
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advancement on photocatalytic plastic upcycling
Jingrun
Ran†
,
Amin
Talebian-Kiakalaieh†
,
Shuai
Zhang
,
Elhussein M.
Hashem
,
Meijun
Guo
and
Shi-Zhang
Qiao
 *
*
School of Chemical Engineering, University of Adelaide, Adelaide, SA 5005, Australia. E-mail: s.qiao@adelaide.edu.au
First published on 27th December 2023
Abstract
More than 8 billion tons of plastics have been generated since 1950. About 80% of these plastics have been dumped in landfills or went into natural environments, resulting in ever-worsening contamination. Among various strategies for waste plastics processing (e.g., incineration, mechanical recycling, thermochemical conversion and electrocatalytic/photocatalytic techniques), photocatalysis stands out as a cost-effective, environmentally benign and clean technique to upcycle plastic waste at ambient temperature and pressure using solar light. The mild reaction conditions for photocatalysis enable the highly selective conversion of plastic waste into targeted value-added chemicals/fuels. Here, we for the first time summarize the recent development of photocatalytic plastic upcycling based on the chemical composition of photocatalysts (e.g., metal oxides, metal sulfides, non-metals and composites). The pros and cons of various photocatalysts have been critically discussed and summarized. At last, the future challenges and opportunities in this area are presented in this review.
| Jingrun Ran Dr Jingrun Ran has received his PhD degree in Chemical Engineering from the University of Adelaide under the supervision of Professor Shi-Zhang Qiao. Currently he is appointed as a Senior Lecturer in the School of Chemical Engineering in the University of Adelaide and working in Professor Qiao's group. In 2020–2023, he has been recognised as a Clarivate Highly Cited Researcher. In 2023, he was awarded the ARC Future Fellowship. His current research is focused on the atomic-scale design and synthesis of photocatalysts for producing energy fuels and value-added chemicals using renewable solar energy. |
| Amin Talebian-Kiakalaieh Amin Talebian-Kiakalaieh received his bachelor's degree in petroleum engineering in 2007. Then he changed his major to Chemical Engineering and received his master's degree in chemical engineering from Universiti Teknologi Malaysia (UTM) in 2012. He joined the Prof. Shi-Zhang Qiao group in 2021 as a PhD candidate under the supervision of Dr Jingrun Ran at the University of Adelaide. Currently he is working on synthesizing advanced photocatalysts for renewable energy applications. |
| Shuai Zhang Shuai Zhang received his BS degree from the Beijing University of Chemical Technology and his MS degree from the Technical Institute of Physics and Chemistry, Chinese Academy of Sciences. He is now a PhD candidate under the supervision of Prof. Shi-Zhang Qiao and Dr Jingrun Ran at the University of Adelaide. Currently, Shuai is working on solar-driven chemical conversions using advanced catalysts. |
| Elhussein M. Hashem Elhussein M. Hashem is currently pursuing his PhD degree in the School of Chemical Engineering at the University of Adelaide as a part of Prof. Shi-Zhang Qiao's esteemed research group. Under the supervision of Dr Jingrun Ran, his research is primarily focused on the development and rational design of advanced atomic-level photocatalysts that can efficiently produce solar fuels and value-added chemicals across a wide range of applications. |
| Meijun Guo Meijun Guo received her Bachelor's and Master's degrees in Environmental Science from Heilongjiang University. She is currently a PhD candidate in Prof. Shi-Zhang Qiao's group at the University of Adelaide, focusing on the atomic level design and synthesis of highly efficient photocatalysts for solar energy conversion and value-added chemical production. |
| Shi-Zhang Qiao Shi-Zhang Qiao is currently a chair professor at the School of Chemical Engineering of The University of Adelaide, Australia. His research expertise is in functional materials for new energy technologies. He has co-authored more than 530 papers in refereed journals with over 120, 000 citations and an h-index of 177. Dr Qiao is a Thomson Reuters/Clarivate Analytics Highly Cited Researcher in three categories (Chemistry, Materials Science, and Environment and Ecology). He is an elected fellow of the Australian Academy of Science (FAA) and has been awarded several prestigious awards, including the inaugural ARC Industry Laureate Fellow, South Australian Scientist of the Year (2021), ARC Australian Laureate Fellow, ExxonMobil Award, and ARC Discovery Outstanding Researcher Award among others. He is the founding Editor-in-Chief of EES Catalysis. |
1. Introduction
Up till now, more than 8 billion tons of plastics have been synthesized, in which less than 20% of them are incinerated or recycled. Around 80% of used plastics are accumulated in the natural environment or ended up in landfills.1–33 Recently, various dealing strategies have been developed for treating plastic wastes, such as incineration, mechanical recycling, thermochemical conversion and electrocatalytic/photocatalytic techniques.1–33 Among them, large-scale incineration and landfills are the two most general routes because they are inexpensive, facile and adaptable to various feedstock.1–33 Unfortunately, these two traditional routes are susceptible to deleterious environmental impacts and negligible added values.1–33 Low value products are manufactured by mechanical recycling, which down-cycles plastic wastes into low value products and is also restricted to single-component and few types of clean thermoplastics.1–33 In contrast, upcycling conversion of plastic wastes, e.g., thermocatalytic/photocatalytic/electrocatalytic techniques, has attracted significant attention recently, since value-added chemicals/fuels or materials with extra economic value can be acquired via these appealing techniques.1–33 Among these techniques, photocatalysis stands out as a cost-effective, environmentally-benign and green strategy able to upcycle plastic wastes at ambient temperature and pressure utilizing renewable sunlight.34–79 A photocatalysis reaction conducted in mild conditions is anticipated to accurately activate target chemical bonds, while reserving the other functional groups, thus realizing high selectivity to desirable products.34–49Recently, various photocatalysts, such as metal oxides (e.g., Pt loaded P25 TiO2 (ref. 34 and 35) and Co doped Ga2O3 (ref. 37)), metal sulphides (e.g., MoS2 tipped CdS nanorods39), non-metals (e.g., graphitic carbon nitride)41–44 and composites (e.g., ZnO encapsulated in UiO66-NH2 (ref. 46) and Ag2O encapsulated in Fe based MOFs47), have been developed for catalytic upcycling of plastics. To date, many reviews have covered upcycling of plastic waste,1–31,80,81 including various upcycling techniques such as the photo-electrochemical (PEC) technique, photo-thermal technique, photocatalysis technique, thermo-catalysis technique, bio-catalysis technique, pyrolysis technique, hydrogenolysis technique, solvolysis technique, hydrolysis technique, microwave-assisted technique, plasma-assisted technique, mechanical technique and combination of these techniques.3,5–17,19–33,81 Among these reviews, the introduction/discussion of the photocatalysis technique is not insightful and comprehensive. Some reviews only introduce the mechanisms and applications of various photocatalysts for plastic upcycling.1,2,4,6,18,80 But the advantages/disadvantages of various photocatalysts are rarely discussed and highlighted. Besides, the structure–performance relationship of these photocatalysts is much less summarized. Moreover, the insightful reaction mechanism of photocatalytic plastic upcycling under various reactions conditions is still not very clear at this stage. Especially, oxygen plays a key role in plastic upcycling, which should be explained more clearly and thoroughly.
In this review, we for the first time summarize and review recently reported photocatalysts for plastic upcycling based on their chemical compositions including metal oxides, metal sulphides, non-metals and composites. The accurate advantages/disadvantages of photocatalysts are critically analysed and discussed according to their chemical compositions. Especially, the perceptive reaction mechanisms for various photocatalysts under varied reaction conditions are also introduced and summarized in this review.
2. Fundamentals of photocatalytic plastic upcycling
Owing to the ultrastability of most plastics, such as polyolefins and polyesters, it is very challenging to directly convert these plastics into valuable chemicals via photocatalysis at ambient temperature and pressure using sunlight only. Thus, in most cases, these plastics are usually pre-treated by various routes to release monomers/oligomers or to be converted into short-chain carbon-based molecules before photocatalysis treatment.34,38–43,49 For example, polyesters, e.g., polyethylene terephthalate (PET), polylactic acid (PLA) and polyurethane (PUR), are usually pre-treated in alkaline solution at slightly raised temperature (e.g., 40–70 °C) to be hydrolysed, thus releasing abundant monomers/oligomers.38–43,49 Additionally, polyolefins, e.g., polyethylene (PE), are usually hydrothermally treated in nitric acid aqueous solution, to be transformed into various short-chain carboxylic acids, e.g., succinic acid, glutaric acid, acetic acid, adipic acid and propanoic acid.34,39 On the other hand, in some rare cases, these plastics, e.g., PE, polyvinyl chloride (PVC), polypropylene (PP) and polystyrene (PS), are directly added into the reaction system for photocatalytic plastic upcycling, which can yield value-added chemicals/fuels, e.g., H2, CO, formic acid, acetic acid, benzoic acid, acetophenone and benzaldehyde.36,37,44–46,48 In some of these cases, metal oxide photocatalysts with strongly oxidative photo-induced holes are applied.36,37,46 In the other case, an air/O2 atmosphere and raised temperature/pressure are adopted to boost the oxidative cleavage of the strong C–C/C–H bonds in these untreated plastics.44 Another key reaction condition for photocatalytic plastic upcycling is the existence/absence of oxygen (O2) in the reaction system. If photocatalytic plastic upcycling is conducted under aerobic conditions, abundant reactive oxygen species (ROS), such as ˙O2−, ˙HO2− and ˙OH radicals, will be produced by the following reactions:36,37,44,46–48| e− + O2 → ˙O2− | (1) |
| ˙O2− + e− + 2H+ → H2O2 | (2) |
| H2O2 + hν → 2˙OH | (3) |
| H2O2 + ˙O2− → ˙OH + O2 + OH− | (4) |
| e− + H2O2 → ˙OH + OH− | (5) |
These ROSs will significantly boost the oxidative cleavage of the C–C/C–H/C–O/C–N/C–Cl bonds in the robust plastics and the production of short-chain carbon-based molecules as valuable chemicals/fuels.36,37,44,46–48 But O2 in the reaction system can compete with H+/H2O to obtain photo-induced electrons, resulting in a much lower H2 yield under aerobic conditions compared to that under anaerobic conditions. In contrast, if photocatalytic plastic upcycling is performed under anaerobic conditions, these ROSs are much more challenging to form in the reaction system unless the photo-induced holes with sufficient oxidation ability can oxidize OH−/H2O to yield ˙OH radicals. Without the competition from O2, many more photo-excited electrons can be utilized for H2 evolution, resulting in usually a much higher H2 evolution rate under anaerobic conditions compared to that under aerobic conditions.
On the basis of the above introductions, the reaction mechanisms for photocatalytic plastic upcycling are categorized into three different types: (i) photocatalytic upcycling with pre-treated plastics and under anaerobic conditions;34,38–43,49 (ii) photocatalytic upcycling with untreated plastics and under aerobic conditions;36,37,44,46,48 (iii) photocatalytic upcycling with untreated plastics and under anaerobic conditions45 (Fig. 1a–c). Besides, we have also discussed the reaction thermodynamics and kinetics for plastic upcycling as well as engineering routes on photocatalysts.
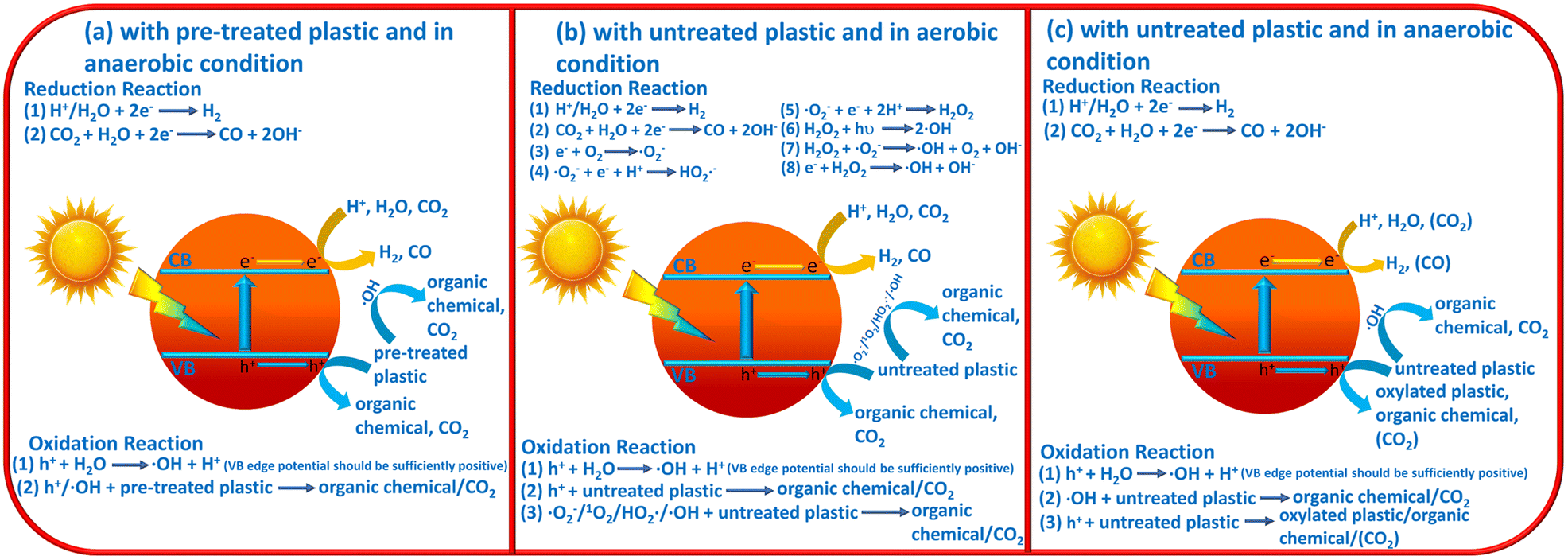 | ||
| Fig. 1 Schematic images for (a) photocatalytic plastic upcycling using pre-treated plastics and under anaerobic conditions, (b) photocatalytic plastic upcycling using untreated plastics and under aerobic conditions and (c) photocatalytic plastic upcycling using untreated plastics and under anaerobic conditions. | ||
2.1 Plastic upcycling with pre-treated plastics and under anaerobic conditions
As the most reported reaction condition for plastic upcycling,34,38–43,49 the reaction mechanism is summarized in Fig. 1a: (i) monomers/oligomers/short-chain carbon-based molecules/surface oxygenated plastics are first generated from the pre-treatment of various plastics; (ii) under anaerobic conditions, photo-excited electrons mainly involve in H2 evolution, thus resulting in a relatively high photocatalytic H2 evolution rate under these reaction conditions; (iii) photo-induced holes can oxidize the formed monomers/oligomers/short-chain carbon-based molecules to yield value-added chemicals/fuels; (iv) in some cases, if metal oxide photocatalysts with strongly oxidative holes are utilized, these monomers/oligomers/short-chain carbon-based molecules could be over-oxidized to yield CO2, which could be further reduced by photo-generated electrons to form CO.2.2 Plastic upcycling with untreated plastics and under aerobic conditions
Another often-reported reaction condition is plastic upcycling with untreated plastics and under aerobic conditions. The reaction mechanism is shown in Fig. 1b: (i) plastic powder is directly added into the reaction system; (ii) photocatalysts with strongly oxidative photo-induced holes, e.g., Nb2O5 atomic layers,36 Co doped Ga2O3 (ref. 37) and ZnO coupled UiO66-NH2,46 are usually adopted in this reaction system, which can oxidize H2O/OH− to form highly oxidative ˙OH radicals; (iii) the photo-induced electrons can involve in the reaction with O2 to form various ROSs, e.g., ˙O2−, 1O2, ˙HO2− and ˙OH; (iv) all the formed ROSs and strongly oxidative holes can oxygenate the plastics to yield hydroxyl, carbonyl and carboxyl groups, thus polarizing and greatly weakening the inert C–C/C–O/C–H/C–N bonds in plastics; (v) these C–C/C–O/C–H/C–N bonds of oxygenated plastics can be further oxidatively cleaved to yield short-chain carbon-based molecules or even over-oxidized to form CO2; (vi) photo-induced electrons can reduce the formed CO2 to yield CO or even carbon-based molecules, e.g., acetic acid;36 (vii) photo-induced electrons can also reduce H+/H2O to yield H2; (viii) in some cases, non-metal photocatalysts with moderate oxidation ability, e.g., g-C3N4, and organic solvent,44e.g., acetonitrile, instead of aqueous solution, are applied in the reaction system with raised temperature (e.g., 70 °C) rather than room temperature (25 °C).2.3 Plastic upcycling with untreated plastic and under anaerobic conditions
Photocatalytic plastic upcycling under these reaction conditions is rarely reported.45 And the reaction mechanism is not very clear. So we propose a possible mechanism in Fig. 1c as follows: (i) for the reduction reaction, under anaerobic conditions, photo-excited electrons mainly involve in H2 evolution via reducing H+/H2O in the reaction system. In some cases, CO2 could be generated due to the overoxidation of untreated plastic by strongly oxidative ˙OH and holes. Thus, generated CO2 can be further reduced by photo-excited electrons to produce some carbon-based fuels/chemicals; (ii) for the oxidation reaction, the photo-induced holes with sufficient oxidation ability can react with the H2O molecule to generate strongly oxidative ˙OH. Thus, ˙OH can react with untreated plastics to generate organic chemicals and CO2. In contrast, photo-excited holes with weak oxidative capacity can hardly cleave the C–C bond in untreated plastics to produce organic chemicals. And no CO2 would be generated under these conditions. Overall, photocatalytic plastic upcycling under these reaction conditions is rarely reported and more studies under these reaction conditions should be conducted to understand the reaction mechanism.2.4 Reaction thermodynamics and kinetics for plastic upcycling
Reaction thermodynamics and kinetics for photocatalytic upcycling of plastic wastes into fuels/valuable chemicals are complex. Nevertheless, compared to photocatalytic water splitting with a theoretical thermodynamic requirement of 1.23 eV (ΔG0 = +237 kJ mol−1), the thermodynamic requirement for photocatalytic plastic upcycling is usually much lower. This is because the oxidation of plastics, especially for small molecules from pre-treated plastics, is much more facile than water oxidation for O2 evolution. For instance, reforming ethylene glycol and lactic acid, from pre-treated PET and PLA, shows the Gibbs free energy changes of +9.2 and +27 kJ mol−1, respectively. These are much lower than that of water splitting (G0 = +237 kJ mol−1). Thus, the much less thermodynamics requirement for plastic oxidation compared to water splitting enables the utilization of a semiconductor photocatalyst with a smaller band gap, which exhibits a much broader light absorption range for the solar spectrum. Thus, nitride, sulphide, phosphate, arsenide, selenide and even tellurite catalysts, which show deficient valence band potential for water oxidation, could be suitable for photocatalytic plastic oxidation, especially for small molecules from pre-treated plastics. Thus, abundant and undesirable plastic wastes serve as an efficient hole scavenger to increase the electron–hole separation/transfer efficiency, thus boosting the reduction reaction (H2 evolution or CO2 reduction). Simultaneously, value-added chemicals/fuels can be generated via plastic oxidation.Reaction kinetics in photocatalytic plastic upcycling are very complex and are affected by many factors, such as plastic type, pre-treatment route, reaction solution and reaction atmosphere. First, the types of plastics obviously affect the reaction kinetics. Polyolefins (e.g., PE, PP, PS and PVC), which account for 57% of the total plastics, consist of inert C–C and C–H bonds with high dissociation energies. Thus, reaction kinetics for breaking these C–C/C–H bonds and upcycling polyolefins are very sluggish. Besides, the hydrophobic nature of polyolefins makes their dispersion in aqueous solution and adsorption on photocatalysts very challenging. In contrast, polyesters/polyamides (e.g., PET, PLA, PUR) with ester/amide bonds are much easier to decompose to yield the corresponding monomers, which are facilely adsorbed on photocatalysts for further forming value-added chemicals.
Second, the pre-treatment route also greatly affects the reaction kinetics. Without pre-treatment, the reaction kinetics for upcycling most plastics are rather slow, especially for inert polyolefins. The hydrothermal pre-treatment of polyolefins (e.g., PE) in nitric acid could cleave the C–C bond and convert most of them into various carboxylic acids (e.g., succinic acid, glutaric acid, acetic acid, adipic acid and propanoic acid). These short-chain water-soluble carboxylic acids could be more easily adsorbed onto photocatalysts and further yield value-added chemicals/fuels more efficiently. Furthermore, the hydrolysis pre-treatment of polyesters/polyamides (e.g., PET and PUR) in alkaline solution at elevated temperature could yield the corresponding monomers, which can be easily dissolved in aqueous solution and adsorbed on photocatalysts for more efficient conversion into valuable chemicals/fuels. Moreover, the plasma treatment of plastics at room temperature and atmosphere could graft oxygen-containing functionalities on the PE/PP/PVC surface, leading to increased hydrophilicity in water and more intimate interaction with photocatalysts. Thus, reaction kinetics for yielding fuels/valuable chemicals from pre-treated PE/PP/PVC is improved to some extent. Nevertheless, since this plasma treatment doesn't convert PE/PP/PVC into short-chain monomers/oligomers, the enhancement of reaction kinetics for plastic upcycling is limited.
