
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper-catalyzed trichloromethylative carbonylation of ethylene†
Youcan
Zhang‡
bc,
Bing-Hong
Teng‡
ac and
Xiao-Feng
Wu
 *cd
*cd
aSchool of Chemistry and Chemical Engineering, Liaoning Normal University, 850 Huanghe Road, Dalian 116029, China
bCollege of Chemistry and Chemical Engineering, Shanghai University of Engineering Science, Shanghai 201620, China
cDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 116023 Dalian, Liaoning, China. E-mail: xwu2020@dicp.ac.cn
dLeibniz-Institut Für Katalyse e.V., Albert-Einstein-Straβe 29a, 18059 Rostock, Germany
First published on 14th December 2023
Abstract
Difunctionalization of alkenes is an efficient strategy for the synthesis of complex compounds from readily available starting materials. Herein, we developed a copper-catalyzed visible-light-mediated trichloromethylative carbonylation of ethylene by employing commercially available CCl4 and CO as trichloromethyl and carbonyl sources, respectively. With this protocol, various nucleophiles including amines, phenols, and alcohols can be rapidly transformed into β-trichloromethyl carboxylic acid derivatives with good functional-group tolerance. Bis-vinylated γ-trichloromethyl amides can also be obtained by adjusting the pressure of carbon monoxide and ethylene. In addition, this photocatalytic system can be successfully applied in the late-stage functionalization of bioactive molecules and pharmaceutical derivatives as well.
Polychloroalkyl motifs are well known as key components to alter the biological activity of organic compounds, which are widely distributed in natural products, pharmaceuticals, and bioactive molecules.1–3 During the last few decades, thousands of natural products containing C–Cl bonds have been discovered, especially those embedding the trichloromethyl skeleton, such as desenamide A,4,5 desenpyridine,6,7 sintokamide A,8,9 and callyspongiamide B,10 which have shown excellent biological activities in antibiotics and antitumor applications (Fig. 1a). Additionally, the trichloromethyl motif can also be employed as a versatile building block for organic transformations.11–13 Therefore, the incorporation of the trichloromethyl group into organic compounds has attracted considerable attention from chemists. Conventionally, trichloromethylation strategies employ base-promoted deprotonation of CHCl3 (ref. 14–16) or deprotection of TMS-CCl3 (ref. 17–19) to form trichloromethyl anions, which are then applied as coupling partners. Meanwhile, the Kharasch reaction considered as an alternative method of trichloromethylation proceeds through the addition of trichloromethyl radicals to alkenes via C–H cleavage of CHCl3 or C–X bond activation of CXCl3 (X = Cl, Br)20,21 (Fig. 1b). In recent years, the catalytic functionalization of C–Cl or C–H bonds of polychloromethanes (CCl4 and CHCl3) with alkenes and other coupling partners has emerged as a powerful trichloromethylation protocol to construct trichloromethyl functionalized compounds in a single step (Fig. 1b).22–25 However, these methods often reacted at high temperatures and require a stoichiometric amount of oxidant. Accordingly, redesign of redox-neutral reactions for preparing functionalized trichloromethyl-containing compounds under mild conditions remains urgently needed.
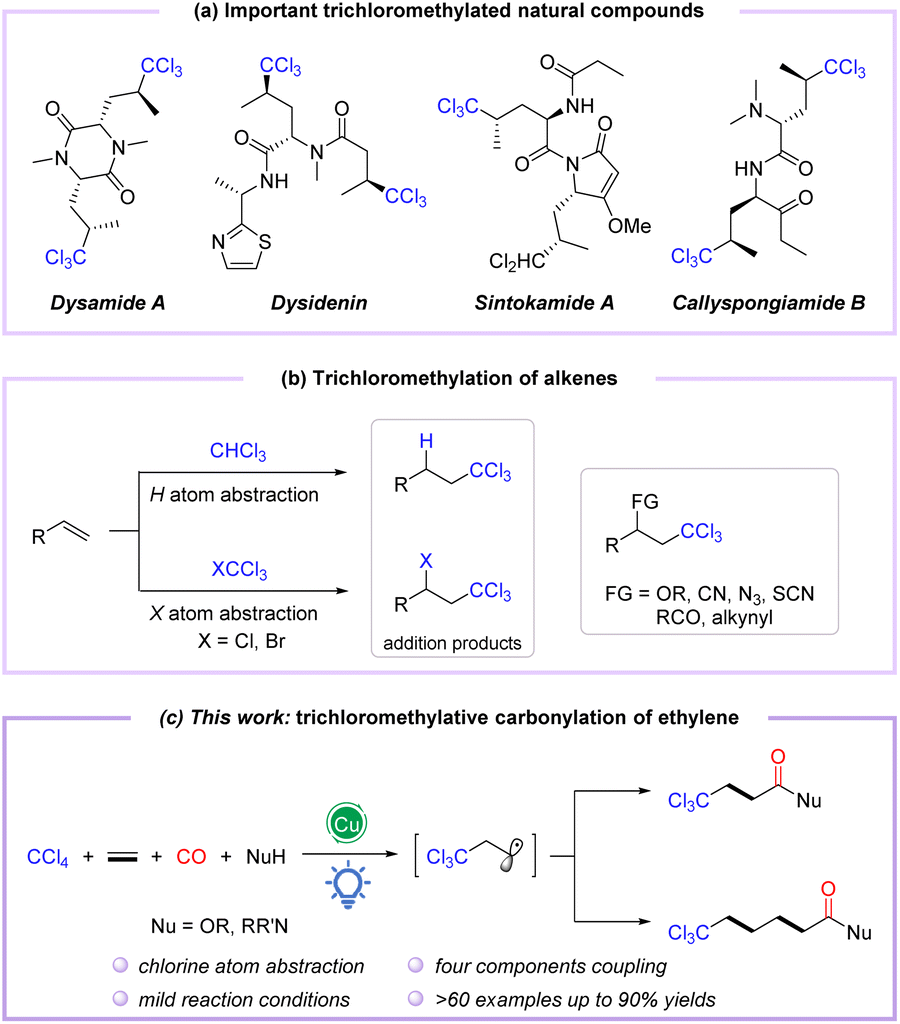 | ||
| Fig. 1 (a) Important trichloromethylated natural compounds. (b) Trichloromethylation of alkenes. (c) Copper-catalyzed visible-light-induced trichloromethylation/carbonylation of alkenes. | ||
