
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputational design of an imine reductase: mechanism-guided stereoselectivity reversion and interface stabilization†
Kai
Wu
a,
Jinrong
Yan
bd,
Qinde
Liu
ac,
Xiaojing
Wang
a,
Piaoru
Wu
a,
Yiyang
Cao
a,
Xiuhong
Lu
 a,
Yixin
Xu
a,
Junhai
Huang
*bd and
Lei
Shao
*ad
a,
Yixin
Xu
a,
Junhai
Huang
*bd and
Lei
Shao
*ad
aSchool of Pharmacy, Shanghai University of Medicine & Health Sciences, 279 Zhouzhu Highway, Pudong New Area, Shanghai 201318, China. E-mail: shaolei00@gmail.com
bCollege of Chemistry and Chemical Engineering, Shanghai University of Engineering Science, 333 Longteng Road, Shanghai 201620, China. E-mail: huang_junhai@163.com
cShanghai University of Traditional Chinese Medicine, 1200 Cailun Road, Shanghai 201203, China
dState Key Laboratory of New Drug and Pharmaceutical Process, Shanghai Institute of Pharmaceutical Industry, 285 Gebaini Rd., Shanghai 200040, China
First published on 15th December 2023
Abstract
Imine reductases (IREDs) are important biocatalysts in the asymmetric synthesis of chiral amines. However, a detailed understanding of the stereocontrol mechanism of IRED remains incomplete, making the design of IRED for producing the desired amine enantiomers challenging. In this study, we investigated the stereoselective catalytic mechanism and designed an (R)-stereoselective IRED from Paenibacillus mucilaginosus (PmIR) using pharmaceutically relevant 2-aryl-substituted pyrrolines as substrates. A putative mechanism for controlling stereoselectivity was proposed based on the crucial role of electrostatic interactions in controlling iminium cation orientation and employed to achieve complete inversion of stereoselectivity in PmIR using computational design. The variant PmIR-Re (Q138M/P140M/Y187E/Q190A/D250M/R251N) exhibited opposite (S)-stereoselectivity, with >96% enantiomeric excess (ee) towards tested 2-aryl-substituted pyrrolines. Computational tools were employed to identify stabilizing mutations at the interface between the two subunits. The variant PmIR-6P (P140A/Q190S/R251N/Q217E/A257R/T277M) showed a nearly 5-fold increase in activity and a 12 °C increase in melting temperature. The PmIR-6P successfully produced (R)-2-(2,5-difluorophenyl)-pyrrolidine, a key chiral pharmaceutical intermediate, at a concentration of 400 mM with an ee exceeding 99%. This study provides insight into the stereocontrol elements of IREDs and demonstrates the potential of computational design for tailored stereoselectivity and thermal stability.
Introduction
Imine reductase (IRED) plays an important role as a biocatalyst in the asymmetric synthesis of optically pure amines, and it exhibits the ability to catalyze various transformations, including cyclic imine asymmetric reduction, direct asymmetric reductive amination, and stepwise conjugate reduction followed by reductive amination.1–3 Despite the diverse reactions catalyzed by IREDs, they all involve a pivotal step: the asymmetric reduction of the iminium cation intermediate.4,5 This step is crucial for determining the stereoconfiguration of the final amine product.The understanding of the specific roles of varying active-site residues in stereocontrol remains limited. To gain insight into the correlation between active site residues and stereoselectivity, multiple studies have compared IREDs exhibiting stereocomplementary activity towards 2-methyl-1-pyrroline and other cyclic imines.6–8 These investigations have led to the categorization of IREDs into two groups based on their opposite stereoselectivities and the putative catalytic residues involved. Y-type IREDs possess tyrosine as a potential proton-donating amino acid, whereas D-type IREDs feature aspartate at equivalent position. Although it seemed that the different putative catalytic residues, tyrosine and aspartate, led to opposite stereoselectivity of the IREDs, there remain exceptions. In 2015, Wetzl and coworkers reported IREDs that possessed phenylalanine or asparagine residues at equivalent positions.9 In 2016, Turner et al. reported an intriguing finding with AoIRED, which demonstrated higher activity and stereoselectivity in asymmetric imine reduction despite the absence of tyrosine or aspartate residues.10 These discoveries suggest that potentially different stereocontrol mechanisms are formed by distinct active sites in IREDs. The varying active-site residues and unidentified key stereocontrol elements make the rational design of an IRED for the desired stereoselectivity challenging. Additionally, stability has been emphasized as a crucial factor in the practical application of evolved IREDs.2,11,12 The conventional approach for mass screening in directed evolution is time-consuming. However, a rational design approach for improving stereoselectivity and stability remains underdeveloped.
In this study, we investigated the possible stereocontrol mechanism of IRED in Paenibacillus mucilaginosus (PmIR). Furthermore, by employing rational design with catalytic constraints based on a putative stereocontrol mechanism, we successfully reversed the stereoselectivity of PmIR towards 2-(2,5-difluorophenyl)pyrroline (2-DFPL) and other 2-aryl-substituted pyrrolines in the PmIR-Re variant. PmIR-Re represents the first instance of overcoming the boundary between Y-type- and D-type IREDs, provides valuable insights into the key stereocontrol elements of IREDs, and demonstrates an effective mechanism-guided approach for manipulating stereoselectivity. Furthermore, a combinational method including salt-bridge construction and ΔΔGfold prediction was applied to enable the rapid identification of interface mutations for stability enhancement. Finally, the engineered variant PmIR-6P was used to successfully produce (R)-2-(2,5-difluorophenyl)-pyrrolidine (2-DFPD), a key chiral intermediate of larotrectinib, demonstrating its potential for practical applications.
Results and discussion
A putative stereoselective control mechanism in PmIR
Although the imine reduction step is crucial for determining the stereoconfiguration of the product in IRED-catalyzed reactions, the role of active-site residues in stereoselective catalysis remains elusive. Several studies have proposed that protic residues, including conserved aspartate and tyrosine located near the NADPH cofactor, serve as proton donors and facilitate the transfer of protons necessary for the reduction of unsaturated imine bonds.5,8 Furthermore, most IREDs possess either aspartate or tyrosine residues at equivalent positions in the active site, which frequently display opposite stereoselectivity.6–8 This suggests that these residues not only serve as proton donors but also play a role in controlling stereoselectivity. However, it should be noted that there are many exceptions to this hypothesis. For instance, AoIRED exhibited high activity and stereoselectivity towards 2-methyl-1-pyrroline despite having an N171 residue in a position equivalent to aspartate or tyrosine, suggesting that a protic residue at this position is not essential for catalysis.9 Conservation analysis of the IRED database (https://ired.biocatnet.de/) also revealed that neither aspartate nor tyrosine residues are completely conserved among IREDs.13,14 This indicates that a proton donor may not be necessary for catalysis, and that the residues may play different roles. From the perspective of the imine substrate, when the buffer pH is lower than the pKa, the nitrogen atom of the imine is protonated to form an iminium cation in the solution. Therefore, we speculated that the active-site residues in certain IREDs could merely play a role in binding the protonated iminium cation in a reactive conformation, positioning the specific C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N face at an appropriate distance from NADPH. Subsequently, the chirality of the product was determined by transferring a hydride ion from NADPH to a prochiral carbon atom on the CN face (Scheme 1).
N face at an appropriate distance from NADPH. Subsequently, the chirality of the product was determined by transferring a hydride ion from NADPH to a prochiral carbon atom on the CN face (Scheme 1).
