
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolar-driven CO2-to-ethanol conversion enabled by continuous CO2 transport via a superhydrophobic Cu2O nano fence†
Hailing
Huo
a,
Hua
He
 b,
Chengxi
Huang
a,
Xin
Guan
b,
Fang
Wu
c,
Yongping
Du
a,
Hongbin
Xing
a,
Erjun
Kan
*a and
Ang
Li
*a
b,
Chengxi
Huang
a,
Xin
Guan
b,
Fang
Wu
c,
Yongping
Du
a,
Hongbin
Xing
a,
Erjun
Kan
*a and
Ang
Li
*a
aMIIT Key Laboratory of Semiconductor Microstructure and Quantum Sensing, Nanjing University of Science and Technology, Nanjing 210094, P. R. China. E-mail: liang2100@njust.edu.cn; ekan@njust.edu.cn
bState Key Laboratory of Heavy Oil Processing and College of Chemical Engineering, China University of Petroleum (East China), Qingdao 266580, P. R. China
cCollege of Information Science and Technology, Nanjing Forestry University, Nanjing 210037, P. R. China
First published on 27th November 2023
Abstract
The overall photocatalytic CO2 reduction reaction presents an eco-friendly approach for generating high-value products, specifically ethanol. However, ethanol production still faces efficiency issues (typically formation rates <605 μmol g−1 h−1). One significant challenge arises from the difficulty of continuously transporting CO2 to the catalyst surface, leading to inadequate gas reactant concentration at reactive sites. Here, we develop a mesoporous superhydrophobic Cu2O hollow structure (O–CHS) for efficient gas transport. O–CHS is designed to float on an aqueous solution and act as a nano fence, effectively impeding water infiltration into its inner space and enabling CO2 accumulation within. As CO2 is consumed at reactive sites, O–CHS serves as a gas transport channel and diffuser, continuously and promptly conveying CO2 from the gas phase to the reactive sites. This ensures a stable high CO2 concentration at reactive sites. Consequently, O–CHS achieves the highest recorded ethanol formation rate (996.18 μmol g−1 h−1) to the best of our knowledge. This strategy combines surface engineering with geometric modulation, providing a promising pathway for multi-carbon production.
Introduction
The global consumption of fossil fuels and the excessive emission of greenhouse gas CO2 necessitate the development of effective technologies to address energy and environmental problems.1 The overall photocatalytic CO2 reduction reaction (PCRR) offers an environmentally friendly approach to converting CO2 and H2O into valuable products without requiring hole scavengers.2 Of particular interest are multi-carbon products with two or more carbons (C2+), which possess higher energy density and economic potential compared to one-carbon (C1) products.3 Ethanol, a liquid C2 solar fuel that is easily transportable, holds significant importance due to its versatile applications in the production of disinfectants and other organic chemicals.4,5However, ethanol production still faces efficiency issues, typically with formation rates below 605 μmol g−1 h−1.6–11 The formation of ethanol involves a difficult C–C coupling process, which is hindered by a high energy barrier.12 Researchers have discovered that the concentration of CO2 on the catalyst surface plays a crucial role in this C–C coupling process.13,14 A high surface CO2 concentration not only accelerates the reaction rate but also increases the selectivity of C2+ products by facilitating the C–C coupling process.13 Unfortunately, the low solubility and diffusion coefficient of CO2 in aqueous solutions result in a low ratio of CO2 to H2O molecules (approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1300 at 1 atm pressure) in the reaction system.15 This leads to an extremely inadequate supply of CO2 molecules reaching the reactive sites on the catalyst surface, further resulting in the suppressed PCRR and promoted side reaction of the hydrogen evolution reaction (HER). Therefore, a significant challenge in improving ethanol production efficiency arises from the difficulties of efficiently transporting CO2 to the catalyst surface and maintaining a stable high CO2 concentration there.
:1300 at 1 atm pressure) in the reaction system.15 This leads to an extremely inadequate supply of CO2 molecules reaching the reactive sites on the catalyst surface, further resulting in the suppressed PCRR and promoted side reaction of the hydrogen evolution reaction (HER). Therefore, a significant challenge in improving ethanol production efficiency arises from the difficulties of efficiently transporting CO2 to the catalyst surface and maintaining a stable high CO2 concentration there.
At present, various strategies have been developed to create hydrophobic materials aiming at accumulating more CO2 molecules near the catalyst surface.16–21 These strategies typically involve modifying catalysts with hydrophobic modifiers16–18 or supporting catalysts on hydrophobic substrates.19–21 However, the developed systems still face limitations in effectively transporting CO2. As shown in Scheme 1a, in system I, the material is suspended in the aqueous solution. Although hydrophobicity provided by hydrophobic modifiers or hydrophobic substrates aids in aggregating CO2 on the catalyst surface, CO2 still needs to diffuse through water to the catalyst surface, which is slow due to the low diffusion coefficient. In system II (Scheme 1b), the material can float on the aqueous solution and directly contact gaseous CO2, but diffusion of CO2 to the catalyst surface within the aqueous solution still needs to occur through water. Consequently, both system I and system II suffer from slow CO2 diffusion, making it difficult to continuously and promptly supply the constantly consumed surface CO2.
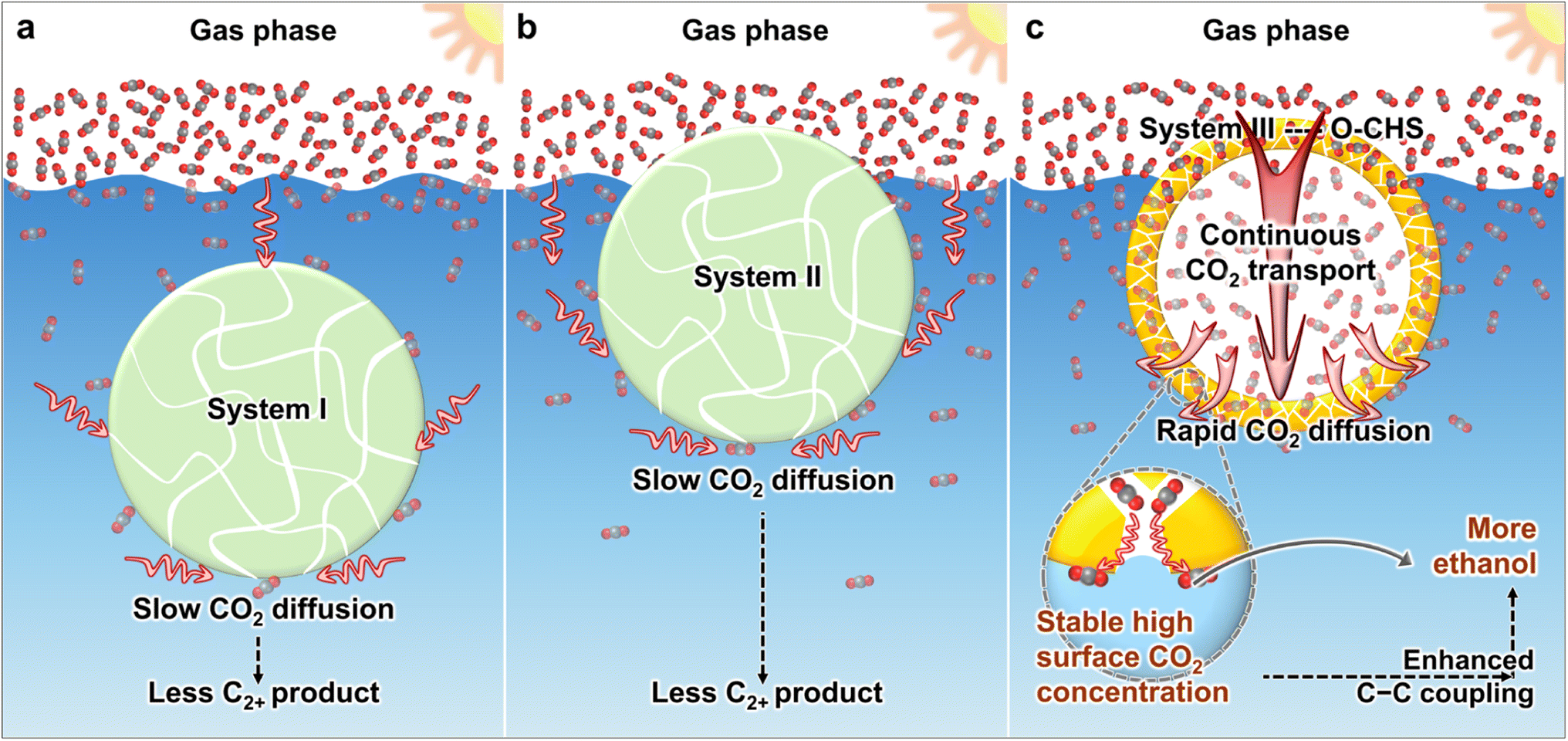 | ||
| Scheme 1 Schematics of CO2 diffusion in systems involving reported hydrophobic materials and the designed O–CHS. (a) System I with hydrophobic materials suspended in the aqueous solution. (b) System II with hydrophobic materials floating on the aqueous solution. (c) System III with the designed O–CHS. The gas phase of CO2 is depicted in white, and the aqueous solution is depicted in blue. The gray and red balls represent C and O atoms, respectively. | ||