The radical relay-type vicinal difunctionalization of alkenes is one of the most attractive strategies to rapidly incorporate diversely functionalized molecular backbones.26–29 However, examples of trichloromethylative difunctionalization of alkenes employing CCl4 as a versatile trichloromethyl radical precursor under external oxidant-free conditions are still rarely reported.30,31 On the other hand, amides and esters are powerful molecular backbones ubiquitous in compounds that display biological properties,32–35 especially in complex molecules with a trichloromethyl group. In this context, divergent syntheses from simple and readily available substrates to obtain diverse trichloromethyl-containing carbonylated compounds have received much attention. To date, only one report on palladium-catalyzed trichloromethylative carbonylation of alkenes in ethanol to synthesize β-trichloromethyl esters in 12–60% yields has been reported by Tsuji and coworkers in 1985.36
Catalytic carbonylation represents a direct and efficient tool; in particular, difunctionalized carbonylation based on alkenes allows the simultaneous introduction of two building units, including the carbonyl group, enabling the synthesis of functionalized carbonylated compounds.37–40 Among them, visible light-induced radical carbonylation has received much attention because it can avoid certain disadvantages of conventional thermal based methods and promote the formation of radicals under relatively mild conditions.41–44 The trichloromethyl group has been well installed in alkenes as a trichloromethyl radical equivalent, generated by visible-light-induced Cl-atom abstraction of CCl4.45–47 Nevertheless, trichloromethylative difunctionalization is still challenging, primarily due to the weak C–X bond of CCl4, which induces side reactions such as addition, coupling, and elimination to compete with the target reaction. In view of our continuing research interest in transition metal-catalyzed and photo-induced difunctionalization of alkenes by the radical pathway,48–50 we envisioned the trichloromethylative carbonylation of alkenes to construct a series of trichloromethyl-modified carboxylic acid derivatives under mild photocatalytic conditions in which CCl4 was selected as the trichloromethyl radical precursor (Fig. 1c). In this strategy, the insertion of CO (carbon monoxide) and the oxidation of acyl radicals are the key steps during the photocatalytic process.
To explore this multicomponent reaction, screening studies were initiated in ethylene and CO atmospheres with CCl4 and aniline as the trichloromethyl precursor and nucleophile, respectively (Table 1). After systematic screening of reaction conditions, the desired β-trichloromethyl amide 3a could be smoothly provided in 77% isolated yield when the reaction was performed in the presence of a photoredox catalyst in situ generated from Cu(OTf)2 and bpy (bipyridine), using K2CO3 as the base in MeCN upon irradiation with 456 nm blue LEDs at room temperature (25–30 °C) for 24 h under ethylene (1 bar) and CO (50 bar) atmospheres (Table 1, entry 1). Other N-ligands were subsequently examined, such as 1,10-phen (1,10-phenanthroline) and 4,4′-diOMe-2,2′-bpy (4,4′-dimethoxy-2,2′-bipyridine) delivered similar yields, and lower conversion was found when 6,6′-diMe-2,2′-bpy (6,6′-dimethyl-2,2′-bipyridine) was selected as the ligand (Table 1, entries 2–4). Undesirable, none of the other copper precursors could improve the yield of the targeted product (Table 1, entries 5–6). Next, different bases were also investigated; among them with the inorganic bases Na2CO3 and K3PO4, slightly reduced yields were observed (Table 1, entries 7–8), whereas the organic base triethylamine resulted in a drastic decrease in yield (Table 1, entry 9). In addition, reaction solvent screening revealed that MeCN was the best solvent among the evaluated solvents (Table 1, entries 10–12). Using well prepared copper complexes with different ligands instead of Cu(TfO)2 and bpy were tested as well, and the desired carbonylation product can be obtained in all the cases (Table S2,† entries 7–10).51–53 Control experiments indicated that a copper salt, ligand, base, and blue light were all indispensable, although the desired product was still detectable in the absence of the ligand at elevated temperature (Table 1, entries 13–16, for more details, see the ESI†).
|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yield (%)a |
| a Reaction conditions: CCl4 (1.0 mmol), ethylene (1 bar), PhNH2 (0.2 mmol), Cu(OTf)2 (1 mol%), bpy (2 mol%), and K2CO3 (2.0 equiv.) in MeCN (1.0 mL) at 25–30 °C for 24 h under CO (50 bar). Yields were determined by GC-FID analysis using n-hexadecane as the internal standard. b Isolated yield. n.d.: not detected. | ||
| 1 | None | 80 (77)b |
| 2 | 1,10-Phen instead of bpy | 78 |
| 3 | 4,4′-diOMe-2,2′-bpy instead of bpy | 76 |
| 4 | 6,6′-diMe-2,2′-bpy instead of bpy | 60 |
| 5 | Cu(OAc)2 instead of Cu(OTf)2 | 75 |
| 6 | CuI instead of Cu(OTf)2 | 76 |
| 7 | Na2CO3 instead of K2CO3 | 79 |
| 8 | K3PO4 instead of K2CO3 | 76 |
| 9 | Et3N instead of K2CO3 | 22 |
| 10 | PhCF3 instead of MeCN | 53 |
| 11 | 1,4-Dioxane instead of MeCN | 65 |
| 12 | DCE instead of MeCN | 57 |
| 13 | No bpy | 24 |
| 14 | No Cu(OTf)2, no K2CO3, or no light | n.d. |
| 15 | No light, 40 °C | 56 |
| 16 | No light, 80 °C | 76 |
With the optimal reaction conditions in hand, the general substrate scope of this copper-catalyzed photo-induced multicomponent trichloromethylation/carbonylation was systematically explored. As shown in Scheme 1, various nucleophiles including amines, phenols, and alcohols were successively investigated. Arylamines with different functional groups on the benzene ring, including electron-donating (isopropyl, methoxy, methylthio, phenoxy, and acetamido) and electron-withdrawing (trifluoromethyl, trifluoromethoxy, acetyl, and cyano), smoothly participate in carbonylation, delivering the corresponding products 3a–3m in 25–90% yields. Among them, the scope of arylamines was sensitive to steric and electronic changes of functional groups, and large steric hindrance and electron-donating functional groups significantly inhibit the reaction; in contrast, electron-withdrawing functional groups promote efficient conversion of substrates. Notably, aryl halides (3g and 3h) or aryl nitriles (3l) are well compatible with catalytic systems. Moderate yields were obtained when this protocol was extended to substrates with the pyridine motif (3n and 3o). Secondary arylamines such as N-methylaniline and indoline also proceeded smoothly, while 3q was obtained in lower yield, likely due to its steric hindrance. Benzylamine, even NH3 were converted successfully as well, afforded the desired β-trichloromethylamides 3r and 3s in 39% and 70% yields, respectively.
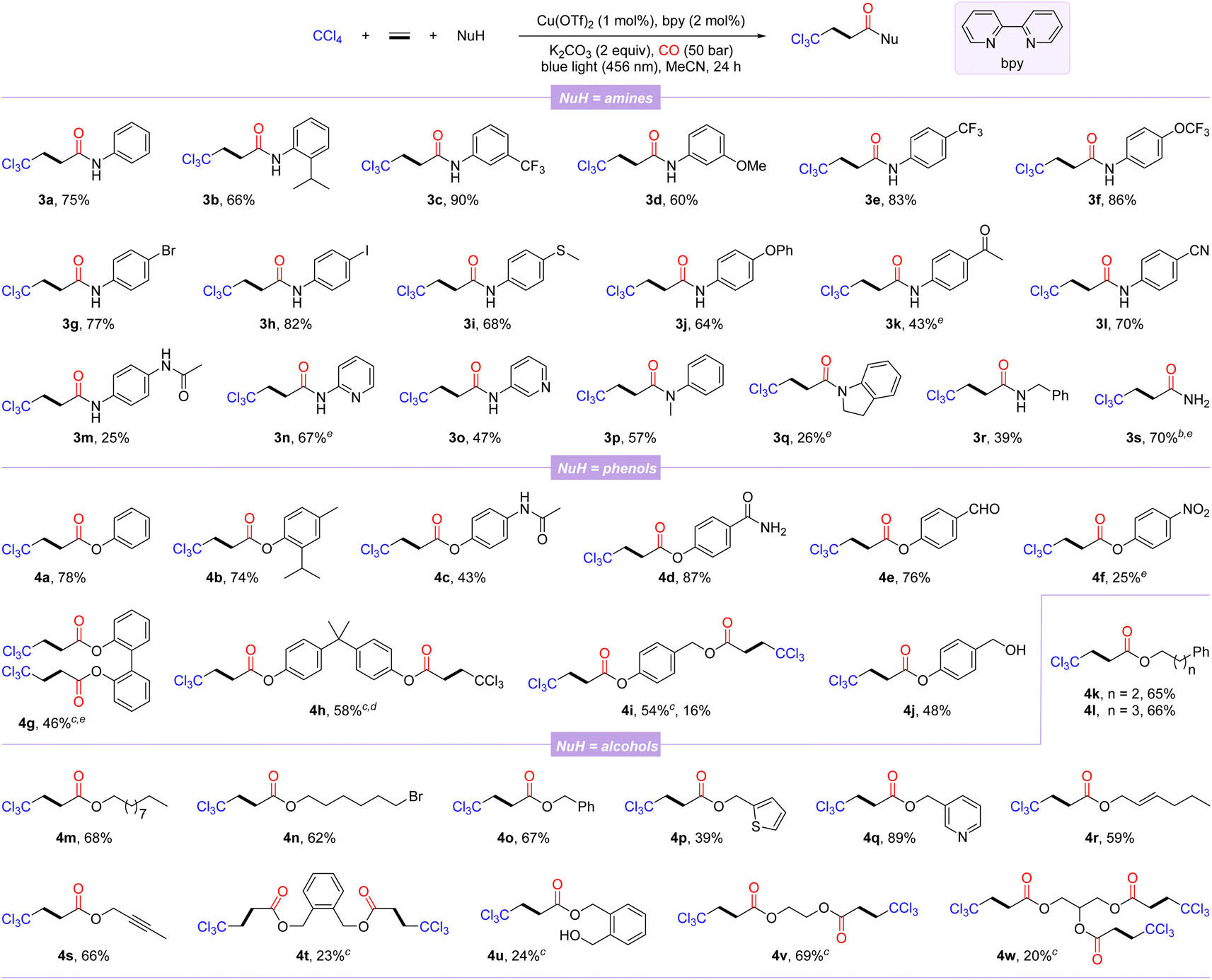 | ||
| Scheme 1 Scope of nucleophiles. [a] Reaction conditions: CCl4 (2.0 mmol, 5.0 equiv.), ethylene (1 bar), NuH (0.4 mmol, 1.0 equiv.), Cu(OTf)2 (1 mol%), bpy (2 mol%), and K2CO3 (0.8 mmol, 2.0 equiv.) in MeCN (2.0 mL) at 25–30 °C for 24 h under CO (50 bar); isolated yields. [b] NH3 (0.4 mmol, 0.4 M in dioxane). [c] NuH (0.2 mmol). [d] MeCN (3.0 mL). [e] 95% purity. | ||