Third, different reaction solutions (e.g., pure water, alkaline aqueous solution and organic solvent) also obviously affect the reaction kinetics for plastic upcycling. As pure water is applied as the reaction solution, the reaction kinetics for upcycling most plastics in pure water are very slow. This is because that most of the plastics are hydrophobic, which float on the surface of pure water or precipitate at the bottom rather than suspend in the water, making their adsorption or interaction with photocatalysts challenging. Besides, the undissolved plastics also hinder light absorption by photocatalysts to some extent. In contrast, as alkaline aqueous solution (e.g., NaOH and KOH) is applied as the reaction solution, some kinds of plastics, such as polyesters/polyamides, could be dissolved/hydrolysed in alkaline aqueous solution, yielding monomers easily adsorbed on photocatalysts for upcycling reactions. Thus, the corresponding reaction kinetics for these plastics are obviously improved. Nevertheless, this alkaline environment could corrode the photocatalysts and reduce their activity/selectivity/stability. As organic solvents (e.g., acetonitrile, tetrahydrofuran, cyclohexane and toluene) are utilized as the reaction solution, hydrophobic plastics (e.g., PE, PP, PS and PVC) can be dissolved in these organic solvents with stirring and elevated temperature. These will boost their adsorption on photocatalysts and further transformation into valuable chemicals/fuels. The activity/selectivity/stability of photocatalysts will also be affected. Moreover, by-products from these organic solvents might also be generated.
Fourth, the reaction atmosphere considerably influences the reaction kinetics for plastic upcycling. Under aerobic conditions, various ROS species (e.g., ˙O2−, 1O2, ˙HO2−, ˙OH and H2O2) could be generated to greatly boost the oxidation and conversion of plastics into value-added chemicals/fuels. Nevertheless, strongly oxidative and non-selective ROS could over-oxidize these plastics and generate CO2. In contrast, under anaerobic conditions, only photo-excited holes (and ˙OH) involve in the oxidation and conversion of plastics into valuable chemicals/fuels. Thus, it is much easier to regulate the oxidation ability of photo-excited holes for controlling the selectivity of plastic upcycling. Nevertheless, due to the lack of massive ROS species for oxidation of plastics, the reaction kinetics of plastic upcycling is reduced.
2.5 Engineering routes on photocatalysts
A range of engineering routes have been developed for optimizing the physicochemical features of photocatalysts to achieve increased photocatalytic activity/selectivity/stability for plastic upcycling. For instance, researchers have reported various strategies, such as loading co-catalysts,34,35,39–43,45 doping engineering,37 morphology engineering,36 surface engineering,38 band structure engineering40 and forming heterojunctions,45–49 which can significantly increase the photocatalytic performances. The accurate structure–performance relationship and reaction mechanism for these photocatalysts will be introduced in Section 3 as follows.3. Photocatalysts for plastic upcycling
Currently, the reported photocatalysts for plastic upcycling are categorized into metal oxide based photocatalysts,34–37 metal sulphide based photocatalysts,38–40 non-metal based photocatalysts41–44 and composite photocatalysts.45–49 We will introduce the research in these fields according to the above four categories.3.1 Metal oxide based photocatalysts for plastic upcycling
Various metal oxides, such as TiO2,34,35 Nb2O5 (ref. 36) and Ga2O3,37 have been applied for photocatalytic plastic upcycling. Owing to the strong oxidation abilities of metal oxide based photocatalysts arising from their deep O 2p derived valence band (VB), they are often adopted to upcycle polyolefins with strong C–C bonds, such as high density polyethylene (HDPE), low density polyethylene (LDPE), polypropylene (PP) and polyvinyl chloride (PVC). With light irradiation, they can generate strongly oxidative photo-induced holes and yield exceptionally oxidative hydroxyl radicals (˙OH) in aqueous solution under aerobic/anaerobic conditions (h+ + OH− → ˙OH). If applied in an air/O2 atmosphere, the photo-induced electrons in the CB of these metal oxide based photocatalysts usually can react with O2 to form a series of oxidative reactive oxygen species (ROSs), such as ˙O2− and ˙HO2− radicals. These ROSs (e.g., ˙OH, ˙O2− and ˙HO2−) can first oxidize polyolefins to form hydroxyl, carbonyl and carboxyl functionalities. Then, the strong C–C/C–H bonds of these polyolefins will be polarized and significantly weakened, resulting in the much easier cleaving of these C–C/C–H bonds and upcycling of these polyolefins into value-added chemicals/fuels.First, we will introduce two studies using the most extensively studied metal oxide photocatalyst, TiO2, for photocatalytic plastic upcycling under anaerobic conditions with pre-treated polyolefins.34,35 Both these studies adopt commercial P25 TiO2 loaded with the Pt cocatalyst. The Reisner34 group first used 6% nitric acid and a hydrothermal reaction at 180 °C for 4 hours to convert ∼40% polyethylene (PE) into a variety of liquid chemicals including succinic acid (44%), glutaric acid (22%), acetic acid (21%), adipic (9%) and propanoic acid (4%). Succinic acid and glutaric acid are identified as the major products from PE conversion (Fig. 2a). Then, they synthesized 1 wt% Pt nanoparticle (NP) loaded P25 TiO2 using a chemical reduction route. After a 96 hour reaction using the PE decomposition solutions, the as-synthesized 1 wt% Pt NP loaded P25 TiO2 exhibits photocatalytic performance for the evolution of ethylene (0.017 mmol g−1), ethane (0.25 mmol g−1), propylene (0.007 mmol g−1), propane (0.14 mmol g−1), H2 (6.3 mmol g−1) and CO2 (5.9 mmol g−1). Thus, ethane and propane are detected as the major alkane products from photo-reforming of PE decomposition solution (Fig. 2a). They found that only a small amount of ethylene is generated in this reaction, attributed to the efficient transfer of adsorbed hydrogen to the intermediate radical on 1 wt% Pt NP loaded P25 TiO2. Some of the generated CO2 and H2 arise from the decarboxylation reaction. Besides, due to the strong oxidation abilities of photogenerated holes in TiO2, mineralization of the acquired chemicals (e.g., succinic and glutaric acid) after pre-treatment can occur, also leading to CO2 and H2 evolution. 13C-labelled succinic acid using 1 wt% Pt NP loaded TiO2via1H-nuclear magnetic resonance (1H NMR) spectroscopy confirms that the evolved ethane is generated from the succinic acid. They found that without Pt as the co-catalyst, larger amounts of ethylene and smaller amounts of ethane are observed. Ethane became the major product again as MoS2 was adopted as the co-catalyst. They have also synthesized Pt loaded cyanamide-regulated carbon nitride powder (NCNCNx|Pt). After a 72 hour photocatalytic reaction, the major alkane products from P25|Pt and NCNCNx|Pt are ethane at 56.3 and 7.2 μmol g−1 h−1 (Fig. 2b and c). Then, they have set up a flow photocatalytic reactor system (Fig. 2d), in which a continuing generation of ethane and propane was achieved for both P25|Pt and NCNCNx|Pt (Fig. 2e and f), together with the constant production of ethylene and propylene for NCNCNx|Pt (Fig. 2f). In another study, a plasma pre-treatment strategy was reported to treat polyolefins to partially cleave the C–C/C–H bonds and generate oxygenated functional groups on the backbones of PE, PP or PVC.35 FTIR and XPS spectra together confirm the formation of hydroxyl, carboxyl and carbonyl functionalities on the surface of PE. Contact angle measurement further reveals the gradually reduced contact angle of PE with water as plasma treatment time increases, suggesting the increased hydrophilicity of treated PE. This will lead to better dispersion of treated PE in aqueous solution and more intimate contact between the photocatalyst and treated PE. Molecular dynamics computations reveal a stronger interaction between the TiO2 surface and plasma treated PE compared to that between the TiO2 surface and untreated PE.
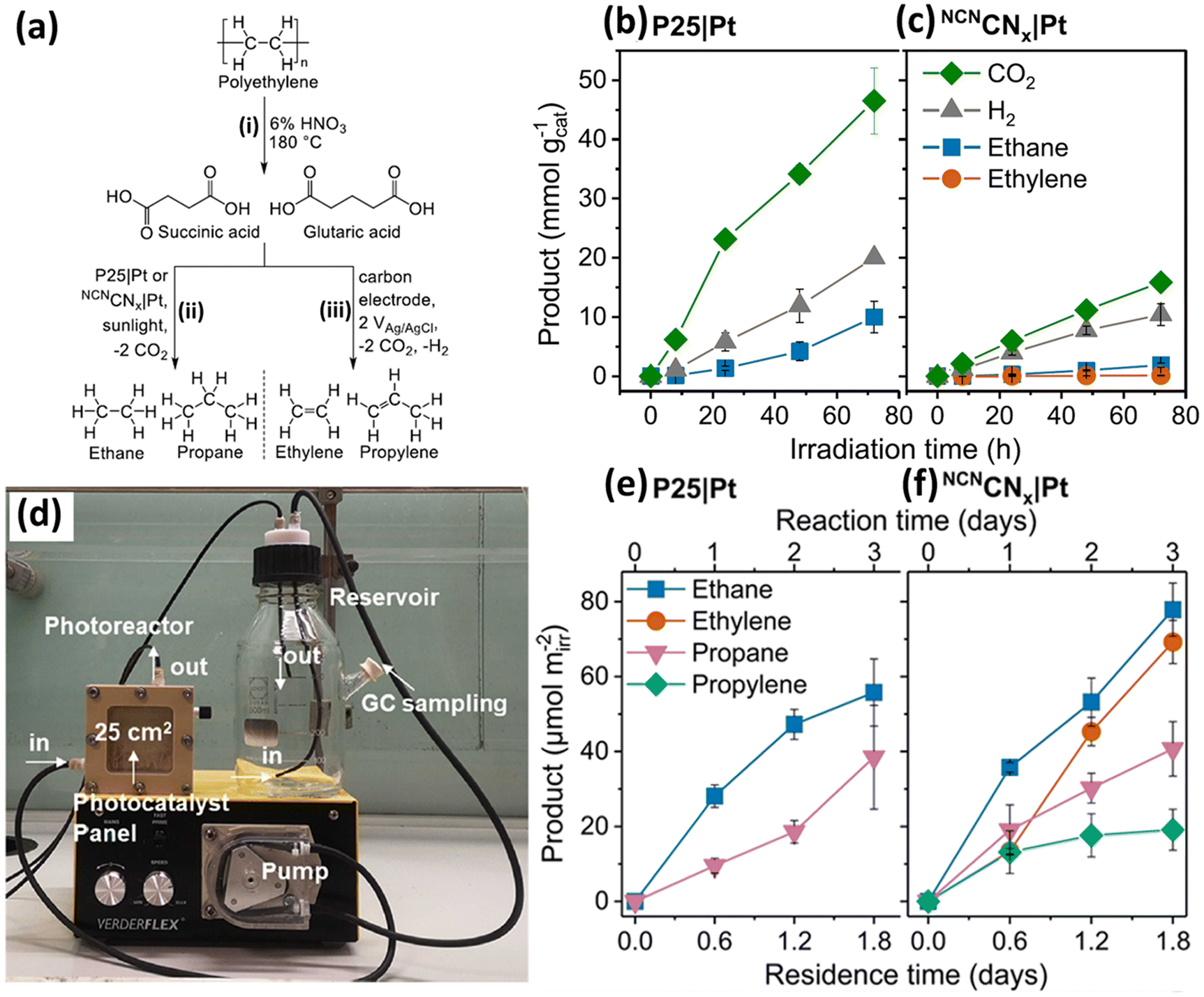 | ||
| Fig. 2 (a) Hydrothermal pre-treatment of PE to form dicarboxylic acid (i) followed by conversion into gaseous hydrocarbon using photocatalysis to yield alkanes (ii) or electrolysis to yield alkenes (iii). (b) Photocatalytic reforming of succinic acid in 0.1 M HNO3 using (b) P25|Pt or (c) NCNCNx|Pt. Reaction conditions: AM 1.5G (100 mW cm−2), 25 °C, 2 ml of 10 mg ml−1 succinic acid in 0.1 M HNO3 (pH set to be 4), and 2 mg ml−1 photocatalyst. (d) Image of the photocatalytic flow setup. The pre-treated PE solution (not displayed in this image) in the reservoir is continuously pumped with a peristaltic pump into the photoreactor with an irradiation area of 25 cm2. Then, the solution is pumped into the reservoir again; the generated gaseous products are sampled and studied by gas chromatography. Photocatalytic product generation using a flow setup with (e) P25|Pt and (f) NCNCNx|Pt. Reproduced with permission from copyright 2021, American Chemical Society.34 | ||
Pt NPs with sizes of 5–15 nm are loaded on P25 TiO2via photo-deposition to synthesize a Pt–TiO2 photocatalyst. Photocatalytic experiments show that H2 evolution first rises and then decreases with increasing plasma treatment time on PE using Pt–TiO2. This is because the first activity increase arises from the generation of abundant –OH functionalities on the surface of PE. The subsequent activity decrease is due to the formation of massive carbonyl and carboxyl functionalities on treated PE. Reforming of PE also generates CH4, C2H4 and C2H6. But no liquid products were found by 1H NMR for short-/long-term photocatalysis tests. This plasma treatment effect also increased the H2 evolution activity on treated PP or PVC compared to untreated PP or PVC, suggesting the universality of this route. When comparing the above two studies using the same photocatalyst (Pt loaded P25 TiO2) under similar reaction conditions (Table 1), we can find that the H2/C2H4/C2H6/CO2 evolution rates of PE upcycling using the hydrothermal pre-treatment are much higher than those of PE upcycling using the plasma pre-treatment. These are easy to understand because the hydrothermal treatment has already converted most of the PE into C2−6 based carboxylic acids, which are much easier to upcycle into short-chain molecule chemicals/fuels. In comparison, plasma treatment only oxygenated PE with –OH, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and O–CO functionalities without effectively cleaving the backbones of PE to form small molecules. Nevertheless, the hydrothermal pre-treatment using nitric acid is energy-intensive with potential environmental concerns. Therefore, it is desirable to develop an efficient strategy for photocatalytic upcycling of polyolefins with cost-effective and environmentally benign pre-treatment routes or even without pre-treatment.
O and O–CO functionalities without effectively cleaving the backbones of PE to form small molecules. Nevertheless, the hydrothermal pre-treatment using nitric acid is energy-intensive with potential environmental concerns. Therefore, it is desirable to develop an efficient strategy for photocatalytic upcycling of polyolefins with cost-effective and environmentally benign pre-treatment routes or even without pre-treatment.
| Metal oxide based catalyst | Pre-treatment | Product after pre-treatment | Reaction conditions | Activity and stability | Reference (year) |
|---|---|---|---|---|---|
| 1 wt% Pt loaded P25 TiO2 | 180 °C, 4 h hydrothermal reaction using 6% HNO3 aqueous solution | PE converted to succinic acid (44%), glutaric acid (22%), acetic acid (21%) adipic acid (9%), propanoic acid (4%) | Solar simulator (AM 1.5G, 100 mW cm−2) with a water filter (removing infrared light), 0.2 ml PE decomposition solution and 1.8 ml H2O, pH = 4, 4 mg catalyst, N2 atmosphere, 25 °C | H2 (6.3 mmol g−1), C2H4 (0.017 mmol g−1), C2H6 (0.25 mmol g−1), C3H6 (0.007 mmol g−1), C3H8 (0.14 mmol g−1), CO2 (5.9 mmol g−1), 96 h reaction | 34 (2021) |
| 1 wt% Pt loaded P25 TiO2 | 30 min plasma treatment at room temperature and atmospheric pressure with a 100 ml min−1 air flow | Grafting oxygen-containing functionalities on the PE/PP/PVC surface to achieve better hydrophilicity and stronger interaction between PE/PP/PVC and the catalyst | Simulated solar light (AM 1.5G, 100 mW cm−2), 30 mg catalyst, 30 mg PE powder, 30 ml water, N2 atmosphere, 25 °C | H2 (1056.10 μmol g−1), CH4 (93.56 μmol g−1), C2H4 (2.25 μmol g−1), C2H6 (4.25 μmol g−1), CO (10.99 μmol g−1), CO2 (452.43 μmol g−1), 24 h reaction | 35 (2023) |
| Simulated solar light (AM 1.5G, 100 mW cm−2), 30 mg catalyst, 30 mg PP powder, 30 ml water, N2 atmosphere, 25 °C | H2 (225.27 ± 6.06 μmol g−1), 4 h reaction | ||||
| Simulated solar light (AM 1.5G, 100 mW cm−2), 30 mg catalyst, 30 mg PVC powder, 30 ml water, N2 atmosphere, 25 °C | H2 (278.56 ± 11.84 μmol g−1), 4 h reaction | ||||
| Nb2O5 atomic layers | — | — | 300 W Xe lamp (AM 1.5G, 100 mW cm−2), 50 mg catalyst, 150 mg PE, 50 ml water, in air, 25 °C | Acetic acid (47.4 μg g−1 h−1), CO (0.4 μg g−1 h−1), 35 h reaction | 36 (2020) |
| — | — | 300 W Xe lamp (AM 1.5G, 100 mW cm−2), 50 mg catalyst, 150 mg PP, 50 ml water, in air, 25 °C | Acetic acid (40.6 μg g−1 h−1), CO (0.3 μg g−1 h−1), 35 h reaction | ||
| — | — | 300 W Xe lamp (AM 1.5G, 100 mW cm−2), 50 mg catalyst, 300 mg PVC, 50 ml water, in air, 25 °C | Acetic acid (39.5 μg g−1 h−1), CO (0.5 μg g−1 h−1), 35 h reaction | ||
| Co-Ga2O3 | — | — | 300 W Xe lamp (AM 1.5G, 100 mW cm−2), 50 mg catalyst, 100 mg PE, 100 ml H2O, in air, 25 °C | H2 (647.8 μmol g−1 h−1), CO (158.3 μmol g−1 h−1), CO2 (419.3 μmol g−1 h−1), 24 h reaction | 37 (2022) |
| — | — | 300 W Xe lamp (AM 1.5 G, 100 mW cm−2), 50 mg catalyst, 100 mg PP, 100 ml H2O, in air, 25 °C | H2 (603.5 μmol g−1 h−1), CO (147.2 μmol g−1 h−1), CO2 (389.1 μmol g−1 h−1), 24 h reaction | ||
| — | — | 300 W Xe lamp (AM 1.5 G, 100 mW cm−2), 50 mg catalyst, 100 mg PET, 100 ml H2O, in air, 25 °C | H2 (384.2 μmol g−1 h−1), CO (100.6 μmol g−1 h−1), CO2 (258.9 μmol g−1 h−1), 24 h reaction |
The Xie group36,37 is a pioneer in researching on photocatalytic upcycling of polyolefins and polyethylene terephthalate (PET) without a pre-treatment step. They have used two engineering strategies, i.e. structure regulation and element incorporation, on two metal oxide photocatalysts, Nb2O5 (ref. 36) and Ga2O3 (ref. 37), respectively, for directly photocatalytic upcycling of polyolefins and PET in an air atmosphere without pre-treatment. In one study,36 the authors have designed a general strategy of converting different plastic wastes (PE, PP and PVC) into CO2 followed by photo-reduction to form acetic acid as a C2 fuel under simulated natural environmental conditions (Fig. 3a). First, they designed and synthesized Nb2O5 atomic layers using the as-synthesized niobic acid atomic layers as the precursor followed by annealing in air. The earth-abundant and robust Nb2O5 is chosen due to its suitable conduction band (CB) and valence band (VB) positions (+2.5 V vs. SHE for the CB and −0.9 V vs. SHE for the VB at pH = 7). Thus, Nb2O5 can generate highly oxidative ˙OH radicals (+2.32 V vs. SHE at pH = 7) to degrade plastics and photo-generated electrons to reduce CO2 (−0.6 V vs. SHE at pH = 7). Nb2O5 atomic layers can degrade PE, PP and PVC with identical numbers of carbon in 40, 60 and 90 hours, respectively. The generated CO2 amounts increase gradually and reach the highest values in the corresponding time (Fig. 3b). They found that the overall numbers of moles of carbon in the generated CO2 gas and CO2 dissolved in solution are almost equivalent to that in pure PE, PP or PVC. These results confirm that plastics are completely degraded to form CO2 gas. Furthermore, the generated CH3COOH amounts are also gradually increased (Fig. 3c) and averaged CH3COOH generation rates on PE, PP and PVC are ∼47.4, 40.6 and 39.5 μg g−1 h−1, respectively (Fig. 3d). To obtain insightful understanding of the reaction mechanism on photocatalytic conversion of plastics into CH3COOH, in situ electron spin resonance (ESR) spectra were collected. The results confirm the generation of ˙OH and ˙O2− radicals in photocatalytic PE conversion. Furthermore, to study the accurate origin of generated CO2, synchrotron-radiation vacuum ultraviolet photoionization mass spectrometry (SVUV-PIMS) was conducted to study the reaction products in photocatalytic PE conversion under simulated natural conditions with a low amount of H218O. The results reveal that H2O can participate in both the photo-oxidation procedures to generate CO2 and O2, respectively. To seek the origin of generated CH3COOH, in situ FTIR spectroscopy was conducted. The results exhibit the formation of ˙COOH intermediates in photocatalytic PE conversion. Then, the C–C coupling of neighbouring ˙COOH leads to the formation of HOOC–COOH intermediates. Then, the continuous protonation of HOOC–COOH occurs to form HOOC–CO˙, HOOC–CHO˙ and HOOC–CH2O˙ intermediates as well as CH3COOH finally. Based on the above results, as shown in Fig. 3e and f, they propose the mechanism for photoconversion of pure PE into CH3COOH as follows: (i) the O2 and ˙OH radical mediated oxidative C–C bond cleavage to yield CO2 using Nb2O5 atomic layers, whilst photo-reduction of O2 occurs to generate ˙O2−, H2O2 and H2O. (ii) Light induced C–C coupling of ˙COOH intermediates generates CH3COOH, whilst H2O is oxidized to form O2. In another study, the Xie group37 has synthesized Co doped Ga2O3 nanosheets (Co-Ga2O3) for photocatalytic conversion of pulverized powder from PE bags, PP boxes or PET bottles into syngas (CO and H2) along with CO2 in the presence of water under ambient conditions. First, they reveal the CB and VB edge positions for both Co-Ga2O3 and Ga2O3, confirming that both of their photo-induced electrons and holes can drive some pivotal reactions, e.g., H2O oxidation or CO2/O2/H2O reduction. Then PE bags, PP boxes and PET plastic bottles were crushed into powders using a pulveriser. Afterwards, Co-Ga2O3 or Ga2O3 was utilized to convert these powders in pure water with simulated solar light (AM 1.5G, 100 mW cm−2) at ambient temperature and pressure via a photocatalysis reaction. 1H NMR spectroscopy reveals no detectable liquid product and H2, CO and CO2 were found by gas chromatography (GC). Especially, Co-Ga2O3 exhibits the photocatalytic evolution activities of H2 (647.8 μmol g−1 h−1) and CO (158.3 μmol g−1 h−1) from converting PE powders, about 160 and 190% times larger than those of Ga2O3. After 48 hour irradiation, the weight loss of PE bags can reach 81%. Co-Ga2O3 also exhibits excellent stabilities for photocatalytic conversion of these plastic powders. Control experiments show that the generated H2 arises from H2O rather than plastics; both O2 and H2O participated in the oxidation of PE to CO2, which is further reduced to form CO. The in situ electron spin resonance (ESR) technique confirms the existence of ˙OH and ˙O2− radicals with Co-Ga2O3 or Ga2O3 in photocatalytic reactions. Combining with isotope-labelling experiments, they deduce that both the ˙OH radical and O2 are involved in the photocatalytic oxidation of PE to CO2. In situ FTIR spectra further confirm that Co-Ga2O3 can reduce CO2 to CO via a photocatalytic reaction. Based on the revealed results, they propose the photocatalytic mechanism as below: (i) with light illumination, the photo-induced electrons/holes in Co-Ga2O3 or Ga2O3 can react with water to form H2 and O2, respectively; (ii) ˙OH radicals and O2 are involved in converting plastics into CO2, whilst O2 was also reduced to form ˙O2− radicals, H2O2 and H2O; (iii) the formed CO2 is further reduced to generate CO and O2 is also formed via oxidation of H2O. Furthermore, the reasons for the increased activities of Co-Ga2O3 compared to those of Ga2O3 are summarized as follows: (i) the enhanced light absorption of Co-Ga2O3 from d–d internal transition; (ii) the increased density of states (DOS) at the VB edge for Co-Ga2O3; (iii) the obviously reduced charge recombination for Co-Ga2O3; (iv) the stronger adsorption towards CO2 for Co-Ga2O3; (v) the lower energy barrier for CO2 reduction or H2 evolution on Co-Ga2O3. Both of the above two studies have adopted a one-step photocatalytic upcycling route to directly convert polyolefins into value-added chemicals in an air atmosphere. With the existence of O2 in air, various ROSs can be generated to participate in the reactions, which can help convert these plastics into oxygenated value-added chemicals/fuels and CO2. The generated CO2 can be further reduced by photo-induced electrons to form value-added chemicals, e.g., CO and acetic acid. All the performances and reaction conditions of the studies in this section are summarized in Table 1.