Herein, we utilize cuprous oxide (Cu2O) as a model photocatalyst and construct a mesoporous superhydrophobic Cu2O hollow structure (O–CHS) to enable continuous and rapid CO2 transport. Cu2O, with a band gap of approximately 2.2 eV, is considered a highly promising material for CO2 photoreduction. It can efficiently utilize visible light, which accounts for a large proportion of the solar spectrum.22 Moreover, the surface of Cu2O exhibits remarkable potential for facilitating the activation of CO2.23,24 Additionally, the low-coordinated CuI surface atoms promote the adsorption of *CO, which is a crucial intermediate in the production of multi-carbon compounds.5,23 O–CHS is designed to float on an aqueous solution as depicted in Scheme 1c. Its superhydrophobic surface acts like a fence, preventing water from passing through the mesoporous shell to the internal space, thus allowing CO2 to enter and accumulate within. In system III with the designed O–CHS, as CO2 is consumed at reactive sites on the O–CHS surface, a concentration gradient of CO2 forms in the direction from the gas phase to the O–CHS surface. Therefore, O–CHS serves as a gas transport channel and diffuser, continuously transporting and dispersing CO2 from the gas phase to its surface. This concentration gradient-driven diffusion process occurs in the gas phase, where the diffusion coefficient of CO2 is significantly higher than in the liquid phase. Therefore, CO2 can rapidly disperse onto the catalyst surface, promptly replenishing its consumption. This design ensures a stable high CO2 concentration at reactive sites. Mechanistic investigations based on in situ Fourier transform infrared spectroscopy (FTIR) and density functional theory (DFT) calculations reveal that the concentrated CO2 on the catalyst surface lowers the energy barrier of C–C coupling, thereby promoting ethanol generation. Through this innovative strategy, we have achieved a superior ethanol generation rate of up to 996.18 μmol g−1 h−1 (Table S1†) and a solar-to-ethanol conversion efficiency (STE) of 0.152%. The ethanol selectivity of 59.59% is also significantly higher compared to the designed Cu2O catalysts lacking the ability to continuously transport and disperse gas. This strategy combines catalyst surface engineering with geometric modulation, providing a promising pathway for producing C2+ products.
Results and discussion
Morphological characterization
The Cu2O hollow structure (CHS) was first synthesized using an in situ one-pot chemical transformation procedure (Fig. 1a–c).25 The as-synthesized CHS exhibits a hydrophilic property with a water contact angle (WCA) of 43.96° (Fig. 1d). Subsequently, the target catalyst O–CHS was obtained by hydrophobically modifying CHS with tiny amounts of 1-dodecanethiol (DDT) (Fig. 1e–g). Following the modification, O–CHS exhibits a superhydrophobic property with a WCA of 152.24° (Fig. 1h).26 To investigate the continuous gas transport and dispersion effect of O–CHS, four additional reference catalysts were synthesized. The first catalyst was the hydrophilic Cu2O solid sphere (CSS) with a WCA of 40.71°, which was synthesized using a modified method based on previous studies (Fig. S1a–d†).27 The second catalyst was the superhydrophobic Cu2O solid sphere (O–CSS) with a WCA of 150.02°, which was also obtained through DDT modification of CSS (Fig. S1e–h†). The third catalyst was the hydrophilic Cu2O nanosheets (CNS) with a WCA of 40.08°, which was obtained after prolonged stirring treatment during CHS preparation (Fig. S2a–d†). Essentially, CNS represents the fragmented CHS, with the main difference between CNS and CHS being the absence of the hollow structure in the former. Lastly, the fourth catalyst was the superhydrophobic Cu2O nanosheet (O–CNS) with a WCA of 152.39°, which was also obtained through DDT modification of CNS (Fig. S2e–h†). Notably, the key distinction between O–CHS and O–CNS is the loss of the hollow space in the latter. Photographs of the six samples are displayed in Fig. S3† and no significant macroscopic differences are observed among these six samples. | ||
| Fig. 1 Morphologies and wettability of CHS and O–CHS. (a and c) TEM images of CHS. (b) HRTEM image of CHS. (d) WCA of CHS. (e and g) TEM images of O–CHS. (f) HRTEM image of O–CHS. (h) WCA of O–CHS. (i–k) EDS mapping images of Cu, O, and S for O–CHS, respectively. (l) Overlapping EDS mapping signals of Cu and S for O–CHS. | ||
The transmission electron microscopy (TEM) image of CHS presented in Fig. 1a illustrates a typical hollow sphere structure. The lattice fringe of 0.246 nm displayed in the high-resolution TEM (HRTEM) image of CHS conforms with the (111) plane of Cu2O (Fig. 1b), indicating the composition of CHS as Cu2O. In combination with the TEM image of a larger view in Fig. 1c, CHS is demonstrated to have been successfully synthesized. Following DDT modification, the images of O–CHS depicted in Fig. 1e–g closely resemble the corresponding images of CHS, indicating the preservation of the hollow structure during the modification process. In addition, compared to CHS, O–CHS displays an additional lattice fringe of 0.212 nm in Fig. 1f, which aligns with the (200) plane of Cu2O, further confirming the composition of O–CHS as Cu2O. The scanning electron microscopy (SEM) image of O–CHS shown in Fig. S4† also showcases the uniform morphology of O–CHS. The energy-dispersive X-ray spectroscopy (EDS) mapping images of O–CHS in Fig. 1i–l show that the signal of sulfur (S) from DDT overlaps with copper (Cu), affirming the uniform distribution of DDT on the O–CHS surface. Additionally, TEM and HRTEM images of CSS (Fig. S1a–c†), O–CSS (Fig. S1e–g†), CNS (Fig. S2a–c†), and O–CNS (Fig. S2e–g†) validate the successful synthesis of the corresponding structures.