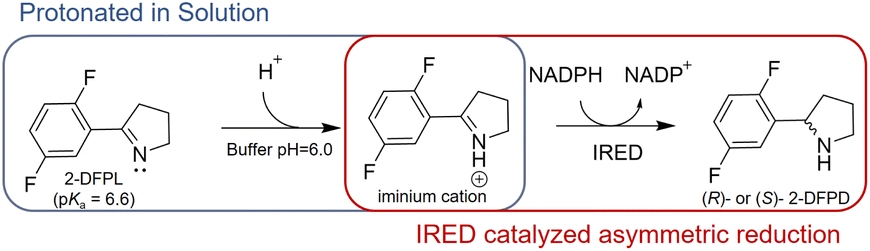 | ||
| Scheme 1 Asymmetric Reduction of Protonated 2-DFPL by IRED. | ||
After screening IREDs stored in our laboratory, PmIR from Paenibacillus mucilaginosus emerged as one of the few enzymes capable of catalyzing the reduction of 2-DFPL to yield (R)-2-DFPD, which displayed the highest ee, exceeding 96%. Owing to the conserved binding position of NADPH in IREDs,5,7,13 the PmIR-NADPH complex model was constructed by superimposing the PmIR crystal structure (PDB ID: 8KFK) on the NADPH-bound BcSIRED crystal structure (sequence identity 75.8%). To investigate the key residues of PmIR controlling the asymmetric reduction, 18 residues around Y187 within 10 Å, located at the subunit interface forming the binding pocket, were targeted for alanine scanning (Fig. S1 and Table S2†). Next, key residues with potential catalytic roles that dramatically affected the activity or stereoselectivity were subjected to directed mutagenesis to further investigate their role in asymmetric reduction (Table 1). Alanine scanning results indicated that the M194A, F194A, F232A, and F234A mutations led to negligible activity. Both D250A and W195A mutations resulted in a complete loss of activity. The alanine mutation of the potential proton donor Y187, which is often considered catalytic and conserved in IREDs, displayed a low yet detectable conversion with an almost racemic ee of 4%, suggesting the possible role of a bulky aromatic side chain in activity and stereoselectivity. To further elucidate the contribution of the phenolic hydroxyl group of Y187 to imine reduction, the Y187F mutant was constructed. The Y187F mutant exhibited a conversion of 34%, slightly lower than that observed for the WT (46.5%), but stereoselectivity increased from 96% to >99%. Enzymatic kinetic analysis of the Y187F mutant revealed a 3.6-fold increase in KM but only a 24% reduction in kcat compared with those in the WT (Table 2). The predicted pKa of 2-DFPL is 6.60 ± 0.20 (CAS Scifinder), suggesting that at least 75% of the substrate exists in the form of the iminium cation in the reaction pH of 6.0. In summary, the catalytic activity of the Y187F mutant and its decreased substrate affinity suggest that Y187 in PmIR primarily contributes to substrate binding during catalysis, rather than serving as a proton donor, which differs from the previously reported role of tyrosine at equivalent positions in other IREDs. This indicates that Y187 is a modifiable residue that can be used for further enzyme design.
| Variants | Conversiona (%) | ee (%) | Stereoselectivity |
|---|---|---|---|
| a The reaction mixture (500 μL) included 275 μL crude enzyme extract from 50 mg mL−1 resting cells, 50 μL of 2.5 mM NADP stock, 50 mM glucose, 10 mM substrate, 100 μL crude enzyme of recombinant glucose dehydrogenase, and potassium phosphate buffer (100 mM, pH 6.0). The reaction mixture was stirred at 30 °C for 40 min and analyzed using chiral HPLC. b Not detected. | |||
| Wild type | 46.5 ± 3.6 | 96 | R |
| Y187A | 4.3 ± 1.2 | 4 | R |
| Y187F | 34.8 ± 0.9 | >99 | R |
| Y187D | 2.3 ± 0.9 | 58 | R |
| Y187E | 4.5 ± 0.8 | 33 | S |
| D250A | n.d.b | n.d. | n.d. |
| D250E | 38.1 ± 4.4 | 97 | R |
| Variants | K M (mM) | V max (μmol min−1 mg−1) | k cat (s−1) | k cat/KM (s−1 mM−1) |
|---|---|---|---|---|
| WT | 0.84 | 0.78 | 0.44 | 0.52 |
| Y187F | 3.09 | 0.67 | 0.38 | 0.12 |
| D250E | 0.68 | 0.72 | 0.41 | 0.60 |
Aspartate is another residue often considered catalytically active in IREDs. A previous report of a fungal IRED catalyzing imine reduction and reductive amination indicated the important role of the Asp active site in binding an imine cation and amine substrate.4 A systematic evaluation of imine-reducing enzymes by Pleiss et al. suggested that most IREDs require a negative electrostatic potential for substrate-binding.8 Residue D250 of PmIR was positioned above Y187, located in the binding pocket, and close to NADPH. A complete loss of activity was observed in the D250A mutant with no detectable products. Saturation mutagenesis was performed to investigate the role of D250. Most mutations resulted in complete inactivation, except for D250E, which featured a carboxyl group with one additional methyl moiety compared with aspartate. The D250E mutant exhibited a comparable ee value, kcat, and slightly reduced KM values (Table 2). The optimal reaction pH for PmIR was determined to be 6.0, corresponding to the deprotonated state of the acidic residues. Therefore, the complete inactivation of the alanine mutation suggests that the loss of activity is primarily due to the absence of a negatively charged deprotonated carboxyl group. Furthermore, using the previously reported (R)-selective BcSIRED17 catalyzing 2-DFPL with 89% conversion and 88% ee, negligible conversion (1.7%) and relatively high conversion (19%) were observed with BcSIRED mutants D251A and Y188F, respectively. These results were consistent with the findings observed for PmIR. Multiple sequence alignments of eight IREDs displaying (R)-stereoselectivity towards 2-aryl-substituted pyrrolines also revealed that D250 is conserved in certain IRED subgroups (Fig. 1A). Electrostatic interactions between the negatively charged D250 and iminium cation during catalysis cannot be excluded in PmIR.
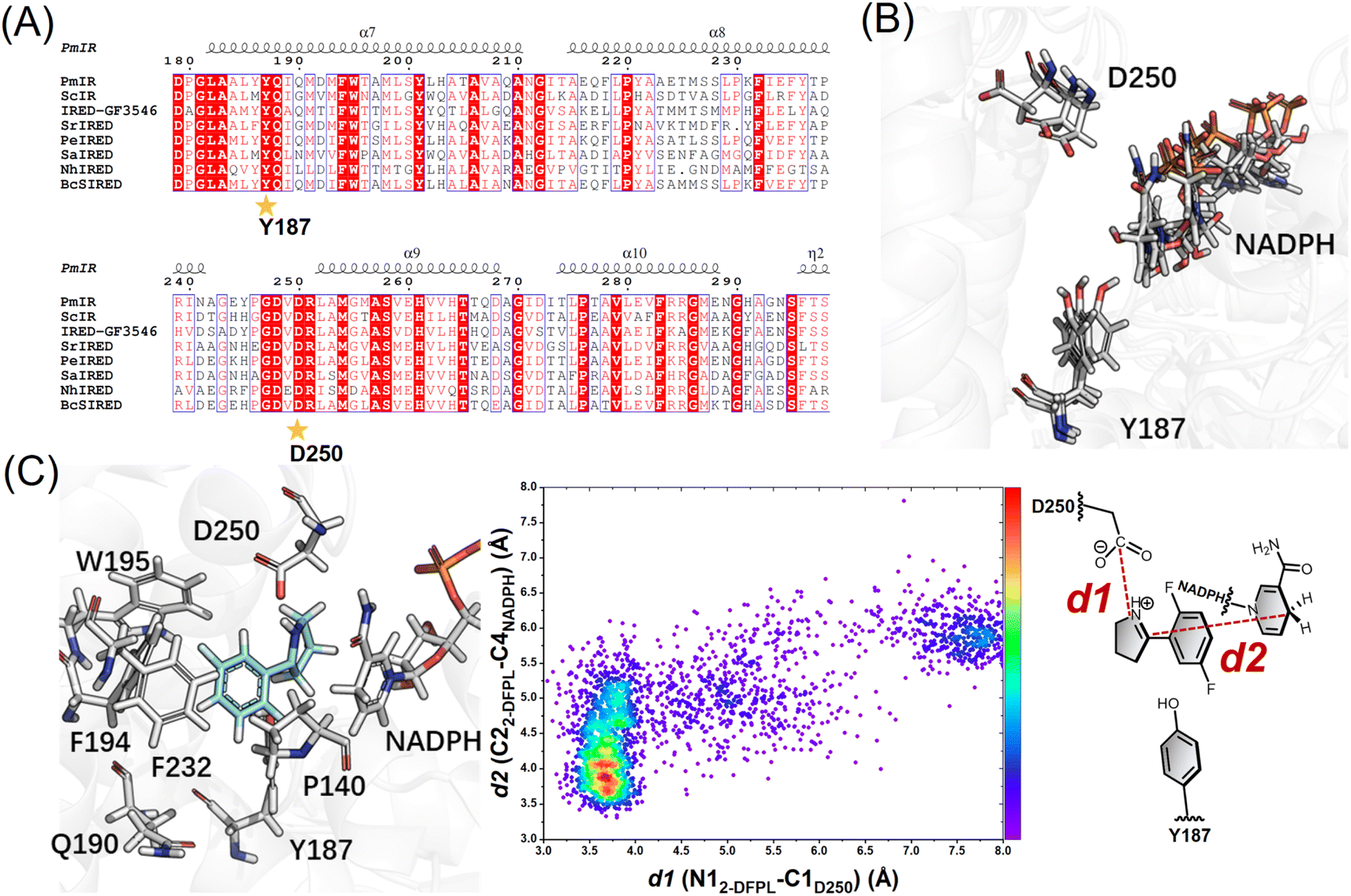 | ||