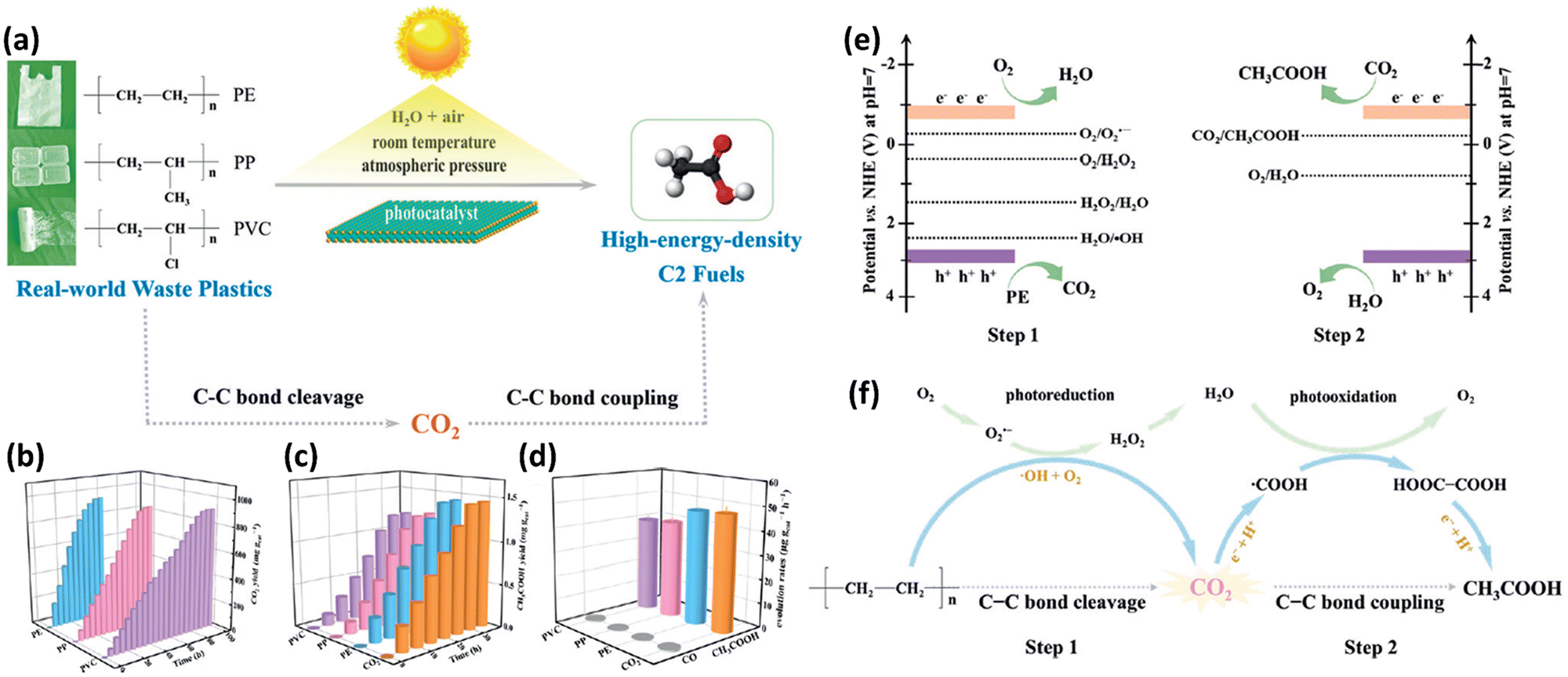 | ||
Fig. 3 (a) Schematic figure showing the conversion of various plastic wastes into C2 fuels via a designed two-step reaction pathway under simulated natural environmental conditions. (b) Generation of CO2 in photocatalytic oxidation of pure PE, PP and PVC using Nb2O5 atomic layers. In this reaction, the molar ratio of carbon in each plastic and Nb2O5 atomic layers is about 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. (c) The production amounts of CH3COOH and (d) generation rates of CH3COOH and CO in photocatalytic conversion of pure PE, PP and PVC, together with the photocatalytic reduction of pure CO2 in water. Schematic illustration for (e) the band edge potentials of Nb2O5 atomic layers as well as the potentials for CO2, H2O, H2O2 and O2 redox couples at pH = 7. (f) The increased two-step C–C bond cleavage and coupling mechanism for conversion of PE into CH3COOH under simulated natural environmental conditions. Reproduced with permission from copyright 2020, Wiley-VCH.36 :1. (c) The production amounts of CH3COOH and (d) generation rates of CH3COOH and CO in photocatalytic conversion of pure PE, PP and PVC, together with the photocatalytic reduction of pure CO2 in water. Schematic illustration for (e) the band edge potentials of Nb2O5 atomic layers as well as the potentials for CO2, H2O, H2O2 and O2 redox couples at pH = 7. (f) The increased two-step C–C bond cleavage and coupling mechanism for conversion of PE into CH3COOH under simulated natural environmental conditions. Reproduced with permission from copyright 2020, Wiley-VCH.36 | ||
3.2 Metal sulphide based photocatalysts for plastic upcycling
Up till now, most metal sulphide based photocatalysts reported for plastic upcycling are based on Cd-based catalysts,38–40 such as CdS QD38 and CdxZn1−xS solid solution,40 because of their strong light absorption arising from the appropriate band gap, together with their excellent reduction abilities due to the favourable CB edge positions.38–40 Thus, they usually exhibit outstanding H2 evolution rates under visible light irradiation under anaerobic conditions when applied on photocatalytic plastic upcycling. On the other hand, these metal sulphides possess moderate oxidation abilities owing to the S 3p derived VB. Thus, their photo-induced holes can oxidize the pre-treated plastics without over-oxidation to form CO2 under anaerobic conditions. However, owing to their moderate oxidation abilities, it is challenging for metal sulphide based photocatalysts to directly upcycle the plastics without pre-treatment. The following studies in this section all focus on photocatalytic upcycling of polyesters (e.g., PET, PLA and PUR) and polyolefins (e.g., PE) after pre-treatment, in which these polymers with large molecular weights are first transformed into monomers/oligomers or short-chain carbon-based molecules.38–40 Several strategies, such as surface engineering,38 loading cocatalysts39 and band structure engineering,40 have been adopted to increase the photocatalytic efficiencies.The Reisner38 group has presented a strategy to photo-reform plastic wastes to yield H2 and value-added organics using photocatalysts in water under sunlight (Fig. 4a). They have designed and synthesized a CdS/CdOx quantum dot (QD) photocatalyst. They found that when CdS QDs were added into aqueous NaOH, a thin cadmium oxide/hydroxide (CdOx) is generated to impede photo-corrosion. Ligand-free QDs are found to work with most substrates due to their exposed surfaces. In comparison, oleic acid-capped QDs can only work with PET, probably owing to the hydrophobic effect benefiting the substrate–QD interaction. First, CdS/CdOx was adopted for photocatalytic reforming of various polymers including polylactic acid (PLA), PET, polyurethane (PUR), polyvinylpyrrolidone (PVP), polyethylene glycol (PEG), LDPE, PVC, poly(methyl methacrylate) (PMMA), polystyrene (PS) and polycarbonate (PC), respectively. Among them, only PLA, PET and PUR are found to achieve higher photocatalytic H2 evolution performances while much less photocatalytic H2 evolution is observed on the other substrates. So these three polymers are selected for photocatalytic reforming. CdS/CdOx QDs were used for photocatalytic reforming of PLA, PET and PUR in NaOH aqueous solution in a N2 atmosphere. With optimised reaction conditions, CdS/CdOx QDs exhibit the photocatalytic H2 evolution activities of 64.3 ± 14.7, 3.42 ± 0.87 and 0.85 ± 0.28 mmol g−1 h−1, respectively. Isotope-labelling experiments reveal that the H2 produced arises from water, not the substrate. As a comparison, 5% Pt loaded TiO2 only exhibits the H2 evolution rates of 0.011 ± 0.004 and 0.074 ± 0.029 mmol g−1 h−1 under identical reaction conditions. And without expensive Pt as the co-catalyst, bare TiO2 exhibits no H2 evolution. Besides, ZnSe QDs as a Cd-free catalyst show no H2 production under the same reaction conditions. These highlight the advantages of strong visible-light absorption, no use of co-catalysts and fast oxidation of complicated substrates for CdS/CdOx QDs. To further increase the rate, a pre-treatment route was developed to hydrolyse PET, PUR and PLA in 10 M NaOH aqueous solution for 24 hours at 40 °C to release the monomers. After pre-treatment and removing the undissolved polymer by centrifugation, the supernatant was used to reduce the absorbance and scattering, thus leading to more photons absorbed by CdS/CdOx QDs and higher rates. Pre-treated PET and PUR solutions were found to obviously increase the activities of CdS/CdOx QDs, compared to Raw PET and PUR, respectively (Fig. 4b). Compared with Raw PLA, pre-treated PLA exhibits almost no influence on the efficiency of CdS/CdOx QDs (Fig. 4b). This is because PLA is easily dissolved in NaOH aqueous solution. Another advantage of CdS/CdOx QDs is that they can function even in highly alkaline solution. Then, 1H-NMR spectroscopy is adopted to study the reaction solutions and organic oxidation products. It was found that PLA is first hydrolysed to form sodium lactate followed by oxidation to generate a pyruvate-based compound (Fig. 4c). As for PET, it is first hydrolysed to generate terephthalate, ethylene glycol and isophthalate, followed by the formation of formate, glycolate, ethanol, acetate and lactate (Fig. 4d). Photo-reforming terephthalic acid doesn't generate H2, indicating that only the aliphatic component of PET yields the oxidation products. And terephthalic precipitates as a disodium salt to be easily recovered as a valuable chemical. As shown in Fig. 4e, PUR is hydrolysed to form an aliphatic component (propylene glycol) and aromatic component (2,6-diaminotoluene). While propylene glycol is oxidized to form formate, acetate, pyruvate and lactate (Fig. 4e), 2,6-diaminotoleune remains intact. The overall conversion of all polymers is lower than 40%, since CdS/CdOx QDs cannot completely mineralise these polymers into CO2. And no CO32− or CO2 is detected. These polymers are just partially oxidized to form various chemicals. CdS/CdOx QDs are also shown to achieve photo-reforming of a PET bottle and pre-treated PET bottle to generate H2 and value-added chemicals (Fig. 4f).
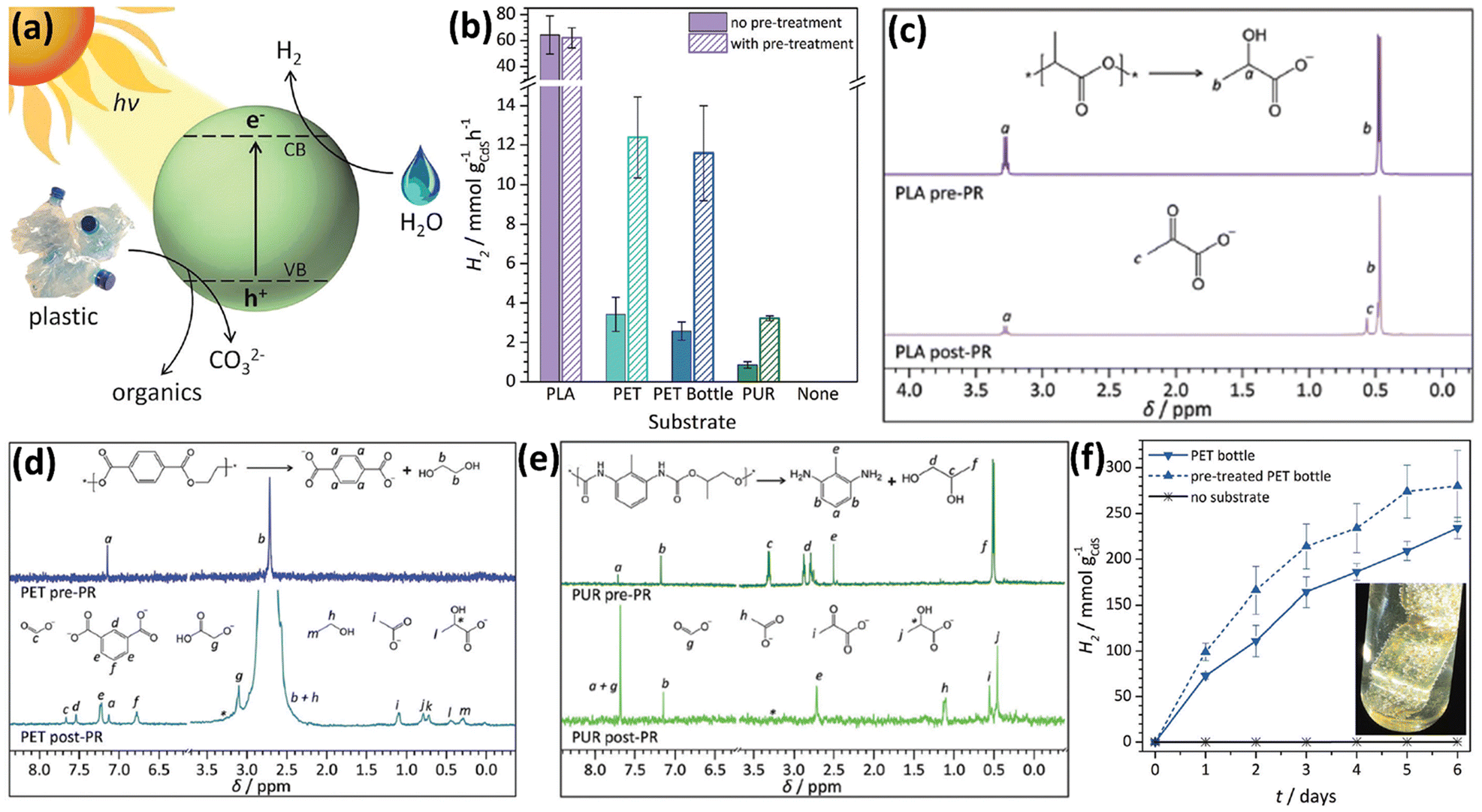 | ||
| Fig. 4 (a) Schematic illustration for photo-reforming various plastic wastes using CdS/CdOx QDs in alkaline aqueous solution. (b) Photocatalytic reforming of various plastics by CdS/CdOx QDs. Reaction conditions: 1 nmol CdS/QDs, plastic powders (50 mg ml−1 PLA, 25 mg ml−1 PET, PET bottle or PUR), without pre-treatment or pre-treated in 2 ml 10 M NaOH aqueous solution. 1H-NMR spectra for (c) PLA, (d) PET and (e) PUR prior to (pre-PR) and after (post-PR) 24 hour light illumination using 1 nmol CdS/CdOx QDs in 2 ml 10 M NaOD in D2O. (f) Photocatalytic reforming of the PET bottle to generate H2 using CdS/CdOx QDs. Reaction conditions: 1 nmol CdS/CdOx QDs, ground PET bottle (25 mg ml−1) directly used or pre-treated in 2 ml 10 M NaOH aqueous solution. (f) Inset shows the image of the PET bottle and H2 bubbles on the surface of plastics. Reproduced with permission from copyright 2018, Royal Society of Chemistry.38 | ||
In another study, a cocatalyst/photocatalyst MoS2/CdS system was synthesized for photocatalytic upcycling of pre-treated polyester/polyolefin.39 The Qiu group39 designed this system, MoS2-tipped CdS nanorod (MoS2/CdS), to reform the pre-treated PE, PLA or PET to generate various value-added chemicals and H2, as shown in Fig. 5a. The as-synthesized MoS2/CdS composite exhibits the accumulation of photo-generated electrons and holes on the MoS2 tip and sidewalls of CdS, respectively (Fig. 5a). The TEM image (Fig. 5b and c) and HRTEM image (Fig. 5d) directly reveal the intimate coupling of MoS2 on the tip of CdS NSs. The strong electronic coupling between MoS2 and CdS is confirmed by the surface-sensitive high-resolution XPS technique, showing that electrons are transferred from CdS to MoS2. Results of steady-state PL, photocurrent response and electrochemical impedance spectroscopy (EIS) all confirm the more efficient charge separation/migration in MoS2/CdS. Furthermore, selective deposition reactions indicate that MnOx nanosheets and Pt nanoparticles are selectively loaded on the sidewalls of CdS and the MoS2 tip, respectively. These results also indicate that photo-generated electrons and holes are accumulated on the MoS2 tip and sidewalls of CdS, respectively. Linear sweep voltammetry (LSV) curves further indicate the significantly higher HER activity of MoS2/CdS compared to MoS2 alone. Then, the electron spin resonance (ESR) technique is further utilized to detect the active species on MoS2/CdS, which confirms the existence of photo-generated holes and ˙OH with light illumination. Nevertheless, a photoluminescence test using terephthalic acid (TPA) as the ˙OH scavenger only shows a negligible characteristic PL peak arising from ˙OH, suggesting the minimal role of ˙OH in photo-reforming. In contrast, photo-generated holes are deemed as the major active species in photo-reforming. The MoS2/CdS photocatalyst was utilized for photo-reforming of pre-treated PLA, PET and PE. The largest H2 evolution of 6.68 ± 0.10 mmol g−1 h−1 was realized by MoS2/CdS from photo-reforming of pre-treated PLA in 10 M NaOH aqueous solution (Fig. 5e). Fig. 5f shows the influence of various MoS2 loading amounts on the tip of CdS, with 21.8 wt% MoS2 loading reaching the highest H2 evolution activity. To highlight the unique benefit of the MoS2/CdS structure, MoS2 nanosheets selectively loaded on the sidewalls of CdS nanorods were synthesized as the control sample, denoted as CdS@MoS2. In contrast, CdS@MoS2 with a similar loading of 21.1 wt% only exhibits a much lower photocatalytic H2 evolution rate of 2.21 ± 0.25 mmol g−1 h−1, compared with that of MoS2/CdS under identical reaction conditions (Fig. 5f). MoS2/CdS exhibits 200 hour long-term stability for photocatalytic H2 evolution in pre-treated PLA solution, with 72% of the activity in the 8th cycle compared with that in the 1st cycle. Furthermore, isotope labelling experiments confirm that the source of generated H2 is water splitting, but not the pre-treated PLA substrate. Both 1H NMR spectroscopy and high-performance liquid chromatography (HPLC) together indicate the generation of formate (5.37 ± 0.67 mmol l−1) after 5 hour light illumination (Fig. 5g). 