Structural characterization
The chemical composition of each sample, namely CHS, O–CHS, CSS, O–CSS, CNS, and O–CNS, was first analyzed using X-ray diffraction (XRD). Fig. 2a illustrates that all six samples exhibit highly comparable XRD patterns, which are well-matched with the cubic phase Cu2O (JCPDS Card no. 78-2076). The chemical composition was further determined using X-ray photoelectron spectroscopy (XPS). The Cu 2p XPS spectra (Fig. 2b) exhibit two prominent peaks at ∼932.4 and ∼952.4 eV, attributable to CuI, as supported by the Cu LMM Auger spectra (Fig. S5a†).28,29 Although weaker signals at ∼933.7 and ∼953.6 eV, associated with CuII, are also observed, their intensity is considerably lower than that of CuI. This indicates that the primary components of CHS and O–CHS are indeed Cu2O. Notably, the Cu 2p peak positions show no obvious changes between CHS and O–CHS, suggesting the preservation of the electronic structures of the catalyst surface after the DDT modification.30 Furthermore, the Cu 2p XPS spectra and Cu LMM Auger spectra of CSS, O–CSS, CNS, and O–CNS depicted in Fig. S5† exhibit similarities to those of CHS and O–CHS, indicating comparable compositions among all six samples.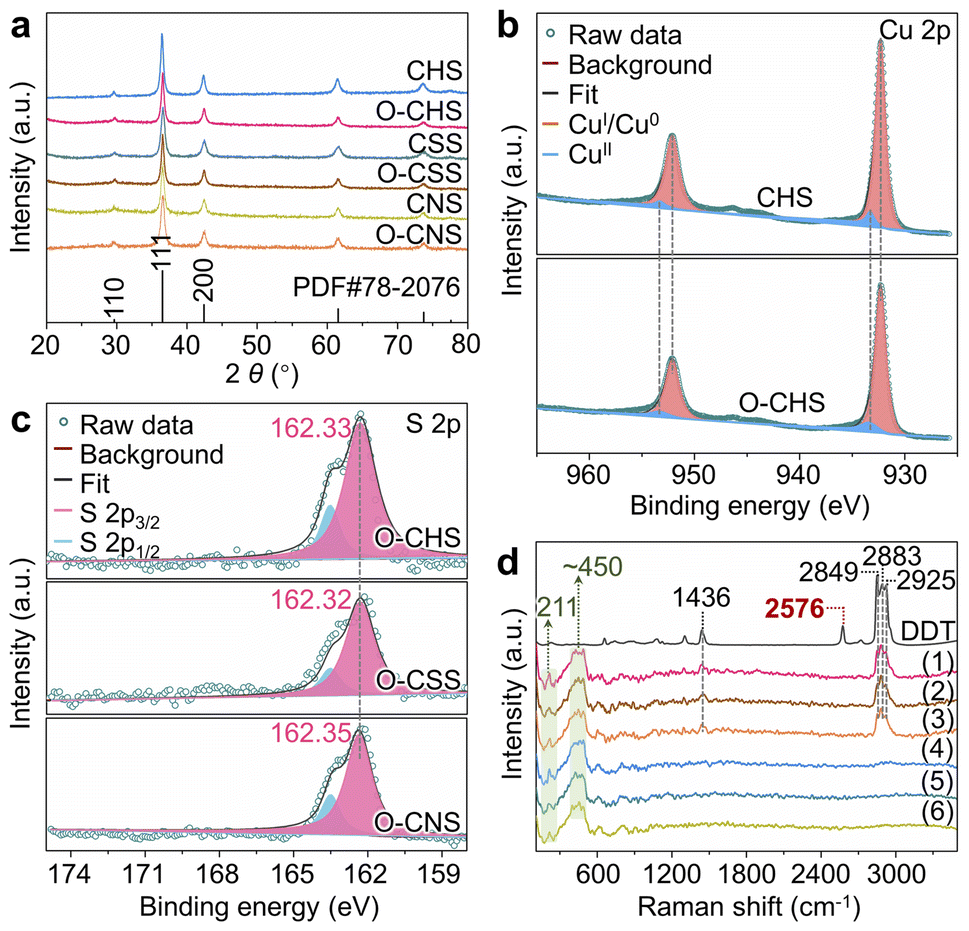 | ||
| Fig. 2 Structural characterizations. (a) XRD patterns of CHS, O–CHS, CSS, O–CSS, CNS, and O–CNS. (b) High-resolution Cu 2p XPS spectra of CHS and O–CHS. (c) High-resolution S 2p XPS spectra of O–CHS, O–CSS, and O–CNS. (d) Raman spectra of DDT, O–CHS (1), O–CSS (2), O–CNS (3), CHS (4), CSS (5), and CNS (6). | ||
Moreover, the signals of S originating from DDT are observed in the XPS survey spectra (Fig. S6a†) for O–CHS, O–CSS, and O–CNS, allowing inference of the existing state of DDT on the Cu2O surface based on the S 2p XPS spectra (Fig. 2c). The binding energies for the S 2p3/2 core level observed on O–CHS, O–CSS, and O–CNS exhibit a prominent peak at 162.33, 162.32, and 162.35 eV, respectively, which falls well within the expected range for a surface thiolate species (RS-Cu).31,32 This finding implies the formal dissociative adsorption of the S–H bond in DDT on the Cu2O surface,31,33 whereas S 2p1/2 peaks at 163.6 eV are attributed to residual unbound thiols.34,35 This primarily demonstrates that chemical adsorption occurs during the DDT modification. Moreover, the S 2p3/2 binding energies exhibit negligible disparity among the three catalysts, indicating that there is no significant difference in the electronic structure of the surface of the three catalysts due to the introduction of S.36 Additionally, the Raman spectra provide additional confirmation of the chemical adsorption between DDT and the Cu2O surface of the catalysts. As depicted in Fig. 2d, DDT shows strong characteristic peaks at 1436, 2576, 2849, 2883, and 2925 cm−1, corresponding to the vibrations of C–H in –CH2– (1436, 2849, and 2925 cm−1), –S–H (2576 cm−1), and C–H in –CH3 (2883 cm−1), respectively.37 In contrast, the peaks at 1436, 2849, 2883, and 2925 cm−1 still remain for O–CHS, O–CSS, and O–CNS, while the peak at 2576 cm−1 disappears. This indicates that the –S–H in DDT is broken upon adsorption on the surfaces of O–CHS, O–CSS, and O–CNS, as illustrated in Fig. S7,† which coincides with the findings obtained from the S 2p XPS spectra.38 In addition, all six samples display characteristic peaks of Cu2O at 211 and ∼450 cm−1, further confirming their composition.39 Therefore, it is reasonable to conclude that DDT is attached to the catalyst surface through chemisorption. Compared to physical adsorption, chemisorption renders DDT more difficult to desorb, ensuring the excellent hydrophobic stability of the catalysts, which is crucial for the PCRR. Consequently, even after 6 hours of Xe lamp irradiation or prolonged storage for 20 days, the hydrophobicity can still be well maintained (Fig. S8†). Additionally, the amount of chemisorbed DDT is sufficiently small (Fig. S6b†), thereby avoiding the hindrance to surface charge transfer during the reaction. This is well supported by the Ag+ probe experiments (for details see experimental procedures in the ESI, Fig. S9 and S10†).16
Subsequently, additional characterizations were conducted to further investigate the properties of the six samples. The ultraviolet-visible (UV-vis) diffuse reflectance spectra in Fig. S11a† demonstrate the ability of all samples to absorb both ultraviolet and visible light. Moreover, the estimated bandgaps from the Tauc plots (Fig. S11b†) show similar values at ∼2.19 eV, manifesting comparable light absorption ability among the samples. The ability of charge carrier separation was investigated through steady-state photoluminescence (PL). The lower PL intensity of CHS, O–CHS, CNS, and O–CNS than that of CSS and O–CSS indicates their better charge-separation efficiency (Fig. S12a†). This could be attributed to the shortened charge migration distance resulting from the provided thin-walled structures of hollow spheres or nanosheets.40 To obtain a deeper understanding of the charge transfer dynamics, time-resolved PL (TRPL) measurements were conducted for O–CHS, O–CNS, and O–CSS using an excitation wavelength of 350 nm (Fig. S12b†). Charge carriers with longer lifetimes would show slower PL decay, indicating more efficient separation.41 By fitting the decay curves with a model involving three exponential functions (Fig. S12b and Table S2†), the average PL lifetimes (τa) for O–CHS, O–CNS, and O–CSS were determined to be 1.97, 1.85, and 1.49 ns, respectively. The prolonged lifetimes observed for O–CHS and O–CNS compared to O–CSS further suggest the advantages of the thin-walled structures for charge separation. Furthermore, the similar lifetimes for O–CHS and O–CNS indicate that both structures exhibit comparable efficiency in separating charge carriers. Additionally, photoelectrochemical measurements were employed to further reveal the ability of different samples to separate and transfer charge carriers. The photocurrent response analysis reveals obvious photocurrent signals in all samples and excellent reproducibility of the response intensity in the process of on–off cycles (Fig. S13a†). Electrochemical impedance spectroscopy (EIS) provides further evidence of charge-transfer resistance (Fig. S13b†). The higher transient photocurrent intensity and smaller semicircle radii of CHS, O–CHS, CNS, and O–CNS than those of CSS and O–CSS imply their more efficient charge separation and transfer.42 These photoelectrochemical test results are consistent with the PL results, collectively indicating the structural advantages of CHS, O–CHS, CNS, and O–CNS in charge carrier separation and transfer. Furthermore, a comparison of the PL intensity, photocurrent intensity, and charge-transfer resistance of the samples before and after DDT modification reveals a minor impact of DDT modification on carrier behavior. To gain further insights into the porous structure of the six samples, N2 adsorption–desorption isotherms and pore diameter distributions are presented in Fig. S14 and Table S3.