| Fig. 1 (A) Multisequence alignment of reported IREDs with (R)-stereoselectivity towards 2-aryl-substituted pyrrolines. The IREDs used for sequence alignment include PmIR (this study), ScIR (WP_003961038.1),15 IRED-GF3546 (BAM99301),16BcsIRED (WP_000841637),17SrIRED (WP_012890722),7PeIRED (WP_010497949.1),18SaIRED (WP_016644884.1),16 and NhIRED (WP_017538739.1).17 The figure was prepared using ESPript19 (numbering as PmIR). Y187 and D250 were conserved in the (R)-selective enzyme. (B) Representative snapshots showing the reorientation of the D250 side chain. (C) Docking conformation in pro-R pose and following conformation analysis with molecular dynamics (MD) simulations. The distance distributions indicate the high proportion of reactive conformation in the pro-R pose. | ||
Several studies speculated that conformational changes and domain motion of IREDs can affect stereoselectivity.9,10,17 The crystal structure alone may have limitations in that it cannot reflect important motions. Although the crystal structure indicated that the carboxyl group of D250 was not ideally positioned to interact with the iminium cation, MD simulations revealed that D250 was in a flexible binding loop (residues 242–251) (Fig. S3†), and conformational reorientation of the D250 side chain was observed during 100 ns simulations of substrate-free PmIR (Fig. 1B). This reorientation suggests that the carboxyl group could dynamically adjusts to accommodate the positive iminium cation, stabilizing it in a reactive conformation and promoting hydride transfer. Hence, D250 was selected as a flexible residue for molecular docking. The docking results indicate preferential binding of the substrate in the pro-R pose with electrostatic interaction to the D250 side chain (Fig. 1C), as a lower interaction energy of pro-R pose than pro-S pose (−31.9 vs. −27.2 kcal mol−1) was observed. The Y187, F232, F194, and W195 form a hydrophobic region that stabilizes 2-DFPL aromatic ring binding, which could explain why alanine mutations at these sites resulted in inactive or low activities. These interactions could position the specific CN face at an appropriate distance from NADPH.
Based on the importance of electrostatic interactions and hydride transfer during reduction, the distances between the imine nitrogen atom and the D250 carboxyl carbon atom (d1), and the distance between the imine carbon and NADPH-C4 (d2) are critical for catalysis. Electrostatic interactions can be strong, even at relatively long distance.20,21 The ternary complex of AoIRED has been previously reported, which revealed a binding site for the amine product that placed the chiral carbon within 4.0 Å of the C4 atom of NADPH.9 In addition, studies of enzymes such as short-chain dehydrogenase, which catalyzes bio-reduction using NADPH as a cofactor, reported that hydride transfer could happen when the distance between the NADPH-C4 atom and electrophile carbon is ≤ 4.5 Å,22–24 and a few of them show moderate imine-reducing activity.8 Therefore, conformations of d1 less than 5.0 Å and d2 less than 4.0 Å were considered reactive. Based on these two distance criteria, MD simulations of the docking complex demonstrated the high occurrence of a reactive conformation in the pro-R pose of the protonated 2-DFPL (Fig. 1C). These findings suggest that negatively charged acidic residues play a role in promoting the formation of reactive pro-R positions during catalysis. Therefore, based on the results of site-directed mutagenesis and MD simulations, we inferred that D250 is a crucial residue for stereocontrol in PmIR.
Based on the above findings, we propose a putative mechanism for stereocontrol, in which the electrostatic interaction between the negatively charged side chains of Asp or Glu and the iminium cation governs the orientation of the pyrroline face, thereby determining the stereoconfiguration of the product. To manipulate the stereoselectivity, we hypothesized that the orientation of the 1-pyrroline face could be reversed by simply introducing new electrostatic interactions in the opposite direction, considering that a proton transfer system or any flanked residues that lower the pKa value of the proton donor are unnecessary. Because the residues at positions 250 and 187 are adjacent to each other and close to the nicotinamide moiety of NADPH, we speculated that replacing Y187 with an acidic amino acid could create an electrostatic interaction in the opposite direction, thereby altering the orientation of the 1-pyrroline face. The side chains of aspartate and glutamate in the model compounds, with the titratable residue in the middle and two alanines on each side, are determined experimentally by previous study in water, yielding pKa values of 3.6 and 4.2, respectively.25 The protonation state of these residues was taken into consideration to ensure that most of the side chain was negatively charged in the protein. Using a pKa predicting module in Discovery Studio 4.0,26 the pKa value of Y187E was predicted as 5.4 with a 1.2 shift compared with glutamate in water, and Y187D was predicted as 4.7 with a 1.1 shift compared with aspartate in water, indicating a negative electrostatic potential at pH 6.0. As anticipated, the Y187D mutation resulted in a dramatically reduced ee to 58%, but a negligible conversion. The Y187E mutation successfully resulted in slightly reversed (S)-stereoselectivity with an ee of 33% (Table 1), providing further evidence for the crucial role of electrostatic interactions in stereoselective control. The docking results were in accordance with the proposed stereoselective control mechanism, as a pro-S pose was observed in the Y187E variant complex using the 2-DFPL iminium as the substrate, showing an opposite orientation to the 1-pyrroline face (Fig. 2).