13C NMR spectroscopy and the self-built route together confirm the production of CO32− arising from lactate oxidation by MoS2/CdS. Additionally, DMPO and Na2S/Na2SO3 are added as scavengers of ˙OH radicals and Na2S/Na2SO3, respectively. No obvious reduction of formate was observed after adding DMPO. In contrast, apparent reduction of formate was found after adding Na2S/Na2SO3, indicating the principal role of holes in lactate oxidation. This is in accordance with the ESR and PL results. Then, it is proposed that lactate is oxidized by holes to form acetaldehyde followed by acetate, methanol and formate. But the other products are not observed by 1H NMR spectroscopy and HPLC except formate. They have also tested the photocatalytic reforming of PET on MoS2/CdS, displaying stable H2 evolution in a 25 hour test, with a rate of up to 3.90 ± 0.07 mmol g−1 h−1. The 200 hour test further indicates the structural and composition stability of MoS2/CdS. Isotope-labelling experiments suggest that the generated H2 arises from water splitting, but not from PET constituent monomers. Furthermore, photocatalytic reforming of a pre-treated PET bottle shows a H2 evolution rate of 3.53 ± 0.07 mmol g−1 h−1, further confirming the potential realistic application. Formate, acetate and glycolate can also be detected by 1H NMR spectroscopy in photo-reforming of pre-treated PET. The gradual increase of formate and acetate amounts can be observed in a 5 hour reaction (Fig. 5h). The 1H NMR test shows that the concentration of terephthalate is not changed but the concentration of ethylene glycol is altered, suggesting that these formed carboxylate chemicals arise from ethylene glycol but not from terephthalate. Holes were confirmed to be the major active species for ethylene glycol oxidation via control experiments. MoS2/CdS was also utilized for photo-reforming of the pre-treated PE by nitric acid in a hydrothermal reaction (Fig. 5i). HPLC tests confirm the existence of formic acid, succinic acid, glutaric acid, acetic acid, propionic acid and adipic acid, with the major products of succinic acid and glutaric acid (Fig. 5i). Then, MoS2/CdS was utilized to photo-reform the pre-treated PE, generating a H2 evolution rate of 1.13 ± 0.06 mmol g−1 h−1 (Fig. 5j). Notably, MoS2/CdS still exhibits robust H2 evolution after a 200 hour test, with good PET compositional/structural stability for the reacted MoS2/CdS. Isotope-labelling experiments also reveal that most of the generated H2 arises from water splitting. Besides, MoS2/CdS also shows a CH4 generation rate of up to 196.2 ± 1.76 μmol g−1 h−1 (Fig. 5k) and a CO2 generation rate of 2.75 ± 0.05 μmol g−1 h−1. Control experiments reveal that CH4 originates from the Kolbe photo-oxidation decarboxylation of carboxylic acid, not from CO2 reduction. Due to the existence of abundant carboxylic acids in substrates, other gaseous alkanes, such as ethane, propane and n-pentane, are also generated with rates of 1.86 ± 0.04, 0.78 ± 0.20 and 7.6 ± 0.60 μmol g−1 h−1 (Fig. 5k), respectively, via decarboxylation with a hydrogen transfer mechanism. An in situ ESR test confirms the production of carbon-centred radical species, deemed as the pivotal intermediates in the Kolbe decarboxylation reaction (Fig. 5l). These results confirm that MoS2/CdS can induce the decarboxylation reaction. Control experiments further indicate that holes play a key role in photocatalytic decarboxylation. Finally, they propose a mechanism: (i) photo-induced holes lead to the oxidation of acetic acid to generate methane via decarboxylation; (ii) photocatalytic decarboxylation of succinic acid and glutaric acid leads to generation of ethane and propane, respectively. This successful photocatalytic decarboxylation reaction is for the first time reported on CdS based photocatalysts. Similarly, Li et al.40 combined the strategies of band structure engineering and loading cocatalysts to synthesize 4.3 wt% MoS2 coupled Cd0.5Zn0.5S (M4.3/C0.5Z0.5S). The strong electronic coupling between MoS2 and Cd0.5Zn0.5S was confirmed by the obvious peak shifts in Raman and XPS spectra. Transient surface photovoltage (TR-SPV) results show the 250% higher SPV value of M4.3/C0.5Z0.5S compared to that of Cd0.5Zn0.5S, as well as the elongated SPV signal of M4.3/C0.5Z0.5S. These results indicate the much more efficient charge separation and longer lifetimes of photo-induced charge carriers in M4.3/C0.5Z0.5S. These are further corroborated by M4.3/C0.5Z0.5S via the electrochemical impedance spectra (EIS) and photoelectrochemical current (PEC) densities. The polarization curves of M4.3/C0.5Z0.5S exhibit increased HER activity compared to that of Cd0.5Zn0.5S, indicating that the loading of MoS2 can increase the activities of M4.3/C0.5Z0.5S. Blank experiments show that without light, a photocatalyst, or NaOH, H2 evolution or degradation of PET cannot occur. M4.3/C0.5Z0.5S shows the largest H2 generation activity (15.9 mmol h−1 g−1) compared to 4.3 wt% MoS2 coupled CdxZn1−xS (X = 0, 0.2, 0.4, 0.8 and 1). An outstanding photocatalytic H2 generation rate was also realized by M4.3/C0.5Z0.5S using PET bottles. Good H2 evolution robustness was also observed in 5 hour irradiation in PET or PET-bottle-based aqueous solution. 1H-NMR spectroscopy was adopted to analyse the degradation products of photocatalytic PET conversion. Ethylene glycol, terephthalic acid and glycolate were detected in the pre-treated solutions before reaction. Finally, the pre-treated PET was oxidized to yield formate, methanol, acetate and ethanol.
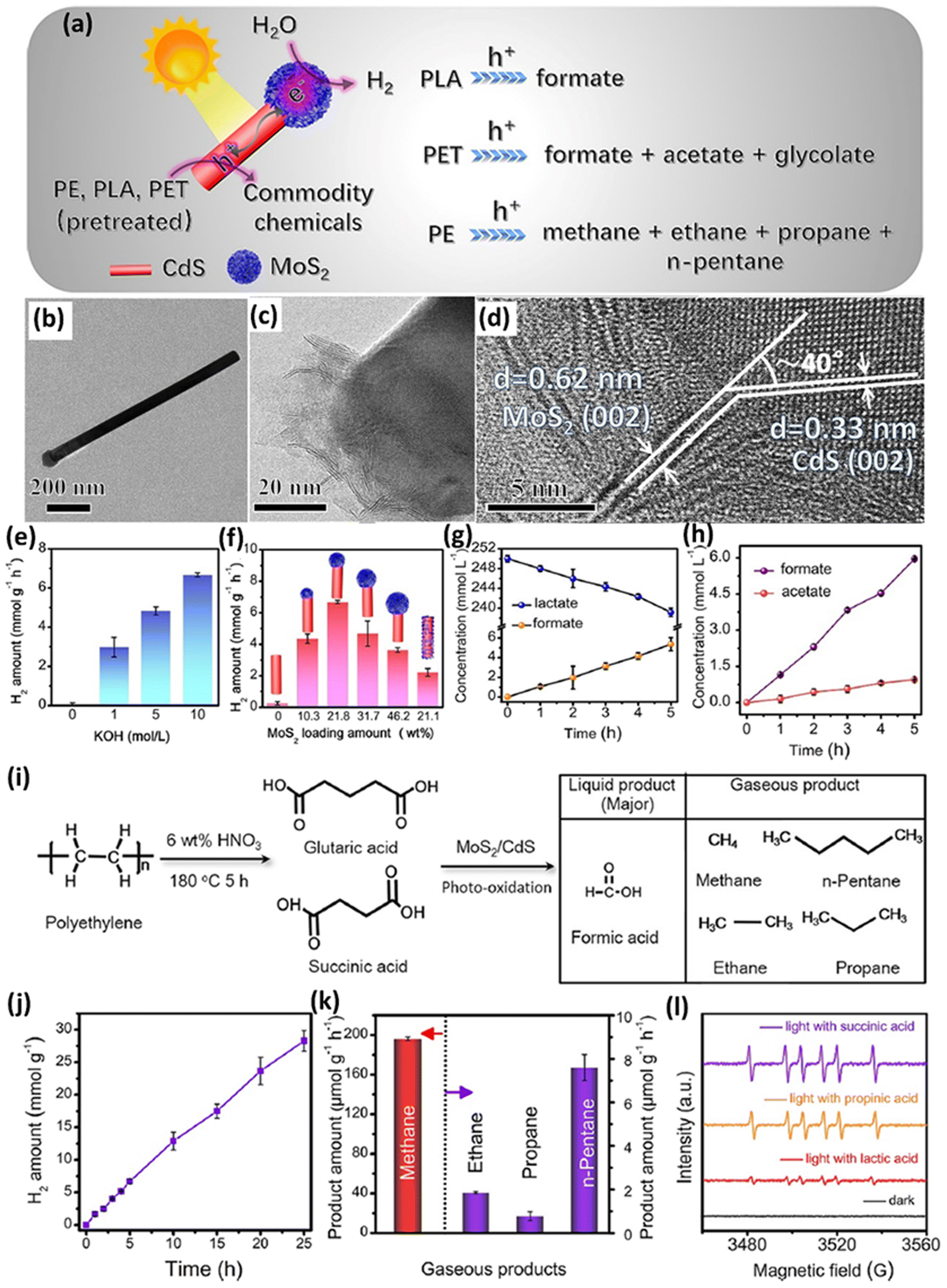 | ||
| Fig. 5 (a) Schematic illustration for photocatalytic reforming of pre-treated plastics using the MoS2/CdS composite. (b and c) TEM images and (d) HRTEM image of the MoS2/CdS composite. (e) Photocatalytic reforming of pre-treated PLA for H2 evolution using 21.8 wt% MoS2 loaded CdS in various KOH concentrations. (f) Effect of MoS2 loading on the H2 evolution from photocatalytic reforming of pre-treated PLA in 10 M KOH using MoS2/CdS composites with different MoS2 contents. (g) The concentrations of lactate and formate in a 5 hour photo-reforming reaction. (h) The concentrations of acetate and formate in a 5 hour photo-reforming reaction. (i) Schematic image showing the conversion of PE into carboxylic acid followed by a photo-reforming reaction on MoS2/CdS. (j) H2 generation from 25 hour photocatalytic reforming of pre-treated PE. (k) Generation activities of alkane from photocatalytic reforming of pre-treated PE in a 5 hour reaction. (l) In situ ESR spectra in the presence of various substrates via using the spin-trapping agent. Reproduced with permission from copyright 2022, American Chemical Society.39 | ||
The above three studies have demonstrated the immense potential of metal sulphide based photocatalysts, especially Cd-based catalysts, to upcycle these pre-treated plastics into valuable chemicals/fuels using simulated solar light. But the toxicity of Cd-based catalysts, together with the insufficient stability and oxidation capacity of metal sulphide based photocatalysts, seriously restricts their realistic applications in industrial scale solar plastic upcycling. All the performances and reaction conditions of the studies in this section are summarized in Table 2.
| Metal sulphide based catalyst | Pre-treatment | Product after pre-treatment | Reaction conditions | Activity and stability | Reference (year) |
|---|---|---|---|---|---|
| CdS/CdOx quantum dots | 50 mg ml−1 PLA, 2 ml 10 M NaOH aqueous solution, 40 °C, 24 h, in air | PLA converted into sodium lactate | Simulated solar light (AM 1.5G, 100 mW cm−2), 2 ml pre-treated solution, 25 °C, N2 atmosphere | H2 (62.1 ± 7.8 mmol g−1 h−1), pyruvate generated, 4 h reaction | 38 (2018) |
| 25 mg ml−1 PET, 2 ml 10 M NaOH aqueous solution, 40 °C, 24 h, in air | PET converted into terephthalate, ethylene glycol and isophthalate | H2 (12.4 ± 2.0 mmol g−1 h−1) formate, glycolate, ethanol, acetate and lactate generated, 4 h reaction | |||
| 25 mg ml−1 PUR, 2 ml 10 M NaOH aqueous solution, 40 °C, 24 h, in air | PUR converted into 2,6-diaminotoluene and propylene glycol | H2 (3.22 ± 0.13 mmol g−1 h−1) formate, acetate, pyruvate and lactate generated, 4 h reaction | |||
| MoS2-tipped CdS nanorod | 1.5 g PLA, 60 ml 10 M KOH solution, 40 °C, 48 h | PLA converted to lactate | 300 W Xe light, simulated solar light (AM 1.5G, 100 mW cm−2), 100 mg catalyst, 5 °C, anaerobic conditions (vacuumed) | H2 (6.20 ± 0.23 mmol g−1 h−1), 25 h reaction, H2 (6.20 ± 0.23 mmol g−1 h−1), 200 h reaction, formate (5.37 ± 0.67 mmol l−1), 5 h reaction | 39 (2022) |
| 1.5 g PET, 60 ml 10 M KOH solution, 40 °C, 4 h | PET converted to ethylene glycol and terephthalate | H2 (3.90 ± 0.07 mmol g−1 h−1), 25 h reaction, formate (5.96 ± 0.02 mmol l−1), acetate (0.95 ± 0.01 mmol l−1), 5 h reaction | |||
| 1.5 g PE, 60 ml 6 wt% HNO3, hydrothermal reaction at 180 °C for 5 h | PE converted to formic acid, succinic acid, glutaric acid, acetic acid, propionic acid, adipic acid | H2 (1.13 ± 0.06 mmol g−1 h−1), 25 h reaction, CH4 (196.2 ± 1.76 μmol g−1 h−1), C2H6 (1.86 ± 0.04 μmol g−1 h−1), C3H8 (0.78 ± 0.2 μmol g−1 h−1), n-pentane (7.60 ± 0.6 μmol g−1 h−1) CO2 (2.75 ± 0.05 μmol g−1 h−1), 5 h reaction | |||
| MoS2–Cd0.5Zn0.5S | 1.5 g PET, 60 ml 10 M NaOH aqueous solution, 40 °C, 24 h | PET converted to ethylene glycol, terephthalic acid, and glycolate | 300 W Xe light (AM 1.5G), 10 mg catalyst, 60 ml pre-treated PET solution, anaerobic conditions (vacuumed) | H2 (15.90 mmol g−1 h−1) formate, methanol, ethanol and acetate generated | 40 (2021) |
3.3 Non-metal based photocatalysts for plastic upcycling
Currently, all the non-metal based photocatalysts are based on carbon nitride (CxNy), which possess non/low toxicity, earth abundance, low cost, good light absorption due to a suitable band gap width, favourable redox abilities arising from appropriate CB/VB edge positions and strong chemical-/photo-stability.41–44 Owing to the moderate oxidation ability of CxNy, it is very challenging for them to directly upcycle plastics into value-added chemicals/fuels at room temperature and under anaerobic conditions. Thus, the Reisner group have developed a cocatalyst/photocatalyst (Ni2P loaded CNx) system to photo-reform various pre-treated plastics to acquire value-added chemicals without generating CO2 or even CO32−.41 They have reported the photo-reforming of plastic wastes to produce value-added organics and H2 using a Ni2P loaded cyanamide-functionalized carbon nitride (H2NCNx|Ni2P) photocatalyst (Fig. 6a). The TEM image of H2NCNx|Ni2P (Fig. 6b) shows the loading of Ni2P NPs on the surface of H2NCNx. A lattice distance of 0.221 nm was observed in the Fig. 6b inset, attributed to the (111) facet of hexagonal N2P. No obvious change is detected between the high resolution XPS spectra of C 1s for H2NCNx and H2NCNx|Ni2P, suggesting that loading the Ni2P co-catalyst doesn't impose obvious influence on the surface features of H2NCNx. The high-resolution XPS spectra of Ni 2p3/2 for H2NCNx|Ni2P obviously shift to the lower binding energy direction, compared to those for Ni2P, suggesting a strong cocatalyst–support interaction with electrons transferring from H2NCNx to Ni2P. First, they pre-treated PET and PLA in KOH aqueous solution at 40 °C for 24 hours to acquire the corresponding monomers (ethylene glycol and terephthalate for PET; lactate for PLA). Quantitative analysis using 1H nuclear magnetic resonance (NMR) spectroscopy indicates that 72% of lactate in PLA as well as 62% of ethylene glycol and 51% of terephthalate in PET are released in pre-treatment. Then, all the reaction conditions, such as the loading of the Ni2P co-catalyst (Fig. 6e) and concentration of KOH (Fig. 6f), were optimized to achieve the highest photocatalytic H2 evolution. 50 hour solar light illumination on H2NCNx|Ni2P leads to photocatalytic H2 evolution of 82.5 ± 7.3 and 178 ± 12 μmol g−1 using pre-treated PET and PLA as the substrates, respectively, in 1 M KOH aqueous solution. CNx|Ni2P without cyanamide functionalization exhibits lower H2 evolution using pre-treated PET or PLA as the substrate under the same conditions. Nevertheless, H2NCNx|Ni2P shows reduced H2 evolution over time; CNx|Ni2P exhibits more stable H2 evolution. 1H NMR spectroscopy combined with 13C NMR spectroscopy indicates that CNx|Ni2P can photo-reform the pre-treated PET to generate formate, glyoxal, glycolate, acetate, glyoxylate and glycolaldehyde. Besides, CNx|Ni2P can photo-reform the pre-treated PLA into formate and acetate. CNx|Ni2P also exhibits the feasibility of photo-reforming real-world waste of polyester microfiber, a PET bottle and an oil-contaminated PET bottle into H2 and a series of chemicals (Fig. 6h). Post-catalysis characterization indicates the good stability of CNx in CNx|Ni2P. However, it should be noted that while 99.1% Ni content of Ni2P still remains on CNx, XPS results show that at least the surface of Ni2P is converted to Ni(OH)2 after reaction in the KOH aqueous solution. Then, they further upscaled the photocatalytic reactor to 120 ml (Fig. 6i). Using this upscaled reactor, a photocatalytic H2 evolution of 53.5 μmol gsub−1 was achieved via photo-reforming of polyester microfibers in five days (Fig. 6j). Furthermore, the Reisner group42 has assembled a photocatalyst panel loaded with CNx|Ni2P to photo-reform various wastes including plastics. They utilized a simple low temperature and drop casting route to synthesize scalable photocatalyst panels with carbon nitride/nickel phosphide (CNx/Ni2P). Ni2P nanoparticles were dispersed on CNx as the co-catalyst. First, they optimised the CNx/Ni2P panels for the highest H2 evolution, light harvesting and recyclability on a scale of 1 cm2. Afterwards, they adopted a 25 cm2 panel to photo-reform PET, α-cellulose and municipal solid waste (MSW), respectively. They found that the illumination configuration (front illumination or back illumination) plays the key role in the H2 evolution activities. To demonstrate the feasibility of “real world” application of this system, it is operated in seawater and 20% sunlight irradiation (20 mW cm−2), and still shows ∼50% H2 evolution activity compared to that in pure water and 1 sunlight irradiation (100 mW cm−2).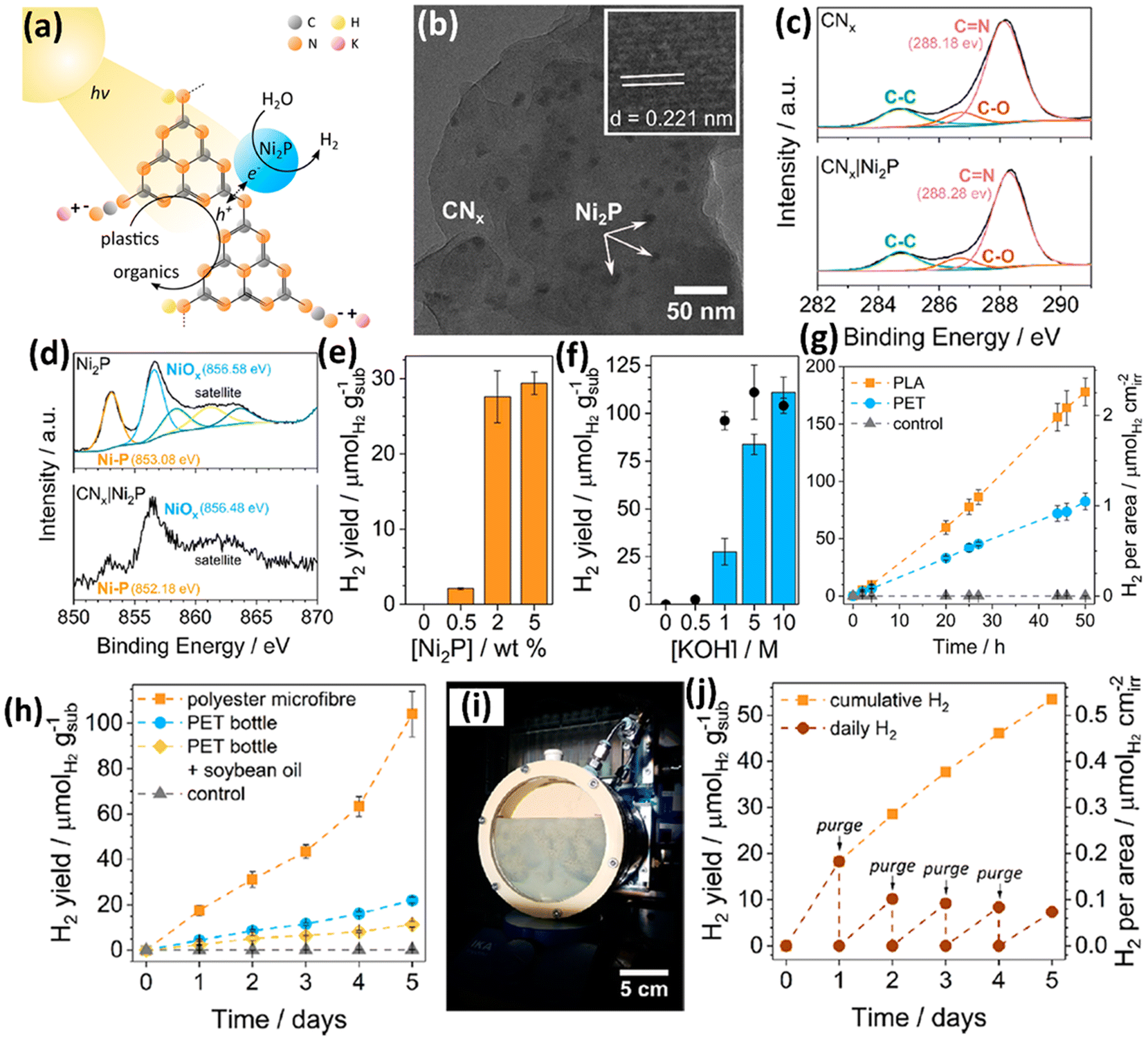 | ||
| Fig. 6 (a) Schematic illustration of plastic photo-reforming using a Ni2P/CNx photocatalyst. (b) TEM image of Ni2P/CNx. (b) Inset shows the lattice distance of Ni2P NPs. (c) High resolution XPS spectra of C 1s for CNx and Ni2P/CNx. (d) High resolution XPS spectra of Ni 2p for Ni2P and Ni2P/CNx. (e) Effect of Ni2P loading on the H2 evolution activities of Ni2P/CNx for 20 hour PET photo-reforming. (f) Effect of KOH concentration on the H2 evolution activities of Ni2P/CNx for 20 hour PET photo-reforming. (g) Long-term photocatalytic reforming of PET and PLA. Reaction conditions: 2 wt% Ni2P/CNx (1.6 mg ml−1), pre-treated PET (25 mg ml−1), 2 ml 1 M KOH aqueous solution, simulated sunlight (AM 1.5G, 100 mW cm−2) and 25 °C. (h) Long term photocatalytic reforming of polyester microfibers, a PET bottle and a PET bottle coated with soybean oil. Reaction conditions: 1.6 mg ml−1 Ni2P/CNx, 2 ml 1 M KOH, 5 mg ml−1 pre-treated microfibers, 25 mg ml−1 pre-treated PET bottle or 25 mg ml−1 pre-treated PET bottle with 5 mg ml−1 soybean oil and simulated sunlight (AM 1.5G, 100 mW cm−2). (i) Image of the upscaled photocatalytic reactor. (j) Upscaled photocatalytic reforming of polyester microfibers. Reaction conditions: 1.6 mg ml−1 Ni2P/CNx, 120 ml 1 M KOH, 5 mg ml−1 pre-treated microfibers and simulated sunlight (AM 1.5G, 100 mW cm−2). Reproduced with permission from copyright 2019, American Chemical Society.41 | ||
The Yang group43 has fabricated a graphitic carbon nitride (CN)/carbon nanotubes (CNTs)/NiMo nanoparticle (CN/CNTs/NM) photocatalyst for photo-reforming of pre-treated PET. The intimate combination between CN and in situ generated CNTs was confirmed by the FTIR, Raman and TGA techniques. CN/CNTs/NM shows the highest activity for H2 evolution from photo-reforming of PET. Carbon nitride (CN), carbon nitride/carbon nanotubes (CN/CNTs) and CN/CNTs/NM were utilized to photo-reform pre-treated PET or PLA. The 1H NMR spectrum confirms the existence of ethylene glycol (EG), terephthalate (TPA) and other small molecules after the pre-treatment of PET. The 1H NMR technique also confirms the formation of glyoxal and glycolate after photo-reforming of pre-treated PET. The highest H2 evolution was observed on CN/CNTs/NM for photo-reforming of PET, about 14 times larger than that of CN. Additionally, CN/CNTs/NM exhibits higher H2 evolution activity for photo-reforming of PLA compared to that for photo-reforming of PET. CN/CNTs/NM also exhibits good robustness for H2 evolution from photo-reforming of pre-treated PET. CN/CNTs/NM was also adopted to photo-reform a pre-treated PET bottle, which shows slightly smaller activity compared to that of pure PET, due to the existence of many different additives in the PET bottle. Single-particle PL tests further confirm that the PL of CN/CNTs/NM is obviously quenched compared to that of CN/CNTs or CN. The PL of CN/CNTs is also quenched compared to CN. These results indicate that the efficient electron transfer from CN to CNTs and further to NM obviously decreases the charge recombination. These are in accordance with the electrochemical impedance spectroscopy (EIS) and photocurrent density measurements. A single-particle PL study further confirms that with the addition of EG, the PL intensity of CN/CNTs/NM is significantly decreased because the photo-generated holes are captured by EG. Based on the above results, they propose the mechanism as follows: after the photo-excitation, the photo-generated electrons transfer from CN to CNTs and further to NM, where photo-generated electrons reduce protons to produce H2 gas. Photo-induced holes oxidize the EG to form glyoxal and carboxylate.
Although it is very challenging to photo-reform untreated plastics at room temperature, in aqueous solution and under anaerobic conditions using CxNy, Cao et al.44 have successfully utilized C3N4 to photocatalytically convert PS into aromatic oxygenates, such as benzoic acid, acetophenone and benzaldehyde in acetonitrile at 80–150 °C, with light irradiation and in air. They have adopted various well-known, environmentally benign and simple-to-synthesize photocatalysts including TiO2, ZnO, ZnS and C3N4 to upcycle PS into aromatic oxygenates at 80 °C, with light illumination and in air. TiO2, ZnO, ZnS and C3N4 show a PS conversion of 13%, 21%, 12% and 55%, 64% and 60%, respectively. TiO2 shows the lowest selectivity of 15%, since the main products for TiO2 are CO2 and CO. This indicates that TiO2 is not a suitable photocatalyst for upcycling PS. Besides, despite that both ZnO and ZnS exhibit good selectivity, their PS conversion is not good. Thus, C3N4 was adopted. A range of metals, such as 0.1% Au, 0.5% Au, 0.5% Pt, 0.5% Fe and 0.5% Cu, were loaded on C3N4, respectively. Although these metal loadings could increase the conversion to some extent, the selectivity was reduced, compared to C3N4 without metal loading. These might be boosted by the overoxidation of intermediates/products. Then, they have investigated the photocatalytic oxidation of PS using g-C3N4. The time-evolution experimental results (Fig. 7a) exhibit an obvious induction period in the first 3 hours, followed by the fast accumulation of various inorganic/organic products (COx, benzoic acid, acetophenone and benzaldehyde) in 24 hours. Based on the spectroscopic result, gel permeation chromatography (GPC) and liquid product analysis, it was found that in the induction period of reaction, reactive oxygen species partially oxidize the PS. The reaction mechanism is disclosed in Fig. 7b as follows: (1) oxidative functionalization of PS with OH groups at Cα or Cphenyl sites as well as OH or CO groups at Cβ sites occurs under both thermo and light illumination conditions using C3N4; (2) with light illumination, C3N4 generates photo-excited electrons/holes to form ˙O2− and possible carbon radical intermediates; (3) reactive oxygen radicals or oxidative photo-generated holes easily attack PS–O to generate the C–C–O˙ intermediate, resulting in the breakage of the C–C bond and scission of the polymer backbone via the β scission procedure. Overoxidation of carbon containing reactants/intermediates/products could occur in any of the above steps, thus generating undesired CO2. As shown in Fig. 7c, they can utilize this strategy to acquire pure chemicals, such as 240 mg benzoic acid, via using column chromatography separation. As presented in Fig. 7d, via appropriately regulating the ratio of substrate/catalyst (5:2) and reaction time (8 hours), they can acquire a stable yield rate of various organics (10 mg g−1 h−1) with a selectivity of 76% in 18 cycles for photocatalytic oxidative conversion of PET plastic pellets (500 mg). Furthermore, they utilized a flow reaction system to optimize the activity and selectivity towards specific products via tuning the weight hourly space velocity (WHSV). They found that better selectivity for benzaldehyde (51%) and acetophenone (31%) is acquired with an optimized WHSV of 0.9 h−1 (Fig. 7f).
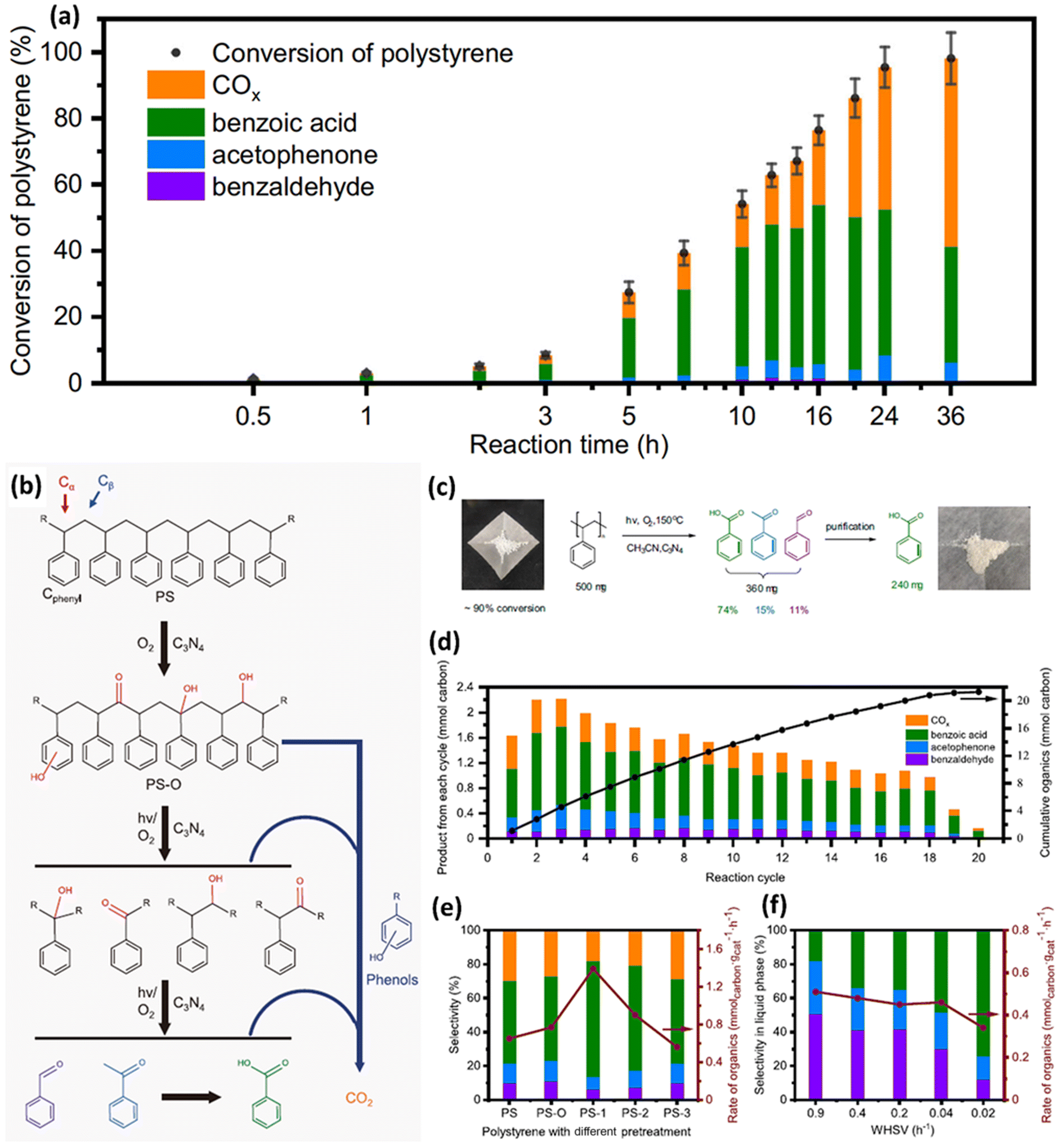 | ||
| Fig. 7 (a) Generation of various products for photocatalytic oxidation of PS. Reaction conditions: 10 mg PS (Mw = ∼50 kDa), 50 mg g-C3N4, 30 ml acetonitrile, 300 W xenon light, 150 °C and under 10 bar O2. Columns with various colours denote different generated products. The standard deviation of conversion in three parallel experiments were denoted by the error bars. (b) Proposed reaction pathways for photocatalytic oxidation of PS. (c) Schematic illustration for the conversion experiments of 500 mg PS pellets. (d) Photocatalytic oxidation of 500 mg PS pellets in 20 reaction cycles. Reaction conditions: 500 mg PS pellets, 200 mg g-C3N4, 40 ml acetonitrile, 150 °C, 10 bar O2 and 300 W xenon light illumination for 8 hours in each cycle. After every cycle, the solution is released and pure solvent is added. (e) Catalytic oxidation of PS after various pre-treatments. Reaction conditions: 20 mg PS, 50 mg g-C3N4, 30 ml acetonitrile, 150 °C and 300 W xenon light illumination for 8 hours. PS–O: thermal treatment at 150 °C in acetonitrile with O2. PS-1: thermal treatment at 220 °C in air. PS-2: thermal treatment at 300 °C in air. PS-3: pyrolysis at 350 °C in N2. (f) Performances of catalytic oxidation of PS at different weight hourly space velocities (WHSVs). Reaction conditions: 30 ml acetonitrile, 100 mg g-C3N4, 120 °C, 300 W xenon light irradiation, and a high pressure syringe pump used to pump PS solution (about 0.3 mg ml−1 in acetonitrile) into the reactor at different rates. 10 ml reaction solution was drained out manually as the PS solution pumped in amounted to the same volume. The standard deviation of conversion in 3 parallel experiments was denoted by the error bars. Reproduced with permission from ref. 44 Springer Nature. | ||
The above four studies show the great potential of using CxNy based photocatalysts to upcycle pre-treated polyester/polyolefin at room temperature and under anaerobic conditions or even untreated polyolefin at raised temperature and in air/O2. Owing to the significant advantages of earth-abundance, cost-effectiveness, excellent absorption of light and suitable redox abilities, further investigation on CxNy based photocatalysts is anticipated. All the performances and reaction conditions of the studies in this section are summarized in Table 3.