† Type IV curves with H3 hysteresis loops are displayed for all samples, confirming their mesoporous nature with a maximum pore size of less than 50 nm, which allows efficient mass transport during the reaction.43
Verification of the nano-fence effect of O–CHS
In order to validate the nano-fence effect of O–CHS resulting from its superhydrophobic surface, which prevents water infiltration into its hollow space, we employed a total internal reflection fluorescence microscope (TIRFM) to directly observe the local distributions of gas and liquid surrounding the catalyst at a single-particle level for the first time. The liquid phase is labeled with a water-soluble Cy5 NHS ester dye, which is a fluorescent molecule that emits a strong red light under 635 nm excitation, enabling the visualization of the boundary morphology of the liquid phase. Therefore, regions exhibiting fluorescence signals indicate the presence of water (Fig. S15†). As depicted in Fig. 3a, the fluorescence signal of O–CHS clearly delineates a distinctive hollow structure, with a significantly weaker signal detected in the hollow region compared to the outer shell region. Conversely, in the case of CHS (Fig. 3b), conspicuous fluorescence signals permeate the entire sample. This observation indicates that water can fully penetrate the inner space of CHS, whereas it is impeded in the case of O–CHS, demonstrating the nano-fence effect of O–CHS.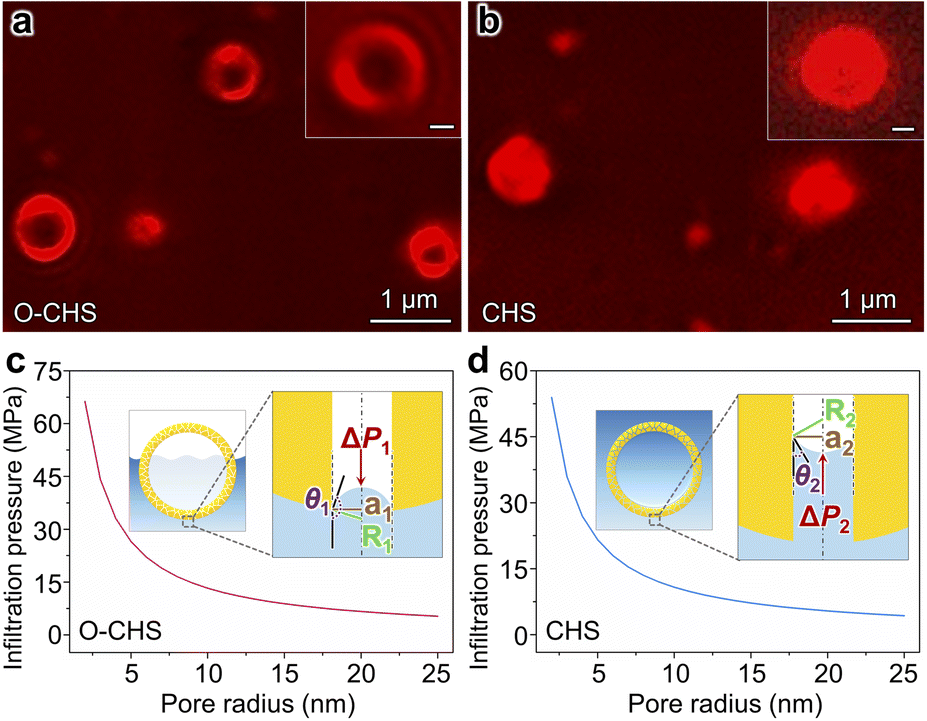 | ||
| Fig. 3 TIRFM images and infiltration pressures of O–CHS and CHS. (a and b) TIRFM images of O–CHS and CHS, respectively. The corresponding insets show the enlarged TIRFM images. The scale bar represents 200 nm. (c and d) Infiltration pressures as a function of the pore radius for O–CHS and CHS, respectively. The corresponding insets depict the schematics of the liquid–gas interface advancing inside the pore for O–CHS and CHS, respectively. ΔP is the infiltration pressure of water pushed into the pores. The red arrow indicates the direction of this pressure. a is the pore radius. R is the radius of curvature of the liquid surface. θ is approximated by the WCA of the catalyst. | ||
To further support this observation, we calculate the infiltration pressure (ΔP) required for water to penetrate the pores on the catalyst based on the Young–Laplace equation (for details see experimental procedures in the ESI†).15 For O–CHS, ΔP1 is calculated as ∼5300 kPa for its largest pore radius of ∼25 nm (Fig. 3c). According to the detailed description of the CO2 photoreduction test process (see experimental procedures in the ESI†), O–CHS, floating on the liquid surface, experiences a maximum pressure of 80 kPa, which is significantly smaller than ΔP1. This indicates that water cannot be forced into the hollow space through the mesopores (Fig. S16a–c†), while for CHS, ΔP2 is calculated as ∼4314 kPa for its largest pore radius of ∼25 nm (Fig. 3d). This pressure is sufficient to allow complete water infiltration into the pores and the interior of the hollow space (Fig. S16d–f†). Identical conclusions can also be drawn from the Cu(OH)2 probe experiments (for details see experimental procedures in the ESI†). The absence of large particles within the hollow structure of O–CHS is displayed in Fig. S17,† whereas the hollow structure of CHS exhibits a noticeable presence of such particles. This observation suggests that Cu2+ ions can permeate the hollow space of CHS when in aqueous solution, but are impeded from entering the interior of O–CHS due to its nano-fence effect.
These findings provide comprehensive evidence that O–CHS can function as a nano fence, effectively segregating water and thereby enabling the entry and accumulation of gas within it. This phenomenon consequently enables a rapid supply of more gaseous CO2 molecules to the O–CHS surface during the PCRR. Furthermore, the accelerated enrichment of CO2 on the O–CHS surface is supported by the CO2 dissolution rate test (for details see experimental procedures in the ESI and Fig. S18†). The results presented in Fig. S19† show that the gas-phase CO2 content decreases more rapidly in the system with floating O–CHS compared to that with CHS, corresponding to a rapid increase in dissolved CO2 in the liquid phase. These findings indicate that the presence of O–CHS promotes the dissolution rate of CO2 due to the accelerated CO2 enrichment on its surface, which is consistent with previous studies highlighting the role of hydrophobic species in promoting gas enrichment.19,21
Overall PCRR performance
The overall PCRR performance for the target catalyst (O–CHS) and all the reference catalysts (CHS, CSS, O–CSS, CNS, and O–CNS) was evaluated in an aqueous solution under visible-light irradiation without hole scavengers. The results are presented in Fig. 4.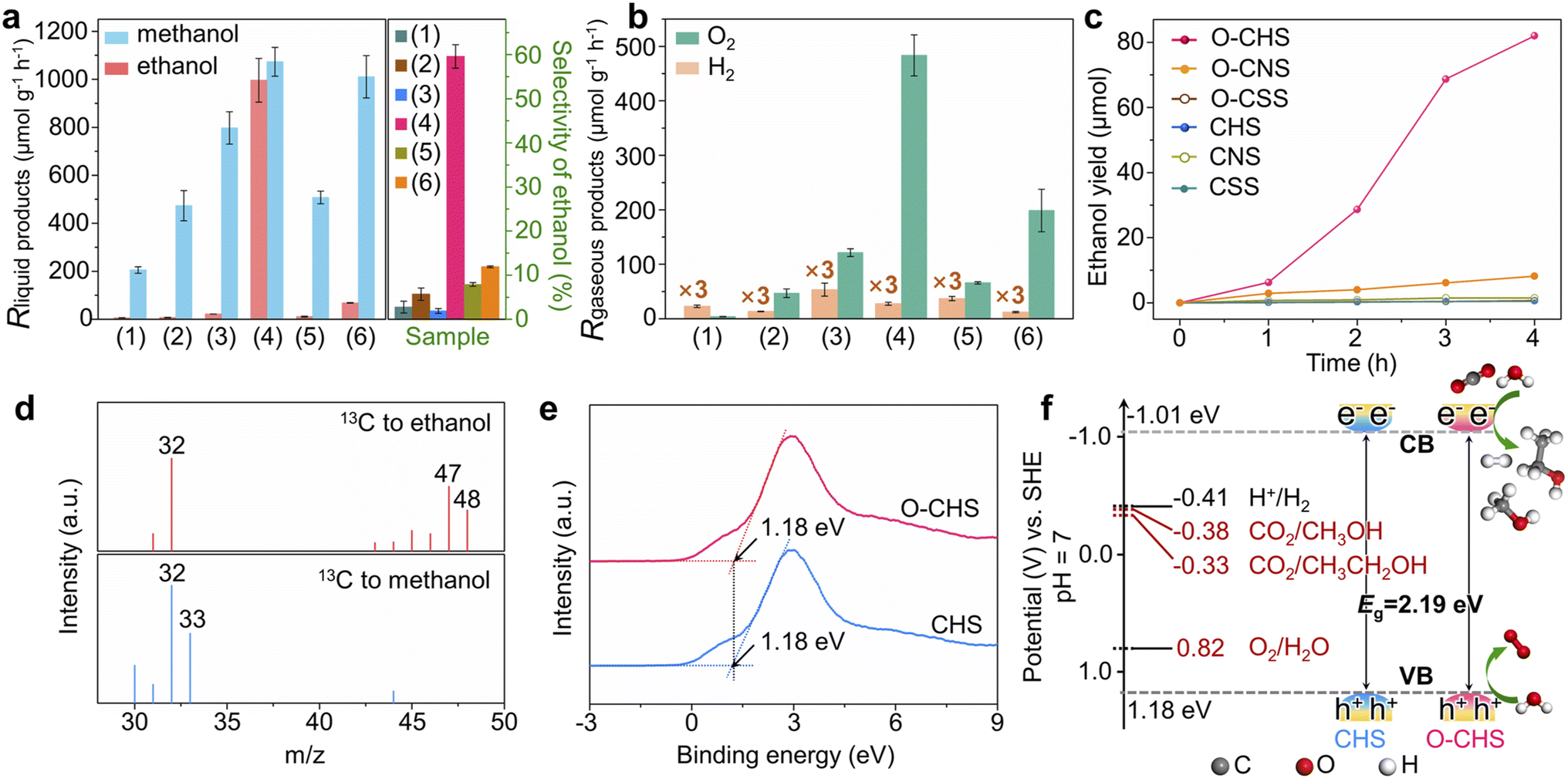 | ||
| Fig. 4 Overall PCRR performance. (a and b) Generation rates of the overall PCRR to liquid products, selectivities towards ethanol, and generation rates of the overall PCRR to gaseous products for CSS (1), O–CSS (2), CHS (3), O–CHS (4), CNS (5), and O–CNS (6). The error bar represents the standard deviation of the measurements. The note ×3 means that the value in the corresponding column has been multiplied by three. (c) Ethanol yield as a function of reaction time. (d) GC-MS spectra of 13CO2 to ethanol (upper panel) and 13CO2 to methanol (lower panel) over O–CHS. (e) Valence-band XPS spectra of O–CHS and CHS. (f) Band-structure diagrams of O–CHS and CHS. | ||
The main liquid product of CSS is methanol with a generation rate of 205.45 μmol g−1 h−1 (Fig. 4a), and the gaseous product of H2 with a generation rate of 7.64 μmol g−1 h−1 (Fig. 4b) is also detected. Upon DDT modification to form O–CSS, the methanol generation rate increases to 473.27 μmol g−1 h−1 with a minor amount of H2 formation (4.5 μmol g−1 h−1). Moreover, the selectivity of carbon derivatives rises from 98.85% to 99.69% (Fig. S20†). This improvement can be attributed to the enhanced enrichment of CO2 facilitated by the superhydrophobicity of O–CSS, which leads to higher CO2 and lower H+ concentrations on the catalyst surface. This, in turn, suppresses the HER and promotes the PCRR.19
CHS also produces methanol as the main liquid product, with a relatively higher generation rate of 797.71 μmol g−1 h−1. This higher rate can be attributed to its structural advantages in charge carrier behavior, as demonstrated by PL and photoelectrochemical measurements. Additionally, as indicated in Fig. 3, although CHS exhibits a hollow structure, it does not possess the nano-fence effect. However, in the case of O–CHS, the nano-fence effect plays a significant role in CO2 ingress and accumulation within the hollow space by effectively segregating water. Furthermore, the CO2 gathered in the cavity can continuously diffuse to the catalyst surface as the reaction progresses, replenishing its consumption. Therefore, a stable high CO2 concentration near reactive sites can be maintained. Consequently, compared to the nearly negligible ethanol generation rate observed for CSS, O–CSS, and CHS, an impressive ethanol generation rate of 996.18 μmol g−1 h−1 is achieved for the target catalyst O–CHS, which is the highest record in PCRR systems to the best of our knowledge (typically ranging from 0.37 to 605 μmol g−1 h−1, as shown in Table S1†). Furthermore, the STE can reach up to 0.152% (for details see experimental procedures in the ESI and Fig. S21†), surpassing the typical range of 0.005% to 0.082% and achieving superior utilization of solar energy for O–CHS.44–49 Moreover, the ethanol selectivity is substantially increased to 59.59%, which is 21, 10, and 29 times higher than that observed for CSS, O–CSS, and CHS, respectively (Fig. 4a). The excellent CO2-to-ethanol performance of O–CHS is further supported by time-dependent activity tests, with the original data presented in Fig. S22.† As shown in Fig. 4c, ethanol yield over O–CHS increases rapidly with reaction time, surpassing the rates observed over all the reference catalysts. Additionally, the methanol generation rate of O–CHS is also elevated to 1073.04 μmol g−1 h−1, enabling the highest selectivity of carbon derivatives at 99.99% (Fig. S20†).
In order to illustrate the notable impact of the superhydrophobic hollow structure, which enables the accumulation of CO2 within it due to its nano-fence effect and subsequently functions as a gas transport channel and diffuser during CO2 consumption, we have also investigated the overall PCRR performance of CNS and O–CNS. As mentioned before, CNS is obtained by breaking the hollow structure of CHS, and O–CNS is produced using the same modification method as O–CHS. Therefore, the main distinction between O–CNS and O–CHS lies in the absence of the hollow space in the former. As depicted in Fig. 4a and b, compared to CNS, O–CNS exhibits improved PCRR and suppressed HER performance with methanol, ethanol, and H2 generation rates of 1010.40, 68.45, and 4.12 μmol g−1 h−1, respectively. This further validates the promoting effect of hydrophobic catalysts on the PCRR. However, in comparison to O–CHS, O–CNS shows a 93.13% reduction in ethanol generation rate, confirming the enhancement effect of the superhydrophobic hollow structure on ethanol production.
To investigate the carbon source of generated carbon derivatives, an isotope-labelling test was conducted using 13CO2 as the reactant. The gas chromatography-mass spectrometry (GC-MS) results shown in Fig. 4d exhibit three typical peaks with mass-to-charge ratios (m/z) of 32, 47, and 48, corresponding to the fragments of 13CH2OH, 13CH313CHOH, and 13CH313CH2OH of 13C-ethanol, respectively.12,50 Additionally, the dominant peaks with m/z of 32 and 33 for methanol are assigned to the fragments of 13CH3O and 13CH3OH, respectively.51 These findings indicate that the evolved products originate from the photoreduction of 13CO2 rather than other carbon-containing species present in the reaction system, such as DDT. Furthermore, this conclusion is reinforced by the results of the control experiments shown in Fig. S23.†
Additionally, the simultaneous detection of O2 with carbon derivatives suggests that the designed catalyst can couple H2O oxidation with CO2 reduction reactions. To confirm the suitability of the band structures of the designed catalyst for both CO2 reduction and H2O oxidation reactions, the valence-band (VB) XPS spectra of O–CHS and CHS were examined.52 As depicted in Fig. 4e, the VB positions of both O–CHS and CHS are at 1.18 eV, consistent with the literature.53 In combination with the bandgaps obtained from UV-vis results, the band structure alignments are schematically presented in Fig. 4f. It is evident that the conduction-band (CB) positions exhibit greater negativity compared to the reduction potentials associated with carbonaceous product formation, thereby providing a strong driving force for the PCRR thermodynamically.54 Notably, the VB positions lie below the oxidation potential of H2O (E (O2/H2O) = 0.82 V at pH = 7).53,55 Therefore, the H2O oxidation reaction driven by photogenerated holes is also thermodynamically feasible, coinciding with the observed O2 generation in Fig. 4b, albeit in a sub-stoichiometric ratio.56 Moreover, lattice oxygen could serve as the active site toward O2 evolution due to the presence of hybridization between the oxygen orbital and the Fermi level of the photocatalysts in the highly active oxide.23,57