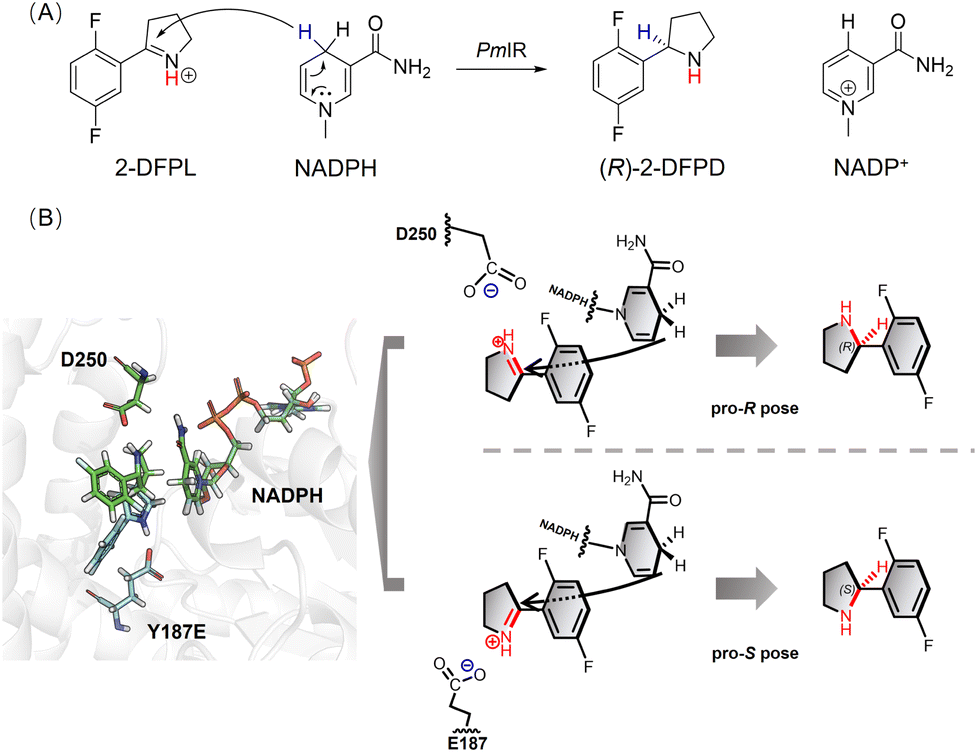 | ||
| Fig. 2 (A) The chirality of the amine produced by PmIR was determined by transferring a hydride ion from NADPH to a prochiral carbon atom on the specific CN face. (B) Superimposition of WT (green) and Y187E (cyan) docking results and a putative mechanism for stereocontrol, in which the electrostatic interaction plays a crucial role in controlling the orientation of the 1-pyrroline face. | ||
Complete reversion of stereoselectivity using computational design
Based on the proposed stereocontrol mechanism, the pro-S pose of 2-DFPL can be generated by inverting the pyrroline face and stabilizing it through electrostatic interactions. To achieve this, a computational design strategy utilizing Rosetta Enzyme Design was employed,27 which is effective in placing the substrate in the enzyme's active site with the desired conformation.28 By applying catalytic geometric constraints, a reactive conformation in the pro-S pose could be achieved and stabilized through the active-site redesign. MD simulations were used to narrow down variants for experimental validation. Variant structures that exhibited a higher occurrence of reactive conformations were selected for experimental validation, thereby reducing the need for laboratory screening (Fig. 3).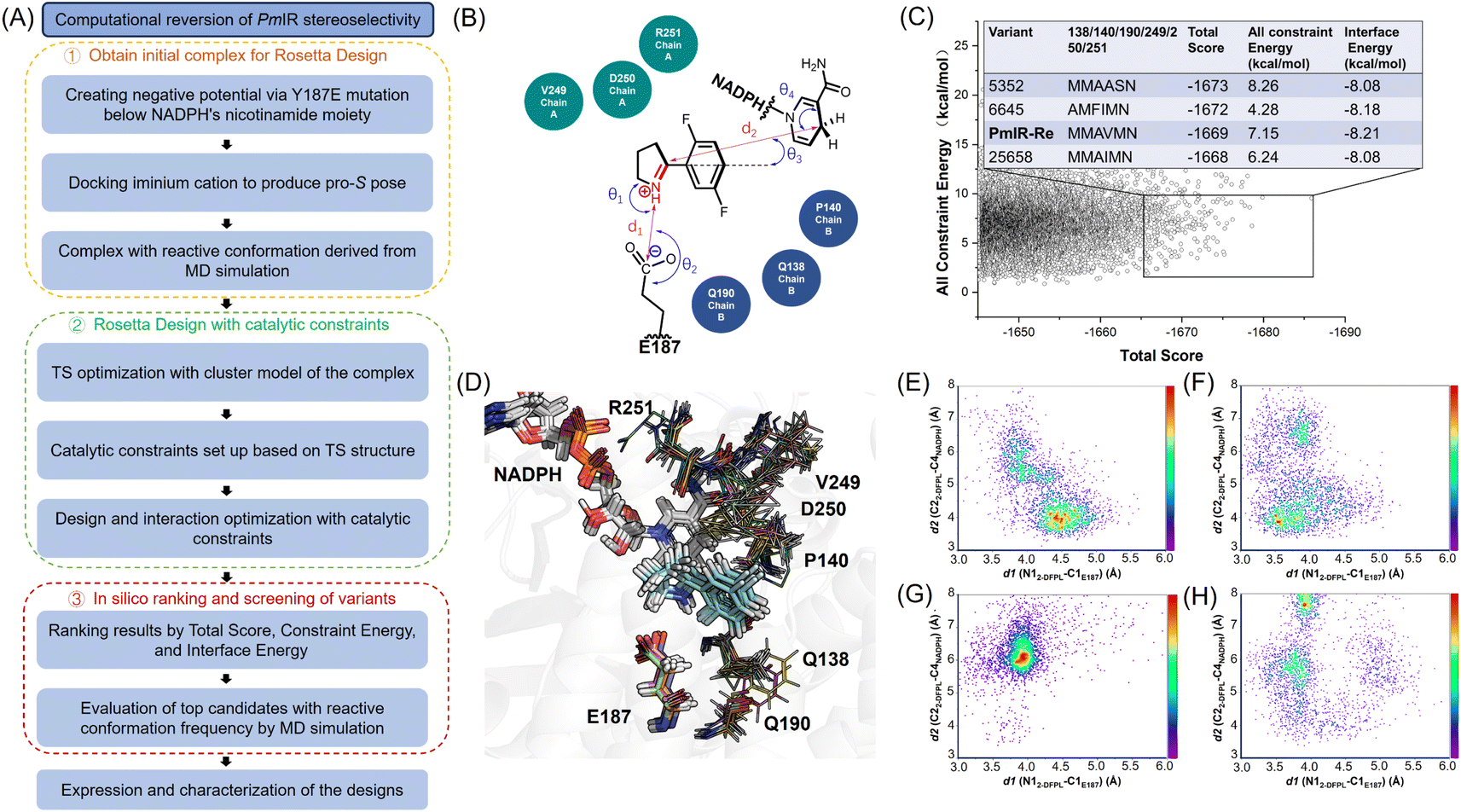 | ||
Fig. 3 Computational reversion of PmIR stereoselectivity. (A) Flowchart illustrating the process of computational stereoselectivity reversion of PmIR (B) the catalytic geometric constraints based on the pro-S pose TS structure were used in design and conformation sampling. (C) In total, 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 variant structures were sorted based on the criteria of a total score <−1645.0 and constraint energy <10.0 kcal mol−1. These structures were further filtered based on an interface energy <−8.0 kcal mol−1, resulting in the selection of the top four non-redundant variant structures. (D) The top variants with different side chain conformations were superimposed to compare their differences. Side-chain conformations of residues generated by virtual mutagenesis are shown in lines. (E) The occurrence of reactive pro-S conformations in the PmIR-Re complex. (F) The occurrence of reactive pro-S conformations in the complex_5352 variant. (G) The occurrence of reactive pro-S conformations in the complex_25658 variant. (H) The occurrence of reactive pro-S conformations in the complex_6645 variant. 000 variant structures were sorted based on the criteria of a total score <−1645.0 and constraint energy <10.0 kcal mol−1. These structures were further filtered based on an interface energy <−8.0 kcal mol−1, resulting in the selection of the top four non-redundant variant structures. (D) The top variants with different side chain conformations were superimposed to compare their differences. Side-chain conformations of residues generated by virtual mutagenesis are shown in lines. (E) The occurrence of reactive pro-S conformations in the PmIR-Re complex. (F) The occurrence of reactive pro-S conformations in the complex_5352 variant. (G) The occurrence of reactive pro-S conformations in the complex_25658 variant. (H) The occurrence of reactive pro-S conformations in the complex_6645 variant. | ||
Encouraged by the putative stereoselective control mechanism and 33% ee observed for the Y187E variant, we used a complex with the pro-S pose in the reactive conformation from MD simulations as the initial structure for the redesign to enhance (S)-stereoselectivity and activity simultaneously. To achieve the distances, angles and dihedrals between Y187E and substrate and NADPH for the catalytic geometric constraints, the cluster model with 2-DFPL in the pro-S pose was optimized to determine the transition state (TS) structure (Fig. S4 and Table S3†). We focused on redesigning the loop residues surrounding 2-DFPL, including V249, D250, and R251 in loops 242–251, and Q138 and P140 in loops 135–140. Additionally, we included Q190 in the design because Q190A displayed a higher conversion than the WT in previous alanine scanning. The hydrophobic region formed by F232, F194, and W195 that stabilizes 2-DFPL aromatic ring binding was not selected for design. A virtual library with 30000 mutant structures was generated using the Rosetta Design to explore the sequence and conformation space and optimize the catalytic interactions. These structures were then sort based on the Rosetta parameters of Total Score, Constraint Energy, and Interface Energy.
The top four non-redundant variants, 5352, 6645, 25658, and PmIR-Re, were further analyzed to determine the occurrence of reactive pro-S conformations using MD simulations. Experimental validations were conducted for 5352 and PmIR-Re, which showed a relatively high probability of adopting the pro-S pose in the reactive conformation. Both variants exhibited significant improvements in stereoselectivity, producing (S)-2-DFPD in 93% ee and >99% ee, respectively. Interestingly, the D250S and D250M mutations, which resulted in a complete loss of activity in the WT, were present in the two active variants, suggesting that E187 replaced D250 to anchor the iminium cation. Importantly, this observation further supports the hypothesis that the inactivation caused by D250S and D250M in the WT can be attributed to the absence of electrostatic interactions rather than destabilization of the protein structure.