| Non-metal based catalyst | Pre-treatment | Product after pre-treatment | Reaction conditions | Activity and stability | Reference (year) |
|---|---|---|---|---|---|
| Carbon nitride combined with a nickel phosphide (Ni2P) cocatalyst (CNx|Ni2P) | 50 mg ml−1 PET soaked in 2 M aqueous KOH, 24 h, 40 °C | PET converted to ethylene glycol, terephthalate | Simulated solar light (AM 1.5G, 100 mW cm−2), 2 ml 1 M aqueous KOH, 1.6 mg ml−1 catalyst, N2 atmosphere, 25 °C | H2 (25.7 ± 2.3 μmol g−1 h−1), 50 h reaction acetate (190 nmol), formate (190 nmol), glyoxal (9300 nmol), 5 day reaction | 41 (2019) |
| 50 mg ml−1 PLA soaked in 2 M aqueous KOH, 24 h, 40 °C | PLA converted to lactate | H2 (55.7 ± 3.7 μmol g−1 h−1), 50 h reaction acetate (100 nmol), formate (95 nmol), 5 day reaction | |||
| Carbon nitride combined with a nickel phosphide (Ni2P) cocatalyst (CNx|Ni2P) | 25 mg ml−1 PET soaked in 0.5 M aqueous KOH, 80 °C, overnight, in air | — | Solar simulator (AM 1.5G, 100 mW cm−2), 2 ml min−1 flow rate, 50 ml 25 mg ml−1 pre-treated PET solution, 25 cm2 catalyst panel, 25 °C, N2 atmosphere | H2 (52 μmol m−2 h−1) | 42 (2021) |
| Carbon nitride–carbon nanotubes–NiMo hybrid (CN–CNT–NiMo) | 50 mg ml−1 PET soaked in 5 M KOH at 70 °C for 24 h | PET converted to ethylene glycol terephthalate and isophthalic acid | 500 W Xe lamp (simulated solar light, 95 mW cm−2), 10 mg catalyst, 10 ml pre-treated PET aqueous solution, argon atmosphere | H2 (90 μmol g−1 h−1) using 10 M KOH aqueous solution | 43 (2022) |
| g-C3N4 | — | — | 300 W xenon lamp, 50 mg catalyst, 20 mg PS, 30 ml acetonitrile solvent, 1 bar air, 150 °C | Selectivity benzoic acid (39%), acetophenone (7%), benzaldehyde (2%), COx (52%), conversion (95 ± 5%), 24 h reaction | 44 (2022) |
3.4 Composite photocatalysts for plastic upcycling
Currently, the reported composite photocatalysts for plastic upcycling are based on two photon absorbers, both of which can absorb photons and generate photo-induced electrons/holes for upcycling plastics. The effectiveness of composite photocatalysts is determined by the junction type and internal interaction between the components: (i) the most common type II heterojunction can increase light absorption and boost charge separation, but the redox abilities are compromised; (ii) Z-scheme heterojunctions can enhance light harvesting, accelerate electron–hole dissociation and reserve strong redox capabilities, simultaneously; (iii) strong and intimate interaction between different components, which are connected by chemical bonds rather than physical bonds, can significantly improve the charge separation efficiency and stability of overall composite photocatalysts, thus leading to greatly increased performances. The following five studies45–49 are categorized into two kinds of composite photocatalysts: (i) inorganic composite photocatalysts45 and (ii) inorganic/organic composite photocatalysts.46–49 An inorganic composite photocatalyst reported is a 0.5 wt% Pt loaded CdOx/CdS/SiC photocatalyst (Pt-CdOx/CdS/SiC).45 The as-synthesized Pt-CdOx/CdS/SiC was adopted to photo-reform various organic wastes (e.g., PE, PS, IR, cellulose, lignin, albumin and keratin) in 10 M NaOH aqueous solution at 70 °C. The photocatalytic H2 evolution activities of 25.0, 19.4 and 36.7 μmol g−1 h−1 were observed in the presence of PE, PS and IR, respectively. It was found the raised temperature and increased basicity of the reaction system could significantly increase the photocatalytic H2 evolution of Pt-CdOx/CdS/SiC in the presence of α-cellulose, albumin or PE. The photocatalytic stabilities for reforming cellulose, albumin and PE were tested, respectively. Pt-CdOx/CdS/SiC exhibits good stability for reforming cellulose and albumin after re-addition of the substrate. In comparison, Pt-CdOx/CdS/SiC shows poor stability for reforming of PE even after re-addition of the substrate, probably because PE gels cover the photocatalysts at raised temperature. The increased photocatalytic performance of CdOx/CdS/SiC is attributed to the formation of a type II heterojunction between CdS and SiC resulting in enhanced charge separation/transfer.Reported inorganic/organic composite photocatalysts are categorized into inorganic/MOF based composite photocatalysts46,47 and inorganic/C3N4 based composite photocatalysts,48,49 respectively. The Zhang group have reported two research studies on inorganic/MOF based composite photocatalysts.46,47 The Zhang group47 have synthesized a zinc oxide (ZnO)/UiO66-NH2 composite via a partial calcination route, with ultra-small-sized ZnO nanoparticles (NPs) confined into the framework of UiO66-NH2. The as-synthesized ZnO/UiO66-NH2 composite photocatalyst is utilized for photocatalytic valorisation of PLA and PVC. First, they utilized a post-synthesis route to acquire Zn-UiO66-NH2via coordinating Zn2+ with the –NH2 group in Zn-UiO66-NH2 (Fig. 8a). Then, ZnO/UiO66-NH2 with a porous structure was acquired via annealing Zn-UiO66-NH2 in air at 350 °C (Fig. 8a). The SEM image of ZnO/UiO66-NH2 exhibits a uniform rhombic octahedral morphology exposed with a smooth surface (Fig. 8b), indicating that the raw structure of UiO66-NH2 is reserved after the synthesis. The TEM image of ZnO/UiO66-NH2 shows a uniform particle size of about 200 nm (Fig. 8c). The STEM-HAADF image and corresponding elemental mapping images of ZnO/UiO66-NH2 (Fig. 8d) show the homogeneous distribution of O, Zn and Zr elements on ZnO/UiO66-NH2. As for PLA valorisation, the ZnO/UiO66-NH2 composite exhibits a higher acetic acid yield of 14.4%, compared to those of ZnO (3.3%) and UiO66-NH2 (4.7%), respectively (Fig. 8e). They found that the acetic acid generation in the preliminary stage is low followed by gradual enhancement with increasing time (Fig. 8f). This is attributed to the small exposed surface area and low hydrophilicity of big-sized PLA particles in the beginning. Nevertheless, as reaction time increases, these big-sized PLA particles are gradually transformed into small-sized PLA particles with increased hydrophilicity, leading to a larger exposed surface area and higher activity. The ZnO/UiO66-NH2 composite shows an excellent selectivity of 91.6% for acetic acid via valorisation of PLA (Fig. 8g). As for the control experiments, the ZnO/UiO66-NH2 composite shows a much higher acetic acid yield (14.4%) than ZnO (3.3%) and physically mixed ZnO/UiO66-NH2 (2.0%), respectively. These results indicate the great importance of intimate interaction between ZnO and UiO66-NH2. Fig. 8h exhibits the total organic carbon (TOC) concentrations of ZnO/UiO66-NH2 (2.1 g l−1), ZnO (0.6 g l−1) and UiO66-NH2 (0.9 g l−1). The obviously larger TOC concentration for ZnO/UiO66-NH2 suggests its capability of continuously transforming PLA into soluble organic chemicals, e.g., acetic acid, boosted by the synergistic effect between UiO66-NH2 and ZnO. Control experiments (Fig. 8i) exhibit much lower acetic acid generation for physically mixed ZnO/UiO66-NH2 (2.0%) and negligible acetic acid generation without light, a catalyst or PLA, suggesting the key role of strong interaction between ZnO and UiO66-NH2. Further study shows that no acetic acid was detected as a N2 atmosphere is adopted for photocatalytic PLA conversion, indicating the key role of O2 in photocatalytic plastic conversion. Additionally, ZnO/UiO66-NH2 also exhibits excellent stability for photocatalytic PLA valorisation with no obvious change observed on the compositions, structures and morphologies. Besides, ZnO/UiO66-NH2 also exhibits the generation of H2 in photocatalytic PLA valorisation, with the TON (26.36) and TOF (0.75 h−1) observed for H2 evolution (Fig. 8j). The ZnO/UiO66-NH2 composite was also utilized for photocatalytic PVC valorisation, with an acetic acid yield of 9.2% (Fig. 8k) as well as TON of 33.13 and TOF of 0.95 h−1 for H2 evolution (Fig. 8l). Compared to ZnO and UiO66-NH2, the increased photocatalytic activity of the ZnO/UiO66-NH2 composite arises from the broad light absorption, rapid charge dissociation/migration and highly exposed active sites. Then, they use FTIR spectroscopy to test the intermediate products in PLA valorisation by the ZnO/UiO66-NH2 composite. As time increases, the rising intensities of two peaks at 1760 and 3400 cm−1, ascribed to the CO and –OH of the carboxylic acid, respectively, are observed (Fig. 8m). These results also confirm the capability of the ZnO/UiO66-NH2 composite to transform PLA into carboxylic acid containing substances in photocatalysis. Furthermore, they also confirm that the developed ZnO/UiO66-NH2 composite could convert LDPE and PET through a photocatalysis reaction. They also demonstrate that the ZnO/UiO66-NH2 composite can valorise a commercial PLA bag and PLA straw via photocatalysis. Photoelectrochemical (PEC) current density measurement (Fig. 8n) shows the largest PEC current density for ZnO/UiO66-NH2, again confirming the key role of intimate interaction between ZnO and UiO66-NH2. Based on the above results, the excellent PLA conversion of the ZnO/UiO66-NH2 composite is attributed to the following reasons: (i) partial annealing route reserves the highly porous structure of ZnO/UiO66-NH2, thus supplying numerous active centres; (ii) the combination of porous UiO66-NH2 and ZnO leads to efficient interfacial charge separation/migration; (iii) the combination of ZnO with UiO66-NH2 optimises the electronic structure. ESR results and quenching experiments together reveal that the radicals of ˙O2− and ˙OH together play a key role in photocatalytic PLA conversion (Fig. 8o). Besides, Z-scheme charge separation/transfer is increased for the ZnO/UiO66-NH2 composite (Fig. 8o). Finally, a possible reaction pathway is proposed for photocatalytic PLA conversion, and active radicals preferentially cleave the C–O bond of the PLA chain in photocatalysis, leading to the gradual cracking of the PLA carbon chain. Then, PLA is transformed into PLA plastic fragments or oligomers by these active radicals. At last, these PLA plastic fragments or oligomers are transformed into acetic acid by the ZnO/UiO66-NH2 composite. For the conversion of PVC, they propose that the active radicals first cleave the C–Cl bond, followed by further oxidation to oxygen-containing organic intermediates released into the reaction solution. Finally, active radicals further oxidize these organic intermediates to generate acetic acid. In another study, the Zhang group47 have fabricated Ag2O NPs embedded in an Fe-based MOF via in situ conversion of unstable Ag sites in a Fe–Ag bimetallic MOF. First, they synthesized the Fe-based MOF followed by a post-synthesis technique to acquire the Fe–Ag bimetallic MOF. Then, the as-synthesized Fe–Ag bimetallic MOF was subjected to light irradiation to form the Ag2O NP enclosed Fe-based MOF, denoted as Ag2O/Fe-MOF. The MOF structure impedes the growth of Ag2O NPs and renders better dispersion of Ag2O NPs. XRD results confirm that the addition of Ag sites and Ag2O generates defects in the structure of the Fe based MOF. XPS results reveal strong electronic coupling between added Ag sites and the Fe-based MOF in the Fe–Ag bimetallic MOF. XPS results also disclose the defects generated in the structure of the Fe–Ag bimetallic MOF and Ag2O/Fe-MOF. The XPS results also indicate the oxygen vacancies formed in Ag2O/Fe-MOF, due to the cleaving of Fe–O bonds following light illumination. Besides, the etching XPS test of Ag2O/Fe-MOF shows the increasing peak intensity as the etching time increases, further confirming that Ag2O NPs are incorporated into the pores of the Fe based MOF. The incorporation of Ag2O NPs in the pores of Fe-MOF is also confirmed by the N2 sorption analyses, which reveal the obviously reduced surface area of Ag2O/Fe-MOF (110 m2 g−1) compared with that of the Fe–Ag bimetallic MOF (268 m2 g−1). Nevertheless, this surface area of Ag2O/Fe-MOF (110 m2 g−1) is still much higher than that of bare Ag2O (17 m2 g−1), highlighting the advantage of the structure for Ag2O/Fe-MOF. As the porous Ag2O/Fe-MOF can supply a much larger surface area with increased active centres, compared with the Fe based MOF, Ag2O/Fe-MOF exhibits an obviously widened light absorption range, which will boost the photocatalytic performance.
Furthermore, MS plots further indicate that the Fe based MOF and Ag2O are p-type and n-type semiconductors, respectively. These results reveal the formation of a p–n junction in Ag2O/Fe-MOF. Then, transient photocurrent (TPC) density measurements were conducted on bare Ag2O, the Fe based MOF and Ag2O/Fe-MOF. The results show that the TPC density values are as follows: Ag2O/Fe-MOF > Fe-MOF > Ag2O. The highest TPC density value of Ag2O/Fe-MOF is attributed to the formation of a p–n junction in Ag2O/Fe-MOF, leading to increased dissociation/transportation of electrons/holes. Besides, the existence of defects in the structures of Ag2O/Fe-MOF leads to more open frameworks and active centres, thus inducing enhanced photo-generated electrons/holes. Additionally, the results of EIS, steady-state PL spectroscopy and bode-phase plots also confirm the highest efficiency of charge separation/transfer for Ag2O/Fe-MOF among these samples, in accordance with the TPC density measurement results. Then, the as-synthesized Ag2O/Fe-MOF is adopted for photocatalytic H2 evolution coupled with upcycling of PEG. With the introduction of 0.2 wt% Ag2O in Fe-MOF, Ag2O/Fe-MOF (0.2 wt%) exhibits the highest photocatalytic PET MP weight loss (27.5 mg in 3 hours) and H2 evolution (6.2 mmol g−1 in 2.5 hours). In contrast, with lower (0.05 wt%) or higher (1 wt%) Ag2O NPs introduced, both Ag2O/Fe-MOF (0.05 wt%) and Ag2O/Fe-MOF (1 wt%) exhibit inferior photocatalytic PET MP weight loss and H2 evolution. This is because the introduction of Ag2O NPs could boost the catalytic activity of Ag2O/Fe-MOF, whilst excessively introduced Ag2O NPs induce some destruction in the structure of Fe-MOF, thus reducing the number of active sites. In contrast, bare Ag2O and the Fe MOF exhibit inferior PEG conversion efficiency owing to their unfavourable band gap and insufficient light absorption. Additionally, Ag2O shows no H2 evolution since the CB of Ag2O (0.12 V vs. NHE) is lower than the H2 evolution potential (0 V vs. NHE). The physically mixed sample of Ag2O and Fe-MOF (Ag2O@Fe-MOF) exhibit a low PEG conversion (6.1 mg) and H2 evolution (2.3 mmol g−1). These results reveal that the developed photochemistry route induces intimate interaction between Ag2O and Fe-MOF for efficient charge transport. The small sizes of Ag2O NPs incorporated in the pores of Fe-MOF ensure the exposure of abundant active sites, leading to enhanced conversion of MPs. Total organic carbon (TOC) analysis results disclose a remarkable increase in TOC for Ag2O/Fe-MOF and Fe-MOF, since the photocatalyst starts to transform the MPs into water-dissolvable long-chain fragments. This also results in the obvious reduction of the weight on PEG in the beginning of photocatalysis. Further analysis indicates the formation of small amounts of acetic acid in 0–5 hours of photocatalytic reaction, suggesting that in the beginning of photocatalysis radicals convert PEG MPs into long-chain fragments. The generation of ethanol and formic acid is also disclosed. The increased TOC concentration is much higher than the formation of formic acid, ethanol and acetic acid in total, indicating that some of the PEG MPs were transformed into soluble MPs and other intermediates. They also tested the photocatalytic reforming of PE and PET MPs for all the samples. Ag2O/Fe-MOF exhibits much higher PE and PET MP weight loss, compared with bare Ag2O and Fe-MOF, respectively. Ag2O/Fe-MOF also exhibits the increased photocatalytic H2 evolution activities of 1.7 and 1.9 mmol g−1 h−1 for PE and PET MPs, respectively. The above results not only corroborate that the incorporation of Ag2O into porous Fe-MOF can be adopted for photocatalytic reforming of PEG/PE/PET MPs, but also confirm that the active centres arising from structure defects can boost the plastic upcycling. The robustness of the activities and structures for Ag2O/Fe-MOF is also confirmed. ESR experiments confirm the existence of ˙OH radicals from Ag2O/Fe-MOF in photocatalysis. And they propose a possible reaction pathway: PEG → ethylene glycol → glycolaldehyde → glycolate → glyoxylate → acetic acid → formic acid. Polyoxometalates (POMs), or transition metal oxygen anion clusters, are selected since they show outstanding robustness unaffected by pH, solvent and temperature. Besides, these POMs exhibit reversible multi-electron redox conversions while reserving robust structures, leading to their wide application in photo-/electro-catalysis fields. Especially, phosphovanadomolybdate (H5PMo12−nVnO40) is extensively adopted as the active catalyst involving in organic oxidation with O2. More importantly, vanadium atoms with variable valence in Keggin-structured H5PMo10V2O40 exhibits the capability for catalysing C–C bond cleavage reaction via the electron transfer-oxygen transfer reaction. Nevertheless, the rapid recombination of photo-induced electrons/holes and low redox capabilities for VPOM, as decided by its bandgap structure, significantly impedes its application in photocatalysis. Thus, it is desirable to combine VPOM with other materials for constructing VPOM based composite photocatalysts, thus achieving enhanced charge separation/transfer efficiency and increased catalytic abilities.
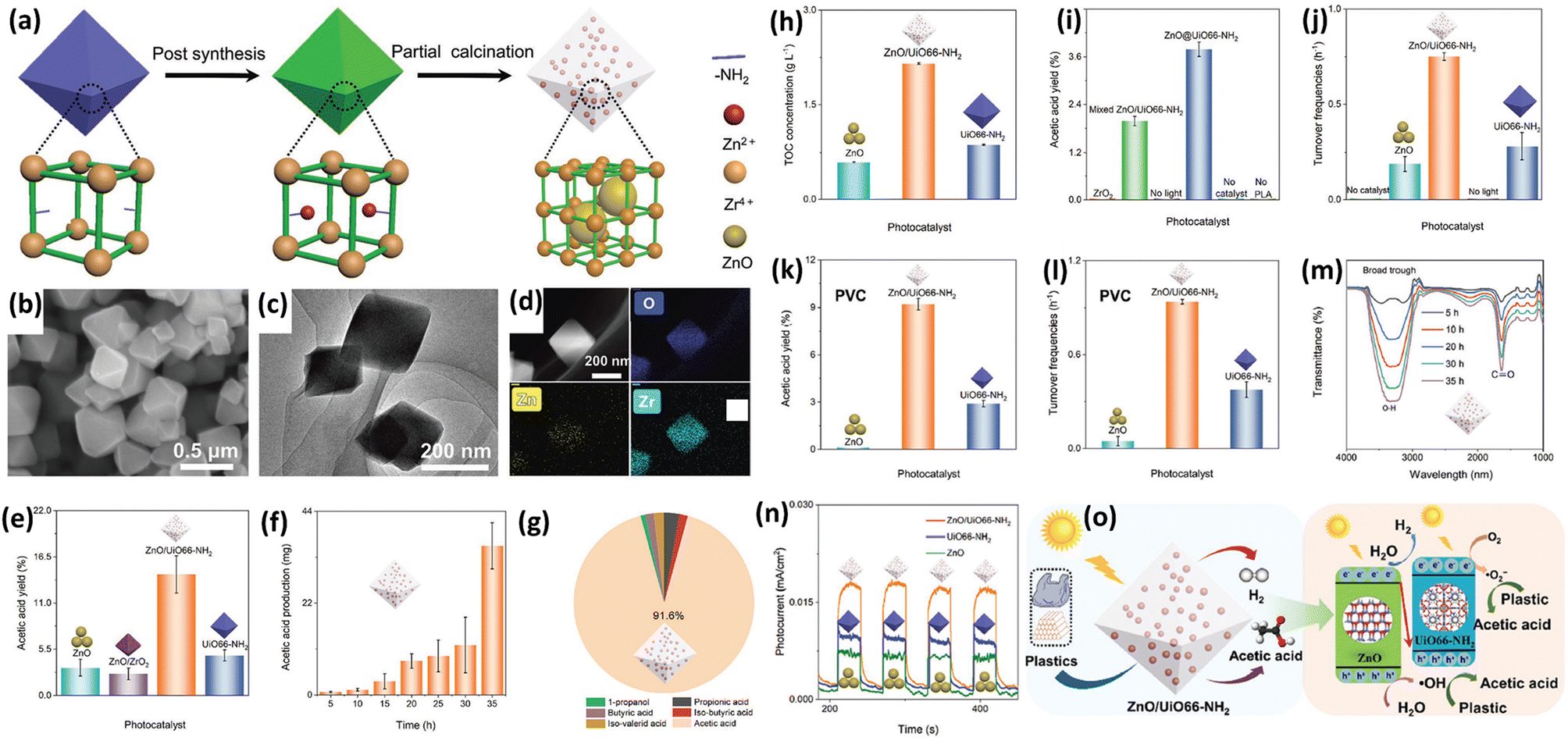 | ||
| Fig. 8 (a) Schematic image showing the synthesis of ZnO/UiO66-NH2 using post-synthesis and partial calcination. (b) SEM image and (c) low magnification TEM images of UiO66-NH2. (d) HAADF-STEM image of the as-synthesized ZnO/UiO66-NH2 and corresponding elemental mapping images. (e) Photocatalytic yields of acetic acid for ZnO, ZnO/ZrO2, UiO66-NH2 and ZnO/UiO66-NH2. (f) Generation of acetic acid for ZnO/UiO66-NH2 at different reaction times. (g) Selectivity of various products for ZnO/UiO66-NH2. (h) Concentration of TOC for ZnO, ZnO/UiO66-NH2 and UiO66-NH2 reaction systems. (i) Yields of acetic acid on ZrO2, mixed ZnO/UiO66-NH2, and ZnO/UiO66-NH2, without light, without a catalyst or without PLA. (j) TOF for H2 evolution on UiO66-NH2, ZnO or ZnO/UiO66-NH2 in PLA reaction systems. (k) Yield of acetic acid and (l) TOF for H2 evolution on ZnO, ZnO/UiO66-NH2 or UiO66-NH2 in PVC reaction systems. (m) FTIR spectra of reaction solution in photocatalytic reforming of PLA using ZnO/UiO66-NH2 at different reaction times. (n) Transient photocurrent density measurements for ZnO, UiO66-NH2 and ZnO/UiO66-NH2. (o) Schematic image for the photo-generated charge dissociation/transfer in ZnO/UiO66-NH2. Reproduced with permission from ref. 47 American Chemical Society. | ||