Typically, sub-stoichiometric O2 production is lower than that of the main carbon derivatives. Some of the generated O2 or the photogenerated holes may be consumed in the formation of H2O2 (Fig. S24a†), the re-oxidation of carbon derivatives, or the photocorrosion of Cu2O nanoparticles, which is a common phenomenon for Cu2O-based photocatalysts.23,53 To ascertain whether the hydrophobically treated catalyst exhibits improved stability, the yields of carbon derivatives over O–CHS and CHS were monitored as a function of reaction time. As shown in Fig. S24b,† O–CHS displays a consistent increase in the yield of carbon derivatives within six hours. However, CHS experiences a decrease in the yield of carbon derivatives after three hours of reaction. To further estimate the stability of O–CHS, activity recycle tests of the overall PCRR using O–CHS were performed for three consecutive cycles, with each cycle lasting 3 h under identical reaction conditions. The results demonstrate that even after three cycles, the yields of ethanol and methanol remain as high as 56.51 and 52.78 μmol, respectively, retaining approximately 82% of the initial activity. This observation serves as evidence for the good stability of O–CHS (Fig. S24c and d†). The investigation of spent O–CHS and CHS after the reaction reveals that the morphology and chemical composition of spent O–CHS are better preserved (Fig. S25†). This fact may be attributed to its significant nano-fence effect, which repels H2O. It is well-known that H2O is involved in the electron-mediated photocorrosion reaction of Cu2O.58,59 Additionally, the faster reaction rate observed over O–CHS prevents the accumulation of charge carriers on the Cu2O surface. Consequently, this further protects O–CHS from photocorrosion, improving its stability.20,60
Mechanism study
In situ FTIR was first employed to detect the reaction intermediates for O–CHS and CHS. As depicted in Fig. 5a and b, there are no obvious peaks prior to light irradiation. However, several peaks gradually emerge and intensify within the 1220 to 2120 cm−1 range over time. Among these, two major peaks at 1647 and 1545 cm−1 can be attributed to the absorption of the carbonyl group in CO2 and *COOH group, respectively.52,61,62 This implies that *COOH acts as a crucial intermediate during the overall PCRR. Additionally, the interaction between CO2 and H2O co-adsorbed on the Cu2O surface leads to the appearance of monodentate carbonate (m-CO32−) peaks at 1507 and 1558 cm−1, which indicates the chemisorption of CO2.63,64 The peak at 2077 cm−1 corresponds to the *CO species, which is generated through a subsequent *COOH protonation process.65 The presence of *CHO is also observed at 1733 and 1760 cm−1, proving evidence for the hydrogenation of *CO during the overall PCRR.37,66 Importantly, two distinct peaks at 1313 and 1520 cm−1 are observed, which could be attributed to the vibrational signature of *OCCHO, implying the occurrence of C–C coupling.67,68 Consequently, it can be inferred that the potential pathway for CO2 photoreduction could proceed as follows.69 (i) The transfer of electrons from the surface of Cu2O to CO2 initiates the single-electron activation process.70 (ii) The activated CO2 interacts with a hydrogen atom to form *COOH. (iii) *COOH then undergoes further reactions involving electrons and hydrogen atoms to form *CO. (iv) *CO then combines with other intermediate species, leading to C–C coupling and subsequent steps involving multiple electron transfers and hydrogenations, ultimately yielding the desired products.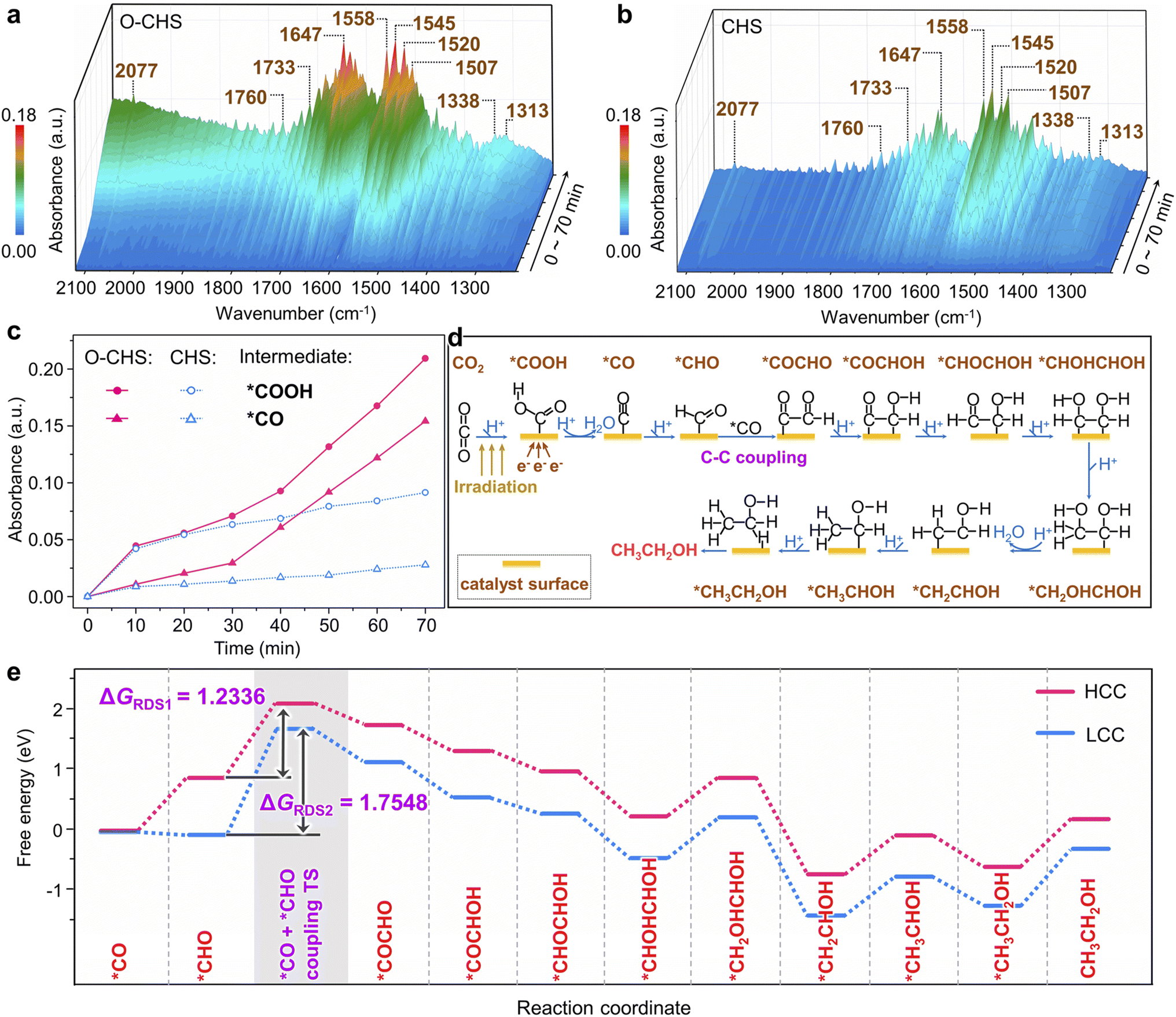 | ||
| Fig. 5 Mechanism insights. (a and b) In situ FTIR spectra obtained under different irradiation times for O–CHS and CHS, respectively. (c) Change of absorbance intensity of two intermediates over time. (d) Possible CO2-to-ethanol reaction pathway. (e) Gibbs free energy diagram for the reaction pathway of ethanol formation on Cu2O (111) surface at high *CO coverage (HCC) and low *CO coverage (LCC). ΔGRDS means the Gibbs free energy change for the rate-determining step of ethanol formation. TS means the transition state. | ||
Notably, peaks present in Fig. 5a also appear in Fig. 5b, indicating the generation of the same type of intermediates on both O–CHS and CHS during the reaction. However, the different intensities imply different concentrations of generated intermediates under the same irradiation time, which can be clearly observed in Fig. 5c. The peak intensity of the characteristic *COOH and *CO increases rapidly within the first 20 minutes for both O–CHS and CHS, but after that, it is nearly stable for CHS. Conversely, for O–CHS, it continues to increase rapidly over time. This indicates that the effective CO2 adsorption in the form of *COOH is indeed enhanced over O–CHS, leading to enhanced *CO formation. This is a consequence of the highly efficient CO2 transport and highly concentrated CO2 on the catalyst surface, which further promotes the overall PCRR.21,52
Based on the analysis results of in situ FTIR spectra, the influence of highly concentrated local CO2 on the catalyst surface is further explored using DFT calculations. It is evident from Fig. 5c that O–CHS exhibits a higher surface *CO coverage compared to CHS. Consequently, distinct *CO coverages are adopted to simulate the reaction processes under different local CO2 concentrations, as referenced in the existing literature.71,72 The different configurations and corresponding energies of each state during the calculation are presented in Fig. S26–S31 and Table S4–S8,† respectively. Regarding the reaction pathway for ethanol formation, DFT calculations reveal that it is identical on the Cu2O surface at both high *CO coverage (HCC) and low *CO coverage (LCC) (Fig. 5d and Table S9†). Notably, the C–C coupling step exhibits the highest energy barrier, denoted as ΔGRDS, which is considered to be the rate-determining step (RDS) during ethanol production (Fig. 5e). It is evident that ΔGRDS1 with a value of 1.2336 eV, is lower by 0.5212 eV in the case of HCC than that in the case of LCC. This result indicates that C–C coupling is more likely to occur on the Cu2O surface under HCC conditions. Similarly, the reaction pathways for methanol formation at the two *CO coverages are also found to be identical (Fig. S32a and Table S10†). The energy comparison of the RDS for methanol formation at the two *CO coverages also demonstrates the preference for HCC in methanol production (Fig. S32b†).
Overall, the DFT calculation results confirm that increasing CO2 concentration on the catalyst surface promotes the generation of carbon derivatives, specifically facilitating the C–C coupling for ethanol production. This finding further supports our conclusion that maintaining a high CO2 concentration on the O–CHS surface improves the efficiency of O–CHS in the overall PCRR, particularly in terms of ethanol production.
Conclusions
In summary, our research successfully achieves efficient ethanol production using a Cu2O photocatalyst by addressing the challenge of efficiently continuous CO2 transport to the catalyst surface. The target catalyst, O–CHS, is designed to be a superhydrophobic hollow structure that can float on an aqueous solution through surface modification and morphological control. Its superhydrophobic surface acts as a nano fence, effectively repelling water from infiltrating its inner space, while its hollow structure provides a space for CO2 molecules to accumulate. As CO2 is consumed during the overall PCRR, O–CHS functions as a gas transport channel and diffuser, continuously and rapidly conveying CO2 from the gas phase to the reactive sites on the O–CHS surface under the driving force of the CO2 concentration gradient. This ensures a stable high CO2 concentration at the reactive sites. Mechanistic investigations indicate that the efficient enrichment of CO2 on the catalyst surface lowers the energy barrier of C–C coupling, thereby enhancing the reaction efficiency of ethanol production. Notably, O–CHS exhibits the highest reported ethanol generation rate of 996.18 μmol g−1 h−1 to the best of our knowledge. The STE and selectivity for ethanol production are also up to 0.152%, and 59.59%, respectively. This breakthrough offers promising prospects for the production of high-value C2+ products and the development of renewable energy technologies.Data availability
The data supporting the findings of this study, including chemicals, sample preparation, instrumentation, supplementary tests, DFT calculations, and corresponding supplementary discussions, are available in ESI.†Author contributions
Conceptualization: Hailing Huo, Ang Li; methodology: Hailing Huo, Chengxi Huang, Fang Wu, Yongping Du, Ang Li, Erjun Kan; investigation: Hailing Huo, Hua He, Xin Guan, Hongbin Xing; supervision: Ang Li, Erjun Kan; validation: Hailing Huo, Hongbin Xing; funding acquisition: Erjun Kan, Ang Li; writing – original draft: Hailing Huo, Ang Li; writing – review & editing: Hailing Huo, Ang Li, Erjun Kan.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22378204, 22008121, and 51790492), National Outstanding Youth Science Fund Project of National Natural Science Foundation of China (T2125004), Funding of NJUST (no. TSXK2022D002), and Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX23_0454).References
- H. Huo, D. Liu, H. Feng, Z. Tian, X. Liu and A. Li, Nanoscale, 2020, 12, 13912–13917 RSC
.
- H. Huo, F. Wu, E. Kan and A. Li, Chem. – Eur. J., 2023, 29, e202300658 CrossRef CAS
- J. Albero, Y. Peng and H. García, ACS Catal., 2020, 10, 5734–5749 CrossRef CAS
- J. Strunk, Nat. Chem., 2023, 15, 1209–1211 CrossRef CAS PubMed
- L. Ding, N. Zhu, Y. Hu, Z. Chen, P. Song, T. Sheng, Z. Wu and Y. Xiong, Angew. Chem., Int. Ed., 2022, 61, e202209268 CrossRef CAS PubMed
- K. Wu, C. Liu, Y. Chen, H. Jiang, Q. Peng, Y. Chen, D. Fang, B. Shen, Q. Wu, L. Zhan, W. Sun, W. Di and H. Sun, Appl. Catal., A, 2023, 650, 118970 CrossRef CAS
- L. Hurtado, R. Natividad and H. García, Catal. Commun., 2016, 84, 30–35 CrossRef CAS
- P. Li, L. Liu, W. An, H. Wang and W. Cui, Appl. Surf. Sci., 2021, 565, 150448 CrossRef CAS
- Q. Jia, J. Zhou, L. Gong, L. Wang, X. Ma and Y. Zhao, Mol. Catal., 2022, 530, 112599 CrossRef CAS
- S. Gao, H. Guan, H. Wang, X. Yang, W. Yang and Q. Li, J. Adv. Ceram., 2022, 11, 1404–1416 CrossRef CAS
- N.-N. Vu, C.-C. Nguyen, S. Kaliaguine and T.-O. Do, Adv. Sustainable Syst., 2017, 1, 1700048 CrossRef
- H. Shi, H. Wang, Y. Zhou, J. Li, P. Zhai, X. Li, G. G. Gurzadyan, J. Hou, H. Yang and X. Guo, Angew. Chem., Int. Ed., 2022, 61, e202208904 CrossRef CAS PubMed
- S. Bai, H. Qiu, M. Song, G. He, F. Wang, Y. Liu and L. Guo, eScience, 2022, 2, 428–437 CrossRef
- Y. C. Tan, K. B. Lee, H. Song and J. Oh, Joule, 2020, 4, 1104–1120 CrossRef CAS
- J. Li, G. Chen, Y. Zhu, Z. Liang, A. Pei, C.-L. Wu, H. Wang, H. R. Lee, K. Liu, S. Chu and Y. Cui, Nat. Catal., 2018, 1, 592–600 CrossRef CAS
- X. Liu, C. Huang, B. Ouyang, Y. Du, B. Fu, Z. Du, Q. Ju, J. Ma, A. Li and E. Kan, Chem. – Eur. J., 2022, 28, e202201034 CrossRef CAS
- Y. Liu, D. Shen, Q. Zhang, Y. Lin and F. Peng, Appl. Catal., B, 2021, 283, 119630 CrossRef CAS
- H. Jian, K. Deng, T. Wang, C. Huang, F. Wu, H. Huo, B. Ouyang, X. Liu, J. Ma, E. Kan and A. Li, Chin. Chem. Lett., 2024, 35, 108651 CrossRef CAS
- H. Huang, R. Shi, Z. Li, J. Zhao, C. Su and T. Zhang, Angew. Chem., Int. Ed., 2022, 134, e202200802 CrossRef
- Y. Xia, K. Xiao, B. Cheng, J. Yu, L. Jiang, M. Antonietti and S. Cao, ChemSusChem, 2020, 13, 1730–1734 CrossRef CAS PubMed
- A. Li, Q. Cao, G. Zhou, B. Schmidt, W. Zhu, X. Yuan, H. Huo, J. Gong and M. Antonietti, Angew. Chem., Int. Ed., 2019, 58, 14549–14555 CrossRef CAS PubMed
- Z.-P. Li, Y.-Q. Wen, J.-P. Shang, M.-X. Wu, L.-F. Wang and Y. Guo, Chin. Chem. Lett., 2014, 25, 287–291 CrossRef CAS
- Y. A. Wu, I. McNulty, C. Liu, K. C. Lau, Q. Liu, A. P. Paulikas, C.-J. Sun, Z. Cai, J. R. Guest, Y. Ren, V. Stamenkovic, L. A. Curtiss, Y. Liu and T. Rajh, Nat. Energy, 2019, 4, 957–968 CrossRef CAS
- R. Xu, H. Xu, S. Ning, Q. Zhang, Z. Yang and J. Ye, Trans. Tianjin Univ., 2020, 26, 470–478 CrossRef CAS
- J. Gao, Q. Li, H. Zhao, L. Li, C. Liu, Q. Gong and L. Qi, Chem. Mater., 2008, 20, 6263–6269 CrossRef CAS
- I. A. Larmour, S. E. Bell and G. C. Saunders, Angew. Chem., Int. Ed., 2007, 46, 1710–1712 CrossRef CAS
- L. Xiong, X. Zhang, L. Chen, Z. Deng, S. Han, Y. Chen, J. Zhong, H. Sun, Y. Lian, B. Yang, X. Yuan, H. Yu, Y. Liu, X. Yang, J. Guo, M. H. Rummeli, Y. Jiao and Y. Peng, Adv. Mater., 2021, 33, e2101741 CrossRef
- X. An, K. Li and J. Tang, ChemSusChem, 2014, 7, 1086–1093 CrossRef CAS PubMed
- C. Kim, K. M. Cho, K. Park, J. Y. Kim, G. T. Yun, F. M. Toma, I. Gereige and H. T. Jung, Adv. Funct. Mater., 2021, 31, 2102142 CrossRef CAS
- H. Wang, Z. Lu, S. Xu, D. Kong, J. J. Cha, G. Zheng, P. C. Hsu, K. Yan, D. Bradshaw, F. B. Prinz and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 19701–19706 CrossRef CAS PubMed
- P. E. Laibinis, G. M. Whitesides, D. L. Allara, Y. T. Tao, A. N. Parikh and R. G. Nuzzo, J. Am. Chem. Soc., 1991, 113, 7152–7167 CrossRef CAS
- D. Wakerley, S. Lamaison, F. Ozanam, N. Menguy, D. Mercier, P. Marcus, M. Fontecave and V. Mougel, Nat. Mater., 2019, 18, 1222–1227 CrossRef CAS PubMed
- V. S. Dilimon, J. Denayer, J. Delhalle and Z. Mekhalif, Langmuir, 2012, 28, 6857–6865 CrossRef CAS PubMed
- Y. Yamamoto, H. Nishihara and K. Aramaki, J. Electrochem. Soc., 1993, 140, 436 CrossRef CAS
- L.-H. Kong, X.-H. Chen, L.-G. Yu, Z.-S. Wu and P.-Y. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 2616–2625 CrossRef CAS
- D. Gao, J. Xu, L. Wang, B. Zhu, H. Yu and J. Yu, Adv. Mater., 2022, 34, 2108475 CrossRef CAS
-
J. G. Speight, in Lange's Handbook of Chemistry, McGraw-Hill Education, 2017, ch. 3, pp. 3.3–3.53 Search PubMed
- L. Jiang, T. You, P. Yin, Y. Shang, D. Zhang, L. Guo and S. Yang, Nanoscale, 2013, 5, 2784–2789 RSC
- Y.-K. Hsu, J.-R. Wu, M.-H. Chen, Y.-C. Chen and Y.-G. Lin, Appl. Surf. Sci., 2015, 354, 8–13 CrossRef CAS
- M. Xiao, Z. Wang, M. Lyu, B. Luo, S. Wang, G. Liu, H. M. Cheng and L. Wang, Adv. Mater., 2019, 31, e1801369 CrossRef
- A. Li, X. Chang, Z. Huang, C. Li, Y. Wei, L. Zhang, T. Wang and J. Gong, Angew. Chem., 2016, 128, 13938–13942 CrossRef
- Z. Y. Tian, L. H. Kong, Y. Wang, H. J. Wang, Y. J. Wang, S. Yao, T. B. Lu and Z. M. Zhang, Small, 2021, 17, 2103558 CrossRef CAS PubMed
- Y. Wei, N. Yang, K. Huang, J. Wan, F. You, R. Yu, S. Feng and D. Wang, Adv. Mater., 2020, 32, e2002556 CrossRef PubMed
- J. Mao, T. Peng, X. Zhang, K. Li, L. Ye and L. Zan, Catal. Sci. Technol., 2013, 3, 1253–1260 RSC
- O. Ola and M. M. Maroto-Valer, Appl. Catal., A, 2015, 502, 114–121 CrossRef CAS
- Q. Han, Y. Zhou, L. Tang, P. Li, W. Tu, L. Li, H. Li and Z. Zou, RSC Adv., 2016, 6, 90792–90796 RSC
- O. Ola and M. M. Maroto-Valer, J. Catal., 2014, 309, 300–308 CrossRef CAS
- J.-P. Zou, D.-D. Wu, J. Luo, Q.-J. Xing, X.-B. Luo, W.-H. Dong, S.-L. Luo, H.-M. Du and S. L. Suib, ACS Catal., 2016, 6, 6861–6867 CrossRef CAS
- L. Tang, L. Kuai, Y. Li, H. Li, Y. Zhou and Z. Zou, Nanotechnology, 2018, 29, 064003 CrossRef PubMed
- B. An, Z. Li, Y. Song, J. Zhang, L. Zeng, C. Wang and W. Lin, Nat. Catal., 2019, 2, 709–717 CrossRef CAS
- Y. Wang, X. Liu, X. Han, R. Godin, J. Chen, W. Zhou, C. Jiang, J. F. Thompson, K. B. Mustafa, S. A. Shevlin, J. R. Durrant, Z. Guo and J. Tang, Nat. Commun., 2020, 11, 2531 CrossRef CAS
- W. Shangguan, Q. Liu, Y. Wang, N. Sun, Y. Liu, R. Zhao, Y. Li, C. Wang and J. Zhao, Nat. Commun., 2022, 13, 1–11 Search PubMed
- L. Yu, G. Li, X. Zhang, X. Ba, G. Shi, Y. Li, P. K. Wong, J. C. Yu and Y. Yu, ACS Catal., 2016, 6, 6444–6454 CrossRef CAS
- L. Yu, X. Ba, M. Qiu, Y. Li, L. Shuai, W. Zhang, Z. Ren and Y. Yu, Nano Energy, 2019, 60, 576–582 CrossRef CAS
- J. Abdul Nasir, A. Munir, N. Ahmad, T. U. Haq, Z. Khan and Z. Rehman, Adv. Mater., 2021, 33, 2105195 CrossRef CAS
- W. C. Chueh, C. Falter, M. Abbott, D. Scipio, P. Furler, S. M. Haile and A. Steinfeld, Science, 2010, 330, 1797–1801 CrossRef CAS PubMed
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y.-L. Lee, L. Giordano, K. A. Stoerzinger, M. T. M. Koper and Y. Shao-Horn, Nat. Chem., 2017, 9, 457–465 CrossRef CAS PubMed
- C. Y. Toe, J. Scott, R. Amal and Y. H. Ng, J. Photochem. Photobiol., C, 2019, 40, 191–211 CrossRef CAS
- R. D. Zhonghai Zhang, L. Zhang, H. Zhu, H. Zhang and P. Wang, ACS Nano, 2013, 7, 1709–1717 CrossRef
- C. Y. Toe, Z. Zheng, H. Wu, J. Scott, R. Amal and Y. H. Ng, Angew. Chem., Int. Ed., 2018, 57, 13613–13617 CrossRef CAS PubMed
- Z.-Z. Yang, D.-e. Jiang, X. Zhu, C. Tian, S. Brown, C.-L. Do-Thanh, L.-N. He and S. Dai, Green Chem., 2014, 16, 253–258 RSC
- X. Li, L. Liang, Y. Sun, J. Xu, X. Jiao, X. Xu, H. Ju, Y. Pan, J. Zhu and Y. Xie, J. Am. Chem. Soc., 2019, 141, 423–430 CrossRef CAS PubMed
- H. Yu, J. Li, Y. Zhang, S. Yang, K. Han, F. Dong, T. Ma and H. Huang, Angew. Chem., Int. Ed., 2019, 58, 3880–3884 CrossRef CAS PubMed
- X. Xiong, Y. Zhao, R. Shi, W. Yin, Y. Zhao, G. I. N. Waterhouse and T. Zhang, Sci. Bull., 2020, 65, 987–994 CrossRef CAS
- J. Wu, X. Li, W. Shi, P. Ling, Y. Sun, X. Jiao, S. Gao, L. Liang, J. Xu, W. Yan, C. Wang and Y. Xie, Angew. Chem., Int. Ed., 2018, 57, 8719–8723 CrossRef CAS PubMed
- Y. Kim, S. Park, S.-J. Shin, W. Choi, B. K. Min, H. Kim, W. Kim and Y. J. Hwang, Energy Environ. Sci., 2020, 13, 4301–4311 RSC
- E. Perez-Gallent, M. C. Figueiredo, F. Calle-Vallejo and M. T. Koper, Angew. Chem., Int. Ed., 2017, 56, 3621–3624 CrossRef CAS PubMed
- H. Ou, G. Li, W. Ren, B. Pan, G. Luo, Z. Hu, D. Wang and Y. Li, J. Am. Chem. Soc., 2022, 144, 22075–22082 CrossRef CAS
- X. Xiong, C. Mao, Z. Yang, Q. Zhang, G. I. N. Waterhouse, L. Gu and T. Zhang, Adv. Energy Mater., 2020, 10, 2002928 CrossRef CAS
- J. Di, B. Lin, B. Tang, S. Guo, J. Zhou and Z. Liu, Small Struct., 2021, 2, 2100046 CrossRef CAS
- X. Y. Zhang, W. J. Li, X. F. Wu, Y. W. Liu, J. Chen, M. Zhu, H. Y. Yuan, S. Dai, H. F. Wang, Z. Jiang, P. F. Liu and H. G. Yang, Energy Environ. Sci., 2022, 15, 234–243 RSC
- X. Wang, A. Xu, F. Li, S. F. Hung, D. H. Nam, C. M. Gabardo, Z. Wang, Y. Xu, A. Ozden, A. S. Rasouli, A. H. Ip, D. Sinton and E. H. Sargent, J. Am. Chem. Soc., 2020, 142, 3525–3531 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc05702j |
| This journal is © The Royal Society of Chemistry 2024 |