The initial complex used for design demonstrated that the lack of tyrosine bulky side chain and electrostatic interaction with Y187E resulted in a lower binding position of the pro-S pose (Fig. S5†). In contrast, the side-chain repacking of Q190A and Q138M in PmIR-Re elevated 2-DFPL binding position, allowing for better interaction with the hydrophobic and aromatic regions formed by F232, F194, and W195. The P140M side chain could also push substrate into the aromatic region and the D250M could form a π–sulphur interaction with 2-DFPL from above. In addition, the PmIR-Re complex displayed a geometric feature that more closely resembled the TS structure than the initial design template, potentially enhancing the hydride transfer process (Table S4†). This observation indicates that the increased S-stereoselectivity and activity achieved by PmIR-Re could be due to the more reactive conformation for hydride transfer while retaining the electrostatic interaction with Y187E. These results further support the idea that the stereoselectivity of PmIR can be directly manipulated by creating electrostatic interactions in opposite directions, thereby reversing the orientation of the 1-pyrroline face.
To the best of our knowledge, only a few studies have investigated the stereoselective control mechanisms of imine reduction reactions. Several studies have compared IREDs that display stereo-complementary activity towards 2-methyl-1-pyrroline and have categorized them into Y-type and D-type IREDs based on their stereoselectivity and putative catalytic residues (tyrosine or aspartate).6,7 In a recent study by Pleiss et al., the stereoselectivity of a D-type IRED from Myxococcus stipitatus towards 2-methyl-1-pyrroline was reversed.29 They obtained a variant with the opposite stereoselectivity and 91% ee through site-saturation mutagenesis. In this case, the stereoselectivity switch was attributed to a difference in the conformation of the nicotinamide moiety of NADPH rather than the substrate-binding conformation. More recently, Himo emphasized the importance of steric clashes in the transition-state energy of the pro-R and pro-S complexes of AoIRED.30 The pro-S conformation, which lacked steric repulsion, resulted in a lower energy state and led to (S)-stereoselectivity towards bulky imine substrates. These results suggest that varying the active sites of IREDs offers multiple possibilities for fine-tuning the relative spatial positions of the imine and nicotinamide groups of the cofactors.
In this study, we crossed the boundary between Y-type and D-type IREDs by redesigning Y-type PmIR into a D/E-type enzyme with opposite stereoselectivity. PmIR-Re represents an engineered IRED with completely reversed stereoselectivity (>99% ee) achieved through mechanism-guided computational design. In contrast to the previously reported residues W210 in AspRedAm31 and W234/F260 in IRED-M5,32 which potentially affect stereoselectivity by steric hindrance of the bulky side chain, the stereoselectivity in PmIR could be controlled by electrostatic interactions between the iminium cation and the acidic amino acids adjacent to the nicotinamide moiety of NADPH. Therefore, the stereoselectivity of IREDs can be tailored through rational reconstruction of electrostatic interactions and reshaping of the binding pocket.
Engineering PmIR with interface stabilization
Only a few engineered IREDs have been reported to meet industrial requirements, with the complete conversion of substrates at concentrations exceeding 50 g L−1.2,12,33–36 Stability is an important factor in determining the overall turnover of imine reduction, making IREDs suitable for industrial processes. IREDs must exhibit high stability and tolerate high substrate concentrations, organic cosolvents, and high reaction temperatures. Furthermore, evidence suggests that proteins that are stable under one type of denaturing condition tend to be stable under other types.37 Therefore, improving the stability of naturally occurring IREDs is crucial for industrial applications. Recently, directed evolution approaches have been employed to engineer ScIR with significantly increased activity and thermal stability towards 2-DFPL.35 This resulted in the development of a practical enzymatic process for the production of (R)-2-DFPD. However, extensive screening is inevitable in directed evolution, which increases the cost and time required. In this study, PmIR was engineered to investigate rational design strategies for the rapid identification of mutations that improve its thermal stability.The PmIR structure is composed of reciprocal homodimers consisting of an N-terminal Rossman fold domain and a C-terminal helical domain connected by a long interdomain helix. The two subunits are held together by interacting helices, and it is noteworthy that the residues from both subunits contribute to the formation of the binding pocket (Fig. S1†). Natural salt bridges were also observed in IREDs on the surface between the two subunits, providing stability to the dimeric structure.38 Based on these observations, we hypothesized that enhanced subunit interactions would result in more stable variants. Herein, we employed a comprehensive approach to obtain a stable variant, PmIR-6P (P140A/Q190S/R251N/Q217E/A257R/T277M), which involved identifying interface mutations through alanine scanning, designing salt bridges between interfaces, and identifying mutations using ΔΔGfold calculations (Table 3).
| Mutation | Origin | Location | T m (°C) | ΔTm (°C) | Relative activitya |
|---|---|---|---|---|---|
| a The activity of PmIR-6P was determined at 35 °C and others were determined at 30 °C. | |||||
| WT | — | — | 52.1 | 0 | 100 ± 0.8 |
| P140A/Q190S/R251N | Alanine scan | Interface | 57.2 | 5.1 | 450.2 ± 2.9 |
| P140A/Q190S/R251N/A257R | Salt bridge | Interface | 59.0 | 1.8 | 533.0 ± 2.5 |
| P140A/Q190S/R251N/Q217E | Salt bridge | Interface | 58.7 | 1.5 | 418.5 ± 1.8 |
| P140A/Q190S/R251N/G143M | ΔΔGfold prediction | Surface | 58.5 | 1.3 | 283.6 ± 2.1 |
| P140A/Q190S/R251N/T277M | ΔΔGfold prediction | Surface | 59.3 | 2.1 | 390.3 ± 3.8 |
| PmIR-6P (P140A/Q190S/R251N/Q217E/A257R/T277M) | Combination of mutations | 64.1 | 12.0 | 453.9 ± 4.6 | |
The previous alanine scanning revealed that the interface residues P140, Q190, and R251 demonstrated higher conversions than the WT (Table S2†). Saturation mutagenesis was conducted to identify the most beneficial mutations at each site. Additive effects of multiple mutations were also investigated (Table S5†). As a result, variant P140A/Q190S/R251N showed a 4.5-fold increase in catalytic efficiency with ee exceeding 99% compared to the WT. The purified variant also exhibited a Tm of 57.2 °C, which was 5.1 °C higher than that of the WT. These findings are consistent with previous reports on evolved IREDs, where mutations in the binding pocket not only increased activity, but also improved stability.29,31 These dual effects may be attributed to enhanced subunit interactions, resulting in a more compact active pocket that stabilizes substrate binding.
Based on the geometric criteria for salt bridges (Fig. S5†) and the crystal structure of PmIR, potential residue pairs that met the requirements for salt bridge formation were identified. Residues with B-factors above the average value were selected as candidates for designing salt bridges. A257 and Q217 were mutated to A257R and Q217E, respectively, which introduced a salt bridge between R257 and D87, and between E217 and R176. The two new salt bridges increased Tm by 1.8 °C and 1.5 °C, respectively.
In silico saturation mutagenesis was performed for each residue of PmIR, and the ΔΔGfold values were calculated using FoldX and Rosetta_ddg to generate point mutation structures. To narrow down the large number of variants for experimental validation, we performed MD simulations and visual inspection on variants below cut-off. The variants exhibited reduced ΔΔGfold values, lower local flexibility than the original residues (ΔZ-score <0), and no obvious structural undesirable mutations were selected for experimental validation (Table S6†). Among the 12 single mutations tested, four variants (G143M, G143W, T277M, and T277L) showed higher residual activity than the template after incubation at 60 °C. G143M and T277M, which had higher residual activity, were further purified to determine their Tm values (Table 3). This in silico workflow allowed for the rapid identification of mutations with a higher likelihood of enzyme stabilization and has been successful in several biocatalysts.39–41 However, there is a risk that certain mutations may increase stability at the expense of reduced activity. Although the G143M mutant exhibited a higher Tm, it also showed a significant reduction in activity. Therefore, it was not included in the final variant. Finally, mutations identified from alanine scanning, salt bridge design, and ΔΔGfold prediction were combined, resulting in the highly stable variant PmIR-6P, which exhibited a 12 °C increase in Tm, from 52.1 to 64.1 °C (Fig. S9†).