The other two studies are based on inorganic/polymerized C3N4 based composite photocatalysts.48,49 One work48 reports a heterostructure composed of V-substituted phosphomolybdic acid clusters coupled with g-C3N4 nanosheets (VPOM/CNNS). The FTIR spectra of VPOM/CNNS exhibit distinctive vibration modes of CNNS and Keggin units of VPOM, confirming the successful combination of CNNS with VPOM. Besides, VPOM/CNNS also shows a similar surface area to CNNS (103.51 m2 g−1), suggesting that CNNS reserving its ultrathin nanosheet morphology after combining with VPOM clusters. TEM results show that the VPOM/CNNS composite reserves the distinct two-dimensional layered structure. Aberration-corrected high-angle annular dark field scanning transmission electron microscopy (AC-HAADF-STEM) combined with elemental mapping analysis confirms the uniform distribution of VPOM clusters on the surface of CNNS. The XPS results confirm the electron transfer from CNNS to VPOM in the VPOM/CNNS composite. The newly formed peaks in the XPS O 1 s spectra indicate the generation of C–O–Mo or C–O–V bonds, again revealing the coupling of VPOM with CNNS. VPOM/CNNS exhibits increased light harvesting in the range of 460–600 nm, in contrast with bare CNNS, again revealing the existence of VPOM in VPOM/CNNS. Via combining the XPS VB and UPS results, they found that VPOM and CNNS construct a type II hetero-junction with a built-in electric field pointing from CNNS to VPOM. Furthermore, in situ XPS results confirm the accumulation of photo-induced electrons and holes in CNNS and VPOM, respectively, with light illumination. These results confirm the Z-scheme charge transfer in the VPOM/CNNS composite. Femtosecond transient absorption spectroscopy (fs-TAS) was adopted to study the photo-induced electron/hole kinetics in VPOM/CNNS. Via applying AgNO3 as the electron scavenger, they found that the peak at ∼686 nm is ascribed to the CNNS˙− absorption and the signal at 550 nm is principally attributed to the photo-induced holes of CNNS. VPOM/CNNS exhibits increased CNNS˙− absorption at ∼686 nm, compared to bare CNNS, suggesting the more effective separation/transfer of photo-generated electrons/holes. Furthermore, they found an additional decay component (τ3 = 19.42 ps) for the hole species of VPOM/CNNS, which is ascribed to the Z-scheme charge transfer pathway in the heterostructure interface. VPOM/CNNS composites all exhibit increased decay lifetimes of electron species, compared to CNNS alone, suggesting the more efficient dissociation/migration of photo-generated electrons in VPOM/CNNS. ESR experiments further show the obvious enhanced signals of DMPO-˙O2− and DMPO-˙OH signals compared with CNNS or VPOM alone, again confirming the increased charge kinetics in the Z-scheme junction. Then, the as-synthesized photocatalysts were adopted for photocatalytic reforming of a range of plastics. First, they were utilized to photo-reform PE as it is extensively applied and not biodegradable. The optimised VPOM-CNNS composite shows an outstanding photocatalytic HCOOH generation rate (24.66 μmol h−1 g−1), about 262 times larger than that of CNNS alone. The optimised VPOM-CNNS composite also exhibits better photocatalytic activity than the mechanically mixed VPOM and CNNS. These further corroborate that the intimate interaction between VPOM and CNNS could obviously increase the charge separation/transfer efficiency. Additionally, a 100 hour stability test was also conducted on the VPOM/CNNS composite. Excellent stability of HCOOH generation was observed on the VPOM/CNNS composite for photocatalytic reforming of PE. After a 100 hour reaction, no apparent alteration can be found in the composition/structure of the VPOM/CNNS composite. Besides, VPOM/CNNS was also used for photocatalytic reforming of PP, PVC, PEG and PAM. HCOOH is identified as the upcycled product. The HCOOH generation rate from photocatalytic upcycling of PEG and PAM is much larger than that of the others. The reasons are as follows: (i) the polarity groups in PEG (hydroxyl and ether groups) and polyacrylamide (–NH2 group) significantly facilitate their dissolution in polar solvents of acetonitrile/water, thus increasing the possibility for the plastic molecules to react with the photo-generated active species; (ii) it is much easier to activate the asymmetric C–O bonds in the PEG, compared to the inert nonpolar C–C bonds. Additionally, VPOM/CNNS also exhibits outstanding photocatalytic HCOOH generation from photocatalytic reforming of real-word PE bags, PVC plastic wrap or PP surgical masks. Species-trapping experiments were also carried out via using nitrogen, p-benzoquinone, oxalic acid and nitrobenzene as the O2, ˙O2−, h+ and e− scavengers, respectively. H+ and ˙O2− were identified as the principal reactive species towards photocatalytic reforming of plastics. 1H NMR spectroscopy was further adopted to study the photocatalytic reforming reaction. After 36 hour visible-light illumination, apart from HCOOH as the principal product, large amounts of long-chain alcohols and a trace amount of formaldehyde were identified in the liquid phase. These are common intermediates in the electron transfer-oxygen transfer oxidation reaction of vanadium compounds. This is further confirmed by the observation of an eight-line signal of VIV after light illumination in an argon atmosphere, suggesting that some of the VV species in VPOM are reduced to form VIV in the photocatalytic reforming of PE reaction. IR spectra reveal the generation of new carbonyl groups in the range of 1710–1760 cm−1 in photocatalytic reforming of PE plastic bags. And the generation of peroxides was also confirmed to arise from the reaction between alkyl radicals and reactive oxygen species. DFT computations were also conducted to acquire the insightful understanding. The computation results indicate that the C and N elements in CNNS and O elements in VPOM serve as the principal reactive sites towards photocatalytic reforming of plastics. On the basis of all the results, they propose a photocatalytic mechanism: with visible-light illumination, both VPOM and CNNS will be excited to generate abundant photo-induced electrons and holes. Via the ligand to metal charge transfer (LMCT), the electrons from the O atom in the HOMO of VPOM is excited to an antibonding orbital of the LUMO in the transition metal centres (V or Mo). For CNNS, with light illumination, electrons are excited from the HOMO or N 2p states to the LUMO or hybridized C 2p and N 2p states. Then, the photo-induced electrons and holes will migrate and dissociate following the Z-scheme scheme. Afterwards, the photo-induced holes remaining in the HOMO (O 2p states) of VPOM will from oxo-centred radicals. Finally, the highly active and photo-excited VPOM clusters would boost the oxidative cleavage of the C–C bond, leading to the formation of formaldehyde and a carbon-centred alkyl radical. Then, reactive oxygen species will oxidize the alkyl radical to generate alkyl peroxide groups, which is transformed into long-chain alcohols. At the same time, formaldehyde will be oxidized to generate formic acid, by the generated ˙O2−. Furthermore, Gong et al.49 have developed a metal-free photocatalyst composed of carbonized polymer dot-graphitic carbon nitride (CPDs-CN). The as-synthesized carbonized polymer dots (CPDs) possess a big conjugated graphitic sp2 carbon combined with sp3 carbons, as confirmed by 1H and 13C NMR spectroscopy. FTIR and XPS spectroscopy techniques together confirm the existence of carboxylic, hydroxyl and amino functional groups in CPDs. The TEM images show the CPDs-CN composite comprising CPDs with sizes of 1.9–2.4 nm loaded on the surface of CN sheets. The XPS results confirm the combination of CPDs with CN sheets via forming amide bonds. The coupling of CPDs with CN sheets also leads to the change of colour from light yellow for CN to dark brown for CPDs-CN, thus increasing the light absorption in the whole visible-light range (400–800 nm). The as-synthesized CPDs-CN was utilized for photocatalytic reforming of PET and PLA. Photocatalytic reforming of the pre-treated PET solution leads to generation of abundant EG-derived chemicals, such as glycolaldehyde, glycolic acid, formic acid, ethanol, acetaldehyde and acetic acid after 8 day photocatalytic reforming using CPDs-CN. 1H and 13C NMR spectroscopy reveal that: (i) PET plastic conversion is increases monotonically with increasing time; (ii) high selectivity is achieved for glycolic acid and acetic acid; (iii) little selectivity change is observed for the intermediates, such as glycolaldehyde, formic acid, ethanol and acetaldehyde. The reaction pathway is revealed: photo-induced holes first oxidize EG to form glycolaldehyde, followed by further oxidation to generate glycolic acid and formic acid. Besides, EG could also be dehydroxylated to generate ethanol, followed by further oxidation to acetaldehyde and acetic acid. The conventional anatase TiO2 exhibits inferior photocatalytic activities for generating the above chemicals, compared to CPDs-CN. The photocatalytic H2 evolution activities coupled with PET/PLA hydrolysis were also determined. Without Pt as the co-catalyst, CPDs-CN exhibits a photocatalytic H2 evolution activity of 298 ± 58 μmol g−1 h−1 using pre-treated PET as the substrate. In comparison, with Pt loaded as the co-catalyst, CPDs-CN exhibits a photocatalytic H2 evolution activity of 1034 ± 134 and 1326 ± 181 μmol g−1 h−1via using pre-treated PET and PLA as the substrate, respectively. As a contrast, anatase TiO2 loaded with Pt only exhibits a photocatalytic H2 evolution activity of 55 ± 4 μmol g−1 h−1 using pre-treated PET as the substrate. The experimental results show that CPDs can obviously increase the light harvesting and boost the dissociation/transportation of photo-excited electrons/holes, thus leading to the increased activities of CPDs/CN. Also, no ˙OH was detected via the fluorescence experiment, suggesting that photo-excited holes play a key role in oxidation of substrates.
The above five studies underscore the appealing prospects of composite photocatalysts for plastic upcycling, which exhibit both an extended absorption range of light and efficient charge separation/transfer.45–49 Especially, MOF based composite photocatalysts show excellent performances for upcycling untreated polyesters/polyolefins in air and in organic solvent (e.g., acetonitrile).46,47 Besides, poly-oxalate based composite photocatalysts also exhibit outstanding activities/selectivity for upcycling untreated polyesters/polyolefins in an O2 atmosphere and in organic solvent (e.g., acetonitrile).48 All the performances and reaction conditions in this section are summarized in Table 4.
| Composite catalyst | Pre-treatment | Products after pre-treatment | Reaction conditions | Activity and stability | Reference (year) |
|---|---|---|---|---|---|
| 0.5 wt% Pt-CdOx/CdS/SiC | — | — | Solar simulator (AM 1.5G), 50 mg catalyst, 5 ml 10 M NaOH aqueous solution, 100 mg PE, AR atmosphere, 70 °C | H2 (25.0 μmol g−1 h−1) | 45 (2022) |
| — | — | Solar simulator (AM 1.5G), 50 mg catalyst, 5 ml 10 M NaOH aqueous solution, 100 mg PE, AR atmosphere, 70 °C | H2 (19.4 μmol g−1 h−1) | ||
| ZnO/UiO66-NH2 | — | — | 300 W Xe lamp, 0.1 g catalyst, 1.0 g PLA, 50 ml water, in air, 25 °C | PLA conversion rate (57.1 mg g−1 h−1), acetic acid evolution (selectivity = 91.6%; yield = 14.4%; TON = 17.92; TOF = 0.51 h−1), H2 evolution (TON = 26.36; TOF = 0.75 h−1) | 46 (2022) |
| — | — | 300 W Xe lamp, 0.1 g catalyst, 1.0 g PVC, 50 ml water, in air, 25 °C | PVC conversion rate (21.4 mg g−1 h−1), acetic acid evolution (yield = 9.2%; TON = 0.90; TOF = 0.03 h−1), H2 evolution (TON = 33.13 and TOF = 0.95 h−1) | ||
| Ag2O nanoparticle encapsulated Fe based MOF (Ag2O/Fe-MOF) | — | — | 300 W Xe lamp (AM 1.5G, 100 mW cm−2), 0.1 g catalyst, 0.5 g PEG MPs, 100 ml water, 25 °C, in air | H2 (3.6 mmol g−1), 2.5 h reaction, acetic acid (11.7 mg l−1), 5 h reaction | 47 (2023) |
| 10 g PET immersed in 1 M NaOH solution at 65 °C, stirred for two weeks followed by centrifuging and freeze-drying the undissolved plastic pieces | PET MPs | 300 W Xe lamp (AM 1.5G, 100 mW cm−2), 0.1 g catalyst, 0.5 g PET MPs, 100 ml water, 25 °C, in air | H2 (1.9 mmol g−1 h−1) | ||
| 10 g PE immersed in 1 M NaOH solution at 65 °C, stirred for two weeks followed by centrifuging and freeze-drying the undissolved plastic pieces | PE MPs | 300 W Xe lamp (AM 1.5G, 100 mW cm−2), 0.1 g catalyst, 0.5 g PE MPs, 100 ml water, 25 °C, in air | H2 (1.7 mmol g−1 h−1) | ||
| V-Substituted phosphomolybdic acid clusters/g-C3N4 nanosheets (VPOM/CNNS) | — | — | 300 W Xe lamp with a 420 nm cut-off optical filter, 10 mg catalyst, 20 mg PE, 10 ml acetonitrile, O2 atmosphere, 20–40 °C | Formic acid evolution (24.66 μmol g−1 h−1), 36 h reaction | 48 (2021) |
| — | — | 300 W Xe lamp with a 420 nm cut-off optical filter, 10 mg catalyst, 20 mg PEG, 10 ml acetonitrile, O2 atmosphere, 20–40 °C | Formic acid evolution (208.65 μmol g−1 h−1), 36 h reaction | ||
| — | — | 300 W Xe lamp with a 420 nm cut-off optical filter, 10 mg catalyst, 20 mg PP, 10 ml acetonitrile, O2 atmosphere, 20–40 °C | Formic acid evolution (26.68 μmol g−1 h−1), 36 h reaction | ||
| — | — | 300 W Xe lamp with a 420 nm cut-off optical filter, 10 mg catalyst, 20 mg PVC, 10 ml acetonitrile, O2 atmosphere, 20–40 °C | Formic acid evolution (29.85 μmol g−1 h−1), 36 h reaction | ||
| — | — | 300 W Xe lamp with a 420 nm cut-off optical filter, 10 mg catalyst, 20 mg PAA, 10 ml water, O2 atmosphere, 20–40 °C | Formic acid evolution (156.57 μmol g−1 h−1), 36 h reaction | ||
| Carbonized polymer dot coupled graphitic carbon nitride (CPD-CN) | Refluxing PET in 5 M KOH aqueous solution at 120 °C for 24 h | PET converted to ethylene glycol terephthalic acid and isophthalic acid | 300 W xenon lamp (AM 1.5G), 20 mg catalyst, 1.25 g PET, 50 ml 1 M KOH aqueous solution, 40 °C, vacuum environment | H2 (1034 ± 134 μmol g−1 h−1), selectivity, glycolaldehyde (4%), glycolic acid (21%), formic acid (12%), ethanol (37%), acetaldehyde (12%), acetic acid (14%) | 49 (2022) |
| Refluxing PLA in 5 M KOH aqueous solution at 100 °C for 2 h | PLA converted to lactate | — | H2 (1326 ± 181 μmol g−1 h−1) |
4. Conclusion and outlook
The above studies introduce the current achievements in various photocatalysts for plastic upcycling. These photocatalysts were categorized into four different types: (i) metal oxide based photocatalysts; (ii) metal sulphide based photocatalysts; (iii) non-metal based photocatalysts and (iv) composite photocatalysts. Usually, metal oxide based photocatalysts (e.g., TiO2) possess very positive valence band potentials and strongly oxidative photo-excited holes, which can generate highly oxidative ˙OH radicals to directly oxidize the inert/robust plastics (e.g., PE, PP and PVC) and cleave the strong C–C/C–O/C–H bonds to form value-added short-chain chemicals/fuels under aerobic/anaerobic conditions and at room temperature. In some work, metal oxide based photocatalysts, e.g. Nb2O5, can even fully oxidize the inert polyolefins (e.g., PE, PP and PVC) to yield CO2 in air and at room temperature, which can be further reduced to form a C2 chemical (acetic acid). But the evolution efficiencies of these chemicals/fuels for metal oxide based photocatalysts are restricted owing to the wide bandgap width and weak absorption of solar light. In contrast, metal sulphide based photocatalysts (e.g., CdS/CdOx and Cd0.5Zn0.5S) with suitable bandgap widths exhibit strong absorption of light and favourable redox abilities. Photo-induced holes of metal sulphide based photocatalysts possess moderate oxidation abilities to upcycle the pre-treated polyester/polyolefin solutions and yield value-added chemicals without their overoxidation to form CO2. On the other hand, photo-excited electrons of metal sulphide based photocatalysts with sufficient reduction capacity could efficiently yield H2 fuel under anaerobic conditions. Even though metal sulphide based photocatalysts exhibit the above advantages, the insufficient stability and notorious toxicity, especially for Cd-based photocatalysts, significantly restrict their industrial scale applications for solar plastic upcycling. Furthermore, non-metal based photocatalysts, e.g., CxNy based catalysts, have shown many attractive edges, such as cheapness, high abundance, suitable bandgap width, adequate redox capacities, strong robustness against photo-/chemical corrosion and regulable structure/composition/functionalities. Actually, CxNy based catalysts coupled with a cocatalyst, such as Ni2P, have displayed some activities/selectivity/stability for yielding valuable chemicals/fuels via upcycling the monomers (e.g., ethylene glycol) released by pre-treating the polyesters (PET and PLA) at room temperature and under anaerobic conditions. Interestingly, CxNy based photocatalysts, e.g., C3N4, even exhibit some activity/selectivity/robustness for photocatalytic upcycling of untreated PS into value-added chemicals at raised temperature, in organic solvent (acetonitrile) and in air. But the efficiencies of non-metal CxNy based photocatalysts are still much lower compared to those of metal-based photocatalysts, making their industrial utilization impossible at the moment. Compared with the above single-photon-absorber based photocatalysts, composite photocatalysts comprising two or even more photo-absorbers are also summarized in this review. They are categorized into inorganic based composite photocatalysts and inorganic/organic based composite photocatalysts. Only one inorganic composite photocatalyst is discussed in this review. The Pt cocatalyst loaded CdOx/CdS/SiC (Pt-CdOx/CdS/SiC) photocatalyst exhibits low H2 evolution activities from upcycling the untreated PE at raised temperature, in concentrated alkaline solution and under anaerobic conditions. These are mainly attributed to the strong C–C/C–H bonds of inert PE and lack of strongly oxidative ROS, e.g., ˙OH radicals, owing to the unfavourable valence band edge potentials of CdS or SiC together with anaerobic reaction conditions. For inorganic/organic composite photocatalysts, two MOF based composites, Ag2O/Fe-MOF and ZnO/UiO66-NH2, are introduced in this review. Ag2O/Fe-MOF exhibits good H2 evolution activities for upcycling the PEG/PET/PE MPs. Especially, Ag2O/Fe-MOF shows some activity in upcycling PEG MPs into acetic acid. Interestingly, ZnO/UiO66-NH2 shows good efficiencies for upcycling untreated PLA or PVC into acetic acid with outstanding selectivity accompanied by some H2 evolution, in water, at room temperature and in air. The good efficiencies of both Ag2O/Fe-MOF and ZnO/UiO66-NH2 are mainly attributed to the rational synthesis strategy to yield Ag2O or ZnO NPs encapsulated in the pores of a MOF structure, which ensures strong interaction between Ag2O or ZnO NPs and the MOF structure for rapid charge separation/transfer, as well as excellent scattering of ultra-little Ag2O or ZnO NPs exposed with a high surface area. Another interesting study about inorganic/organic composite photocatalysts reports the synthesis of poly-oxalate combined polymeric C3N4, i.e., V-substituted phosphomolybdic acid clusters/g-C3N4 nanosheets (VPOM/CNNS), for upcycling untreated PE/PEG/PP/PVC/PAA in water or organic solvent (acetonitrile) to generate a value-added chemical (formic acid) at room temperature and in an O2 atmosphere. This activity arises from the strong oxidation ability on the photo-induced holes from VPOM and the efficient Z-scheme charge transfer in VPOM/CNNS. Currently, although some advancements have been realized in this field, tremendous challenges are required to be overcome in the future, which is anticipated to bring about numerous opportunities in this field. For instance, the realistic application of photocatalytic plastic upcycling is severely restricted by the insufficient activity, selectivity and stability together with the low cost-effectiveness of photocatalysts. First, the inefficient use of the whole solar spectrum seriously restricts the maximum efficiency for solar-to-chemical conversion using the photocatalysis technique. Second, the high recombination rate of photo-excited electrons and holes in photocatalysts further limits the efficiency of photocatalysts seriously. This mainly arises from the different time scales for the generation/recombination of electron–hole pairs (picosecond to nanosecond) and surface redox reactions (millisecond to second). Third, the easy aggregation of photocatalyst powder suspended in aqueous solution significantly impedes the performance of powder-form photocatalysts for large-scale applications. Besides, recycling use of these photocatalysts via regeneration/re-activation is also challenging, since the separation of these powder-form photocatalysts from the suspension reaction system is difficult. Lastly, the long-term use of photocatalysts will lead to the gradual reduction of performance due to the reduced crystallinity and covered active sites by the product. Thus, herein, we summarize these challenges and possible opportunities in the following three sections:4.1 Regulation of the characteristics of photocatalysts for increased activity/selectivity/stability
(1) Currently, no studies report photocatalysts with atomic-scale active sites in this field. So catalysts with atomic-scale reactive sites, such as single atoms, bi-single atoms and clusters, can be screened and developed for photocatalytic upcycling of various types of plastics (e.g., PE, PP, PVC, PS and PET) into targeted value-added chemicals/fuels with excellent activity/selectivity/stability.(2) Only limited engineering methods, e.g., loading cocatalysts, element doping, morphology controlling and constructing Z-scheme/type II junctions, have been applied in this field. Thus, those advanced engineering routes of photocatalysts, e.g., phase engineering, defect engineering, facet engineering and band structure engineering, can also be utilized for photocatalytic plastic upcycling.
(3) The cocatalyst plays a significant role in enhancing the activity/selectivity/stability of the photocatalyst. But currently only a few studies report the loading of cocatalysts (e.g., Pt NPs, Ni2P NPs, MoS2 and NiMo) for photocatalytic plastic upcycling. And no insightful studies on the functional mechanism of these cocatalysts have been performed and reported. So more studies can be focused on engineering the composition/structure of the cocatalyst and its interaction with photocatalysts for tuning their activity/selectivity/stability for specific upcycling reactions.
(4) Currently, all the metal sulphide based photocatalysts reported in this field are based on Cd-based photocatalysts and suffer from notorious toxicity in realistic applications. Thus, Cd-free metal sulphide based photocatalysts can be screened and developed for photocatalytic plastic upcycling.
(5) Certain photocatalysts, e.g., metal sulphides/selenides/phosphides, usually suffer from inferior photo-/thermal-/chemical-stability, compared to those of metal oxides. Their stability can be enhanced by the following strategies: (a) combining with other photocatalysts/co-catalysts (e.g., metal oxides and metals) to boost electron–hole separation/transfer with reductive/oxidative electron/hole transfer to other photocatalysts/co-catalysts for avoiding self-reduction/-oxidation; (b) coating with a metal oxide layer to avoid chemical corrosion from the acidic/alkaline reaction environment.
(6) Cheap and robust CxNy catalysts can be studied more owing to their unique advantages of earth-abundance, strong absorption of light and suitable oxidation abilities.
4.2 Advanced characterization and theoretical computations for revealing the structure–activity relationship and insightful reaction mechanisms
(1) A variety of in situ characterization techniques, such as in situ XPS, in situ ESR, in situ FTIR, in situ Raman, in situ AFM-KPFM, in situ XAS, in situ TAS, in situ SPV and in situ TSPL, can be utilized to reveal the structure–activity relationship and reaction mechanism, especially the in situ time-resolved characterization to study the femtosecond-scale kinetics of electron/hole separation/transfer/trapping/recombination in catalysts.(2) It still remain unknown how the ROS is involved in photocatalytic plastic upcycling reactions. Various in situ characterization techniques, especially in situ ESR and in situ FTIR, can contribute to the study of ROS involved reactions, in which inert and untreated polyester/polyolefins can be efficiently upcycled into valuable short-chain chemicals/fuels.
(3) Online GC-TCD/FID and HPLC systems can be established and utilized to track and monitor the intermediates and products in photocatalytic plastic upcycling for revealing the insightful reaction mechanism under realistic conditions.
(4) Based on experimental results, theoretical computations, especially operando computation approaches, can be utilized to gain further insights into the structure–performance correlation in photocatalysts for plastic upcycling. They can also be applied to study the reaction mechanism via revealing the reaction thermodynamics/kinetics in plastic upcycling.
4.3 Advanced technologies for the realistic application in this area
(1) Currently, most of the photocatalytic reactions were conducted at 25 °C. The infrared region of the solar spectrum should be utilized to raise the reaction temperature for enhancing the reaction rates. Inexpensive and scalable reactors should be developed, which can efficiently utilize infrared light to raise the temperature of the reaction system and reserve the heat inside the reactor to keep the reaction system at a desirable temperature without external heating. Thus, the rates of photocatalysis reactions can be enhanced greatly.(2) Seawater can be utilized to upcycle these plastic wastes to avoid the use of limited fresh water resources.
(3) Flow reactors can be utilized to avoid the overoxidation of chemicals to form tremendous CO2 generated in photocatalytic upcycling.
(4) In photocatalytic plastic upcycling, abundant CO2 might be generated due to the overoxidation of plastics, especially when an air atmosphere is applied. Thus, CO2 concentration should be monitored in photocatalytic plastic upcycling. And the efficient capture of the yielded CO2 and its further conversion into value-added carbon-based chemicals using identical photocatalysts should be studied.
(5) Earth-abundant and cheap cocatalysts can be developed to significantly increase the rate, selectivity and stability of photocatalysts for large-scale and cost-effective plastic upcycling using sunlight. Especially, efficient, low-cost and scalable loading techniques should be explored to atomically disperse these highly effective cocatalysts onto photocatalysts.
(6) Studies should be more focused on one-step photocatalytic upcycling of plastics without any pre-treatment.
(7) Currently, most reactions are conducted in aqueous solution. However, it is very hard for plastics to be suspended well in aqueous solution. More research should be conducted in some organic phase solvent (e.g., acetonitrile and dichloromethane) to better suspend and/or dissolve plastics and ensure better interaction between the catalyst and reactant/intermediate together with more rapid mass transfer.
(8) Currently, all the photocatalytic plastic upcycling is conducted based on one reactor system, which cannot meet the requirements for realistic applications. Reaction systems containing multiple reactors with photocatalysts possessing different functions can be designed and constructed. For examples, one reactor containing metal oxide photocatalysts can be used to cleave the C–C/C–O/C–N/C–H bonds of plastics and yield monomers/oligomers/small molecules. Furthermore, these yielded monomers/oligomers/small molecules can be further transferred to another reactor containing metal sulphides/CxNy, which possess mild oxidation abilities to transform these chemicals to acquire value-added chemicals without over-oxidizing them to yield CO2.
(9) A solar simulator (AM 1.5G, 100 mW cm−2) is utilized in most reactions for photocatalytic plastic upcycling. For realistic applications in the future, solar concentrators can be applied to increase the photon intensity to achieve significantly increased efficiency.
(10) More efficient and cost-effective pre-treatment strategies can be developed and adopted to be combined with the photocatalysis technique for catalytic upcycling of plastics into value-added chemicals/fuels via environmentally benign and cost-effective routes.
(11) In realistic applications, it is very challenging to separate plastics and many of them are mixed with each other. Thus, more studies on photocatalytic upcycling of mixed plastics should be conducted to accelerate the development of realistic plastic upcycling techniques.
Author contributions
S.-Z. Q. conceived the topic and structure of this manuscript. J. R. wrote the major part of the manuscript. A. T.-K. summarized the table, wrote some part of the manuscript and helped revise the manuscript. All the other authors have helped revise the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support from the Australian Research Council (ARC) through the Discovery Project programs (FL170100154, DE200100629, FT230100192, and DP230102027) and Linkage project (LP210301397, CE230100032).References
- S. Chu, B. Zhang, X. Zhao, H. S. Soo, F. Wang, R. Xiao and H. Zhang, Adv. Energy Mater., 2022, 12, 2200435 CrossRef CAS.
- A. Chen, M.-Q. Yang, S. Wang and Q. Qian, Frontal Nanotechnol., 2021, 3, 723120 CrossRef.
- N. Li, H. Liu, Z. Cheng, B. Yan, G. Chen and S. Wang, J. Hazard. Mater., 2022, 424, 127460 CrossRef CAS PubMed.
- K. Su, H. Liu, C. Zhang and F. Wang, Chin. J. Catal., 2022, 43, 589–597 CrossRef CAS.
- X. Chen, Y. Wang and L. Zhang, ChemSusChem, 2021, 14, 4137–4151 CrossRef CAS PubMed.
- S. Kang, T. Sun, Y. Ma, M. Du, M. Gong, C. Zhou, Y. Chai and B. Qiu, SmartMat, 2023, 4, e1202 CrossRef.
- M. R. K. Estahbanati, X. Y. Kong, A. Eslami and H. S. Soo, ChemSusChem, 2021, 14, 4152–4199 CrossRef PubMed.
- Q. Hou, M. Zhen, H. Qian, Y. Nie, X. Bai, T. Xia, M. L. U. Rehman, Q. Li and M. Ju, Cell Rep. Phys. Sci., 2021, 2, 100514 CrossRef CAS.
- K. Hu, Y. Yang, Y. Wang, X. Duan and S. Wang, Chem Catal., 2022, 2, 725–764 Search PubMed.
- Y. Wu, T. Sakurai, T. Adachi and Q. Wang, Nanoscale, 2023, 15, 6521–6535 RSC.
- J. Chen, J. Wu, P. C. Sherrell, J. Chen, H. Wang, W.-X. Zhang and J. Yang, Adv. Sci., 2022, 9, 2103764 CrossRef.
- X. Li, J. Wang, T. Zhang, S. Yang, M. Sun, X. Qian, T. Wang and Y. Zhao, Chem. Eng. Sci., 2023, 276, 118729 CrossRef CAS.
- H. Zhou, Y. Wang, Y. Ren, Z. Li, X. Kong, M. Shao and H. Duan, ACS Catal., 2022, 12, 9307–9324 CrossRef CAS.
- M.-Q. Zhang, M. Wang, B. Sun, C. Hu, D. Xiao and D. Ma, Chem, 2022, 8, 2912–2923 CAS.
- A. Mohanty, R. K. Borah, A. P. Fatrekar, S. Krishnanc and A. A. Vernekar, Chem. Commun., 2021, 57, 10277–10291 RSC.
- X. Jiao, K. Zheng, Z. Hu, S. Zhu, Y. Sun and Y. Xie, Adv. Mater., 2021, 33, 2005192 CrossRef CAS PubMed.
- X. Zhao, M. Korey, K. Li, K. Copenhaver, H. Tekinalp, S. Celik, K. Kalaitzidou, R. Ruan, A. J. Ragauskas and S. Ozcan, Chem. Eng. J., 2022, 428, 131928 CrossRef CAS.
- S. H. Paiman, S. F. M. Noor, N. Ngadi, A. H. Nordin and N. Abdullah, Chem. Eng. J., 2023, 467, 143534 CrossRef CAS.
- S. Gazi, M. Đokić, K. F. Chin, P. R. Ng and H. S. Soo, Adv. Sci., 2019, 6, 1902020 CrossRef CAS PubMed.
- X. Zhao, B. Boruah, K. F. Chin, M. Đokić, J. M. Modak and H. S. Soo, Adv. Mater., 2022, 34, 2100843 CrossRef CAS PubMed.
- T. Uekert, C. M. Pichler, T. Schubert and E. Reisner, Nat. Sustain., 2021, 4, 383 CrossRef.
- L. Wang, S. Jiang, W. Gui, H. Li, J. Wu, H. Wang and J. Yang, Small Struct., 2023, 2300142 CrossRef CAS.
- S. Kang, W. Yuan, W. Chen, M. Du, Y. Zhang and B. Qiu, Nanotechnology, 2023, 34, 462001 CrossRef.
- S. Yue, P. Wang, B. Yu, T. Zhang, Z. Zhao, Y. Li and S. Zhan, Adv. Energy Mater., 2023, 2302008 CrossRef.
- J. Meng, Y. Zhou, D. Li and X. Jiang, Sci. Bull., 2023, 68, 1522–1530 CrossRef CAS PubMed.
- F. Wu, Y. Dou, J. Zhou, J. Qin, T. Jiang, Y. Yao, C. Hélix-Nielsen and W. Zhang, Chem. Eng. J., 2023, 470, 144134 CrossRef CAS.
- K. Zheng, Y. Wu, Z. Hu, S. Wang, X. Jiao, J. Zhu, Y. Sun and Y. Xie, Chem. Soc. Rev., 2023, 52, 8–29 RSC.
- H. Hu, Y. Chen, F. You, J. Yao, H. Yang, X. Jiang and B. Y. Xia, Chin. J. Chem., 2023, 41, 469–480 CrossRef CAS.
- S. Chawla, B. S. Varghese, A. Chithra, C. G. Hussain, R. Keçili and C. M. Hussain, Chemosphere, 2022, 308, 135867 CrossRef CAS.
- K. Lee, Y. Jing, Y. Wang and N. Yan, Nat. Rev. Chem, 2022, 6, 635–652 CrossRef PubMed.
- C. Zhang, Q. Kang, M. Chu, L. He and J. Chen, Trends Chem., 2022, 4, 822–834 CrossRef CAS.
- C. Wang, H. Han, Y. Wu and D. Astruc, Coord. Chem. Rev., 2022, 458, 214422 CrossRef CAS.
- F. Zhang, Y. Zhao, D. Wang, M. Yan, J. Zhang, P. Zhang, T. Ding, L. Chen and C. Chen, J. Cleaner Prod., 2021, 282, 124523 CrossRef CAS.
- C. M. Pichler, S. Bhattacharjee, M. Rahaman, T. Uekert and E. Reisner, ACS Catal., 2021, 11, 9159–9167 CrossRef CAS PubMed.
- Y. Jiang, H. Zhang, L. Hong, J. Shao, B. Zhang, J. Yu and S. Chu, ChemSusChem, 2023, e202300106 CrossRef CAS PubMed.
- X. Jiao, K. Zheng, Q. Chen, X. Li, Y. Li, W. Shao, J. Xu, J. Zhu, Y. Pan, Y. Sun and Y. Xie, Angew. Chem., Int. Ed., 2020, 59, 15497 CrossRef CAS PubMed.
- J. Xu, X. Jiao, K. Zheng, W. Shao, S. Zhu, X. Li, J. Zhu, Y. Pan, Y. Sun and Y. Xie, Natl. Sci. Rev., 2022, 9, nwac011 CrossRef CAS.
- T. Uekert, M. F. Kuehnel, D. W. Wakerley and E. Reisner, Energy Environ. Sci., 2018, 11, 2853–2857 RSC.
- M. Du, Y. Zhang, S. Kang, X. Guo, Y. Ma, M. Xing, Y. Zhu, Y. Chai and B. Qiu, ACS Catal., 2022, 12, 12823–12832 CrossRef CAS.
- Y. Li, S. Wan, C. Lin, Y. Gao, Y. Lu, L. Wang and K. Zhang, Sol. RRL, 2021, 5, 2000427 CrossRef CAS.
- T. Uekert, H. Kasap and E. Reisner, J. Am. Chem. Soc., 2019, 141, 15201–15210 CrossRef CAS PubMed.
- T. Uekert, M. A. Bajada, T. Schubert, C. M. Pichler and E. Reisner, ChemSusChem, 2021, 14, 4190–4197 CrossRef CAS PubMed.
- M. Han, S. Zhu, C. Xia and B. Yang, Appl. Catal., B, 2022, 316, 121662 CrossRef CAS.
- R. Cao, M.-Q. Zhang, C. Hu, D. Xiao, M. Wang and D. Ma, Nat. Commun., 2022, 13, 4809 CrossRef CAS PubMed.
- J. Qin, Y. Dou, F. Wu, Y. Yao, H. R. Andersen, C. Hélix-Nielsen, S. Y. Lim and W. Zhang, Appl. Catal., B, 2022, 319, 121940 CrossRef CAS.
- C. Xing, G. Yu, J. Zhou, Q. Liu, T. Chen, H. Liu and X. Li, Appl. Catal., B, 2022, 315, 121496 CrossRef CAS.
- J. Qin, Y. Dou, J. Zhou, V. M. Candelario, H. R. Andersen, C. Hélix-Nielsen and W. Zhang, Adv. Funct. Mater., 2023, 2214839 CrossRef CAS.
- H. Nagakawa and M. Nagata, ACS Appl. Mater. Interfaces, 2021, 13, 47511–47519 CrossRef CAS PubMed.
- X. Gong, F. Tong, F. Ma, Y. Zhang, P. Zhou, Z. Wang, Y. Liu, P. Wang, H. Cheng, Y. Dai, Z. Zheng and B. Huang, Appl. Catal., B, 2022, 307, 121143 CrossRef CAS.
- J. Ran, L. Chen, D. Wang, A. Talebian-Kiakalaieh, Y. Jiao, M. A. Hamza, Y. Qu, L. Jing, K. Davey and S.-Z. Qiao, Adv. Mater., 2023, 35, 2210164 CrossRef CAS.
- J. Ran, H. Zhang, S. Fu, M. Jaroniec, J. Shan, B. Xia, Y. Qu, J. Qu, S. Chen, L. Song, J. M. Cairney, L. Jing and S.-Z. Qiao, Nat. Commun., 2022, 13, 4600 CrossRef CAS.
- A. Talebian-Kiakalaieh, M. Guo, E. M. Hashem, B. Xia, Y. Jiang, C. Chuah, Y. Tang, P. Kwong, J. Ran and S.-Z. Qiao, Adv. Energy Mater., 2023, 2301594 CrossRef CAS.
- M. Guo, A. Talebian-Kiakalaieh, B. Xia, Y. Hu, H. Chen, J. Ran and S.-Z. Qiao, Adv. Funct. Mater., 2023, 2304912 CrossRef CAS.
- B. Xia, Y. Zhang, B. Shi, J. Ran, K. Davey and S.-Z. Qiao, Small Methods, 2020, 4, 2000063 CrossRef CAS.
- Y. Zhang, B. Johannessen, P. Zhang, J. Gong, J. Ran and S.-Z. Qiao, Adv. Mater., 2023, 2306923 CrossRef CAS PubMed.
- S. Zhang, H. Li, L. Wang, J. Liu, G. Liang, K. Davey, J. Ran and S.-Z. Qiao, J. Am. Chem. Soc., 2023, 145, 6410–6419 CrossRef CAS PubMed.
- T. He, Z. Zhao, R. Liu, X. Liu, B. Ni, Y. Wei, Y. Wu, W. Yuan, H. Peng, Z. Jiang and Y. Zhao, J. Am. Chem. Soc., 2023, 145, 6057–6066 CrossRef CAS PubMed.
- Y. Zhang, C. Pan, G. Bian, J. Xu, Y. Dong, Y. Zhang, Y. Lou, W. Liu and Y. Zhu, Nat. Energy, 2023, 8, 361–371 CrossRef CAS.
- Y. Shen, C. Ren, L. Zheng, X. Xu, R. Long, W. Zhang, Y. Yang, Y. Zhang, Y. Yao, H. Chi, J. Wang, Q. Shen, Y. Xiong, Z. Zou and Y. Zhou, Nat. Commun., 2023, 14, 1117 CrossRef CAS PubMed.
- S. Wang, T. Wu, S. Wu, J. Guo, T. He, Y. Wu, W. Yuan, Z. Zhang, Y. Hua and Y. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202311082 CrossRef CAS.
- Y. Hu, S. Zhang, Z. Zhang, H. Zhou, B. Li, Z. Sun, X. Hu, W. Yang, X. Li, Y. Wang, S. Liu, D. Wang, J. Lin, W. Chen and S. Wang, Adv. Mater., 2023, 35, 2304130 CrossRef CAS.
- C. Qin, X. Wu, L. Tang, X. Chen, M. Li, Y. Mou, B. Su, S. Wang, C. Feng, J. Liu, X. Yuan, Y. Zhao and H. Wang, Nat. Commun., 2023, 14, 5238 CrossRef CAS PubMed.
- F. Kuttassery, Y. Ohsaki, A. Thomas, R. Kamata, Y. Ebato, H. Kumagai, R. Nakazato, A. Sebastian, S. Mathew, H. Tachibana, O. Ishitani and H. Inoue, Angew. Chem., Int. Ed., 2023, 62, e2023089 CrossRef PubMed.
- Y.-W. Han, L. Ye, T.-J. Gong and Y. Fu, Angew. Chem., Int. Ed., 2023, 62, e202306305 CrossRef CAS PubMed.
- W. Soontornchaiyakul, S. Yoshino, T. Kanazawa, R. Haruki, D. Fan, S. Nozawa, Y. Yamaguchi and A. Kudo, J. Am. Chem. Soc., 2023, 145(37), 20485–20491 CrossRef CAS.
- X. Li, C. Li, Y. Xu, Q. Liu, M. Bahri, L. Zhang, N. D. Browning, A. J. Cowan and J. Tang, Nat. Energy, 2023, 8, 1013–1022 CrossRef CAS.
- Y. Yang, H.-Y. Zhang, Y. Wang, L.-H. Shao, L. Fang, H. Dong, M. Lu, L.-Z. Dong, Y.-Q. Lan and F.-M. Zhang, Adv. Mater., 2023, 35, 2304170 CrossRef CAS PubMed.
- K. Wu, X.-Y. Liu, P.-W. Cheng, Y.-L. Huang, J. Zheng, M. Xie, W. Lu and D. Li, J. Am. Chem. Soc., 2023, 145, 18931–18938 CrossRef CAS PubMed.
- Z.-W. Huang, K.-Q. Hu, X.-B. Li, Z.-N. Bin, Q.-Y. Wu, Z.-H. Zhang, Z.-J. Guo, W.-S. Wu, Z.-F. Chai, L. Mei and W.-Q. Shi, J. Am. Chem. Soc., 2023, 145, 18148–18159 CrossRef CAS PubMed.
- M. Zhang, Y. Mao, X. Bao, G. Zhai, D. Xiao, D. Liu, P. Wang, H. Cheng, Y. Liu, Z. Zheng, Y. Dai, Y. Fan, Z. Wang and B. Huang, Angew. Chem., Int. Ed., 2023, 62, e202302919 CrossRef CAS.
- T. Tana, P. Han, A. J. Brock, X. Mao, S. Sarina, E. R. Waclawik, A. Du, S. E. Bottle and H.-Y. Zhu, Nat. Commun., 2023, 14, 4609 CrossRef CAS.
- R. Yanagi, T. Zhao, M. Cheng, B. Liu, H. Su, C. He, J. Heinlein, S. Mukhopadhyay, H. Tan, D. Solanki and S. Hu, J. Am. Chem. Soc., 2023, 145, 15381–15392 CrossRef CAS PubMed.
- X. Guan, Y. Qian, X. Zhang and H.-L. Jiang, Angew. Chem., Int. Ed., 2023, 62, e202306135 CrossRef.
- P. Das, J. Roeser and A. Thomas, Angew. Chem., Int. Ed., 2023, 62, e2023043 Search PubMed.
- M. Liu, G. Zhang, X. Liang, Z. Pan, D. Zheng, S. Wang, Z. Yu, Y. Hou and X. Wang, Angew. Chem., Int. Ed., 2023, 62, e202304694 CrossRef CAS PubMed.
- R. Song, Z. Li, J. Guo, P. N. Duchesne, C. Qiu, C. Mao, J. Jia, S. Tang, Y.-F. Xu, W. Zhou, L. Wang, W. Sun, X. Yan, L. Guo, D. Jing and G. A. Ozin, Angew. Chem., Int. Ed., 2023, 62, e202304470 CrossRef CAS PubMed.
- J. Yu, L. Huang, Q. Tang, S.-B. Yu, Q.-Y. Qi, J. Zhang, D. Ma, Y. Lei, J. Su, Y. Song, J.-C. Eloi, R. L. Harniman, U. Borucu, L. Zhang, M. Zhu, F. Tian, L. Du, D. Lee Phillips, I. Manners, R. Ye and J. Tian, Nat. Catal., 2023, 6, 464–475 CrossRef CAS.
- T. He and Y. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202303086 CrossRef CAS PubMed.
- L. Luo, X. Han, K. Wang, Y. Xu, L. Xiong, J. Ma, Z. Guo and J. Tang, Nat. Commun., 2023, 14, 2690 CrossRef CAS PubMed.
- J. Y. Kim and D. H. Youn, Molecules, 2023, 28, 6502 CrossRef CAS PubMed.
- M. Ashraf, N. Ullah, I. Khan, W. Tremel, S. Ahmad and M. N. Tahir, Chem. Rev., 2023, 123, 4443 CrossRef CAS.
Footnote |
| † J. Ran and A. Talebian-Kiakalaieh contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |