismissed. An analogue [Mg]–TEMPO species has been generated from [Mg]–X (X = alkyl, H) and TEMPO. (D. J. Liptrot, P. M. S. Hill and M. F. Mahon, Angew. Chem., Int. Ed., 2014, 53, 6224). Reactions of 1a with excess TEMPO after 5 days at 100 °C gives a mixture of undetermined species.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2297407 and 2297408. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc05966a |
| This journal is © The Royal Society of Chemistry 2024 |