The highest specific activity of PmIR-6P was observed at an optimal reaction temperature of 35 °C (Fig. S10A†). At this temperature, the kinetic parameters KM and kcat were determined to be 0.92 mM and 2.01 s−1, respectively, showing a 4.7-fold increase in catalytic efficiency compared with the WT. The half-lives of both the WT and PmIR-6P variants were measured to assess their kinetic stability. At 35 °C, PmIR-6P exhibited an 8.4-fold improvement in half-life compared to the WT, increasing from 47.5 h to 396.0 h. Furthermore, PmIR-6P exhibited a half-life of 280.6 h at 40 °C (Fig. S10B†).
MD simulations revealed a significant difference in the radius of gyration and root mean square fluctuation (RMSF) between the WT and PmIR-6P at 308 K (35 °C). PmIR-6P exhibited a more compact structure, which was attributed to mutations at the subunit interface (Fig. 4A). The RMSF of WT and PmIR-6P was calculated and compared (Fig. S11†). It indicated that the most region of PmIR-6P with a less flexibility during simulation compared with WT. Interestingly, the 242–251 binding loop of PmIR -6P was observed with a higher mobility than WT, which suggested the loop motion might affect activity for PmIR.
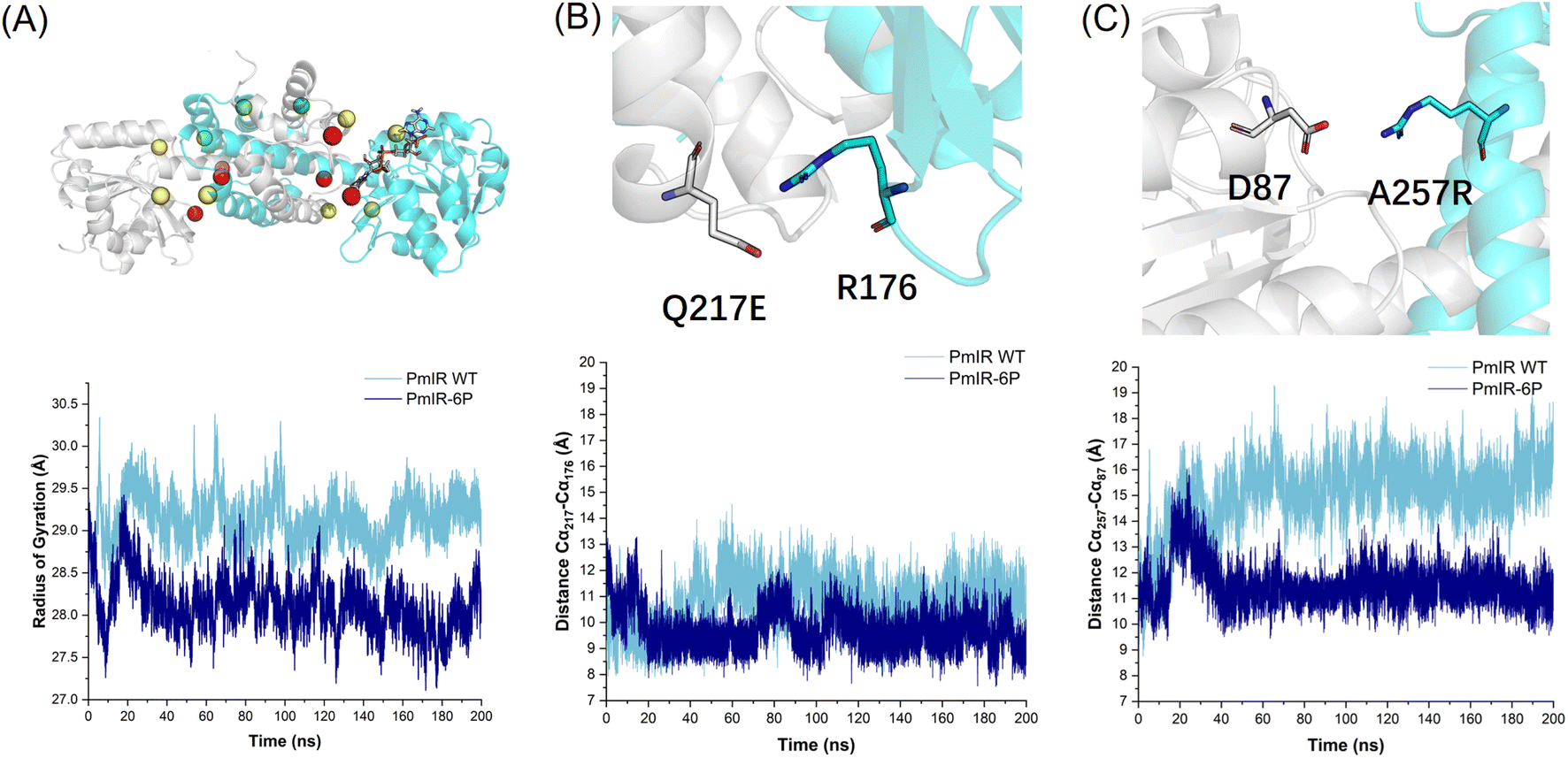 | ||
| Fig. 4 (A) Stabilizing mutations included in PmIR-6P are marked in the sphere. The mutations identified in alanine scanning (P140A/Q190S/R251N) are in red. Residues forming salt bridges are indicated in yellow (R257-D87/E217-R176). The T277M identified using the ΔΔGfold calculation is green. The radius of gyration for WT and PmIR-6P was compared, indicating a more compact structure for PmIR-6P. (B) The salt bridge of E217-R176 and the distance comparison of backbone Cα between E217 and R176 in WT and PmIR-6P. (C) The salt bridge of D87-R257 and the distance comparison of backbone Cα between D87 and R257 in WT and PmIR-6P. | ||
The distance between the backbone Cα atoms of Q217-R176 (Fig. 4B) and D87-R257 (Fig. 4C) clearly indicate that the salt bridges stretched the two subunits more tightly than in the WT. These findings suggest that subunit dissociation plays a crucial role in the irreversible inactivation of IREDs. This characteristic has also been observed in ω-transaminases, where stabilizing mutations are predominantly located at the interface of the two subunits as the active pocket is formed there.41 Therefore, focusing on the subunit interfaces of IREDs may be effective for identifying stabilizing mutations.
Previously reported engineered IREDs were mostly stabilized through directed evolution involving extensive screening. For example, 256 of 296 amino acid positions of IR-46 from Saccharothrix espanaensis were subjected to single-site saturation mutagenesis and screening before the best variant was identified.36 Similarly, SpRedAm from Streptomyces purpureus was engineered to produce cis-cyclobutyl-N-methylamine, with 93 sites (out of 296) selected for single-site saturation mutagenesis and screening.34 Additionally, in the case of ScIR from Streptomyces clavuligerus, significantly increased activity and stabilization were achieved by screening 52 sites.35 These examples demonstrated the power of directed evolution in IRED engineering. However, a rational design approach that requires significantly less screening than the conventional directed evolution still needs to be developed for IREDs. In this study, we combined different computational tools to enhance the thermal stability of IREDs and significantly reduce the library size for experimental testing. Furthermore, our results suggested that the subunit interface, including the binding pocket, plays a crucial role in stabilizing IREDs.
Comparison of substrate scope
To compare the catalytic properties of WT, PmIR-6P, and PmIR-Re, their stereoselectivities and activities towards different 2-aryl-substituted pyrrolines were determined (Fig. 5). Higher activity was observed in the reduction of substrates with meta- and ortho-fluorine substitutions on the benzene ring. In contrast, low activity was observed for substrates with para-substitution. The substrate scope was expanded as the WT showed no activity toward para-fluorine-substituted substrates, whereas PmIR-6P displayed measurable activity. Furthermore, PmIR-6P exhibited higher activity towards all tested substrates than the WT. The stereoselectivity was strictly maintained as (R)-stereoselectivity for all the tested substrates. Engineered PmIR-6P is a promising biocatalyst for the production of bulky substituted pyrrolidines. In contrast, PmIR-Re achieved high (S)-stereoselectivity, with an ee exceeding 96% towards all tested pyrrolidines. It also demonstrated an expanded substrate scope compared with that of the WT. These results indicate that the designed PmIR-Re exhibited complete (S)-stereoselectivity in catalyzing imine reduction towards various 2-aryl-substituted pyrrolines.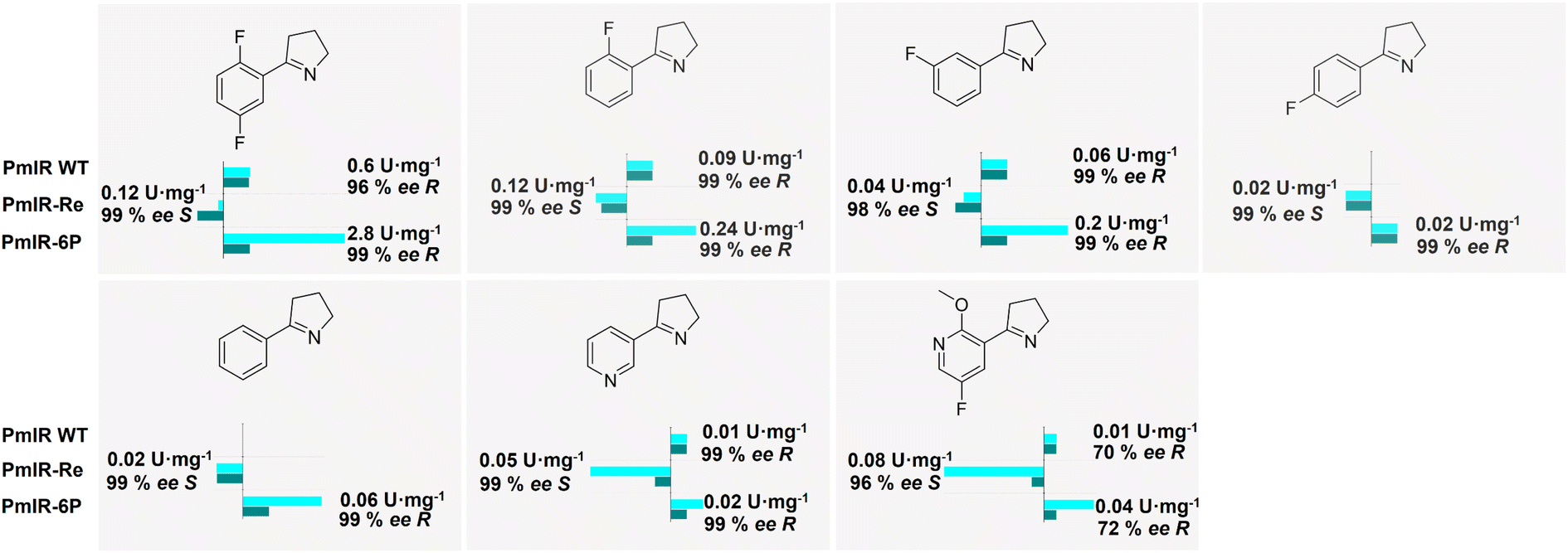 | ||
| Fig. 5 Enzymatic activity and stereoselectivity of WT, PmIR-Re, and PmIR-6P toward different 2-aryl-substituted pyrrolines. The relative activity of WT was set as 100% in bar charts except for 2-phenyl-1-pyrroline and 5-(4-fluorophenyl)-3,4-dihydro-2H-pyrrole which set PmIR-Re as 100%. | ||
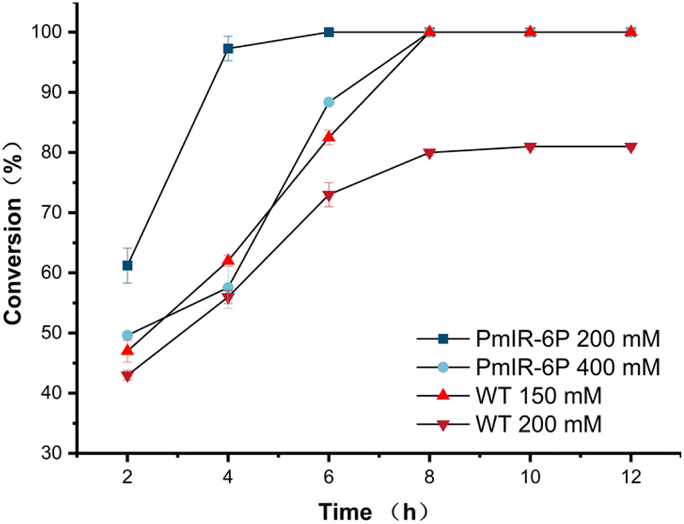 | ||
| Fig. 6 Reaction curve of WT and PmIR-6P toward different substrate loadings. | ||
Conclusions
The putative stereoselective control mechanism proposed in this study enabled the successful reversion of (R)-stereoselective PmIR through computational design. The engineered variant, PmIR-Re (Q138M/P140M/Y187E/Q190A/D250M/R251N), exhibited remarkably opposite stereoselectivities, yielding (S)-2-aryl-substituted pyrrolidines. Through the redesign of Y-type PmIR into a D/E-type variant with opposing stereoselectivity, PmIR-Re stands as the first instance of transcending the boundary between Y-type and D-type IREDs. The electrostatic interaction between the iminium cation and negatively charged residues could be crucial for stereocontrol. Furthermore, computational design of PmIR-Re revealed that, in IREDs, constraining the relative position of the CN moiety between D/E carboxyl group and it between the nicotinamide moiety allows tailoring the pro-S or pro-R pose of the substrate using Rosetta applications. This enables in silico customization of stereoselectivity with reduced screening efforts. Despite the successful revision of stereoselectivity, the practical application of PmIR in the synthesis of chiral amines requires improvement. An approach combining the salt-bridge construction, ΔΔGfold calculation, and alanine scan was employed to enhance stability. Using this approach, a six-point mutant, PmIR-6P (P140A/Q190S/R251N/Q217E/A257R/T277M), was generated, resulting in a significant improvement in stability and an almost 5-fold increase in enzymatic activity. The PmIR-6P exhibited a substantial 12 °C increase in Tm. The optimized PmIR-6P variant was highly effective in preparing a key intermediate for larotrectinib synthesis, achieving high substrate loading. This study provides valuable insights into the mechanisms underlying the stereo control of IREDs. This also demonstrates the potential of computational enzyme design to tailor stereochemical outcomes and optimize the stability of enzymatic reactions.
Data availability
The authors declare that the data supporting the findings of this study are available in the paper and its ESI Files.† All other data are available from the corresponding authors upon reasonable request.Author contributions
K. W., L. S., and J. H. conceptualized and designed the study. K. W. wrote the manuscript. J. Y. synthesized and purified the compounds. J. Y., Q. L., X. W., and P. W. performed mutation construction and screening. Y. C performed enzyme thermostability assays. K. W. performed molecular simulations. J. Y. performed the gram-scale preparation. X. L and Y. X provided important guidance for the experiment. L. S supervised the execution of the experimental plan and reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the Shanghai Sailing Program (No. 20YF1417900), National Natural Science Foundation of China (No. 81773616), and Natural Science Foundation of Shanghai (20ZR1424600). We would like to thank Zeyong Chen for guidance with crystallographic data analysis.References
- J. Mangas-Sanchez, S. P. France, S. L. Montgomery, G. A. Aleku, H. Man, M. Sharma, J. I. Ramsden, G. Grogan and N. J. Turner, Curr. Opin. Chem. Biol., 2017, 37, 19–25 CrossRef CAS PubMed.
- A. K. Gilio, T. W. Thorpe, N. Turner and G. Grogan, Chem. Sci., 2022, 13, 4697–4713 RSC.
- T. W. Thorpe, J. R. Marshall, V. Harawa, R. E. Ruscoe, A. Cuetos, J. D. Finnigan, A. Angelastro, R. S. Heath, F. Parmeggiani, S. J. Charnock, R. M. Howard, R. Kumar, D. S. B. Daniels, G. Grogan and N. J. Turner, Nature, 2022, 604, 86–91 CrossRef CAS PubMed.
- M. Sharma, J. Mangas-Sanchez, S. P. France, G. A. Aleku, S. L. Montgomery, J. I. Ramsden, N. J. Turner and G. Grogan, ACS Catal., 2018, 8, 11534–11541 CrossRef CAS.
- K. Wu, J. Huang and L. Shao, ChemCatChem, 2022, 14, e202200921 CrossRef CAS.
- P. N. Scheller, S. Fademrecht, S. Hofelzer, J. Pleiss, F. Leipold, N. J. Turner, B. M. Nestl and B. Hauer, Chembiochem, 2014, 15, 2201–2204 CrossRef CAS PubMed.
- S. Velikogne, V. Resch, C. Dertnig, J. H. Schrittwieser and W. Kroutil, ChemCatChem, 2018, 10, 3236–3246 CrossRef CAS PubMed.
- P. Stockinger, S. Roth, M. Müller and J. Pleiss, Chembiochem, 2020, 21, 2689–2695 CrossRef CAS PubMed.
- D. Wetzl, M. Berrera, N. Sandon, D. Fishlock, M. Ebeling, M. Müller, S. Hanlon, B. Wirz and H. Iding, Chembiochem, 2015, 16, 1749–1756 CrossRef CAS PubMed.
- G. A. Aleku, H. Man, S. P. France, F. Leipold, S. Hussain, L. Toca-Gonzalez, R. Marchington, S. Hart, J. P. Turkenburg, G. Grogan and N. J. Turner, ACS Catal., 2016, 6, 3880–3889 CrossRef CAS.
- J. Citoler, V. Harawa, J. R. Marshall, H. Bevinakatti, J. D. Finnigan, S. J. Charnock and N. J. Turner, Angew Chem. Int. Ed. Engl., 2021, 60, 24456–24460 CrossRef CAS PubMed.
- J. Zhang, D. Liao, R. Chen, F. Zhu, Y. Ma, L. Gao, G. Qu, C. Cui, Z. Sun, X. Lei and S. S. Gao, Angew Chem. Int. Ed. Engl., 2022, 61, e202201908 CrossRef CAS PubMed.
- S. Fademrecht, P. N. Scheller, B. M. Nestl, B. Hauer and J. Pleiss, Proteins, 2016, 84, 600–610 CrossRef CAS PubMed.
- S. L. Montgomery, A. Pushpanath, R. S. Heath, J. R. Marshall, U. Klemstein, J. L. Galman, D. Woodlock, S. Bisagni, C. J. Taylor, J. Mangas-Sanchez, J. I. Ramsden, B. Dominguez and N. J. Turner, Sci. Adv., 2020, 6, eaay9320 CrossRef CAS PubMed.
- Y. H. Zhang, F. F. Chen, B. B. Li, X. Y. Zhou, Q. Chen, J. H. Xu and G. W. Zheng, Org. Lett., 2020, 22, 3367–3372 CrossRef CAS PubMed.
- T. Huber, L. Schneider, A. Präg, S. Gerhardt, O. Einsle and M. Müller, ChemCatChem, 2014, 6, 2248–2252 CrossRef CAS.
- H. Man, E. Wells, S. Hussain, F. Leipold, S. Hart, J. P. Turkenburg, N. J. Turner and G. Grogan, Chembiochem, 2015, 16, 1052–1059 CrossRef CAS PubMed.
- M. Lenz, P. N. Scheller, S. M. Richter, B. Hauer and B. M. Nestl, Protein Expression Purif., 2017, 133, 199–204 CrossRef CAS PubMed.
- X. Robert and P. Gouet, Nucleic Acids Res., 2014, 42, W320–W324 CrossRef CAS PubMed.
- H. X. Zhou and X. Pang, Chem. Rev., 2018, 118, 1691–1741 CrossRef CAS PubMed.
- X. Pang and H. X. Zhou, Annu. Rev. Biophys., 2017, 46, 105–130 CrossRef CAS PubMed.
- J. An, Y. Nie and Y. Xu, Crit. Rev. Biotechnol., 2019, 39, 366–379 CrossRef CAS PubMed.
- K. Wu, Z. Yang, X. Meng, R. Chen, J. Huang and L. Shao, Catal. Sci. Technol., 2020, 10, 1650–1660 RSC.
- J. Zhou, Y. Wang, G. Xu, L. Wu, R. Han, U. Schwaneberg, Y. Rao, Y. L. Zhao, J. Zhou and Y. Ni, J. Am. Chem. Soc., 2018, 140, 12645–12654 CrossRef CAS PubMed.
- R. L. Thurlkill, G. R. Grimsley, J. M. Scholtz and C. N. Pace, Protein Sci., 2006, 15(5), 1214–1218 CrossRef CAS PubMed.
- V. Z. Spassov and L. Yan, Protein Sci., 2008, 17(11), 1955–1970 CrossRef CAS PubMed.
- F. Richter, A. Leaver-Fay, S. D. Khare, S. Bjelic and D. Baker, PLoS One, 2011, 6, e19230 CrossRef CAS PubMed.
- Y. Cui, Y. Wang, W. Tian, Y. Bu, T. Li, X. Cui, T. Zhu, R. Li and B. Wu, Nat. Catal., 2021, 4, 364–373 CrossRef CAS.
- P. Stockinger, N. Borlinghaus, M. Sharma, B. Aberle, G. Grogan, J. Pleiss and B. M. Nestl, ChemCatChem, 2021, 13, 5210–5215 CrossRef CAS PubMed.
- M. Prejanò, X. Sheng and F. Himo, ChemistryOpen, 2022, 11, e202100250 CrossRef PubMed.
- G. A. Aleku, S. P. France, H. Man, J. Mangas-Sanchez, S. L. Montgomery, M. Sharma, F. Leipold, S. Hussain, G. Grogan and N. J. Turner, Nat. Chem., 2017, 9, 961–969 CrossRef CAS PubMed.
- J. Zhang, Y. Ma, F. Zhu, J. Bao, Q. Wu, S. S. Gao and C. Cui, Chem. Sci., 2023, 14, 4265–4272 RSC.
- F. F. Chen, X. F. He, X. X. Zhu, Z. Zhang, X. Y. Shen, Q. Chen, J. H. Xu, N. J. Turner and G. W. Zheng, J. Am. Chem. Soc., 2023, 145, 4015–4025 CrossRef CAS PubMed.
- R. Kumar, M. J. Karmilowicz, D. Burke, M. P. Burns, L. A. Clark, C. G. Connor, E. Cordi, N. M. Do, K. M. Doyle, S. Hoagland, C. A. Lewis, D. Mangan, C. A. Martinez, E. L. McInturff, K. Meldrum, R. Pearson, J. Steflik, A. Rane and J. Weaver, Nat. Catal., 2021, 4, 775–782 CrossRef CAS.
- Q. Chen, B.-B. Li, L. Zhang, X.-R. Chen, X.-X. Zhu, F.-F. Chen, M. Shi, C.-C. Chen, Y. Yang, R.-T. Guo, W. Liu, J.-H. Xu and G.-W. Zheng, ACS Catal., 2022, 12, 14795–14803 CrossRef CAS.
- M. Schober, C. MacDermaid, A. A. Ollis, S. Chang, D. Khan, J. Hosford, J. Latham, L. A. F. Ihnken, M. J. B. Brown, D. Fuerst, M. J. Sanganee and G.-D. Roiban, Nat. Catal., 2019, 2, 909–915 CrossRef CAS.
- K. M. Polizzi, A. S. Bommarius, J. M. Broering and J. F. Chaparro-Riggers, Curr. Opin. Chem. Biol., 2007, 11, 220–225 CrossRef CAS PubMed.
- T. Meyer, N. Zumbrägel, C. Geerds, H. Gröger and H. H. Niemann, Biomolecules, 2020, 10, 1130 CrossRef CAS PubMed.
- H. J. Wijma, R. J. Floor, P. A. Jekel, D. Baker, S. J. Marrink and D. B. Janssen, Protein Eng., Des. Sel., 2014, 27, 49–58 CrossRef CAS PubMed.
- Y. Cui, Y. Chen, X. Liu, S. Dong, Y. e. Tian, Y. Qiao, R. Mitra, J. Han, C. Li, X. Han, W. Liu, Q. Chen, W. Wei, X. Wang, W. Du, S. Tang, H. Xiang, H. Liu, Y. Liang, K. N. Houk and B. Wu, ACS Catal., 2021, 11, 1340–1350 CrossRef CAS.
- Q. Meng, N. Capra, C. M. Palacio, E. Lanfranchi, M. Otzen, L. Z. van Schie, H. J. Rozeboom, A. W. H. Thunnissen, H. J. Wijma and D. B. Janssen, ACS Catal., 2020, 10, 2915–2928 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods; supplementary tables and figures; HPLC spectra; 1H and 13C NMR spectra. See DOI: https://doi.org/10.1039/d3sc04636b |
| This journal is © The Royal Society of Chemistry 2024 |