Next, several β-trichloromethyl esters 4a–4f were prepared from the corresponding phenols. The steric hindrance of phenols had an insignificant effect on the carbonylation procedure (4a-4b). Phenols with an acetamido, carbamoyl, aldehyde, or nitro group were tolerated well, albeit with relatively low yields of 4c and 4f. The reaction was also amenable to diphenols, furnishing the corresponding double carbonylation products 4g and 4h in 46% and 58% yields, respectively. In addition, 4-(hydroxymethyl)phenol with two hydroxyl groups could selectively lead to the double-carbonylation product 4i or simultaneously to the mono-carbonylated and double-carbonylated products 4i and 4j by controlling the reaction loading. Finally, we also examined a variety of alcohols as the nucleophilic partners, among which alcohols with distinct carbon chain lengths and remote bromine atoms could be successfully converted to the corresponding esterification products 4k–4n in 62–68% yields. Benzyl alcohols and heterocyclic units of thiophene and pyridine replacing phenyl behave well in this reaction (4o–4q). Allyl alcohol and alkynyl alcohol could generate the corresponding esters 4r and 4s in good yields with the C–C unsaturated bonds intact. Notably, alcohols with two reaction sites were suitable substrates, yielding the corresponding products 4t–4v, while the mono-carbonylation product 4u was also observed, probably due to the influence of steric hindrance. Even glycerol with trihydroxyl groups could participate in the reaction, albeit the tricarbonylated product 4w was obtained with a lower yield.
To further validate the practicality and generality of this procedure, the late-stage functionalization of several natural products, pharmaceutical derivatives, and bioactive molecules was performed. As shown in Scheme 2, aminoglutethimide, sulfalene, and anilines modified with menthol and camphorsulfonyl chloride were well matched to furnish the desired products 5a–5d in 39–85% yields, which indicated that the weakly nucleophilic sulfonamide is also a potential reactive site, whereas the amide functional group exhibits no reactivity. Additionally, our protocol could be extended to O-nucleophilic complex molecules with equal ease to advanced β-trichloromethyl esters derived from raspberry ketone (5e), estrogen (5f), fluorescein (5g), diacetonefructose (5h), DL-menthol (5i), epiandrosterone (5j), pregnenolone (5k), and estradiol benzoate (5l) in moderate to high yields (58–81%). As we expected, ezetimibe with two potential hydroxyl reactive sites also successfully accomplished double carbonylation to acquire the trichloromethylated carbonylation product 5m in 65% yield. Satisfactorily, scaling up the carbonylation involving aniline and phenol to 2.0 mmol, respectively, supplied the desired products 3a and 4a smoothly unhindered even with halving the catalytic amounts of copper salt and ligand. Unexpectedly, the scale-up reaction extended to phenylpropanol with a markedly lower yield of the target product 4l. It is also worth mentioning that as analogues of ethylene, but-3-en-1-ylbenzene and styrene were also tested, but only compounds based on a non-carbonylation reaction were detected.
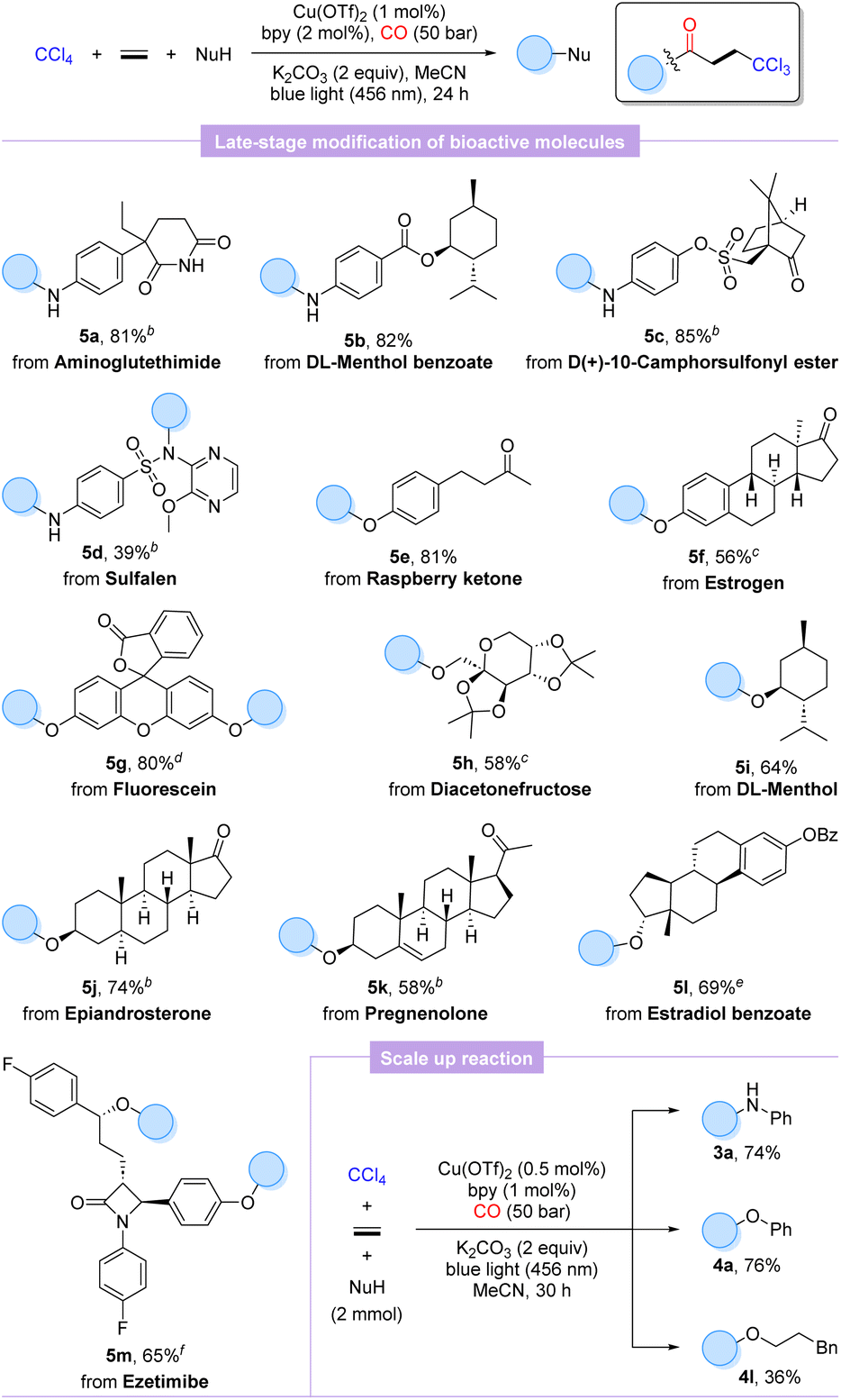 | ||
| Scheme 2 Late-stage modification of bioactive molecules and scale up reaction. [a]Reaction conditions: CCl4 (2.0 mmol, 5.0 equiv.), ethylene (1 bar), NuH (0.4 mmol, 1.0 equiv.), Cu(OTf)2 (1 mol%), bpy (2 mol%), and K2CO3 (0.8 mmol, 2.0 equiv.) in MeCN (2.0 mL) at 25–30 °C for 24 h under CO (50 bar); isolated yields. [b] CCl4 (1.0 mmol), NuH (0.2 mmol), and K2CO3 (0.4 mmol). [c] MeCN (3.0 mL). [d] NuH (0.2 mmol) and MeCN (3.0 mL). [e] CCl4 (0.5 mmol), NuH (0.1 mmol), and K2CO3 (0.2 mmol). [f] CCl4 (1.0 mmol), NuH (0.1 mmol), Cu(OTf)2 (2 mol%), bpy (4 mol%), and K2CO3 (0.4 mmol). Scale up reaction (2.0 mmol level): CCl4 (10.0 mmol, 5.0 equiv.), ethylene (1 bar), NuH (2.0 mmol, 1.0 equiv.), Cu(OTf)2 (0.5 mol%), bpy (1 mol%), and K2CO3 (4.0 mmol, 2.0 equiv.) in MeCN (10.0 mL) at 25–30 °C for 30 h under CO (50 bar); isolated yields. | ||
Inspired by the polymerization reaction of CO with ethylene to produce polyketones,54–56 we are keenly interested in exploring the photo-induced trichloromethylative carbonylation of ethylene for the synthesis of carbon chain-extending remotely trichloromethyl-modified aliphatic carboxylic acid derivatives. We conjectured that radical carbonylation transformations involving bis- or even multi-ethylene relays could be achieved by increasing the amount of ethylene. To confirm our hypothesis, the model reaction was probed in terms of parameters such as ligand, pressure of CO and ethylene (for details, see the ESI†). After identifying the optimal conditions, we then evaluated the substrate scope of this transformation with the access of amines to γ-trichloromethyl amides (6a–6i). As shown in Scheme 3, anilines with diverse functional groups, including methoxy, tert-butyl, trifluoromethyl, methylthio, and trifluoromethoxy, at the meta- or para-positions furnished the desired products 6a–6g in moderate yields. Notably, the radical-relay coupling of anilines bearing a Br or I atom posed no problems, affording 6h and 6i in 44% and 46% yields, respectively.
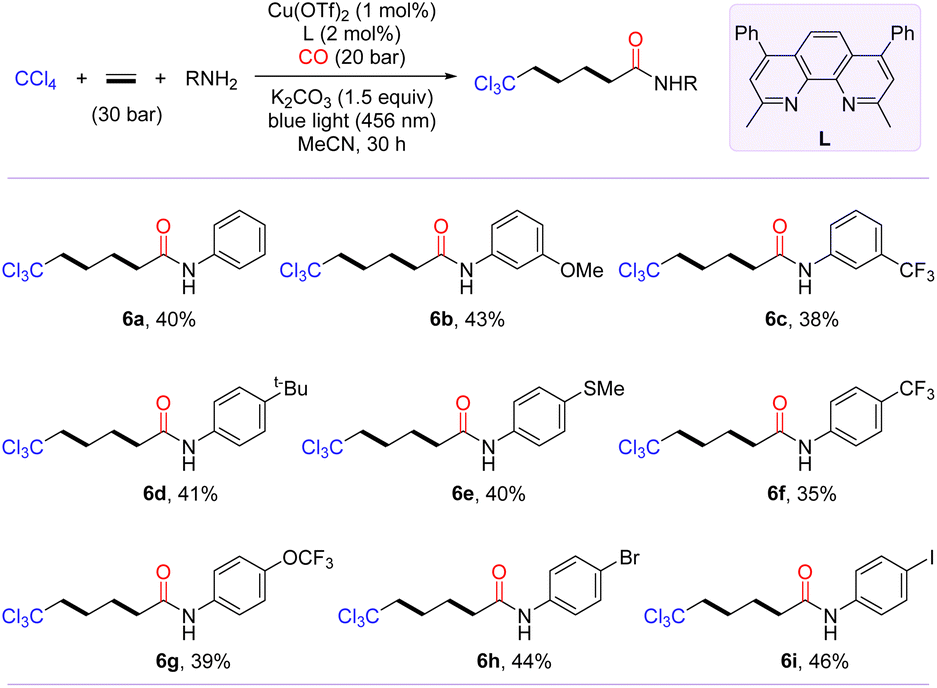 | ||
| Scheme 3 Scope of bis-vinylative carbonylation. [a] Reaction conditions: CCl4 (2.0 mmol, 5.0 equiv.), ethylene (30 bar), NuH (0.4 mmol, 1.0 equiv.), Cu(OTf)2 (1 mol%), bpy (2 mol%), and K2CO3 (0.6 mmol, 1.5 equiv.) in MeCN (2.0 mL) at 25–30 °C for 30 h under CO (20 bar); isolated yields. | ||
Based on the results of the above experiments and previous reports, a plausible radical reaction pathway is proposed, as shown in Scheme 4. The catalytic cycle initiates with the in situ generation of active CuILn species, which reduces CCl4via single electron transfer under light irradiation to form a trichloromethyl radical and CuIILnX species. Then, the trichloromethyl radical is added to ethylene to give the crucial β-trichloromethyl-substituted carbon radical A. Subsequently, radical A can be transformed into acyl copper intermediate D in two routes: (1) carbon radical A traps CO to form acyl intermediate B, which rapidly recombines with the CuIILnX species to provide intermediate D; (2) carbon radical A is captured by CuIILnX to form Cu(III) alkyl complex C, which undergoes coordination/insertion of carbon monoxide to give intermediate D. Finally, intermediate D interacts with a nucleophilic reagent in the presence of a base to obtain the desired β-trichloromethyl carboxylic acid derivatives and the active CuILn species for the next catalytic cycle. On the other hand, β-trichloromethyl-substituted carbon radical A can be further added to ethylene to form γ-trichloromethyl-substituted carbon radical E, which then undergoes the same pathway as β-trichloromethyl-substituted carbon radical A to eventually give γ-trichloromethyl amides. Products based on the oligomerization and polymerization of ethylene can also be detected.
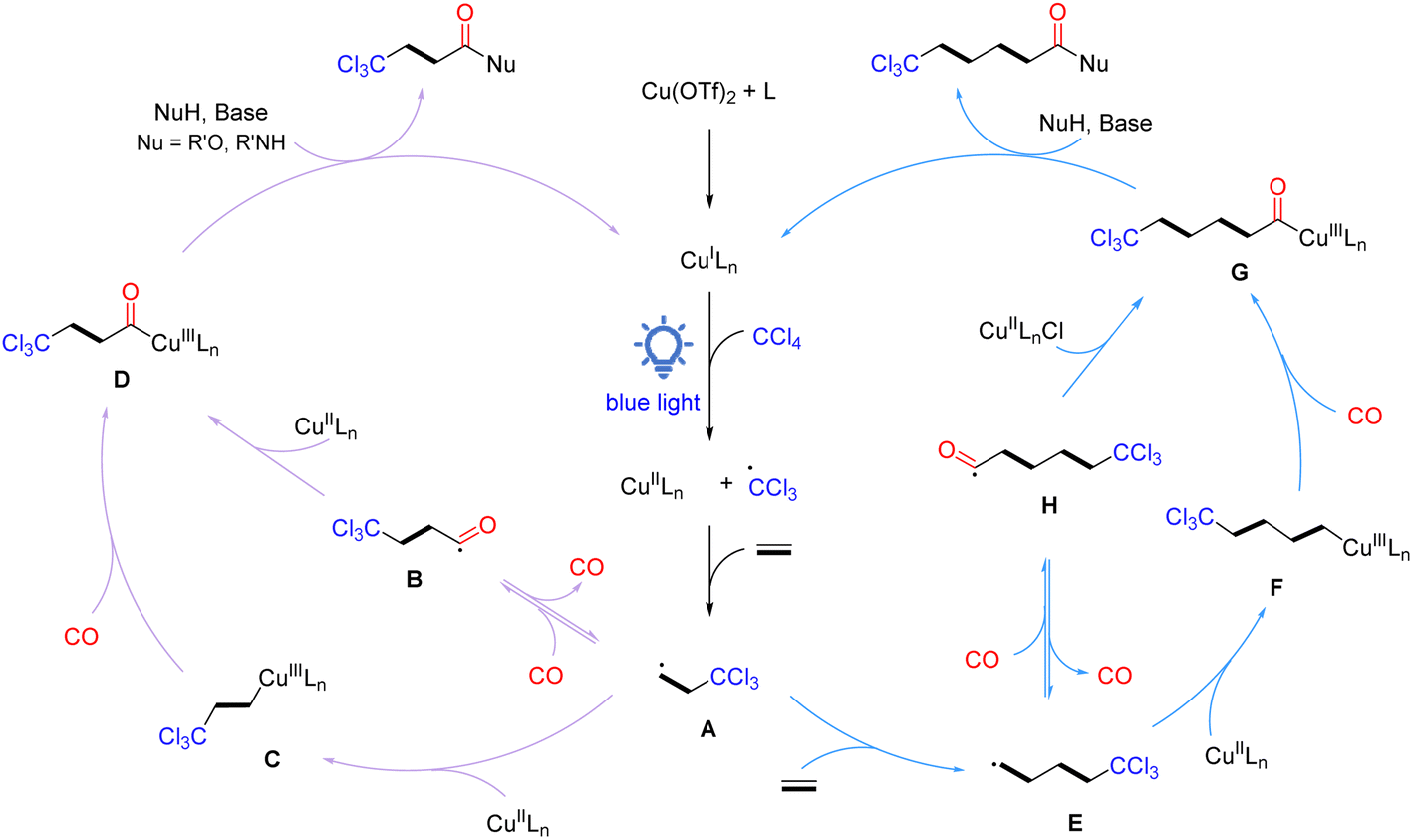 | ||
| Scheme 4 Proposed reaction mechanism. | ||
Conclusions
In summary, we have developed a copper-catalyzed trichloromethylative carbonylation of ethylene under visible light irradiation by employing inexpensive and readily available CCl4 and CO as C1 sources for trichloromethyl and carbonyl, respectively. With this photocatalytic system, a series of functionalized nucleophiles including amines, alcohols, and phenols can be efficiently converted into the corresponding β-trichloromethyl carboxylic acid derivatives. Significantly, this process could be applied for the late-stage functionalization of several complex molecules, in addition to allowing the selectively controlled generation of bis-vinylated γ-trichloromethyl amides.Data availability
The data underlying this study are available in the published article and its ESI.†Author contributions
X. F. W. and Y. Z. conceived and designed the experiments. Y. Z. and B. H. T. performed the experiments. Y. Z. and X. F. W. wrote the manuscript. X. F. W. directed the research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the China Postdoctoral Science Foundation (2022M713087), DICP, and National Key R&D Program of China (2023YFA1507500).References
- F. H. Vaillancourt, E. Yeh, D. A. Vosburg, S. Garneau-Tsodikova and C. T. Walsh, Chem. Rev., 2006, 106, 3364–3378 CrossRef CAS PubMed
.
- C. Wagner, M. E. Omari and G. M. König, J. Nat. Prod., 2009, 72, 540–553 CrossRef CAS PubMed
- P. Jeschke, Pest Manage. Sci., 2010, 66, 10–27 CrossRef CAS PubMed
- J.-Y. Su, Y.-L. Zhong, L.-M. Zheng, S. Wei, Q.-W. Wong, T. C. W. Mak and Z.-Y. Zhou, J. Nat. Prod., 1993, 56, 637–642 CrossRef CAS
- A. C. Durow, G. C. Long, S. J. O'Connell and C. L. Willis, Org. Lett., 2006, 8, 5401–5404 CrossRef CAS PubMed
- R. Kazlauskas, R. O. Lidgard, R. J. Wells and W. Vetter, Tetrahedron Lett., 1977, 18, 3183–3186 CrossRef
- J. B. MacMillan and E. K. Trousdale, Org. Lett., 2000, 2, 2721–2723 CrossRef CAS PubMed
- Z. H. Gu and A. Zakarian, Angew. Chem., Int. Ed., 2010, 49, 9702–9705 CrossRef CAS PubMed
- M. D. Sadar, D. E. Williams, N. R. Mawji, B. O. Patrick, T. Wikanta, E. Chasanah, H. E. Irianto, R. V. Soest and R. J. Andersen, Org. Lett., 2008, 10, 4947–4950 CrossRef CAS PubMed
- M. M. Kapojos, D. B. Abdjul, H. Yamazaki, T. Ohshiro, H. Rotinsulu, D. S. Wewengkang, D. A. Sumilat, H. Tomoda, M. Namikoshi and R. Uchida, Bioorg. Med. Chem. Lett., 2018, 28, 1911–1914 CrossRef CAS PubMed
- H. Shimakoshi, Z. Luo, T. Inaba and Y. Hisaeda, Dalton Trans., 2016, 45, 10173–10180 RSC
- S. Kazuo, K. Hiroshi and Y. Toshiaki, Bull. Chem. Soc. Jpn., 1966, 39, 480–484 CrossRef
- R. M. Joyce, W. E. Hanford and J. Harmon, J. Am. Chem. Soc., 1948, 70, 2529–2532 CrossRef CAS
- V. K. Aggarwal and A. Mereu, J. Org. Chem., 2000, 65, 7211–7212 CrossRef CAS PubMed
- Y. Li, T. Zheng, W. Wang, W. Xu, Y. Ma, S. Zhang, H. Wang and Z. Sun, Adv. Synth. Catal., 2012, 354, 308–312 CrossRef CAS
- M. K. Gupta, Z. Li and T. S. Snowden, Org. Lett., 2014, 16, 1602–1605 CrossRef CAS PubMed
- M. Fujita and T. Hiyama, J. Am. Chem. Soc., 1985, 107, 4085–4087 CrossRef CAS
- N. Wu, B. Wahl, S. Woodward and W. Lewis, Chem. – Eur. J., 2014, 20, 7718–7724 CrossRef CAS PubMed
- J. Kister and C. Mioskowski, J. Org. Chem., 2007, 72, 3925–3928 CrossRef CAS PubMed
- M. S. Kharasch, E. V. Jensen and W. H. Urry, Science, 1945, 102, 128 CrossRef CAS PubMed
- J. Tsuji, K. Sato and H. Nagashima, Tetrahedron, 1985, 41, 393–397 CrossRef CAS
- F. Huang, W. Luo and J. Zhou, Chinese J. Org. Chem., 2023, 43, 2368–2390 CrossRef
- R. K. Nef, Y.-L. Su, S. Liu, M. Rosado, X. Zhang and M. P. Doyle, J. Am. Chem. Soc., 2019, 141, 16643–16650 CrossRef PubMed
- Y.-L. Su, L. Tram, D. Wherritt, H. Arman, W. P. Griffith and M. P. Doyle, ACS Catal., 2020, 10, 13682–13687 CrossRef CAS
- Y.-Y. Liang, J. Huang, X.-H. Ouyang, J.-H. Qin, R.-J. Song and J.-H. Li, Chem. Commun., 2021, 57, 3684–3687 RSC
- P. Sivaguru, Y. Ning and X. Bi, Chem. Rev., 2021, 121, 4253–4307 CrossRef CAS PubMed
- A. Whyte, A. Torelli, B. Mirabi, A. Zhang and M. Lautens, ACS Catal., 2020, 10, 11578–11622 CrossRef CAS
- G. A. Coppola, S. Pillitteri, E. V. Van der Eycken, S.-L. You and U. K. Sharma, Chem. Soc. Rev., 2022, 51, 2313–2382 RSC
- J. Sun, L. Wang, G. Zheng and Q. Zhang, Org. Chem. Front., 2023, 10, 4488–4515 RSC
- Q. Wang, M. Wang, Q. Wu, M. Ma and B. Zhao, Org. Lett., 2022, 24, 4772–4777 CrossRef CAS PubMed
- M. Kusakabe, K. Nagao and H. Ohmiya, Org. Lett., 2021, 23, 7242–7247 CrossRef CAS PubMed
- B. Liu, F. Hu and B.-F. Shi, ACS Catal., 2015, 5, 1863–1881 CrossRef CAS
-
J. Otera and J. Nishikido, Esterification: Methods, Reactions, and Applications, Wiley-VCH, Weinheim, 2010 Search PubMed
- H. Lundberg, F. Tinnis, N. Selander and H. Adolfsson, Chem. Soc. Rev., 2014, 43, 2714–2742 RSC
- J. R. Dunetz, J. Magano and G. A. Weisenburger, Org. Process Res. Dev., 2016, 20, 140–177 CrossRef CAS
- J. Tsuji, K. Sato and H. Nagashima, Tetrahedron, 1985, 41, 5003–5006 CrossRef CAS
-
X.-F. Wu, B. Han, K. Ding and Z. Liu, The Chemical Transformations of C1 Compounds, John Wiley & Sons, 2022 Search PubMed
- J.-B. Peng, F.-P. Wu and X.-F. Wu, Chem. Rev., 2019, 119, 2090–2127 CrossRef CAS PubMed
- J.-X. Xu, Y. Yuan and X.-F. Wu, Coord. Chem. Rev., 2023, 477, 214947 CrossRef CAS
-
B. Gabriele, Carbon Monoxide in Organic Synthesis: Carbonylation Chemistry, Wiley-VCH, Verlag, 2021 Search PubMed
- B. Lu, Z. Zhang, M. Jiang, D. Liang, Z.-W. He, F.-S. Bao, W.-J. Xiao and J.-R. Chen, Angew. Chem., Int. Ed., 2023, 62, e202309460 CrossRef CAS PubMed
- J. Zhu, T. Xu, P. Lu, W. Chen and W. Lu, Mol. Catal., 2022, 532, 112736 CrossRef CAS
- S. Sumino, A. Fusano, T. Fukuyama and I. Ryu, Acc. Chem. Res., 2014, 47, 1563–1574 CrossRef CAS PubMed
- K. P. S. Cheung, S. Sarkar and V. Gevorgyan, Chem. Rev., 2022, 122, 1543–1625 CrossRef PubMed
- C. Wu, X. Hui, D. Zhang, M. Zhang, Y. Zhu and S. Wang, Green Chem., 2022, 24, 1103–1108 RSC
- X. Wu, C. Xiang and J.-T. Yu, Eur. J. Org Chem., 2023, 26, e202300441 CrossRef CAS
- S. Arora, T. Singh, U. Mondal and A. Singh, Eur. J. Org Chem., 2023, 26, e202300469 CrossRef CAS
- Y. Zhang, H.-Q. Geng and X.-F. Wu, Angew. Chem., Int. Ed., 2021, 60, 24292–24298 CrossRef CAS PubMed
- Y. Zhang and X.-F. Wu, Chem. Commun., 2020, 56, 14605–14608 RSC
- Y. Zhang, Y. Yuan, H.-Q. Geng, J.-X. Xu and X.-F. Wu, J. Catal., 2022, 413, 214–220 CrossRef CAS
- K.-O. Keiko, H. Kazuma, Y. Shinya and K. Yoshihiro, Bull. Chem. Soc. Jpn., 2010, 83, 276–278 CrossRef
- H. Michaels, M. Rinderle, R. Freitag, I. Benesperi, T. Edvinsson, R. Socher, A. Gagliardi and M. Freitag, Chem. Sci., 2020, 11, 2895–2906 RSC
- Y. Saygili, M. Söderberg, N. Pellet, F. Giordano, Y. Cao, A. B. Muñoz-Garcia, S. M. Zakeeruddin, N. Vlachopoulos, M. Pavone, G. Boschloo, L. Kavan, J.-E. Moser, M. Grätzel, A. Hagfeldt and M. Freitag, J. Am. Chem. Soc., 2016, 138, 15087–15096 CrossRef CAS PubMed
- T. O. Morgen, M. Baur, I. Göttker-Schnetmann and S. Mecking, Nat. Commun., 2020, 11, 3693 CrossRef CAS PubMed
- S.-Y. Chen, B.-H. Ren, S.-H. Li, Y.-H. Song, S. Jiao, C. Zou, C. Chen, X.-B. Lu and Y. Liu, Angew. Chem., Int. Ed., 2022, 61, e202204126 CrossRef CAS PubMed
- L. Zhu, S. Gaire, C. J. Ziegler and L. Jia, ChemCatChem, 2022, 14, e202200974 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: General comments, general procedure, optimization details, analytical data and NMR spectra. See DOI: https://doi.org/10.1039/d3sc05530b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |