
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aggregate assembly of ferrocene functionalized indium-oxo clusters†
Rong
Zhang
ab,
Jiajing
Lan
ab,
Fei
Wang
 *a,
Shumei
Chen
*b and
Jian
Zhang
*a
*a,
Shumei
Chen
*b and
Jian
Zhang
*a
aState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, People's Republic of China. E-mail: wangfei04@fjirsm.ac.cn; zhj@fjirsm.ac.cn
bCollege of Chemistry, Fuzhou University, Fuzhou, Fujian 350108, People's Republic of China. E-mail: csm@fzu.edu.cn
First published on 4th December 2023
Abstract
In this study, we synthesized multi-nuclear indium oxide clusters (InOCs) using 1,1′-ferrocene dicarboxylic acid (H2FcDCA) as the chelating and surface protection ligand. The obtained clusters include the cubane-type heptanuclear InOCs ([In7]) and the sandwich-type thirteen-nuclear InOCs ([In13]). Notably, [In13] represents the highest nuclear number reported within the InOC family. In addition, the presence of labile coordination sites in these clusters allowed for structural modification and self-assembly. A series of [In7] clusters with adjustable band gaps have been obtained and the self-assembly of [In7] clusters resulted in the formation of an Fe-doped dimer, [Fe2In12], and an imidazole-bridged tetramer, [In28]. Similarly, in the case of [In13] clusters, the coordinated water molecules could be replaced by imidazole, methylimidazole, and even a bridged carboxylic acid, allowing the construction of one-dimensional extended structures. Additionally, part of the H2FcDCA could be substituted by pyrazole. This flexibility in replacing solvent molecules offered diverse possibilities for tailoring the properties and structures of the InOCs to suit specific applications.
Introduction
Indium oxide (In2O3) as an n-type semiconductor possesses excellent electronic and catalytic properties, making it highly promising in various fields.1–3 Many methods have been developed to prepare nano-In2O3,4 high-pressure modified In2O3,5,6 and indium-containing mixed oxides and zeolites,7–9 due to their application in sensors,10 electronics,11 and catalysis.12 In comparison to In2O3, indium oxide clusters (InOCs) provide clear structural information and allow for atomic-level control of cluster size. Consequently, they can be used as molecular mimics to explore functional-oriented structural design and optimize the performance of In2O3 nanomaterials.13,14 However, the research on InOCs remains relatively limited, with reported InOCs having low nuclear numbers such as [In2],15–17, [In4],18, [In5],19–21, [In6],22–24, [In7],25, [In10],20 and [In12].14 In 2006, Neumüller's group synthesized the largest decanuclear InOCs (denoted as [In10]) known at that time by utilizing InMe3 as the indium source.20 Subsequently, the synthesis of high nuclear InOCs has faced stagnation, and their corresponding applications have been scarcely explored. However, the pursuit of crystalline InOCs persisted, driven by the recognition of their promising properties for various technological advancements.To achieve the synthesis of crystalline InOCs, researchers recognized the crucial importance of slowing down the hydrolysis of In3+ ions, as this process greatly influences the formation of well-defined crystal structures. The recently developed coordination delayed hydrolysis (CDH) strategy has shown great potential in the synthesis of crystalline metal-oxo clusters.26,27 Inspired by this, we successfully synthesized a series of bixbyite like In12-oxo clusters by using diethanol amine as the chelating ligand to control the hydrolysis of In3+ ions.14 Despite these initial successes, it is crucial to continue delving into the structural diversity and self-assembly behaviors of InOCs (indium-oxo clusters) to unlock their full potential and broaden their applications. However, research in this field remains severely limited.
Compared to diethanolamine, 1,1′-ferrocene dicarboxylic acid (H2FcDCA) offers several advantageous features that make it an ideal chelating ligand for synthesizing InOCs:
(a) Versatility in coordination: H2FcDCA is widely employed in the construction of coordination compounds due to its flexible conformation and coordination modes.28–30 This flexibility allows for various bonding arrangements with metal ions, which can lead to the formation of diverse InOC structures and potentially higher nuclearity.
(b) Remarkable redox and photoelectrocatalytic activity: the ferrocene unit in H2FcDCA possesses exceptional redox and photoelectrocatalytic activity, which has garnered significant attention in the field of metal-oxo clusters (such as Sn,31 Fe,32–34 Co,35 Zn,36 Mn,37,38 Ti28,29,39–43). The incorporation of such active units into InOCs could introduce intriguing properties and functionalities to InOCs, making them attractive candidates for various electrochemical and catalytic applications.
(c) Strong coordination ability: the presence of multiple carboxylate groups in H2FcDCA facilitates strong coordination with In3+ ions. This strong coordination bond effectively protects InOCs and enhances their stability, making them more robust and durable under various conditions.
Due to these advantages, the combination of H2FcDCA and InOCs holds the potential to yield high-nuclear InOCs with unique properties and enhanced performance. However, no examples of InOCs functionalized with ferrocene have been reported thus far.
Based on the above considerations, H2FcDCA was selected as the chelating ligand to react with InCl3 or In(NO3)3, resulting in the synthesis of two distinct groups of InOCs: cubane-type heptanuclear InOCs ([In7], compounds 1–4) and sandwich-type thirteen-nuclear InOCs ([In13], compounds 5–9) (Table 1). Notably, [In13] represents the highest nuclear number recorded within the InOC family. Furthermore, the terminal coordinated solvents in InOCs exhibit lability, making them easily replaceable by other ligands. Consequently, they can serve as secondary building blocks (SBUs) for the formation of dimers, tetramers, and even a one-dimensional extended structure (Scheme 1). Significantly, the inclusion of ferrocene units within these structures endows them with notable redox activity and excellent photocatalytic performance.
| Complex | Composition | Space group | a [Å] | b [Å] | c [Å] | V [Å3] |
|---|---|---|---|---|---|---|
| a Abbreviations: TPP = tetraphenylphosphine; MPP = N-methylpiperazine; HIm = imidazole; DMF = N,N′-dimethylformamide; 2-mim = 2-methylimidazole; HPy = pyrazol; H2BPDC = 4,4′-biphenyldicarboxylic acid. | ||||||
| 1 | (TPP+)[In7FcDCA6(μ4-O2−)3(μ3-OCH3)(Cl−)3] |
R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) |
16.1506(10) | 16.1506(10) | 65.6859(3) | 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 838.2(2) 838.2(2) |
| 2 | (H+)8[In12FeIIFcDCA10(μ4-O2−)6(μ3-O2−)2(μ2-O2−)6(H2O)6] | P21/n | 15.4037(2) | 20.7536(3) | 25.1730(3) | 8032.26(18) |
| 3 | (H+)[In7FcDCA6(μ4-O2−)3(μ3-OCH3) (MPP−)3] |
R |
25.3175(3) | 25.3175(3) | 29.8382(4) | 16563.2(5) |
| 4 | [In28FcDCA24(μ4-O2−)12(μ3-OCH3)4(Im−)4(OH−)4] | P42/n | 35.1846(4) | 35.1846(4) | 15.8530(4) | 19625.3(7) |
| 5 | (H+)3[In13FcDCA6(μ4-O2−)6(μ2-O2−)6 (μ2-OCH3)6(H2O)6] | C2/c | 32.0443(7) | 14.9493(3) | 28.0793(7) | 13415.8(5) |
| 6 | (H+)3[In13FcDCA6(μ4-O2−)6(μ2-O2−)6(μ2-OCH3)6(HIm)6] |
R |
19.8087(10) | 19.8087(10) | 28.7268(2) | 9761.79(12) |
| 7 | (H+)3[In13FcDCA6(μ4-O2−)6(μ2-O2−)6(μ2-OCH3)6(DMF)2(2-mim)2(H2O)2] | I2/a | 27.37206(19) | 15.00755(12) | 31.8968(3) | 13070.05(17) |
| 8 | (H+)9[In13FcDCA4(μ4-O2−)6(μ2-O2−)6(μ2-OCH3)6(OCH3)4(Cl−)2(Py−)4] | C2/m | 19.6313(8) | 22.9290(6) | 13.3713(4) | 5987.4(3) |
| 9 | (H+)7[In13FcDCA6(μ4-O2−)6(μ2-O2−)6(μ2-OCH3)6(H2BPDC)2(H2O)2] | C2/m | 21.2041(3) | 25.0454(3) | 13.8544(2) | 7145.74(18) |
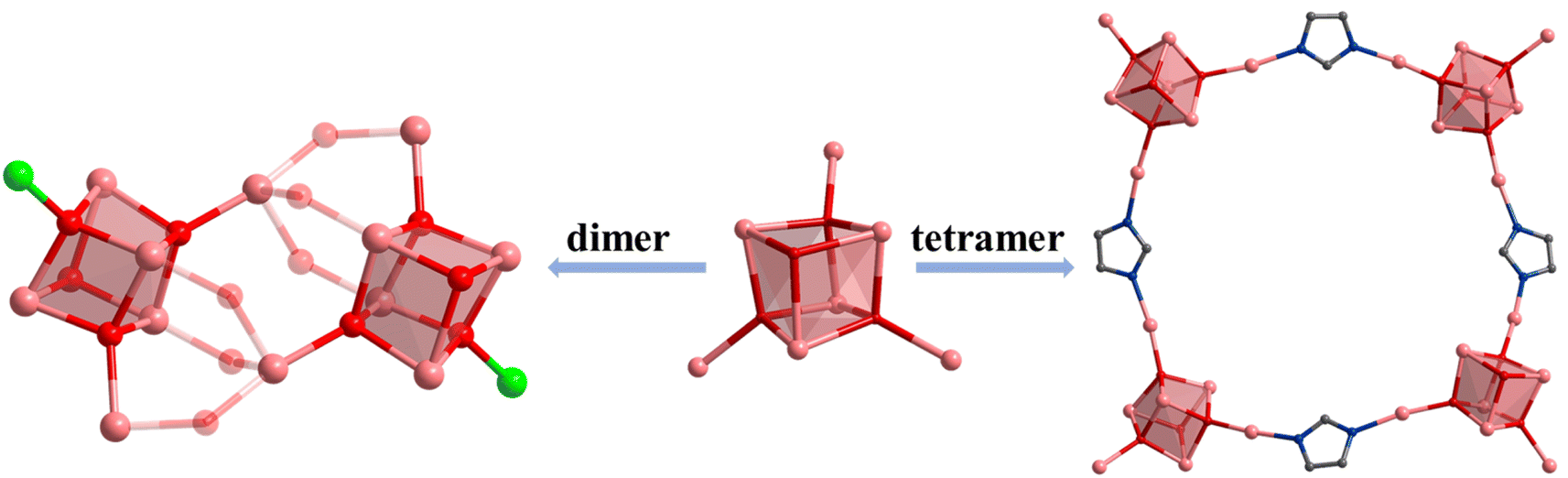 | ||
| Scheme 1 Aggregate assembly of indium oxide clusters. Color: pink, In; red, O; blue, N; gray, C. | ||
Experimental
Materials and instruments
All the reagents and solvents were purchased commercially and were used as received without further purification. 1,1′-ferrocene dicarboxylic acid (H2FcDCA, 99%), InCl3 (99%), In(NO3)3·xH2O (99%), tetraphenylphosphonium bromide (TPPBr, 99%), imidazole (HIm, 99%), acetic acid (AcOH, 99%), 2-methylimidazole (2-mim, 99%), pyrazol (HPy, 99%), and 4,4′-biphenyldicarboxylic acid (H2BPDC, 99%) were acquired from Aladdin Chemical Reagent Shanghai. N,N′-diethylformamide (DEF, 99%), N,N′-dimethylformamide (DMF, 99%), triethylamine (99%), methanol (MeOH, 99%), N-methylformamide (NMF, 99%), and N-methylpiperazine (MPP, 99%) were bought from Sinopharm Chemical Reagent Beijing.IR spectra (KBr pellets) were recorded on an ABB Bomem MB102 spectrometer over the 400–3900 cm−1 range. Powder X-ray diffraction (PXRD) data were collected on a Rigaku Mini Flex II diffractometer using CuKα radiation (λ = 1.54056 Å) under ambient conditions. The UV-vis diffuse reflection data were recorded at room temperature using a powder sample with BaSO4 as a standard (100% reflectance) on a PerkinElmer Lamda-950 UV spectrophotometer and scanned at 200–1200 nm. Metal proportional analyses were performed on an Ultima-2 inductively coupled plasma (ICP) spectrometer. The TGA curves were recorded in the region of 30–800 °C using a heating rate of 10 °C min−1 in a flowing N2 atmosphere on a Mettler Toledo TGA/SDTA 851 analyzer.
:1) was added to 23 mL glass vials respectively, sealed with ultrasound treatment for 5 minutes, and heated in a 100 °C oven for 3 days to generate yellow crystals (yield: 25.0%). FTIR (KBr, cm−1): 3340(v), 3236(w), 2352(s), 1659(s), 1582(s), 1540(m), 1476(m), 1387(s), 1360(w), 1293(w), 1190(s), 1031(s), 1106(v), 1036(s), 963(w), 918(s), 868(m), 812(w), 671(s), 868(m), 592(s), 552(s), 493(s), 446(m).
:1) was added to 23 mL glass vials respectively, sealed with ultrasound treatment for 5 minutes, and heated in a 100 °C oven for 5 days and yellow crystals were obtained (yield: 21.0%). FTIR (KBr, cm−1): 3140(m), 2939(v), 2355(s), 2323(s), 1612(s), 1594(s), 1483(m), 1395(s), 1289(v), 1197(w), 1143(s), 1017(s), 992(w), 925(w), 876(m), 821(s), 777(m), 617(w), 671(s), 587(s), 516(s), 488(s), 451(m).
:1) was added to 23 mL glass vials respectively, sealed with ultrasound treatment for 5 minutes, and heated in a 100 °C oven for 5 days and yellow crystals were obtained (yield: 25.0%). FTIR (KBr, cm−1): 3388(s), 3090(m), 2935(s), 2838(m), 2368(m), 2335(m), 1640(w), 1558(s), 1483(m), 1475(s), 1464(m), 1382(s), 1370(s), 1338(s), 1024(m), 918(w), 826(w), 769(w), 661(w), 617(w), 568(s), 523(s), 493(s), 420(s).
:1) was added to 23 mL glass vials respectively, sealed with ultrasound treatment for 5 minutes, and heated in a 120 °C oven for 7 days and yellow crystals were obtained (yield: 10.0%). FTIR (KBr, cm−1): 3291(v), 3100(m), 2933(m), 2839(m), 2397(m), 1676(w), 1584(v), 1478(s), 1387(s), 1355(w), 1251(w), 1197(w), 1083(s), 1027(w), 980(w), 945(s), 918(w), 824(w), 787(w), 668(s), 617(w), 580(s), 521(s), 483(s), 446(s).
:1) was added to 23 mL glass vials respectively, sealed with ultrasound treatment for 5 minutes, and heated in a 100 °C oven for 2 days and yellow crystals were obtained (yield: 28.0%). FTIR (KBr, cm−1): 3250(m), 2929(m), 2827(m), 1664(s), 1555(s), 1491(m), 1464(m), 1390(w), 1370(w), 1190(w), 1007(w), 925(w), 826(w), 785(s), 573(s), 524(s), 476(w), 422(s).
:1) was added to 23 mL glass vials respectively, sealed with ultrasound treatment for 5 minutes, and heated in a 100 °C oven for 6 days and yellow crystals were obtained (yield: 27.0%). FTIR (KBr, cm−1): 3139(m), 2935(m), 2828(m), 2368(m), 2335(m), 1651(s), 1560(m), 1491(m), 1390(m), 1370(m), 1325(w), 1258(w), 1202(w), 1103(s), 1079(s), 1017(v), 943(s), 923(w), 864(s), 775(m), 649(w), 612(w), 518(s), 419(s).
:1) was added to 23 mL glass vials respectively, sealed with ultrasound treatment for 5 minutes, and heated in a 100 °C oven for 2 days and yellow crystals were obtained (yield: 25.0%). FTIR (KBr, cm−1): 3080(w), 2927(m), 2827(s), 2365(s), 2335(m), 1654(w), 1560(s), 1483(m), 1456(m), 1392(w), 1365(s), 1190(s), 1090(s), 1017(s), 913(w), 824(w), 777(w), 654(w), 580(s), 466(v), 414(s).
:1) was added to 23 mL glass vials respectively, sealed with ultrasound treatment for 5 minutes, and heated in a 100 °C oven for 6 days to generate several yellow plate crystals of 8 and a large amount of unidentified precipitate. Only their crystal structures are described below. (Yield: 26.0%). FTIR (KBr, cm−1): 3280(m), 3095(m), 2975(s), 2930(m), 2825(s), 2335(m), 1646(w), 1548(v), 1496(v), 1392(w), 1372(w), 1300(v), 1202(w), 1100(s), 1014(s), 920(w), 842(w), 780(s), 580(s), 516(s), 474(s), 424(v).
:1) was added to 23 mL glass vials respectively, sealed with ultrasound treatment for 5 minutes, and heated in a 100 °C oven for 14 days and yellow crystals were obtained (yield: 11.0%). FTIR (KBr, cm−1): 3480(v), 3079 (m), 2930(m), 2830(s), 1654(m), 1572(s), 1491(s), 1387(m), 1353(m), 1187(s), 1091(s), 1010(s), 925(s), 834(s), 757(v), 666(m), 582(w), 508(s), 471(s), 424(m).
X-Ray crystallographic analysis
Single crystal X-ray diffraction data of porous materials were collected using a Hybrid Pixel Array detector equipped with Ga Kα radiation (λ = 1.3405 Å) at about 298 K and 100 K. The structures were solved with the dual-direct methods using ShelXT and refined with the full-matrix least-squares technique based on F2 using SHELXL. Non-hydrogen atoms were refined anisotropically. Hydrogen atoms were added theoretically, riding on the concerned atoms and refined with fixed thermal factors. All absorption corrections were performed using the multi-scan program.Photocatalytic measurements
A 25 mL tube was charged with 10 mg of sample powder, 10 mg [Ru(bpy)3]Cl2·6H2O, 8 mL MeCN, 2 mL H2O and 32.1 mg 1,3-dimethyl-2-phenylbenzimidazoline (BIH). It was ultrasonicated for 20 min to obtain a well-dispersed suspension. Then the resulting suspension was transferred into a Pyrex side-irradiation reaction vessel connected to a closed gas system. The reaction mixture was irradiated by visible light generated by a 300 W Xe light-source (PerfectLight, PLS-SXE300/300UV) with a 420 nm cut-off filter. The generated gas products were analyzed by a gas chromatography analyzer (FULI 9790II) equipped with a flame ionization detector (FID) and thermal conductivity detector (TCD). The product evolution rates were determined from a linear regression fit.Results and discussion
Compound 1 was obtained by using InCl3·4H2O as the metal source in DMF and MeOH mixed solvents. Single crystal X-ray diffraction analysis shows that 1 crystallizes in the hexagonal R space group with the formula TPP+[In7FcDCA6(μ4-O2−)3(μ3-OCH3)(Cl−)3]− (TPPBr = tetraphenylphosphonium bromide), which contains an anionic cluster [In7FcDCA6(μ4-O2−)3(μ3-OCH3)(Cl−)3]− and a free cationic guest, TPP+. Its cluster nucleus contains a twisted cubane central ion [In7(μ4-O2−)3(μ3-OCH3)]14+ which was stabilized by six FcDCA2− ligands accompanied by three chlorides (Fig. 1a). In 1, the In3+ ion is six coordinated, and four In3+ ions are linked by three μ4-O2− and a μ3-OCH3 to form a twisted hexahedron (Fig. 1b). The In–O bond distance ranges from 2.120 Å to 2.384 Å (Fig. S1†), which were comparable with those reported in the literature.44,451 is different from the previously reported [Fe7(μ4-O)3(μ3-OCH3)]14+.32 The cluster nuclei are isomorphic. The periphery of the cluster nucleus of 1 is modified by three terminal Cl− ions and six FcDCA2− ligands. The FcDCA2− ligand is divided into two groups: one group in which the carboxyl torsion angle of FcDCA2− ligands is relatively small (8.045°) and which links three In(III) ions in η1:η1:η1:η1:μ3 mode, while the other group (26.361°) links four In(III) ions in η1:η1:η1:η1:μ4 mode (Fig. 1c). Due to the existence of TPP+, the stacking of 1 along the c-axis is relatively dense (Fig. 1d).
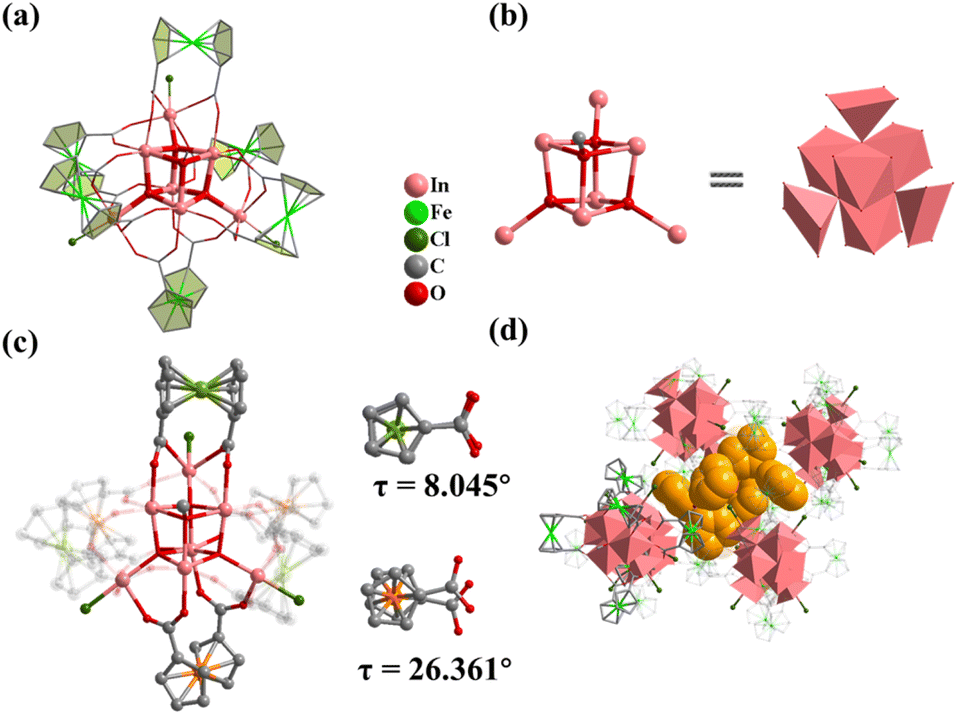 | ||
| Fig. 1 (a) Structure of 1; (b) the cubane central ion is [In7(μ4-O2−)3(μ3-OCH3)]14+; (c) coordination environment of 1; (d) 1 accumulates along the c-axis and is filled with TPP+. Color: pink, In; green, Fe; red, O; blue, N; gray, C; dark green, Cl; yellow, TPP+. | ||
Compound 2 was synthesized in NMF (N-methylformamide) and MeCN (acetonitrile) mixed solvents. Compound 2 contains an anionic cluster, [In12FeII2FcDCA10(μ4-O2−)6(μ3-O2−)2(μ2-O2−)6(H2O)6]8−. Its cluster core contains an [In12FeII2(μ4-O2−)6(μ3-O2−)2(μ2-O2−)6]12+ coordinated by ten FcDCA2− ligands and six water molecules. 2 can be seen as a dimer formed by two [In7] cores of 1. The two outermost In metal centers are replaced by Fe ions which are released by the decomposition of FcDCA2− ligands (Fig. 2a). These structural features and elemental substitutions have been supported by single crystal X-ray diffraction (XRD) and inductively coupled plasma (ICP) analysis results. The two Fe atoms are in the +2 oxidation state, which is speculated by the bond valence sum calculation with a BVS value of ca. 2.0 (Tables S3 and S4†).
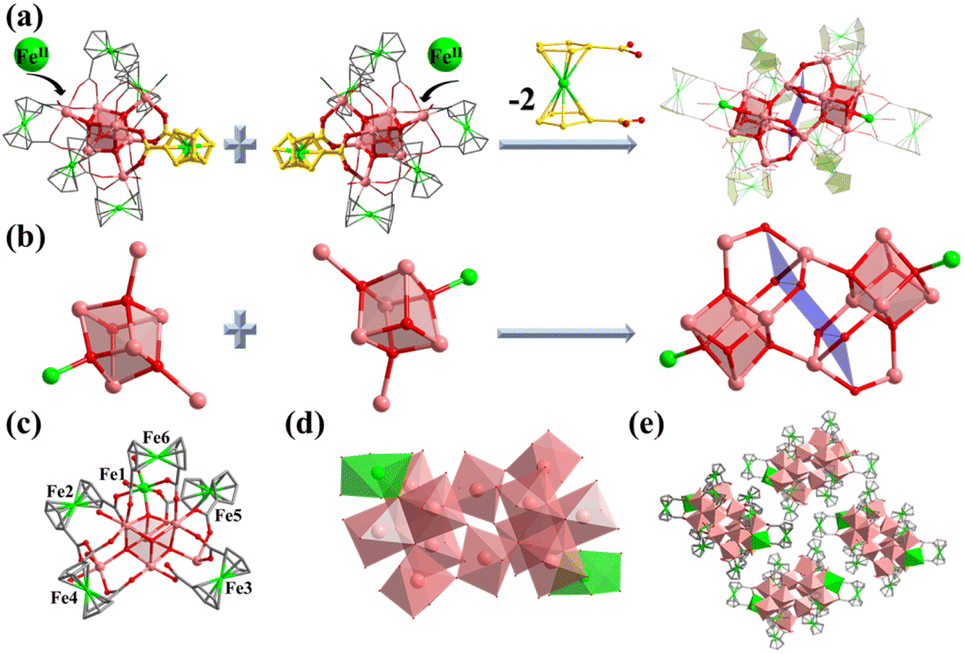 | ||
| Fig. 2 Molecular structure of compound 2. (a) Assembly of the In atom in 2; (b) two cubane central ions [In6Fe(μ4-O2−)3(μ3-O2−)]13+; (c and d) coordination environment in 2 ; (e) packing structure of 2. | ||
[In12FeII2(μ4-O2−)6(μ3-O2−)2(μ2-O2−)6]12+ in 2 contains two cubane central ions [In6FeII(μ4-O2−)3(μ3-O2−)]12+ and six μ2-O2−, where four In3+ ions are linked by three μ4-O2− and one μ3-O2− to form a twisted hexahedron (Fig. 2b–d). The stacking of 2 along the a-axis is relatively loose (Fig. 2e).
Compound 3 was obtained by introducing 1-methylpiperazine (HMPP) in the reaction system of 1, wherein the chlorine terminal sites of 1 (Fig. 3a, site A and site B) are replaced by MPP−. Compound 3 also crystallizes in hexagonal R space group with containing an anionic cluster [In7FcDCA6(μ4-O2−)3(μ3-OCH3)(MPP−)3]−, which contains a cubane central ion [In7(μ4-O2−)3(μ3-OCH3)]14+ and six FcDCA2− ligands and three MPP− ligands (Fig. 3b and c). The stacking of 3 is relatively loose (Fig. 3d).
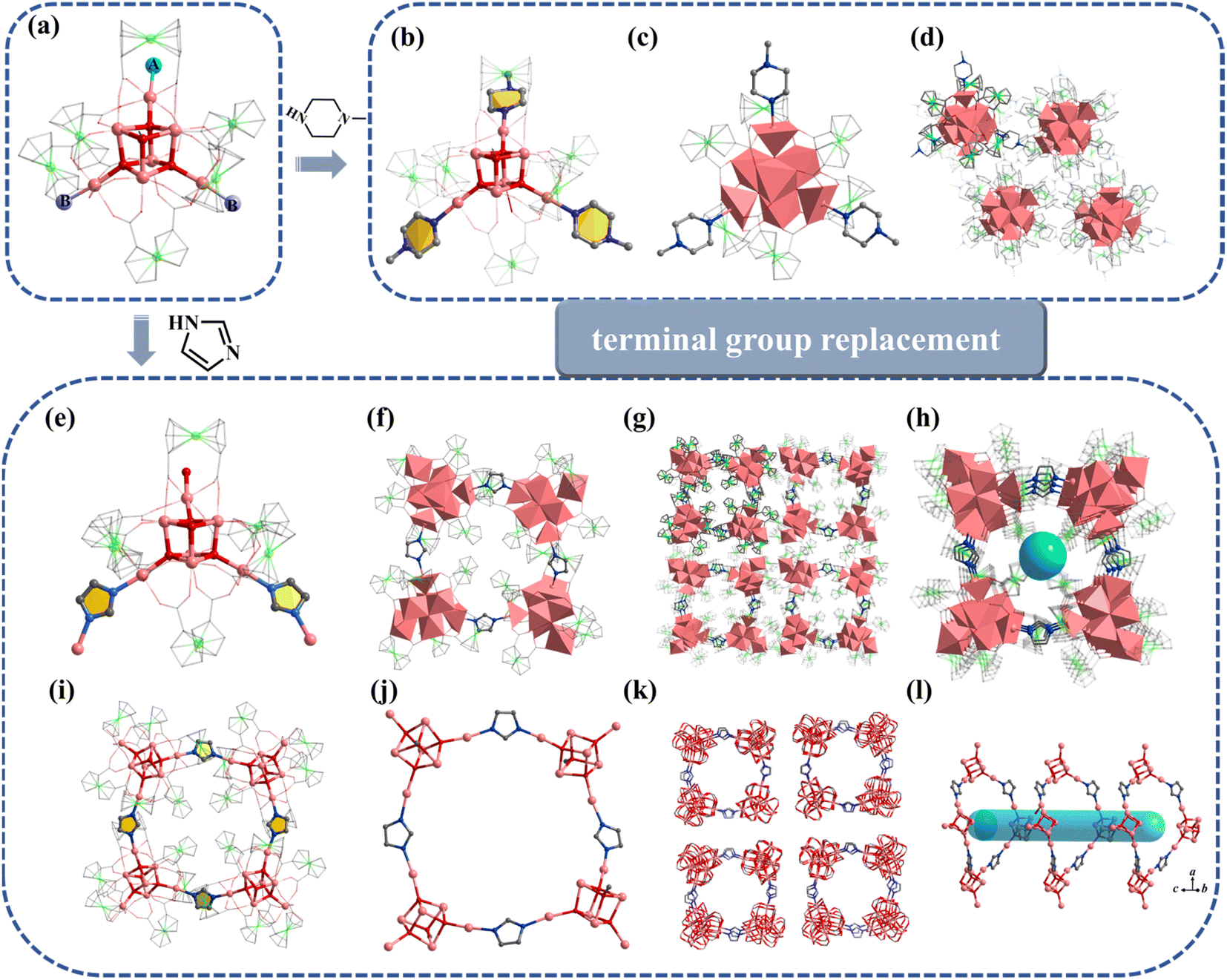 | ||
| Fig. 3 Molecular structure of compound 3. (a) A and B represent substitution sites. (b and c) 3 with MPP ligand. (d) Packing of 3 along the c-axis. (e and f) Molecular structure of 4. (g and h) Packing structure of 4. (i–l) Cluster assembly in 4. The atom color code: pink, In; green, Fe; red, O; blue, N; gray, C; dark green, Cl. | ||
Furthermore, the [In7] core of 1 can be used as SBU to construct a molecular ring. Compound 4 was obtained by adding imidazole (HIm) in the reaction system of 1, resulting in the replacement of site A by an Im− bridged ligand to form a tetramer, [In28] ring (Fig. 3e–l). Compound 4 crystallizes in tetragonal P42/n space group and contains four twisted cubane central ions [In7(μ4-O2−)3(μ3-OCH3)]14+, twenty-eight FcDCA2− ligands, four Im− ions and four OH− ions (Fig. 3e and f). The coordination environment of [In7(μ4-O2−)3(μ3-OCH3)]14+ is similar to 1, except that the two terminal chlorine atom sites are replaced by the Im-ligand (Fig. 3f, i and j). The In–O bond distance ranges from 2.099 Å to 2.290 Å. The In–N bond distance ranges from 2.146 Å to 2.171 Å. Therefore, 4 can be seen as the tetramer of 1. The center of the ring can be stacked along the c-axis to form 1D channels (Fig. 3g, h, k and l). It is worth mentioning that this [In28] ring holds the largest size record among InOCs (Fig. S2 and S9†).
Inspired by the tremendous success of using monodentate carboxylic acid as a regulator in constructing high-nuclear metal–oxygen clusters and high-valence metal–organic frameworks (MOFs),46–53 acetic acid was further introduced into the synthesis system, and a series of 13 nuclear In13-oxo clusters were successfully synthesized. These In13-oxo clusters represent the highest nuclear number in InOCs.
Compound 5 crystallizes in monoclinic C2/c space group and contains an [In13(μ4-O2−)6(μ2-O2−)6]15+ core, which was stabilized by six FcDCA2− ligands accompanied by six deprotonated methanol molecules and six water molecules. [In13(μ4-O2−)6(μ2-O2−)6]15+ can be viewed as a sandwich-type trilayer structure (Fig. 4a–d). The middle part is an Anderson-type [In7(μ4-O2−)6]9+ [In7],54 which is a hexagonal planar ring formed by seven coplanar In(III) ions through six μ4-O2− bridges. Each μ4-O2− is connected to three In(III) ions, forming a coplanar tetrahedron. And the upper and lower parts are approximately equilateral triangle [In3(μ2-O2−)3]6+. The upper and lower layers of [In3] form two equilateral triangles with a side length of 3.85 Å, and the angle of In–O–In is 136.10°. A similar cluster core has been reported in the cobalt(II)-containing arsenomolybdate [Co(H2O)6]K2[As6CoMo6O30],55 but it is the first discovery in the field of indium oxide clusters. Six methanol molecules are uniformly connected around the planar ring of [In13(μ4-O2−)6(μ2-O2−)6]15+ to form an [In13(μ4-O2−)6(μ2-O2−)6(μ3-OCH3)6]9+. The outermost edge of the anionic cluster [In13] is modified by six FcDCA2− ligands, with two carboxyl groups on each FcDCA2− ligand in η1:η1:η1:η1:μ3 modes bridging three In atoms to form [In13FcDCA6(μ4-O2−)6(μ2-O2−)6(μ3-OCH3)6]3− (Fig. 4d). It is similar to the layer previously reported [Mn13].38 It is different from the cluster core configuration of [In13], which may be due to the different coordination modes of FcDCA2− ligands.
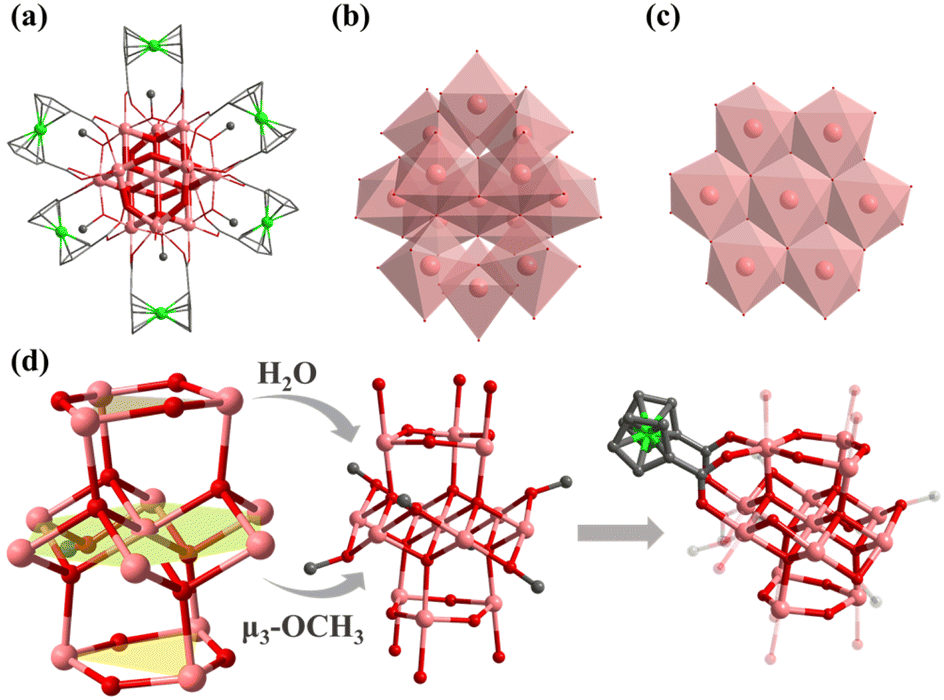 | ||
| Fig. 4 Molecular structure of 5: (a) structure of 5; (b) the cubane central ion is [In13(μ4-O2−)6(μ2-O2−)6]15+; (c) [In7(μ4-O2−)6(μ3-OCH3)6]3+ is the Anderson configuration; (d) coordination environment of [In13(μ4-O2−)6(μ2-O2−)6]15+. Color: pink, In; green, Fe; red, O; blue, N; gray, C; dark green, Cl. | ||
Similar to 1, the terminal coordination sites of [In13] (Fig. 5a, site A, site B and site C) can be replaced by diverse molecules. All sites (A, B and C) are replaced by HIm to obtain compound 6 with the formula [In13FcDCA6(μ4-O2−)6(μ2-O2−)6(μ2-OCH3)6(HIm)6]3− (Fig. 5b). Compound 7 was obtained by using 2-methylimidazole (2-mimH) to replace HIm (Fig. 5c).
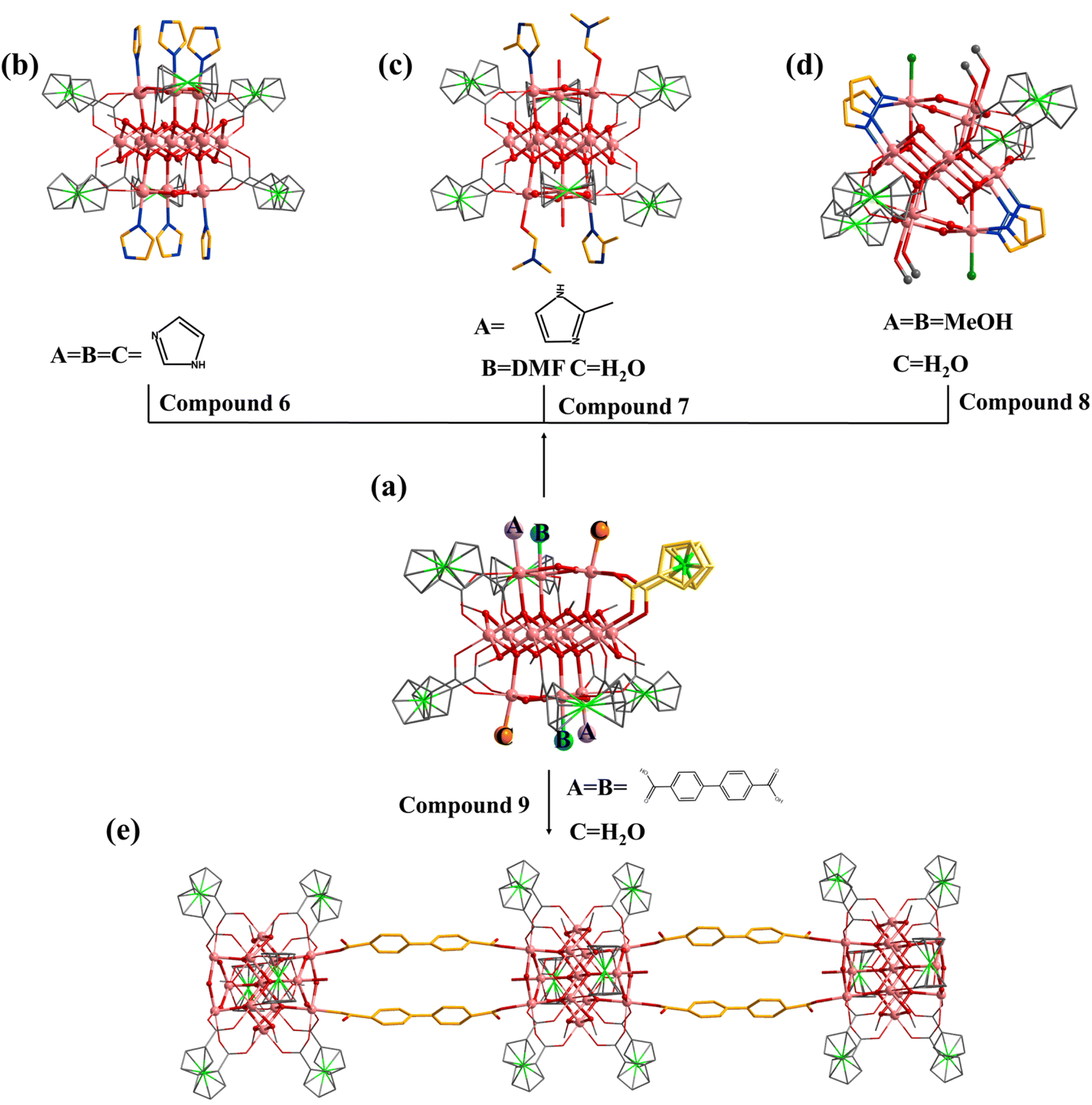 | ||
Fig. 5 (a) [In13] as a secondary building unit: (b) compound 6 (A = B = C = HIm); (c) compound 7 (A= 2-mim, B = DMF, C = ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H2O); (d) compound 8 (A = B = MeOH, C = H2O); (e) compound 9 (A = B = BPDC, C = H2O. A, B and C represent different substitution sites). H2O); (d) compound 8 (A = B = MeOH, C = H2O); (e) compound 9 (A = B = BPDC, C = H2O. A, B and C represent different substitution sites). | ||
Compound 7 contains an anionic [In13FcDCA6(μ4-O2−)6(μ2-O2−)6(μ2-OCH3)6]3− core, and the sites A, B and C are occupied by 2-mim, DMF, and H2O respectively.
Pyrazole (HPy) was added to replace HIm resulting in the formation of compound 8 (Fig. 5d). Compound 8 contains an anionic cluster [In13FcDCA4(μ4-O2−)6(μ2-O2−)6(μ2-OCH3)6(OCH3)4Cl−2(Py−)4]9−. Compared to 5, sites A and B are replaced by methanol molecules in 8. Additionally, due to the similarity of the Py− coordination mode to the carboxyl group, two FcDCA2− ligands are replaced by four chelating Py− ligands.
Interestingly, 4,4′-biphenyldicarboxylic acid (H2BPDC) was utilized as a bridged ligand to construct a one-dimensional extended structure based on [In13], compound 9. Compound 9 contains an anionic cluster [In13FcDCA6(μ4-O2−)6(μ2-O2−)6(μ2-OCH3)6(BPDC)2(H2O)2]7− similar to that of 5 (Fig. 5e). Sites A and B are replaced by 4,4′-biphenyldicarboxylic acid in 9 to form a chain along the a-axis. The description of structural details of compounds 5–9 is provided in the ESI† (Fig. S10–S12). Physical characterizations such as powder X-ray diffraction (PXRD), infrared spectroscopy and thermogravimetry analyses revealed the purity and composition of the samples from a macroscopic perspective, further corroborating the single-crystal characterization results (Fig. S13–S30†).
Here, we summarize the synthesis and structure:
(1) By introducing monodentate carboxylic acid as a regulator, we achieve a metal cluster core design from low to high nuclei;
(2) by introducing auxiliary ligands to obtain dimers, tetramers, and even one-dimensional extended structures, the end coordination solvents of [In7] and [In13] exhibit instability, serving as SBUs.
Electrochemical measurements
Due to its good acid–base stability and thermal stability, 1 has the potential to be used as a photocatalyst material. We investigated the optical properties related to photocatalysis of compounds 1, 3, and 4 with similar structures. The UV absorption spectrum obtained from the solid UV diffuse reflection spectrum conversion shows a very obvious strong absorption peak at around 600 nm in the UV band (Fig. S31–S39†). After the Kubelka Munk equation transformation,56 we obtained optical bandgaps of 1.90 eV, 1.85 eV, and 2.25 eV for 1, 3, and 4, respectively (Fig. 6a). We conducted Mott Schottky testing57 (Fig. S40–S42†), and the results show that the test curves of 1, 3, and 4 have a positive slope and are n-type semiconductors. By testing at different frequencies, we get the corresponding lowest unoccupied molecular orbital (LUMO) energies of −1.01 eV, −0.92 eV, and −1.05 eV (V vs. NHE, pH = 7). Therefore, it is calculated that the highest occupied molecular orbital (HOMO) is 0.89 eV, 0.93 eV and 1.20 eV respectively. From the LUMO energy levels, the LUMO positions of 1, 3, and 4 meet the thermodynamic requirements for carbon dioxide reduction.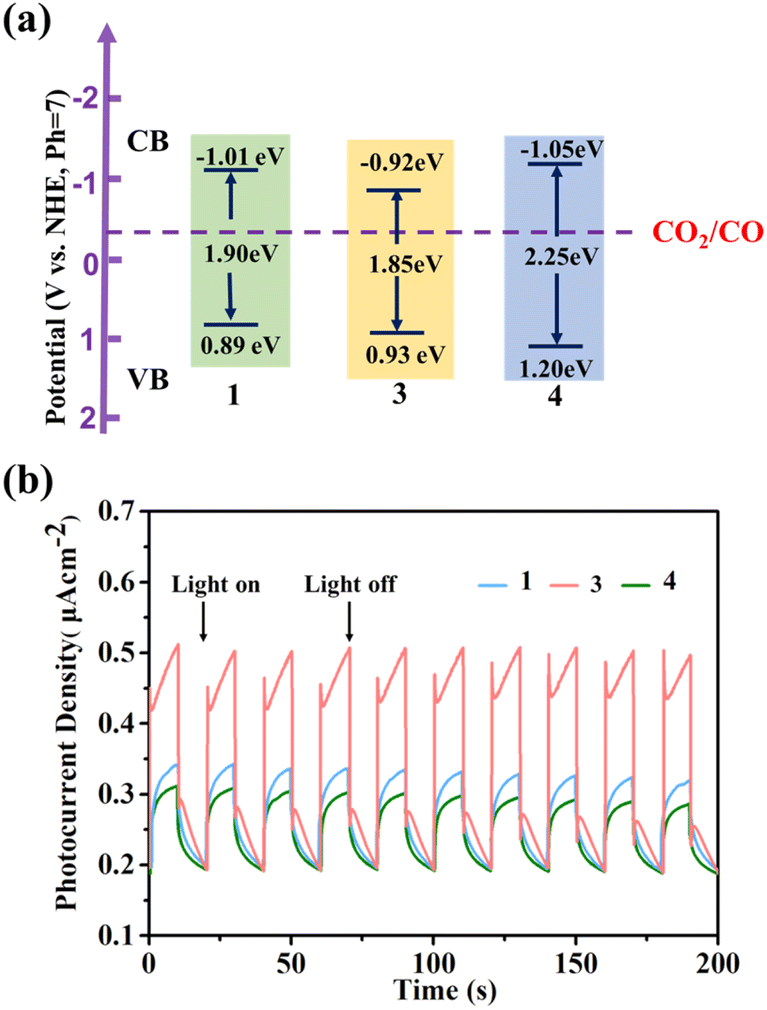 | ||
| Fig. 6 (a) Energy-level diagram of 1, 3 and 4; (b) 0.4 V-bias photocurrent responses of electrodes derived for 1, 3 and 4 in 0.2 M Na2SO4 aqueous solution under repetitive chopped visible light irradiation. | ||
Their photocurrent response was studied under visible light irradiation. The experiment was conducted at room temperature in 0.2 M Na2SO4 electrolyte using a 300 W high-pressure xenon lamp (λ > 420 nm) as a visible light source. To eliminate errors caused by uneven film thickness, a back lighting method is used. During the testing period, the xenon lamp is manually shielded to maintain its on–off cycle illumination (with an interval of 10 seconds), and maintain the voltage at 0.4 V. Visible photocurrent directions were observed for compounds 1, 3, and 4. On the one hand, photocurrent is rapidly generated and remains stable, and the intensity has not significantly decreased, indicating that they have good photoelectric response and stability. On the other hand, we found that the response effect showed the following trend: 3 > 1 > 4, which was consistent with the size trend of the UV band gap (Fig. 6b).
Photocatalytic CO2 reduction
When compounds 1, 3 and 4 were used directly as catalysts for photocatalytic carbon dioxide reduction, discernible photoreduction products were not observed. Consequently, we opted to employ [Ru(bpy)3]Cl2·6H2O as the photosensitizer for the system. Additionally, we utilized 1,3-dimethyl-2-phenylbenzimidazoline (BIH) as the electron sacrificial agent for the reaction. Visible light with wavelengths greater than 420 nm was chosen as the light source with a measured light intensity density of 458 mW cm2 using a light intensity meter for the photocatalytic activity testing. Gas chromatography was employed to monitor the gas phase products. It can be seen that after 5 hours of illumination, the systems employing catalysts 1, 3, and 4 respectively achieved CO generation rates of 3477 μmol g−1 h−1, 11 μmol g−1 h−1, and 10 μmol g−1 h−1. Correspondingly, the rates of H2 reached 967 μmol g−1 h−1, 571 μmol g−1 h−1, and 500 μmol g−1 h−1 (Fig. 7a and b). The selectivity of 1 pair of CO products reached 82%.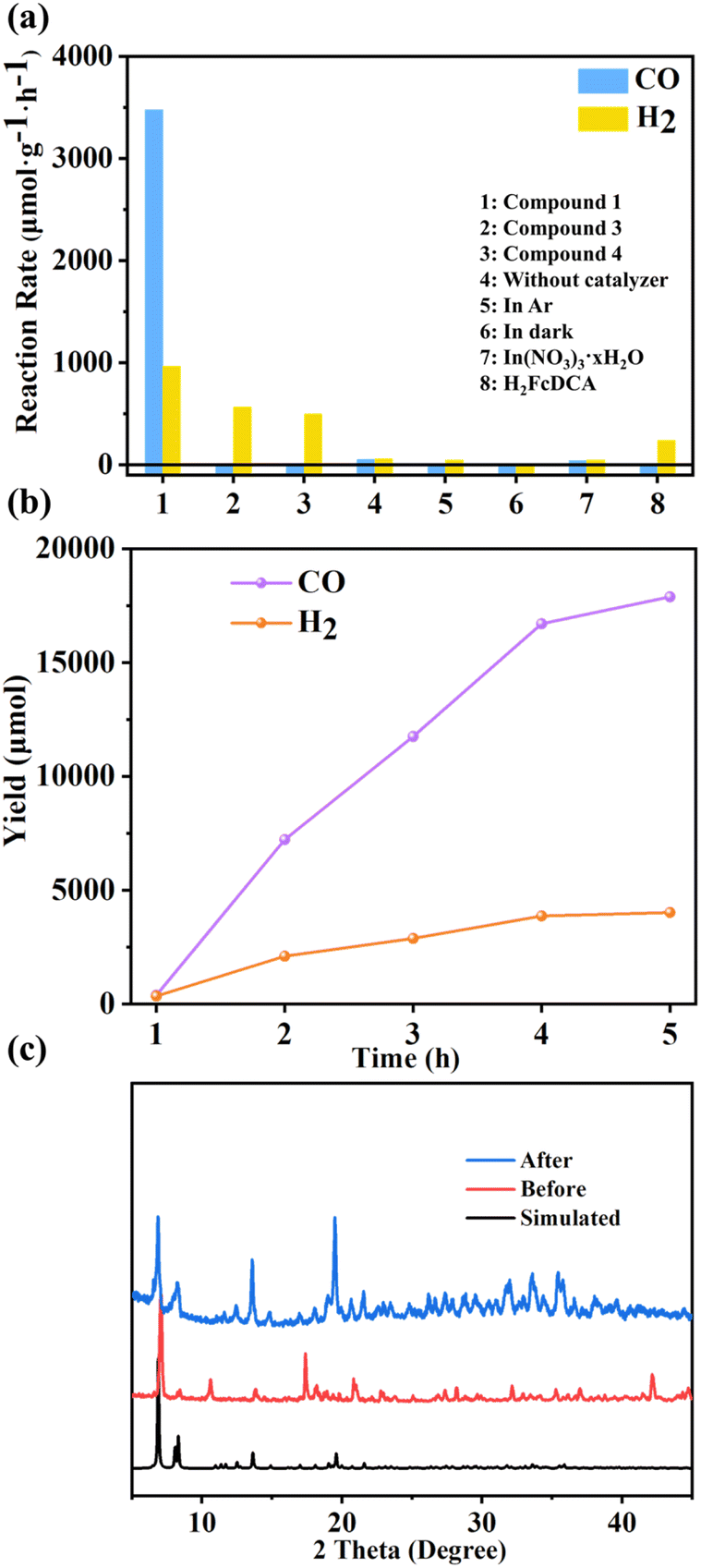 | ||
| Fig. 7 (a) Reaction rates under different conditions; (b) photocatalytic performance test chart of compound 1; (c) powder XRD patterns for compound 1 before and after photocatalytic reaction. | ||
A series of control experiments were also conducted to elucidate the roles of various components within the photocatalytic system. The lack of photosensitizers has led to a significant reduction in products, highlighting the role of photosensitizers in helping catalysts improve light utilization efficiency in the reaction (Fig. S43†). In instances where our cluster was not added, marginal amounts of CO and H2 were detected, likely attributed to the inherent photocatalytic activity of the photosensitizer alone. Substituting CO2 with Ar yielded negligible carbon product generation, confirming the origin of CO from CO2 reduction. In the absence of light, no gas-phase products were detected, confirming the photocatalytic nature of the reaction. Similarly, when BIH was omitted, product formation was not observed, highlighting the essential nature of electron sacrificial agents. Further investigations encompassed the photocatalytic activity of metal salts and ligand H2FcDCA. Metal salts exhibited similarity to the scenario involving solely photosensitizers without catalysts, suggesting the absence of significant catalytic activity in metal salts. The system incorporating ligand H2FcDCA yielded a hydrogen generation rate of 239.1 μmol g−1 h−1, along with an average CO production rate of 0.8 μmol g−1 h−1. This indicates the marginal catalytic impact of H2FcDCA in photocatalytic CO2 reduction.
Following the reaction, compounds 1, 3, and 4 were recovered through filtration, washing, and drying processes, and their structures were characterized. Post-reaction PXRD data indicated a loss of crystalline states in 3 and 4, evidenced by the broadening and disappearance of peak patterns. In contrast, the PXRD pattern of 1 remained nearly unchanged (Fig. 7c), signifying its stability after the reaction. Compared to the other two materials, the pronounced photocatalytic CO2 effect of 1 may be attributed to its superior stability. Notably, 3 and 4 displayed a propensity for hydrogen gas production akin to the catalytic outcomes of H2FcDCA. The selectivity of compounds 3 and 4 (pair of H2 products) increases, which is probably attributed to the decomposition of compounds 3 and 4 to produce FcDCA2− ligands, mainly for photocatalytic hydrogen production. This trend could potentially stem from the instability of the In–N bond within 3 and 4, leading to decomposition within the catalytic system and resulting in FcDCA2− formation.
On the basis of the above discussion, a feasible mechanism of photocatalytic CO2RR can be explained as follows. In3+ with d10 structure and H2FcDCA are extraordinary light-trapping elements.58–60 First, many electron–hole pairs are generated in compounds 1, 3, and 4 driven by visible light, and the In3+ ions of InOCs can simultaneously obtain the photoexcited electrons migrated from the FcDCA2− ligands and photosensitizers to become reduced indium ions.58,61 At the same time, the BIH molecules behave as sacrificial agents to quench the remaining photogenerated holes.62 At last, the accepted photoexcited electrons in reduced indium ions further move to the absorbed CO2 molecules for the CO2 reduction reaction, while reduced indium ions are oxidized to the original In3+ ions. Due to the lack of significant catalytic activity in metal salts, the ligand H2FcDCA generates H2 and trace amounts of CO. That is a good reflection for the effectiveness of our initial design for InOCs as a visible light catalyst by selecting an In(III) center with d10 properties and H2FcDCA ligand with strong light absorption.
Conclusions
In this study, we successfully synthesized InOCs using 1,1′-ferrocene dicarboxylic acid (H2FcDCA) as the chelating ligand and surface protection ligand. The cubane-type heptanuclear InOCs ([In7]) and sandwich-type thirteen-nuclear InOCs ([In13]) were obtained for the first time. Notably, [In13] represents the highest nuclear number. The self-assembly of these InOCs results in the formation of a series of dimers, tetramers, and one-dimensional extended structure. The inclusion of ferrocene units within these clusters resulted in remarkable redox activity and exceptional photocatalytic performance. These findings highlight the potential of H2FcDCA as a versatile chelating ligand for synthesizing InOCs with enhanced properties. The ability to control the size and structure of InOCs opens up new avenues for their utilization in various applications such as catalysis, optoelectronics, and nanotechnology. Further exploration and application of InOCs with structural diversity are necessary to uncover their full potential and promote their broad range of applications.Data availability
Data will be available on request.Author contributions
All authors contributed extensively to the work presented in this paper. F. Wang, J. Zhang and S.-M. Chen conceived the research project. R. Zhang performed the synthesis and characterizations and catalytic experiments. J.-J. Lan assisted with the data collection. F. Wang and R. Zhang wrote the manuscript and ESI with input from the other authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (21935010 and 21971241).Notes and references
- A. Tsoukalou, P. M. Abdala, D. Stoian, X. Huang, M. G. Willinger, A. Fedorov and C. R. Muller, J. Am. Chem. Soc., 2019, 141, 13497–13505 CrossRef CAS PubMed.
- L. Wang, Y. Dong, T. Yan, Z. Hu, F. M. Ali, D. M. Meira, P. N. Duchesne, J. Y. Y. Loh, C. Qiu, E. E. Storey, Y. Xu, W. Sun, M. Ghoussoub, N. P. Kherani, A. S. Helmy and G. A. Ozin, Nat. Commun., 2020, 11, 2432 CrossRef CAS PubMed.
- Z. Wang, Y. Zhou, D. Liu, R. Qi, C. Xia, M. Li, B. You and B. Y. Xia, Angew. Chem., Int. Ed., 2022, 61, e202200552 CrossRef CAS PubMed.
- S. Al-Resheedi, N. S. Alhokbany and R. M. Mahfouz, Mater. Res., 2015, 18, 931–938 CrossRef.
- M. Epifani, P. Siciliano, A. Gurlo, N. Barsan and U. Weimar, J. Am. Chem. Soc., 2004, 126, 4078–4079 CrossRef CAS PubMed.
- S.-q. Guo, X. Zhang, Z.-w. Hao, G.-d. Gao, G. Li and L. Liu, RSC Adv., 2014, 4, 31353–31361 RSC.
- Z. Maeno, S. Yasumura, C. Liu, T. Toyao, K. Kon, A. Nakayama, J.-y. Hasegawa and K.-i. Shimizu, Phys. Chem. Chem. Phys., 2019, 21, 13415–13427 RSC.
- H. Mengwen, S. Yasumura, T. Toyao, K.-i. Shimizu and Z. Maeno, Phys. Chem. Chem. Phys., 2023, 25, 10211–10230 RSC.
- I. Serykh, J. Phys. Chem. C, 2016, 120, 21436–21440 CrossRef.
- S. M. Majhi, S. T. Navale, A. Mirzaei, H. W. Kim and S. S. Kim, Inorg. Chem. Front., 2023, 10, 3428–3467 RSC.
- K. Yap, Z. Zhang, G. S. H. Thien, K.-Y. Chan and C. Y. Tan, Appl. Surf. Sci. Adv., 2023, 16 Search PubMed.
- N. Rui, F. Zhang, K. Sun, Z. Liu, W. Xu, E. Stavitski, S. D. Senanayake, J. A. Rodriguez and C.-J. Liu, ACS Catal., 2020, 10, 11307–11317 CrossRef CAS.
- Z. L. Mensinger, W. Wang, D. A. Keszler and D. W. Johnson, Chem. Soc. Rev., 2012, 41, 1019–1030 RSC.
- X. Yi, D. Wang, F. Li, J. Zhang and L. Zhang, Chem. Sci., 2021, 12, 14414–14419 RSC.
- J. Bortoluzzi, A. Neves, I. Vencato, C. Zucco and M. Hörner, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1999, 55, 1634–1636 CrossRef.
- Y.-P. Tong, S.-L. Zheng and X.-M. Chen, Aust. J. Chem., 2006, 59, 653–656 CrossRef CAS.
- K. Wieghardt, M. Kleine-Boymann, B. Nuber and J. Weiss, Inorg. Chem., 1986, 25, 1654–1659 CrossRef CAS.
- S. S. Al-Juaid, N. H. Buttrus, C. Eaborn, P. B. Hitchcock, A. T. L. Roberts, J. D. Smith and A. C. Sullivan, J. Chem. Soc., Chem. Commun., 1986, 12, 908–909 RSC.
- C. Bradley, H. Chudzynska, D. M. Frigo, M. B. Hursthouse and M. A. Mazid, J. Chem. Soc., Chem. Commun., 1988, 18, 1258–1259 RSC.
- N. N. Chamazi, M. M. Heravi and B. Neumüller, Z. Anorg. Allg. Chem., 2006, 632, 2043–2048 CrossRef CAS.
- S. Chitsaz and B. Neumüller, Z. Anorg. Allg. Chem., 2001, 627, 2451–2459 CrossRef CAS.
- H. Kaspar, G. Michele, T. F. Fassler and N. Reinhard, Angew. Chem., Int. Ed., 1993, 105, 1514–1515 Search PubMed.
- S. Mishra, V. Mendez, E. Jeanneau, V. Caps and S. Daniele, Eur. J. Inorg. Chem., 2012, 2013, 500–510 CrossRef.
- R. W. Saalfrank, C. Deutscher, H. Maid, A. M. Ako, S. Sperner, T. Nakajima, W. Bauer, F. Hampel, B. A. Hess, N. J. van Eikema Hommes, R. Puchta and F. W. Heinemann, Chem, 2004, 10, 1899–1905 CrossRef CAS PubMed.
- N. N. Chamazi, M. M. Heravi and B. Neumüller, Z. Anorg. Allg. Chem., 2007, 633, 2154–2158 CrossRef CAS.
- W. H. Fang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2018, 47, 404–421 RSC.
- L. Zhang, X. Fan, X. Yi, X. Lin and J. Zhang, Acc. Chem. Res., 2022, 55, 3150–3161 CrossRef CAS PubMed.
- J.-J. Liu, N. Li, J.-W. Sun, J. Liu, L.-Z. Dong, S.-J. Yao, L. Zhang, Z.-F. Xin, J.-W. Shi, J.-X. Wang, S.-L. Li and Y.-Q. Lan, ACS Catal., 2021, 11, 4510–4519 CrossRef CAS.
- J. J. Liu, S. N. Sun, J. Liu, Y. Kuang, J. W. Shi, L. Z. Dong, N. Li, J. N. Lu, J. M. Lin, S. L. Li and Y. Q. Lan, J. Am. Chem. Soc., 2023, 145, 6112–6122 CrossRef CAS PubMed.
- Z. Huang, H. Yu, L. Wang, X. Liu, T. Lin, F. Haq, S. Z. Vatsadze and D. A. Lemenovskiy, Coord. Chem. Rev., 2021, 430 Search PubMed.
- G. L. Zheng, J. F. Ma, Z. M. Su, L. K. Yan, J. Yang, Y. Y. Li and J. F. Liu, Angew. Chem., Int. Ed., 2004, 43, 2409–2411 CrossRef CAS PubMed.
- A. Masello, Y. Sanakis, A. K. Boudalis, K. A. Abboud and G. Christou, Inorg. Chem., 2011, 50, 5646–5654 CrossRef CAS PubMed.
- V. Mereacre, M. Nakano, J. Gomez-Segura, I. Imaz, C. Sporer, K. Wurst, J. Veciana, C. Turta, D. Ruiz-Molina and P. Jaitner, Inorg. Chem., 2006, 45, 10443–10445 CrossRef CAS PubMed.
- V. Mereacre, D. Prodius, A. M. Ako, S. Shova, C. Turta, K. Wurst, P. Jaitner and A. K. Powell, Polyhedron, 2009, 28, 3551–3555 CrossRef CAS.
- M. L. Ospina Castro, A. Briceño, T. González, A. Reiber, G. Jorge and M. Helena Duran, Inorg. Chim. Acta, 2015, 432, 275–282 CrossRef CAS.
- Y. S. Kim, J. Kim, D. Kim and H. K. Chae, Chem. Lett., 2007, 36, 150–151 CrossRef CAS.
- A. Masello, K. A. Abboud, W. Wernsdorfer and G. Christou, Inorg. Chem., 2013, 52, 10414–10423 CrossRef CAS PubMed.
- A. Masello, M. Murugesu, K. A. Abboud and G. Christou, Polyhedron, 2007, 26, 2276–2280 CrossRef CAS.
- Z. Liu, J. Lei, M. Frasconi, X. Li, D. Cao, Z. Zhu, S. T. Schneebeli, G. C. Schatz and J. F. Stoddart, Angew. Chem., Int. Ed., 2014, 53, 9193–9197 CrossRef CAS PubMed.
- Y. Fan, Y. Cui, G. D. Zou, R. H. Duan, X. Zhang, Y. X. Dong, H. T. Lv, J. T. Cao and Q. S. Jing, Dalton Trans., 2017, 46, 8057–8064 RSC.
- Y. Fan, H. M. Li, R. H. Duan, H. T. Lu, J. T. Cao, G. D. Zou and Q. S. Jing, Inorg. Chem., 2017, 56, 12775–12782 CrossRef CAS PubMed.
- J. L. Hou, W. Luo, Y. Guo, P. Zhang, S. Yang, Q. Y. Zhu and J. Dai, Inorg. Chem., 2017, 56, 6451–6458 CrossRef CAS PubMed.
- M. Han, W. D. Yu, L. J. Li, X. Y. Yi, J. Yan and C. Liu, Chem. Commun., 2021, 57, 2792–2795 RSC.
- R. Gao, S. M. Chen, F. Wang and J. Zhang, Inorg. Chem., 2021, 60, 239–245 CrossRef CAS PubMed.
- R. Zhang, B. Wang, F. Wang, S.-M. Chen and J. Zhang, Inorg. Chem. Front., 2023, 10, 201–210 RSC.
- M. Y. Gao, F. Wang, Z. G. Gu, D. X. Zhang, L. Zhang and J. Zhang, J. Am. Chem. Soc., 2016, 138, 2556–2559 CrossRef CAS PubMed.
- W. H. Fang, L. Zhang and J. Zhang, J. Am. Chem. Soc., 2016, 24, 7480–7483 CrossRef PubMed.
- T. Tsuruoka, S. Furukawa, Y. Takashima, K. Yoshida, S. Isoda and S. Kitagawa, Angew. Chem., Int. Ed., 2009, 48, 4739–4743 CrossRef CAS PubMed.
- A. Schaate, P. Roy, A. Godt, J. Lippke, F. Waltz, M. Wiebcke and P. Behrens, Chem.–Eur. J., 2011, 17, 6643–6651 CrossRef CAS PubMed.
- S. Yuan, T.-F. Liu, D. Feng, J. Tian, K. Wang, J. Qin, Q. Zhang, Y.-P. Chen, M. Bosch, L. Zou, S. Teat, S. Dalgarno and H.-C. Zhou, Chem. Sci., 2015, 6, 3926–3930 RSC.
- S. Yuan, L. Feng, K. Wang, J. Pang, M. Bosch, C. Lollar, Y. Sun, J. Qin, X. Yang, P. Zhang, Q. Wang, L. Zou, Y. Zhang, L. Zhang, Y. Fang, J. Li and H. C. Zhou, Adv. Mater., 2018, 30, e1704303 CrossRef PubMed.
- Y. Sun, D.-F. Lu, Y. Sun, M.-Y. Gao, N. Zheng, C. Gu, F. Wang and J. Zhang, ACS Mater. Lett., 2021, 3, 64–68 CrossRef CAS.
- Q. Yan, J. Wang, L. Zhang, J. Liu, M. Wahiduzzaman, N. Yan, L. Yu, R. Dupuis, H. Wang, G. Maurin, M. Hirscher, P. Guo, S. Wang and J. Du, Nat. Commun., 2023, 14, 4189 CrossRef CAS PubMed.
- J. S. Anderson, Nature, 1937, 140, 850 CrossRef CAS.
- J. Martin-Frere, Y. Jeannin, F. Robert and J. Vaissermann, Inorg. Chem., 1991, 30, 3635–3639 CrossRef CAS.
- H. Yang, M. J. Hao, Y. H. Xie, X. L. Liu, Y. F. Liu, Z. S. Chen, X. K. Wang, G. I. N. Waterhouse and S. Ma, Angew. Chem., Int. Ed., 2023, 62, e202303129 CrossRef CAS PubMed.
- S. Bi, Z. X. Zhang, F. C. Meng, D. Q. Wu, J. S. Chen and F. Zhang, Angew. Chem., Int. Ed., 2021, 61, e202111627 CrossRef PubMed.
- F. C. Leng, H. Liu, M. Ding, Q.-P. Lin and H. L. Jiang, ACS Catal., 2018, 8, 4583–4590 CrossRef CAS.
- H. G. Zhang, S. H. Si, G. Y. Zhai, Y. J. Li, Y. Y. Liu, H. F. Cheng, Z. Y. Wang, P. Wang, Z. K. Zheng, Y. Dai, T. X. Liu and B. B. Huang, Appl. Catal., B, 2023, 337, 1–10 Search PubMed.
- C. X. Lu, D. Xiong, C. Chen, J. Wang, Y. Kong, T. Liu, S. Ying and F.-Y. Yi, Inorg. Chem., 2022, 61, 2587–2594 CrossRef CAS PubMed.
- J. Wei, S. Zhang, J. Sun, T. Liang, Z. Li, Z. Li, X. Yi, R. Xiong, J. Deng, Z. Yu, S. Wang and Y. Hou, J. Colloid Interface Sci., 2023, 629, 92–102 CrossRef CAS PubMed.
- H. Lin, Y. Liu, Z. Wang, L. Ling, H. Huang, Q. Li, L. Cheng, Y. Li, J. Zhou, K. Wu, J. Zhang and T. Zhou, Angew. Chem., Int. Ed., 2022, 134, e202214142 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details, crystallographic studies, ICP, additional figures, general characterization, pH stability and solvent stability, and photoelectrochemical testing. CCDC 2281558–2281566. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc05824g |
| This journal is © The Royal Society of Chemistry 2024 |