DOI:
10.1039/D3SC05356C
(Edge Article)
Chem. Sci., 2024,
15, 1409-1417
Two active species from a single metal halide precursor: a case study of highly productive Mn-catalyzed dehydrogenation of amine-boranes via intermolecular bimetallic cooperation†
Received
9th October 2023
, Accepted 6th December 2023
First published on 7th December 2023
Abstract
Metal–metal cooperation for inert bond activation is a ubiquitous concept in coordination chemistry and catalysis. While the great majority of such transformations proceed via intramolecular mode in binuclear complexes, to date only a few examples of intermolecular small molecule activation using usually bimetallic frustrated Lewis pairs (Mδ+⋯M′δ−) have been reported. We introduce herein an alternative approach for the intermolecular bimetallic cooperativity observed in the catalytic dehydrogenation of amine-boranes, in which the concomitant activation of N–H and B–H bonds of the substrate via the synergetic action of Lewis acidic (M+) and basic hydride (M–H) metal species derived from the same mononuclear complex (M–Br). It was also demonstrated that this system generated in situ from the air-stable Mn(I) complex fac-[(CO)3(bis(NHC))MnBr] and NaBPh4 shows high activity for H2 production from several substrates (Me2NHBH3, tBuNH2BH3, MeNH2BH3, NH3BH3) at low catalyst loading (0.1% to 50 ppm), providing outstanding efficiency for Me2NHBH3 (TON up to 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200) that is largely superior to all known 3d-, s-, p-, f-block metal derivatives and frustrated Lewis pairs (FLPs). These results represent a step forward towards more extensive use of intermolecular bimetallic cooperation concepts in modern homogeneous catalysis.
200) that is largely superior to all known 3d-, s-, p-, f-block metal derivatives and frustrated Lewis pairs (FLPs). These results represent a step forward towards more extensive use of intermolecular bimetallic cooperation concepts in modern homogeneous catalysis.
Introduction
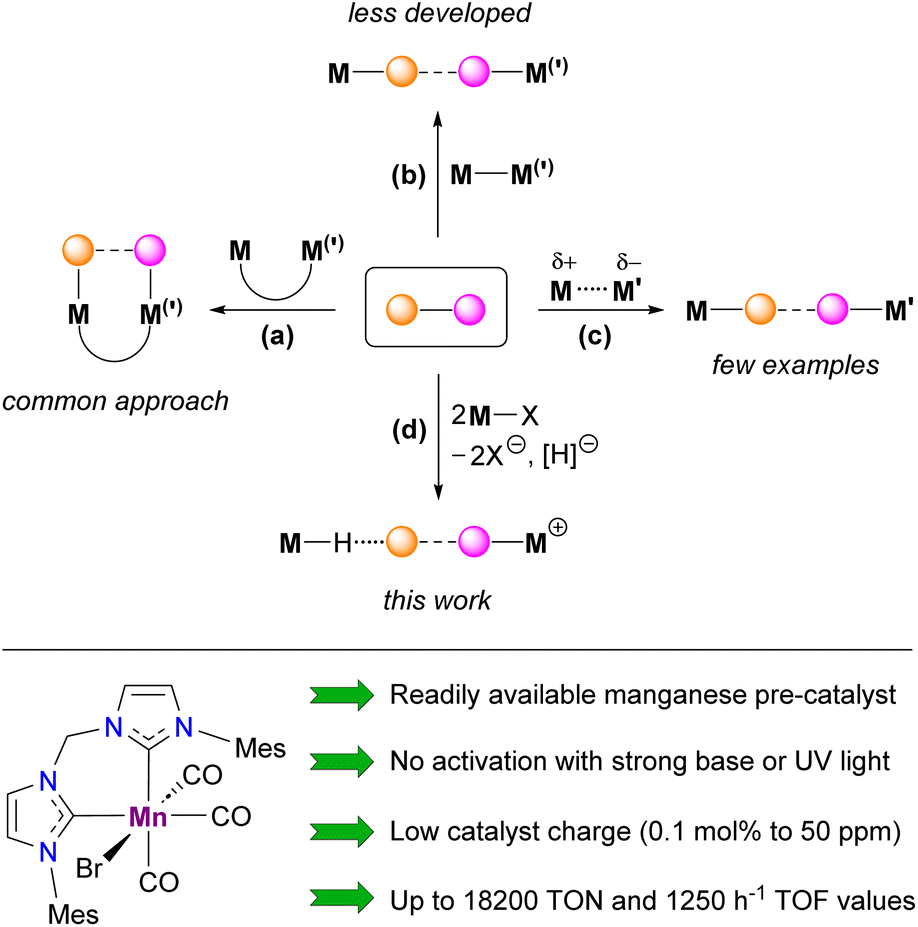
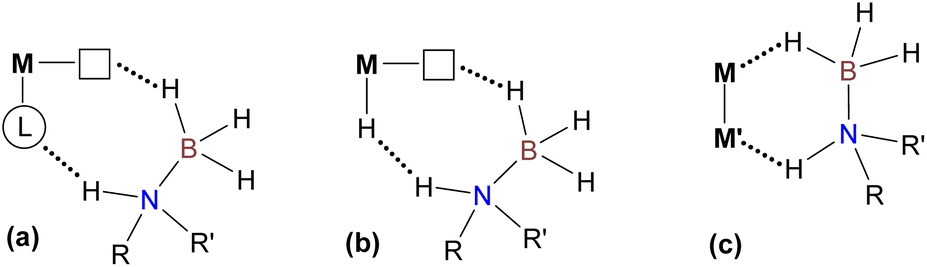
Cooperative action of two metal atoms for the activation of organic molecules often accompanied by inert bond cleavage is omnipresent in heterogeneous catalysts,1 metalloenzymes2 and related biomimetic systems.3 In coordination chemistry, these processes generally take place intramolecularly within well-defined homo- or heterobimetallic complexes (Scheme 1(a)), in which two metal atoms are kept in close proximity by bridging ligand(s)3 occasionally supported by additional metal–metal interaction.4 Similar reactivity has also been observed for binuclear derivatives with a highly polarized metal–metal single bond (Scheme 1(b)), also known as metal-only Lewis pairs (MOLPs).5,6 In the latter case, if the inert bond of the substrate is completely cleaved, the two resulting monometallic species can participate in a single catalytic cycle with a synergetic effect.7 In sharp contrast, the intermolecular cooperative activation of small molecules mediated by two mononuclear transition metal species (Scheme 1(c)) remains rare.8 The most pertinent examples of such behavior deal with the interaction of bimetallic frustrated Lewis pairs (FLPs) with dihydrogen,9 acetylene,9c amine-boranes10 or formic acid.11 Conceptually related intermolecular activation of CO2 and epoxide by a radical pair arising from the spontaneous homolytic cleavage of a Fe–Al bond in a bimetallic complex prior to interaction with a substrate was also recently reported.12 Despite remarkable results already achieved in the area of homogeneous catalysis exploiting bimetallic cooperation, in most cases either quite sophisticated binuclear transition metal complexes (Scheme 1(a and b))3–7 or a carefully selected combination of two monometallic species (Scheme 1(c))10 were employed. In this contribution, we provide experimental and theoretical evidence for a novel type of intermolecular bimetallic cooperativity using Lewis acidic and hydride components (Scheme 1(d)) generated in situ from a single mononuclear metal precursor, halide abstractor and bifunctional substrate. Beyond the apparent simplicity of this approach, its potential usefulness was illustrated by the development of a convenient procedure for dehydrogenation of various amine-boranes catalyzed by an air-stable Mn(I) bis(NHC) complex (Scheme 1, bottom), showing, in particular, state-of-the-art performance for H2 production from Me2NHBH3 (DMAB) that largely surpasses those of all known 3d metal catalysts.
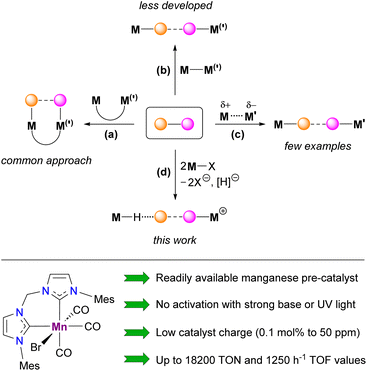 |
| | Scheme 1 Different types of cooperative bimetallic activation of organic molecules (top, dashed and dotted lines refer to activated/cleaved bonds in the substrate and non-covalent interactions, respectively) and the Mn(I) precursor used for catalytic intermolecular amine-borane dehydrogenation (bottom). | |
Results and discussion
Discovery and optimization of amine-borane dehydrogenation catalyzed by Mn(I) fac-[(CO)3(L–L′)MnBr] (L, L′ = phosphine, NHC) complexes
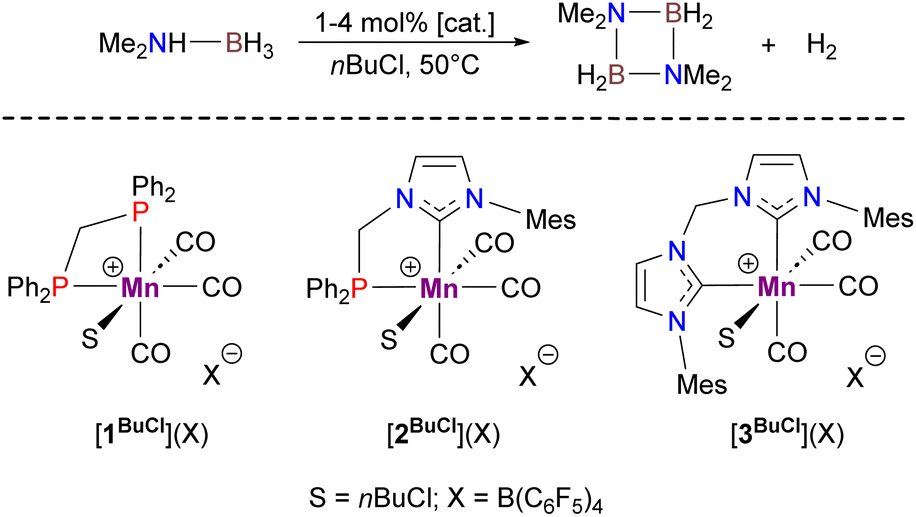
In the frame of our research dealing with Mn(I) organometallic chemistry relevant to homogeneous catalysis, we have unexpectedly observed that the cationic complex [1BuCl]+ (Scheme 2)13 obtained by reaction of the corresponding hydride 1H with [Ph3C](B(C6F5)4) in nBuCl is capable of catalyzing DMAB dehydrogenation at 50 °C to produce H2 with a turnover frequency (TOF) of ca. 0.6 h−1. Further studies have revealed that parent cationic species [2BuCl]+ and [3BuCl]+,14 in which Ph2P moieties are progressively replaced with more donating N-heterocyclic carbene (NHC) fragments, are much more active, affording TOFs of 9 h−1 and 143 h−1, respectively. Encouraged by the latter result being more superior to the performance of previously known Mn-based catalysts for this substrate (TOF 0.7–11 h−1),15 we proceeded to further optimization of this catalytic system.
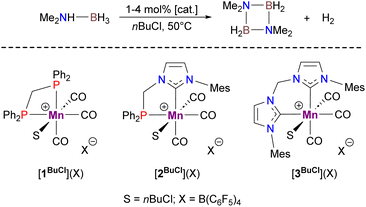 |
| | Scheme 2 DMAB dehydrogenation catalyzed by cationic Mn(I) complexes [1–3BuCl](B(C6F5)4). | |
Looking for a more convenient source of cationic Mn(I) species, we first prepared the corresponding MeCN complex [3MeCN](BF4) from AgBF4 and the readily available bromide precursor 3Br.16 This compound was isolated in 85% yield and fully characterized, including single-crystal X-ray diffraction (Fig. S1†). While this compound showed very low activity in nBuCl, the use of a more chemically inert chlorinated solvent such as PhCl allowed almost quantitative DMAB conversion at only 0.1 mol% catalyst charge (Table 1, entry 1). Gratifyingly, the catalyst can also be generated in situ from 3Br and NaBF4 (Table 1, entry 2) even if its activity is lower than that of the isolated [3MeCN](BF4) complex. The application of sodium salts with non-coordinating anions boosted the catalyst performance (Table 1, entries 3 and 4) being better than for the initially tested complex [3BuCl]+ in nBuCl (vide supra). Solvent screening revealed that PhF provided slightly inferior results (Table 1, entry 5), whereas the use of coordinating THF led to a serious drop in the reaction rate (Table 1, entry 6). The reaction even slowly proceeds in dichloromethane at 30 °C (Table 1, entry 7), albeit with a ten-fold higher catalyst loading. The addition of mercury did not alter the dehydrogenation process (Table 1, entry 8), thus ruling out the well-known catalysis of amine-borane dehydrogenation by metal nanoparticles.17,18 Finally, it was observed that neutral 3Br and 3H without any additives displayed only modest catalytic activity (Table 1, entries 9 and 10).
Table 1 Optimization of DMAB dehydrogenation catalyzed by bis(NHC) Mn(I) complexesa
| No. |
Pre-catalyst |
Solv. |
Additive |
Time,h |
TONb |
TOFc, h−1 |
|
DMAB (1.5 mmol), 0.1 mol% of pre-catalyst, 1.0 mol% of sodium salt, 2 mL of solvent in a closed vessel at 50 °C.
Estimated from the pressure changes due to H2 release under constant volume conditions.
TOF values were calculated based on a 10–20 min induction period required for catalyst activation.
1 mol% of 3Br and 10 mol% of NaBPh4 at 30 °C.
Reaction in the presence of 250 equivalents of mercury.
|
| 1 |
[3MeCN](BF4) |
PhCl |
— |
26 |
981 |
38 |
| 2 |
3Br
|
PhCl |
NaBF4 |
69 |
902 |
13 |
| 3 |
3Br
|
PhCl |
NaBPh4 |
3.9 |
1000 |
259 |
| 4 |
3Br
|
PhCl |
NaB(C6F5)4 |
4.1 |
1000 |
244 |
| 5 |
3Br
|
PhF |
NaBPh4 |
6.7 |
1000 |
149 |
| 6 |
3Br
|
THF |
NaBPh4 |
26 |
1000 |
38 |
| 7d |
3Br
|
DCM |
NaBPh4 |
11 |
100 |
9 |
| 8e |
3Br
|
PhCl |
NaBPh4 |
4.0 |
1000 |
256 |
| 9 |
3Br
|
PhCl |
— |
118 |
535 |
5 |
| 10 |
3H
|
PhCl |
— |
69 |
253 |
4 |
Then, we focused on the evaluation of the catalytic performance of this system (Table 2). At 60 °C, the reaction rate is significantly accelerated (Table 2, entry 1) and the catalyst charge may be decreased to 50 ppm, resulting in turnover number (TON) and turnover frequencies (TOF) values of 16221, and 661 h−1, respectively (Table 2, entries 2–4). Surprisingly, we noted that the reaction was even faster in the dark, providing a TON of 18242 at a 50 ppm charge for 3Br (Table 2, entry 5), probably preventing photo-induced catalyst decomposition. Background DMAB dehydrogenation in the presence of NaBPh4 under these conditions was not observed at all (Table 2, entry 6). Remarkably, the catalytic performance of this substrate is more than 50 times higher than those previously observed for 3d metal catalysts (20–330 TONs),15,19 especially considering that activation by strong bases (nBuLi, tBuOK)19a,e,f or UV-light15b,c,19c,g was required for most of these systems. The results obtained for catalyst 3Br also surpass significantly the TON values obtained for noble metal derivatives (up to a TON of 2240),20 s-, p- and f-block metal complexes (up to a TON of 485)21–25 as well as FLP-based systems (up to a TON of 500).26 The only comparable precedent in terms of productivity was reported for Ru(acac)3/oleylamine combination (TON = 15000),27 however, the reaction rate in this case was much slower (TOF 78 h−1 at 60 °C).
Table 2 Dehydrogenation of various amine-boranes catalyzed by bis(NHC) Mn(I) complex 3Bra
| No. |
Substrate |
3Br, mol% |
Time, h |
H2, eq.b,c |
TON (TOF, h−1)b,c |
|
Amine-borane (0.8–1.5 mmol), 0.5–1.0 mol% of NaBPh4, 2 mL of PhCl in a sealed vessel at 60 °C.
Average from two independent runs.
Estimated from the pressure changes due to H2 release under constant volume conditions and 11B NMR spectra of the crude products.
In the dark.
Background dehydrogenation experiments were carried out in the presence of 1.0 mol% of NaBPh4 under strictly identical conditions to those of the Mn-catalyzed process.
|
| 1 |
DMAB |
0.1 |
2.1 |
>0.99 |
1000 (476) |
| 2 |
DMAB |
0.02 |
4.8 |
>0.99 |
5000 (1058) |
| 3 |
DMAB |
0.01 |
8.7 |
>0.99 |
10000 (1157) |
| 4 |
DMAB |
0.005 |
24.6 |
0.81 |
16221 (661) |
| 5 |
DMAB |
0.005d |
14.4 |
0.91 |
18242 (1267) |
| 6 |
DMAB |
— |
60 |
<0.01e |
— |
| 7 |
tBuNH2BH3 |
0.1d |
48.6 |
1.53 |
1527 (31) |
| 8 |
tBuNH2BH3 |
— |
60 |
0.36e |
— |
| 9 |
MeNH2BH3 |
0.1d |
28.4 |
1.34 |
1343 (47) |
| 10 |
MeNH2BH3 |
— |
60 |
0.2e |
— |
| 11 |
NH3BH3 |
0.1d |
60 |
0.61 |
611 (10) |
| 12 |
NH3BH3 |
— |
60 |
0.07e |
— |
We found that our system is also suitable for amine-boranes with a higher hydrogen content relevant for hydrogen storage materials.28 Preliminary results without any additional optimization showed that tBuNH2BH3 can release ca. 1.53 equivalents of H2 with a catalyst charge of only 0.1 mol% (Table 2, entry 7). Though this substrate can be slowly dehydrogenated under these conditions without any catalyst (Table 2, entry 8), the Mn-catalyzed reaction was much faster, producing ca. 1.3 equivalents of hydrogen after 10 h (TOF of 130 h−1 for this period) followed by a more sluggish process releasing a further 0.2 equivalents in ca. 40 h (Fig. S13†). To the best of our knowledge, this value represents the best result ever obtained for this substrate among all other metal complexes.10,23b,25a,2911B NMR analysis of the crude reaction mixture (Fig. S15†) revealed the presence of substituted borazine [tBuNBH]3, cyclic trimer [tBuNHBH2]3 and oligomers [tBu3NHBH2]n as the main dehydrogenation products. We supposed that the drop in the reaction rate after evolution of the first equivalent of hydrogen was due to the more difficult uptake of partially dehydrogenated products by our catalytic system. Dehydrogenation of MeNH2BH3 (Table 2, entry 9) was found to be slightly slower than in the case of tBuNH2BH3 (TOF of 123 h−1 for a 10 h period) and less efficient in terms of the amount of produced H2 (1.2 equivalents, Fig. S13†), probably due to the lower solubility of this substrate in PhCl and the extensive formation of insoluble polymeric products [MeNHBH2]n. However, the catalytic efficiency attained in this case (TON of 1343) is competitive compared to the majority of known 3d metal catalysts (TONs of 20–250)30 and remains inferior only to highly active Co-based complexes (TONs of up to 5000).31 Finally, the reaction of NH3BH3 afforded 0.61 equivalents of H2 in 60 h (Table 2, entry 11) leading to the formation of an insoluble polymer [NH2BH2]n. The TON value was estimated to be in the range of 500–550 considering the background substrate dehydrogenation process (Table 2, entry 12). However, despite slow reaction kinetics and a low amount of produced dihydrogen compared to the best catalytic systems based on first row transition metal complexes32 or purely organic molecules33 working at 1–5 mol% catalyst charge (1.7–2.5 equiv. of H2 per NH3BH3), these numbers are still superior in terms of overall performance to previously reported Mn-based catalysts (TON ca. 50–70).34 Considering the remarkable progress recently achieved in formic acid dehydrogenation using Mn(I) complexes,35 our results further highlight the privileged position of this metal in the future design of sustainable homogeneous catalysts for H2 production.
Spectroscopic and kinetic studies of the reaction mechanism for DMAB dehydrogenation catalyzed by the Mn(I) complex fac-[(CO)3(bis(NHC))MnBr] (3Br)
Next, we turned our attention to a deeper understanding of this catalytic system, as all conventional known mechanisms for transition metal-catalyzed amine-borane dehydrogenation36 based on either metal–ligand cooperation (Scheme 3(a)), reactivity of coordinatively unsaturated hydrides (Scheme 3(b)) or bimetallic activation (Scheme 3(c)) are a priori inappropriate in this case. Firstly, regarding possible metal–ligand cooperation, it should be noted that the non-innocent character of dppm37 and NHC-phosphine38 ligands due to the deprotonation of the CH2 bridge in the corresponding Mn(I) bromide complexes may only be observed in the presence of strong base, which is not required in the present case. Furthermore, no evidence for the cooperative behavior of the bis(NHC) ligand incorporated into the most active catalyst has been reported so far, which thus permitted us to exclude path (a). Then, despite extensive applications of different manganese bis(NHC) complexes in hydrosilylation,16a,39 electrochemical CO2 reduction,16b,40 hydrogenation41 and hydrogen-borrowing processes42 systematically involving hydride fac-[(CO)3(bis(NHC))MnH] intermediates, the dissociation of a CO ligand in such species has never been proved experimentally. Moreover, the formation of the dicarbonyl complex fac-[(CO)2(η2-H2)(bis(NHC))MnH] from H2 and a tricarbonyl hydride precursor was recently proposed in a Mn-catalyzed ester hydrogenation mechanism,42a but the calculated barrier for the CO-to-H2 substitution of 38.5 kcal mol−1 seems too high for our reaction conditions, thus making path (b) very unlikely (vide infra). Finally, the bimetallic activation pathway43 (Scheme 3(c)) can also be ruled out because of the insufficient reducing ability of amine-boranes to form Mn(0) derivatives from 3Br and the apparent impossibility of metal–metal bond formation to generate a [(CO)3(bis(NHC))Mn] radical due to steric hindrance.40b
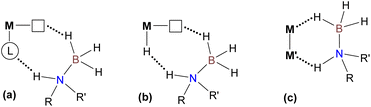 |
| | Scheme 3 An overview of the main known approaches for amine-borane dehydrogenation catalyzed by transition metal complexes. | |
We started our mechanistic investigations from the operando spectroscopic monitoring of the catalytic process in CH2Cl2. This solvent was chosen instead of PhCl because of slower reaction kinetics (Table 1), its full transparency in νCO region and the availability of its deuterated version for NMR studies. Similar trends were also observed in PhCl (Fig. S19†) albeit that a full set of spectroscopic data for reaction intermediates cannot be obtained in this solvent (see the ESI† for details). The analysis of the IR spectra of 3Br/NaBPh4/DMAB solution in the νCO region (Fig. 1) revealed an initial transformation of the starting bromide into the cationic species [3DMAB](BPh4) with a coordinated substrate (Fig. 1(a)). Subsequent evolution of the latter into the hydride 3H (Fig. 1(b)) is consistent with the induction period observed by volumetric H2 evolution studies (vide infra). The concomitant conversion of DMAB into the dimer [Me2NBH2]2 was evident from characteristic changes in the νBH region (Fig. S18†). After full DMAB consumption, 3H rapidly decayed to give a mixture of neutral chloride compound 3Cl and the cationic complex  bearing a coordinated [Me2NBH2]2 ligand (Fig. 1(c), see Fig. S17† for νCO band deconvolution). 11B NMR monitoring of the reaction mixture in CD2Cl2 (Fig. S23†) showed the appearance of the characteristic signal of Me2N
bearing a coordinated [Me2NBH2]2 ligand (Fig. 1(c), see Fig. S17† for νCO band deconvolution). 11B NMR monitoring of the reaction mixture in CD2Cl2 (Fig. S23†) showed the appearance of the characteristic signal of Me2N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) BH2 at δB 37.4 ppm, consistent with an “off-metal” dimerization mechanism for the formation of the final product.36 The 1H NMR spectra (Fig. S21†) confirmed the presence of the hydride complex 3H at δH −7.04 ppm and additionally revealed a broad signal at δH −3.38 ppm. We were able to retrieve a full set of NMR data for this intermediate (see the ESI†), attributed to a cationic Mn(I) σ-borane complex [3DMAB](BPh4).
BH2 at δB 37.4 ppm, consistent with an “off-metal” dimerization mechanism for the formation of the final product.36 The 1H NMR spectra (Fig. S21†) confirmed the presence of the hydride complex 3H at δH −7.04 ppm and additionally revealed a broad signal at δH −3.38 ppm. We were able to retrieve a full set of NMR data for this intermediate (see the ESI†), attributed to a cationic Mn(I) σ-borane complex [3DMAB](BPh4).
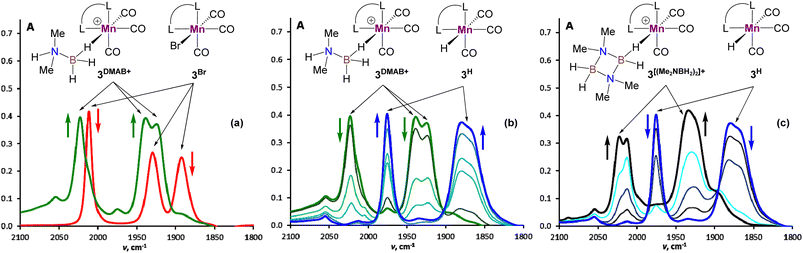 |
| | Fig. 1 IR spectroscopy monitoring in the νCO range for DMAB dehydrogenation in the presence of 3Br (3.3 mol%) and NaBPh4 (16.5 mol%) in CH2Cl2 at ca. 30 °C (L–L = bis(NHC), c(DMAB) = 0.30 M, l = 0.01 cm). (a) IR spectra of the initial 3Br solution (red) and reaction mixture after 2 min upon DMAB addition (green). (b) IR spectra between 2 min (green) and 20 min (blue) of reaction time. (c) IR spectra between 20 min (blue) and 40 min (black) of reaction time. | |
In order to confirm this assignment, we prepared the model Mn(I) cationic complex 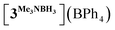 from Me3NBH3 that is unreactive towards dehydrogenation, 3Br and NaBPh4. Its structure elucidated from X-ray diffraction measurements (Fig. 2) features a η1-BH coordination mode previously observed in half-sandwich Mn(I) analogues.44 The BH3 moiety in the NMR spectra of
from Me3NBH3 that is unreactive towards dehydrogenation, 3Br and NaBPh4. Its structure elucidated from X-ray diffraction measurements (Fig. 2) features a η1-BH coordination mode previously observed in half-sandwich Mn(I) analogues.44 The BH3 moiety in the NMR spectra of 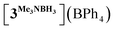 appears as broad signals at δB −11.9 ppm and δH −3.30 ppm. The latter value matches well with that observed during the NMR mechanistic investigation, clearly indicating the participation of such species in the catalytic process. According to IR and NMR data, all transformations occurring in catalytic DMAB dehydrogenation involved only tricarbonyl Mn(I) complexes and we did not find any spectroscopic evidence for the formation of the neutral dicarbonyl hydride species fac-[(CO)2(DMAB)(bis(NHC))MnH] essential for the realization of the route (b), as depicted in Scheme 3.
appears as broad signals at δB −11.9 ppm and δH −3.30 ppm. The latter value matches well with that observed during the NMR mechanistic investigation, clearly indicating the participation of such species in the catalytic process. According to IR and NMR data, all transformations occurring in catalytic DMAB dehydrogenation involved only tricarbonyl Mn(I) complexes and we did not find any spectroscopic evidence for the formation of the neutral dicarbonyl hydride species fac-[(CO)2(DMAB)(bis(NHC))MnH] essential for the realization of the route (b), as depicted in Scheme 3.
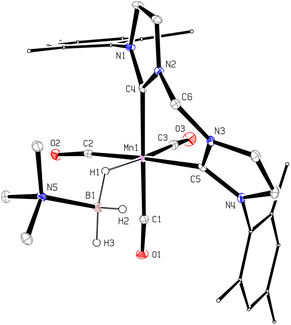 |
| | Fig. 2 Molecular geometry of the complex 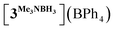 (20% probability ellipsoids, Mes groups represented as a wireframe, most hydrogen atoms, BPh4− anion and CH2Cl2 solvate molecules are not shown). (20% probability ellipsoids, Mes groups represented as a wireframe, most hydrogen atoms, BPh4− anion and CH2Cl2 solvate molecules are not shown). | |
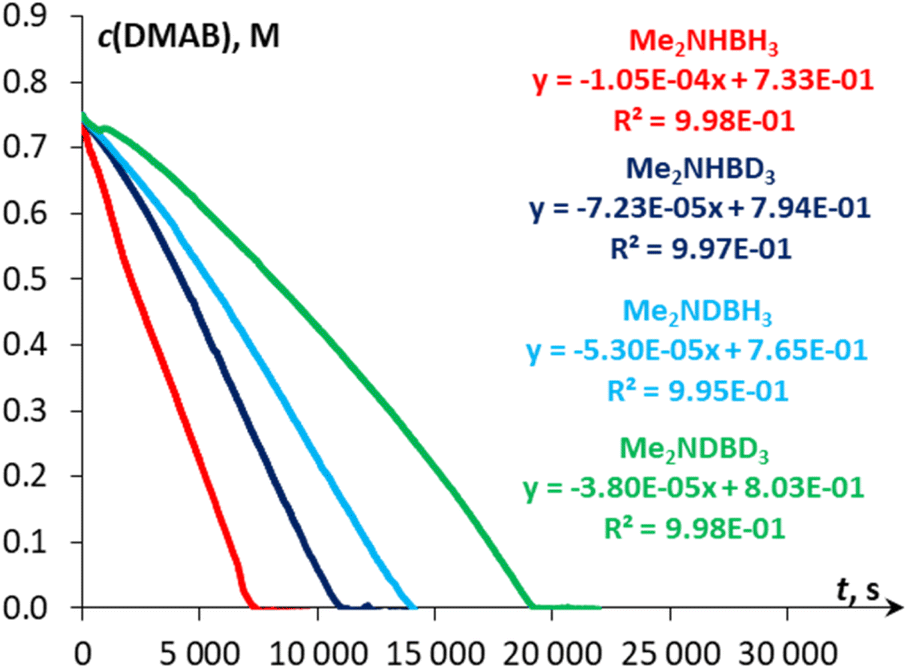
The kinetic studies revealed [pseudo]zero order for the substrate (Fig. S27†) and first order for catalyst concentration (Fig. S28†). The variation in the amount of NaBPh4 (0.125–8 mol%) showed virtually no influence on the reaction rate (Fig. S29†), thus ruling out the occurrence of a conceptually similar DMAB dehydrogenation process that proceeds via a Na+/Mn–H couple. While the reversible coordination of a sodium cation with DMAB cannot be excluded, we supposed that this interaction is too weak to activate the substrate towards deprotonation with the Mn(I) hydride complex 3H (vide infra). Variable-temperature experiments in the 30–60 °C range (Fig. 3) allowed estimation of the activation enthalpy ΔH‡ = 14.5 ± 0.4 kcal mol−1 (Fig. S26 and Table S6†), which is typical for proton transfer to transition metal hydrides.45 The detailed description of the proposed kinetic model is provided in the ESI.† Finally, kinetic isotope effects (kH/kD, Fig. 4) measured at 60 °C for Me2NHBD3 (1.5 ± 0.1), Me2NDBH3 (2.1 ± 0.1) and Me2NDBD3 (2.9 ± 0.1) are consistent with the involvement of B–H and N–H bond cleavage in the rate determining step.32a,c These data taken together with spectroscopic results show the implication of cationic and hydride Mn(I) species in the catalytic process, allowing us to propose an unconventional intermolecular DMAB activation, which was then studied by theoretical means.
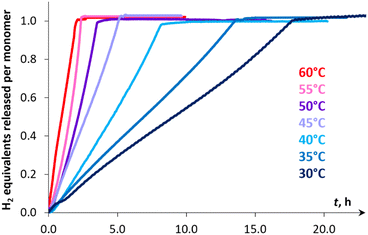 |
| | Fig. 3 Hydrogen evolution kinetic curves of DMAB dehydrogenation in PhCl catalyzed by 3Br (0.1 mol%) and NaBPh4 (1 mol%) at various temperatures. The initial parts of the curves corresponding to the induction period (10–20 min) are not shown. | |
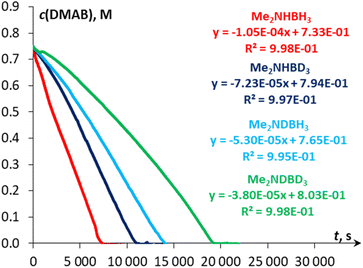 |
| | Fig. 4
c(DMAB) vs. time plots for the initial reaction rate determination of Me2NHBH3, Me2NHBD3, Me2NDBH3 and Me2NDBD3 (c0 = 0.75 M) dehydrogenation catalyzed by 3Br (0.1 mol%) in the presence of NaBPh4 (1.0 mol%) in PhCl at 60 °C. | |
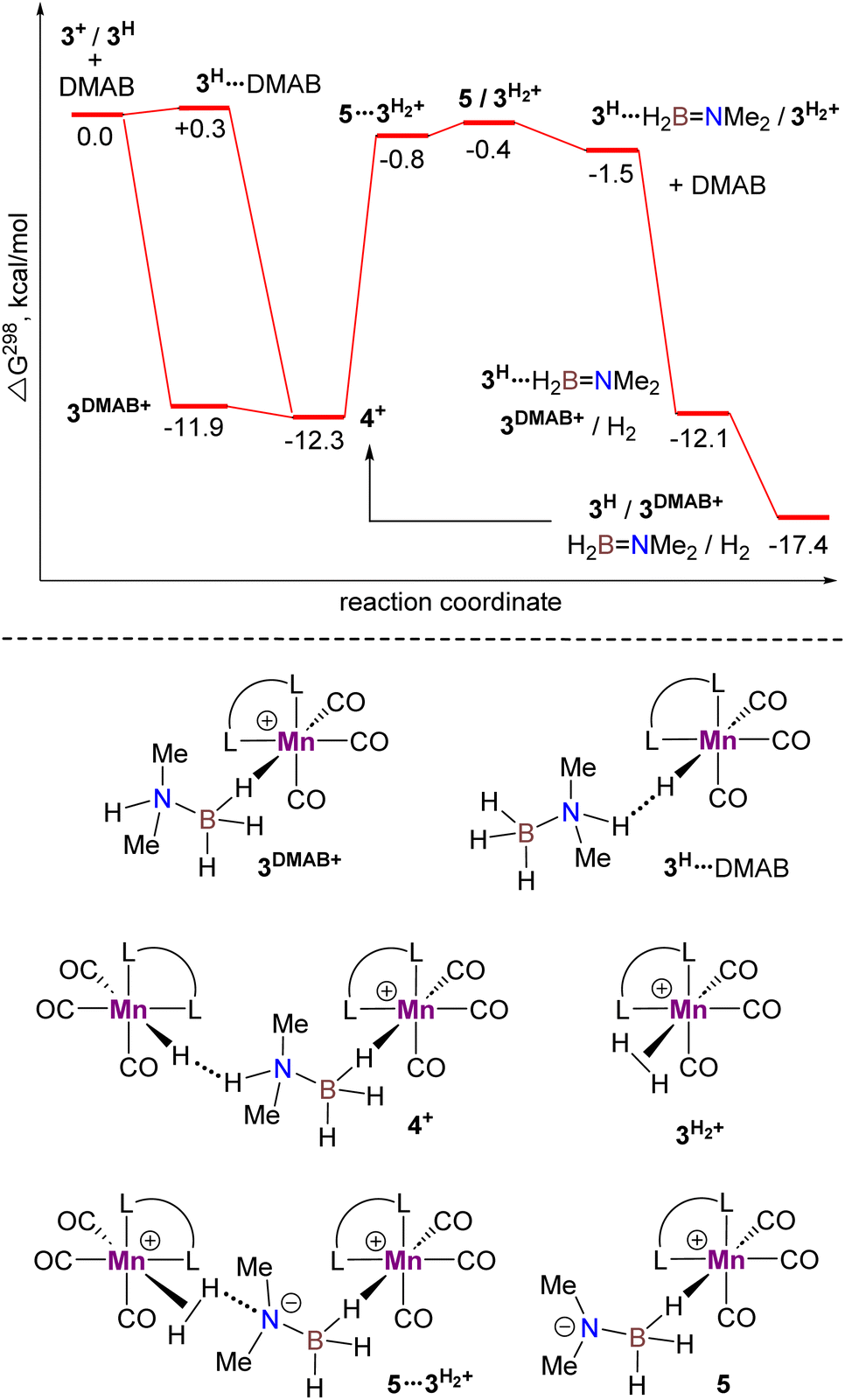
Theoretical investigation of DMAB dehydrogenation catalyzed by the Mn(I) complex fac-[(CO)3(bis(NHC))MnBr] (3Br)
Computational modelling of the catalytic system showed that the monometallic species 3+ and 3H interact with DMAB (Fig. 5) via B–H coordination and dihydrogen bonding, respectively. However, activation barriers for the subsequent hydride and proton transfer steps in these intermediates [3DMAB]+ and 3H⋯DMAB exceed 30–40 kcal mol−1 and no stabilization of the resulting products was observed from DFT calculations (Fig. S31 and S33†). In contrast, proton transfer becomes feasible in the most thermodynamically stable adduct 4+, in which two metal fragments are bridged by a DMAB molecule. Both the N–H (1.040 Å) and B–H (1.264 Å) bonds of the coordinated DMAB moiety in 4+ are slightly longer than those in the corresponding mononuclear species 3H⋯DMAB (1.028 Å) and 3DMAB+ (1.258 Å) thus being consistent with a greater extent of substrate activation. This process leads initially to the formation of bimetallic intermediate 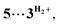 which is composed of the cationic dihydrogen complex
which is composed of the cationic dihydrogen complex 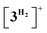 associated with the zwitter-ionic species 5 by hydrogen bonding (2.283 Å), then undergoing dissociation to the individual components. Importantly, the metal-coordinated B–H bond distance in complexes
associated with the zwitter-ionic species 5 by hydrogen bonding (2.283 Å), then undergoing dissociation to the individual components. Importantly, the metal-coordinated B–H bond distance in complexes 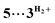 and 5 becomes elongated by 0.093 and 0.234 Å, respectively, relative to that in 4+ with a concomitant shortening of the Mn–H distance by 0.056 and 0.109 Å, respectively. These data are in agreement with the occurrence of a progressive hydride transfer in the sequence of
and 5 becomes elongated by 0.093 and 0.234 Å, respectively, relative to that in 4+ with a concomitant shortening of the Mn–H distance by 0.056 and 0.109 Å, respectively. These data are in agreement with the occurrence of a progressive hydride transfer in the sequence of 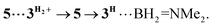 Finally, the essentially barrierless loss of H2 and Me2NBH2 from
Finally, the essentially barrierless loss of H2 and Me2NBH2 from 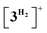 and 3H⋯BH2NMe2, respectively regenerate a 3+/3H couple, thus exhibiting a curious interconversion of both metal species after each catalytic turnover. The key role of the bimetallic adducts 4+ and
and 3H⋯BH2NMe2, respectively regenerate a 3+/3H couple, thus exhibiting a curious interconversion of both metal species after each catalytic turnover. The key role of the bimetallic adducts 4+ and 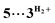 in the DMAB activation process provides additional evidence to consider this reaction mechanism as a viable intermolecular bimetallic cooperation (Scheme 1(d)).
in the DMAB activation process provides additional evidence to consider this reaction mechanism as a viable intermolecular bimetallic cooperation (Scheme 1(d)).
 |
| | Fig. 5 Calculated Gibbs energy profile for DMAB dehydrogenation catalyzed by a 3+/3H couple at the ωB97XD/def2-TZVP level in toluene (dihydrogen bond interactions are indicated as dotted lines, non-interacting molecules are separated by a slash) and structures of the key intermediates (bottom). | |
Proposed catalytic cycle for DMAB dehydrogenation catalyzed by the Mn(I) complex fac-[(CO)3(bis(NHC))MnBr] (3Br)
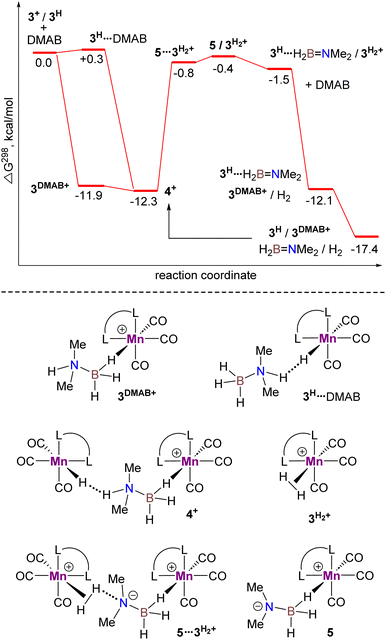
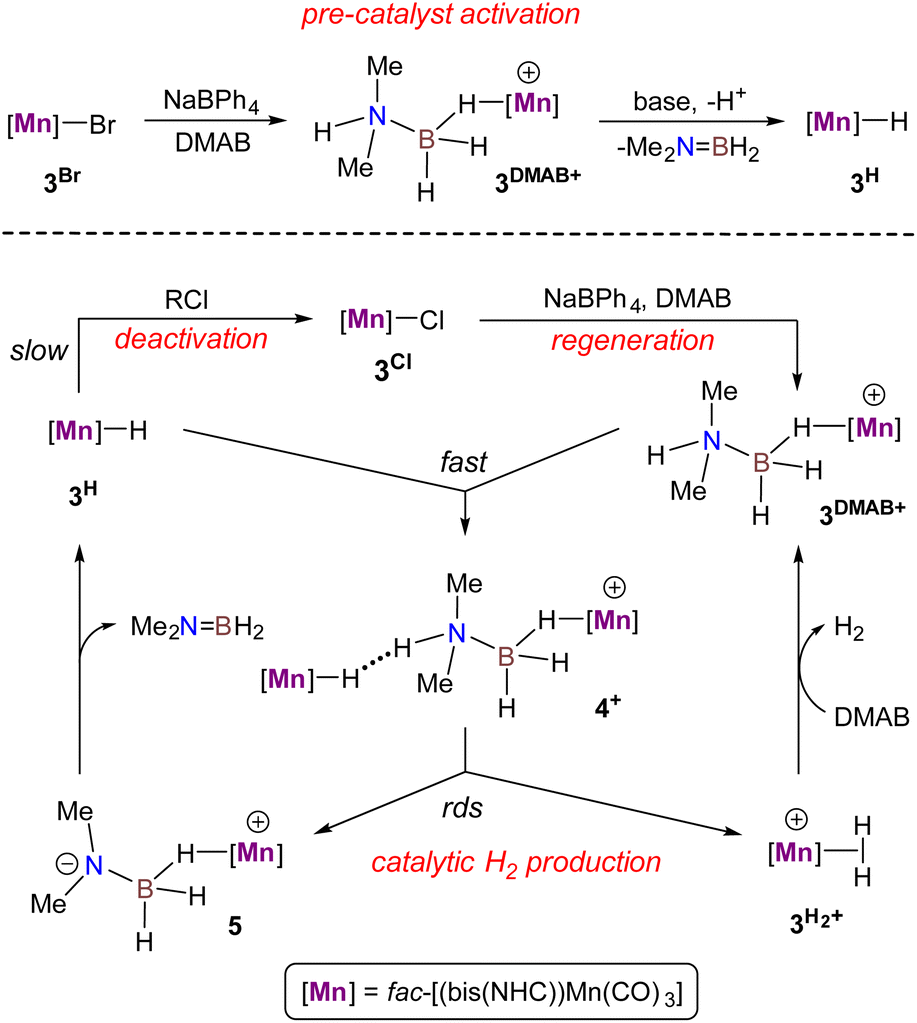
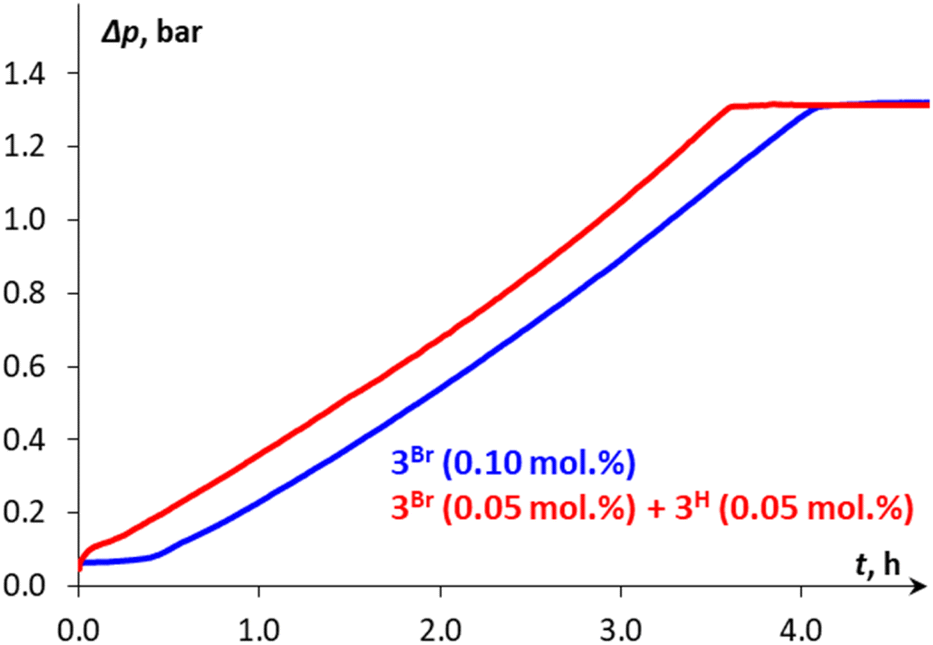
The catalytic cycle for DMAB dehydrogenation emerging from the results of the spectroscopic and kinetic studies combined with the DFT calculations is shown in Scheme 4. The activation steps (Scheme 4, top) include bromide abstraction from 3Br with NaBPh4 in the presence of a substrate to afford the cationic intermediate [3DMAB](BPh4). Its slow deprotonation by traces of amine and/or water followed by hydride transfer presumably leads to the initial accumulation of 3H essential for the catalytic process. Indeed, the addition of isolated 3H to a 3Br/NaBPh4/DMAB mixture resulted in immediate H2 evolution without any induction period (Fig. 6). Interestingly, the formation of a ternary adduct [4](BPh4) is favorable in terms of Gibbs energy regardless of the large entropy effect. This intermediate undergoes a sequence of N–H/B–H bond cleavage steps, ultimately affording 3H, Me2NBH2, and 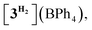 which rapidly exchanges the H2 ligand for DMAB and thus restores the initial [3DMAB](BPh4). The first-order kinetics on the metal, spectroscopic detection of 3H as a resting state and KIE values are consistent with the low steady-state concentration of the cation [3DMAB](BPh4) in the reaction mixture and its deprotonation by 3Hvia the formation of [4](BPh4) as the rate-determining step. Thus, the overall reaction rate is dependent on variable 3DMAB+ concentration, while 3H concentration remains constant and gives the effective reaction rate law: r3 = keff[3DMAB+] (see Scheme S2 and the associated text in the ESI† for a complete kinetic model). For this reason, despite the simultaneous presence of two metal complexes (3H and 3DMAB+) in the reaction mixture, first reaction order in overall metal concentration was actually observed. In this context, the beneficial effect of NHC on the series of Mn(I) complexes with bidentate ligands (Scheme 2) can be rationalized due to the synergy between a better stabilization of the cationic species and a higher basicity of the corresponding hydrides. Importantly, complex 3H may be observed in the reaction mixture uniquely in the presence of an amine-borane substrate, whereas isolated hydride decomposes rapidly in PhCl at 50–60 °C to form mainly chloride derivative 3Cl. We suppose that such counter-intuitive survival of 3H in a variety of chlorinated solvents (nBuCl, PhCl, CH2Cl2) may be explained by the faster dehydrogenation kinetics vs. chlorination and efficient regeneration of the cationic complex [3DMAB]+ from 3Cl by the excess of halide abstractor (Scheme 4), thus explaining the observed trend in the catalytic activity in the order of PhCl ≫ nBuCl > CH2Cl2 and the overall robustness of our catalytic system that results in high TON values.
which rapidly exchanges the H2 ligand for DMAB and thus restores the initial [3DMAB](BPh4). The first-order kinetics on the metal, spectroscopic detection of 3H as a resting state and KIE values are consistent with the low steady-state concentration of the cation [3DMAB](BPh4) in the reaction mixture and its deprotonation by 3Hvia the formation of [4](BPh4) as the rate-determining step. Thus, the overall reaction rate is dependent on variable 3DMAB+ concentration, while 3H concentration remains constant and gives the effective reaction rate law: r3 = keff[3DMAB+] (see Scheme S2 and the associated text in the ESI† for a complete kinetic model). For this reason, despite the simultaneous presence of two metal complexes (3H and 3DMAB+) in the reaction mixture, first reaction order in overall metal concentration was actually observed. In this context, the beneficial effect of NHC on the series of Mn(I) complexes with bidentate ligands (Scheme 2) can be rationalized due to the synergy between a better stabilization of the cationic species and a higher basicity of the corresponding hydrides. Importantly, complex 3H may be observed in the reaction mixture uniquely in the presence of an amine-borane substrate, whereas isolated hydride decomposes rapidly in PhCl at 50–60 °C to form mainly chloride derivative 3Cl. We suppose that such counter-intuitive survival of 3H in a variety of chlorinated solvents (nBuCl, PhCl, CH2Cl2) may be explained by the faster dehydrogenation kinetics vs. chlorination and efficient regeneration of the cationic complex [3DMAB]+ from 3Cl by the excess of halide abstractor (Scheme 4), thus explaining the observed trend in the catalytic activity in the order of PhCl ≫ nBuCl > CH2Cl2 and the overall robustness of our catalytic system that results in high TON values.
 |
| | Scheme 4 Proposed mechanism for DMAB dehydrogenation catalyzed by the bis(NHC) Mn(I) complex 3Br. | |
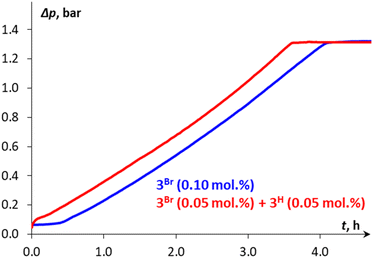 |
| | Fig. 6 Kinetic profile (Δp vs. time) for DMAB dehydrogenation in PhCl at 50 °C catalyzed either by 0.1 mol% 3Br or by a mixture of 3Br (0.05 mol%) and 3H (0.05 mol%) in the presence of NaBPh4 (1 mol%). | |
Conclusions
We have evidenced by spectroscopic, kinetic and theoretical means a new type of intermolecular bimetallic cooperativity between hydride and cationic species, clearly illustrating that a simple mononuclear metal halide precursor can be at the origin of two catalytically active species actually doing two complementary jobs for one price. The potential of this approach for homogeneous catalysis was demonstrated by the development of a practical and efficient system for amine-borane dehydrogenation based on an air-stable bis(NHC) Mn(I) complex and NaBPh4 that is capable of operating at low catalyst charge and providing unprecedented TONs for Me2NHBH3 and tBuNH2BH3 substrates. Our results also show that nucleophilic metal hydrides can successfully operate in chlorinated solvents if the catalytic process is faster than decomposition, thus allowing us to shake up a longstanding paradigm about their incompatibility. Taking into account a plethora of known monometallic halide complexes bearing strong donor ligands, we hope that this contribution will boost the interest of a broader use of intermolecular bimetallic cooperation phenomena in organometallic chemistry and homogeneous catalysis.
Data availability
All relevant experimental data and details of the DFT calculations are provided in the ESI.† Deposition numbers 2262301 (for [3MeCN](BF4)) and 2262302 (for 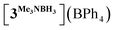 ) contain the supplementary crystallographic data for this paper.
) contain the supplementary crystallographic data for this paper.
Author contributions
O. A. F., N. V. B., E. S. S., Y. C. and D. A. V. planned and guided the research project. E. S. O. and D. A. V. acquired the research funding. E. S. G. carried out the main experimental work and performed initial data analysis. E. S. O. and S. A. K. participated in IR mechanistic investigations. O. A. F. performed the DFT calculations. L. V. and D. A. V. carried out X-ray diffraction experiments. E. S. G., E. S. O., O. A. F., N. V. B., E. S. S., Y. C. and D. A. V. participated in manuscript preparation. All authors have approved the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The work was financially supported by the Russian Science Foundation (grant no. 22-73-00072, spectroscopic mechanistic studies) and by the CNRS. E. S. G. is grateful to the French Embassy in Moscow for a joint PhD fellowship (Vernadski program). Computational studies were performed using HPC resources from CALMIP (grant no. P18038). Spectroscopic data were partially collected using the equipment of the Center for Molecular Composition Studies of INEOS RAS with the support from the Ministry of Science and Higher Education of the Russian Federation (contract no. 075-03-2023-642). We thank Dr Noël Lugan and Dr Olivier Baslé for helpful discussions.
References
-
(a) L. Liu and A. Corma, Chem. Rev., 2023, 123, 4855–4933 CrossRef CAS PubMed;
(b) L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- R. H. Holm, P. Kennepohl and E. I. Solomon, Chem. Rev., 1996, 96, 2239–2314 CrossRef CAS PubMed.
- Selected reviews:
(a) A. C. Ghosh, C. Duboc and M. Gennari, Coord. Chem. Rev., 2021, 428, 213606 CrossRef CAS;
(b) C.-H. Wang and S. DeBeer, Chem. Soc. Rev., 2021, 50, 8743–8761 RSC;
(c) C. Ferousi, S. H. Majer, I. M. DiMucci and K. M. Lancaster, Chem. Rev., 2020, 120, 5252–5307 CrossRef CAS PubMed;
(d) J. Serrano-Plana, I. Garcia-Bosch, A. Company and M. Costas, Acc. Chem. Res., 2015, 48, 2397–2406 CrossRef CAS PubMed;
(e) M. D. Kärkäs, O. Verho, E. V. Johnston and B. Åkermark, Chem. Rev., 2014, 114, 11863–12001 CrossRef PubMed;
(f) M. Costas, K. Chen and L. Que Jr, Coord. Chem. Rev., 2000, 200–202, 517–544 CrossRef CAS.
-
(a) C. Uyeda and C. M. Farley, Acc. Chem. Res., 2021, 54, 3710–3719 CrossRef CAS PubMed;
(b) I. G. Powers and C. Uyeda, ACS Catal., 2017, 7, 936–958 CrossRef CAS;
(c) D. C. Powers and T. Ritter, Acc. Chem. Res., 2012, 45, 840–850 CrossRef CAS PubMed.
- J. Bauer, H. Braunschweig and R. D. Dewhurst, Chem. Rev., 2012, 112, 4329–4346 CrossRef CAS PubMed.
-
(a) R. Govindarajan, S. Deolka and J. R. Khusnutdinova, Chem. Sci., 2022, 13, 14008–14031 RSC;
(b) N. P. Mankad, Chem. Commun., 2018, 54, 1291–1302 RSC;
(c) L. H. Gade, Angew. Chem., Int. Ed., 2000, 39, 2658–2678 CrossRef CAS.
-
(a) H.-C. Yu, S. M. Islam and N. P. Mankad, ACS Catal., 2020, 10, 3670–3675 CrossRef CAS;
(b) L.-J. Cheng and N. P. Mankad, J. Am. Chem. Soc., 2019, 141, 3710–3716 CrossRef CAS PubMed;
(c) M. K. Karunananda and N. P. Mankad, J. Am. Chem. Soc., 2015, 137, 14598–14601 CrossRef CAS PubMed;
(d) S. R. Parmelee, T. J. Mazzacano, Y. Zhu, N. P. Mankad and J. A. Keith, ACS Catal., 2015, 5, 3689–3699 CrossRef CAS;
(e) T. J. Mazzacano and N. P. Mankad, J. Am. Chem. Soc., 2013, 135, 17258–17261 CrossRef CAS PubMed.
-
(a) M. Navarro, J. J. Moreno, M. Pérez-Jiménez and J. Campos, Chem. Commun., 2022, 58, 11220–11235 RSC;
(b) B. Chatterjee, W.-C. Chang, S. Jena and C. Werlé, ACS Catal., 2020, 10, 14024–14055 CrossRef CAS.
-
(a) N. Hidalgo, F. de la Cruz-Martínez, M. T. Martín, M. C. Nicasio and J. Campos, Chem. Commun., 2022, 58, 9144–9147 RSC;
(b) N. Hidalgo, J. J. Moreno, M. Pérez-Jiménez, C. Maya, J. López-Serrano and J. Campos, Chem.–Eur. J., 2020, 26, 5982–5993 CrossRef CAS;
(c) J. Campos, J. Am. Chem. Soc., 2017, 139, 2944–2947 CrossRef CAS PubMed.
- E. S. Osipova, E. S. Gulyaeva, E. I. Gutsul, V. A. Kirkina, A. A. Pavlov, Y. V. Nelyubina, A. Rossin, M. Peruzzini, L. M. Epstein, N. V. Belkova, O. A. Filippov and E. S. Shubina, Chem. Sci., 2021, 12, 3682–3692 RSC.
- E. S. Osipova, D. V. Sedlova, E. I. Gutsul, Yu. V. Nelyubina, P. V. Dorovatovskii, L. M. Epstein, O. A. Filippov, E. S. Shubina and N. V. Belkova, Organometallics, 2023, 42, 2651–2660 CrossRef CAS.
- S. Sinhababu, M. R. Radzhabov, J. Telser and N. P. Mankad, J. Am. Chem. Soc., 2022, 144, 3210–3221 CrossRef CAS PubMed.
- E. S. Osipova, E. S. Gulyaeva, N. V. Kireev, S. A. Kovalenko, C. Bijani, Y. Canac, D. A. Valyaev, O. A. Filippov, N. V. Belkova and E. S. Shubina, Chem. Commun., 2022, 58, 5017–5020 RSC.
- E. S. Osipova, S. A. Kovalenko, E. S. Gulyaeva, N. V. Kireev, A. A. Pavlov, O. A. Filippov, A. A. Danshina, D. A. Valyaev, Y. Canac, E. S. Shubina and N. V. Belkova, Molecules, 2023, 28, 3368 CrossRef CAS PubMed.
-
(a) H. R. Sharpe, A. M. Geer, T. J. Blundell, F. R. Hastings, M. W. Fay, G. A. Rance, W. Lewis, A. J. Blake and D. L. Kays, Catal. Sci. Technol., 2017, 8, 229–235 RSC;
(b) S. Muhammad, S. Moncho, E. N. Brothers and A. A. Bengali, Chem. Commun., 2014, 50, 5874–5877 RSC;
(c) T. Kakizawa, Y. Kawano, K. Naganeyama and M. Shimoi, Chem. Lett., 2011, 40, 171–173 CrossRef CAS.
- Previously described synthesis of 3Br (ref.16) was reoptimized using weak base (K2CO3) in DMF at 120 °C to give target product in 82% yield avoiding the generation of free bis(NHC) with strong bases
(a) Y. Yang, Z. Zhang, X. Chang, Y.-Q. Zhang, R.-Z. Liao and L. Duan, Inorg. Chem., 2020, 59, 10234–10242 CrossRef CAS PubMed;
(b) M. Pinto, S. Friães, F. Franco, J. Lloret-Fillol and B. Royo, ChemCatChem, 2018, 10, 2734–2740 CrossRef CAS.
- W.-W. Zhan, Q.-L. Zhu and Q. Xu, ACS Catal., 2016, 6, 6892–6905 CrossRef CAS.
- For the formation of Mn/Hg intermetallic compounds, see:
(a) Z. Moser and C. Guminski, J. Phase Equilib., 1993, 14, 726–733 CrossRef CAS;
(b) J. F. de Wet, Angew. Chem., 1955, 67, 208 CrossRef CAS;
(c) O. Prelinger, Monatsh. Chem., 1893, 14, 353–370 CrossRef.
-
(a) E. A. LaPierre, B. O. Patrick and I. Manners, J. Am. Chem. Soc., 2019, 141, 20009–20015 CrossRef CAS PubMed;
(b) C. Lichtenberg, L. Viciu, M. Adelhardt, J. Sutter, K. Meyer, B. de Bruin and H. Grützmacher, Angew. Chem., Int. Ed., 2015, 54, 5766–5771 CrossRef CAS PubMed;
(c) J. R. Vance, A. Schäfer, A. P. M. Robertson, K. Lee, J. Turner, G. R. Whittell and I. Manners, J. Am. Chem. Soc., 2014, 136, 3048–3064 CrossRef CAS PubMed;
(d) T.-P. Lin and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 15310–15313 CrossRef CAS PubMed;
(e) M. Vogt, B. de Bruin, H. Berke, M. Trincado and H. Grützmacher, Chem. Sci., 2011, 2, 723–727 RSC;
(f) M. E. Sloan, A. Staubitz, T. J. Clark, C. A. Russell, G. C. Lloyd-Jones and I. Manners, J. Am. Chem. Soc., 2010, 132, 3831–3841 CrossRef CAS PubMed;
(g) Y. Kawano, M. Uruichi, M. Shimoi, S. Taki, T. Kawaguchi, T. Kakizawa and H. Ogino, J. Am. Chem. Soc., 2009, 131, 14946–14957 CrossRef CAS PubMed.
- For noble metal catalysts for DMAB dehydrogenation working at 0.5 mol% charge or below, see:
(a) S. Pal, S. Kusumoto and K. Nozaki, Organometallics, 2018, 37, 906–914 CrossRef CAS;
(b) E. H. Kwan, H. Ogawa and M. A. Yamashita, ChemCatChem, 2017, 9, 2457–2462 CrossRef CAS;
(c) E. U. Barın, M. Masjedi and S. Özkar, Materials, 2015, 8, 3155–3167 CrossRef;
(d) H. C. Johnson, E. M. Leitao, G. R. Whittell, I. Manners, G. C. Lloyd-Jones and A. S. Weller, J. Am. Chem. Soc., 2014, 136, 9078–9093 CrossRef CAS PubMed;
(e) D. F. Schreiber, C. O'Connor, C. Grave, Y. Ortin, H. Müller-Bunz and A. D. Phillips, ACS Catal., 2012, 2, 2505–2511 CrossRef CAS;
(f) R. Dallanegra, A. P. M. Robertson, A. B. Chaplin, I. Manners and A. S. Weller, Chem. Commun., 2011, 47, 3763–3765 RSC;
(g) A. Friedrich, M. Drees and S. Schneider, Chem.–Eur. J., 2009, 15, 10339–10342 CrossRef CAS PubMed.
- For reviews on amine-boranes dehydrogenation using catalysts based on main-group elements, see:
(a) D. H. A. Boom, A. R. Jupp and J. C. Slootweg, Chem.–Eur. J., 2019, 25, 9133–9152 CrossRef CAS PubMed;
(b) R. L. Melen, Chem. Soc. Rev., 2016, 45, 775–788 RSC.
- Alkaline metal catalysts:
(a) R. McLellan, A. R. Kennedy, R. E. Mulvey, S. A. Orr and S. D. Robertson, Chem.–Eur. J., 2017, 23, 16853–16861 CrossRef CAS PubMed;
(b) P. Bellham, M. S. Hill and G. Kociok-Köhn, Dalton Trans., 2015, 44, 12078–12081 RSC.
- Alkaline earth metal catalysts:
(a) L. Wirtz, K. Y. Ghulam, B. Morgenstern and A. Schäfer, ChemCatChem, 2022, 14, e202201007 CrossRef CAS;
(b) L. Wirtz, W. Haider, V. Huch, M. Zimmer and A. Schäfer, Chem.–Eur. J., 2020, 26, 6176–6184 CrossRef CAS PubMed;
(c) A. C. A. Ried, L. J. Taylor, A. M. Geer, H. E. L. Williams, W. Lewis, A. J. Blake and D. L. Kays, Chem.–Eur. J., 2019, 25, 6840–6846 CrossRef CAS PubMed;
(d) X. Zheng, J. Huang, Y. Yao and X. Xu, Chem. Commun., 2019, 55, 9152–9155 RSC;
(e) D. J. Liptrot, M. S. Hill, M. F. Mahon and D. J. MacDougall, Chem.–Eur. J., 2010, 16, 8508–8515 CrossRef CAS PubMed.
- Group 3 metal catalysts:
(a) P. Xu and X. Xu, Organometallics, 2019, 38, 3212–3217 CrossRef CAS;
(b) E. Lu, Y. Yuan, Y. Chen and W. Xia, ACS Catal., 2013, 3, 521–524 CrossRef CAS;
(c) H. J. Cowley, M. S. Holt, R. L. Melen, J. M. Rawson and D. S. Wright, Chem. Commun., 2011, 47, 2682–2684 RSC;
(d) M. S. Hill, G. Kociok-Köhn and T. P. Robinson, Chem. Commun., 2010, 46, 7587–7589 RSC.
- Lanthanide- and actinide-based catalysts:
(a) V. A. Kirkina, A. A. Kissel, A. N. Selikhov, Y. V. Nelyubina, O. A. Filippov, N. V. Belkova, A. A. Trifonov and E. S. Shubina, Chem. Commun., 2022, 58, 859–862 RSC;
(b) K. A. Erickson and J. L. Kiplinger, ACS Catal., 2017, 7, 4276–4280 CrossRef CAS;
(c) P. Cui, T. P. Spaniol, L. Maron and J. Okuda, Chem.–Eur. J., 2013, 19, 13437–13444 CrossRef CAS PubMed.
-
(a) M. Boudjelel, E. D. Sosa Carrizo, S. Mallet-Ladeira, S. Massou, K. Miqueu, G. Bouhadir and D. Bourissou, ACS Catal., 2018, 8, 4459–4464 CrossRef CAS;
(b) C. Appelt, J. C. Slootweg, K. Lammertsma and W. Uhl, Angew. Chem., Int. Ed., 2013, 52, 4256–4259 CrossRef CAS PubMed;
(c) A. M. Chapman, M. F. Haddow and D. F. Wass, J. Am. Chem. Soc., 2011, 133, 8826–8829 CrossRef CAS PubMed.
- S. Duman, M. Masjedi and S. Özkar, J. Mol. Catal. A: Chem., 2016, 411, 9–18 CrossRef CAS.
-
(a) A. Staubitz, A. P. M. Robertson and I. Manners, Chem. Rev., 2010, 110, 4079–4124 CrossRef CAS PubMed;
(b) C. W. Hamilton, R. T. Baker, A. Staubitz and I. Manners, Chem. Soc. Rev., 2009, 38, 279–293 RSC.
-
(a) K. A. Erickson, J. P. W. Stelmach, N. T. Mucha and R. Waterman, Organometallics, 2015, 34, 4693–4699 CrossRef CAS;
(b) D. García-Vivó, E. Huergo, M. A. Ruiz and R. Travieso-Puente, Eur. J. Inorg. Chem., 2013, 2013, 4998–5008 CrossRef.
-
(a) F. Anke, S. Boye, A. Spannenberg, A. Lederer, D. Heller and T. Beweries, Chem.–Eur. J., 2020, 26, 7889–7899 CrossRef CAS PubMed;
(b) T. M. Maier, S. Sandl, I. G. Shenderovich, A. J. von Wangelin, J. J. Weigand and R. Wolf, Chem.–Eur. J., 2019, 25, 238–245 CrossRef CAS PubMed;
(c) T. Jurca, T. Dellermann, N. E. Stubbs, D. A. Resendiz-Lara, G. R. Whittell and I. Manners, Chem. Sci., 2018, 9, 3360–3366 RSC;
(d) F. Anke, D. Han, M. Klahn, A. Spannenberg and T. Beweries, Dalton Trans., 2017, 46, 6843–6847 RSC;
(e) C. Lichtenberg, M. Adelhardt, T. L. Gianetti, K. Meyer, B. de Bruin and H. Grützmacher, ACS Catal., 2015, 5, 6230–6240 CrossRef CAS.
- T. M. Boyd, K. A. Andrea, K. Baston, A. Johnson, D. E. Ryan and A. S. Weller, Chem. Commun., 2020, 56, 482–485 RSC.
-
(a) P. Bhattacharya, J. A. Krause and H. Guan, J. Am. Chem. Soc., 2014, 136, 11153–11161 CrossRef CAS PubMed;
(b) R. T. Baker, J. C. Gordon, C. W. Hamilton, N. J. Henson, P.-H. Lin, S. Maguire, M. Murugesu, B. L. Scott and N. C. Smythe, J. Am. Chem. Soc., 2012, 134, 5598–5609 CrossRef CAS PubMed;
(c) R. J. Keaton, J. M. Blacquiere and R. T. Baker, J. Am. Chem. Soc., 2007, 129, 1844–1845 CrossRef CAS PubMed.
-
(a) M. Hasenbeck, J. Becker and U. Gellrich, Angew. Chem., Int. Ed., 2020, 59, 1590–1594 CrossRef CAS PubMed;
(b) Z. Mo, A. Rit, J. Campos, E. L. Kolychev and S. Aldridge, J. Am. Chem. Soc., 2016, 138, 3306–3309 CrossRef CAS PubMed;
(c) Z. Lu, L. Schweighauser, H. Hausmann and H. A. Wegner, Angew. Chem., Int. Ed., 2015, 54, 15556–15559 CrossRef CAS PubMed;
(d) W. C. Ewing, A. Marchione, D. W. Himmelberger, P. J. Carroll and L. G. Sneddon, J. Am. Chem. Soc., 2011, 133, 17093–17099 CrossRef CAS PubMed.
-
(a) K. Sarkar, K. Das, A. Kundu, D. Adhikari and B. Maji, ACS Catal., 2021, 11, 2786–2794 CrossRef CAS;
(b) M. Gediga, C. M. Feil, S. H. Schlindwein, J. Bender, M. Nieger and D. Gudat, Chem.–Eur. J., 2017, 23, 11560–11569 CrossRef CAS PubMed.
-
(a) D. Wei, X. Shi, P. Sponholz, H. Junge and M. Beller, ACS Cent. Sci., 2022, 8, 1457–1463 CrossRef CAS PubMed;
(b) D. Wei, R. Sang, P. Sponholz, H. Junge and M. Beller, Nat. Energy, 2022, 7, 438–447 CrossRef CAS;
(c) A. Léval, A. Agapova, C. Steinlechner, E. Alberico, H. Junge and M. Beller, Green Chem., 2020, 22, 913–920 RSC.
-
(a) A. Rossin and M. Peruzzini, Chem. Rev., 2016, 116, 8848–8872 CrossRef CAS PubMed;
(b) S. Bhunya, T. Malakar, G. Ganguly and A. Paul, ACS Catal., 2016, 6, 7907–7934 CrossRef CAS.
- N. V. Kireev, O. A. Filippov, E. S. Gulyaeva, E. S. Shubina, L. Vendier, Y. Canac, J.-B. Sortais, N. Lugan and D. A. Valyaev, Chem. Commun., 2020, 56, 2139–2142 RSC.
- R. Buhaibeh, O. A. Filippov, A. Bruneau-Voisine, J. Willot, C. Duhayon, D. A. Valyaev, N. Lugan, Y. Canac and J.-B. Sortais, Angew. Chem., Int. Ed., 2019, 58, 6727–6731 CrossRef CAS PubMed.
-
(a) S. C. A. Sousa, S. Realista and B. Royo, Adv. Synth. Catal., 2020, 362, 2437–2443 CrossRef CAS;
(b) B. Royo, C. J. Carrasco, S. C. A. Sousa and M. F. Pinto, ChemCatChem, 2019, 11, 3839–3843 CrossRef.
-
(a) S. Fernández, F. Franco, M. Martínez Belmonte, S. Friães, B. Royo, J. M. Luis and J. Lloret-Fillol, ACS Catal., 2023, 13, 10375–10385 CrossRef;
(b) F. Franco, F. P. Mara, B. Royo and J. Lloret-Fillol, Angew. Chem., Int. Ed., 2018, 57, 4603–4606 CrossRef CAS PubMed.
-
(a) N. F. Both, A. Spannenberg, H. Jiao, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2023, 62, e202307987 CrossRef CAS PubMed;
(b) K. Azouzi, L. Pedussaut, R. Pointis, A. Bonfiglio, R. Kumari Riddhi, C. Duhayon, S. Bastin and J.-B. Sortais, Organometallics, 2023, 42, 1832–1838 CrossRef CAS.
-
(a) K. Ganguli, A. Mandal and S. Kundu, ACS Catal., 2022, 12, 12444–12457 CrossRef CAS;
(b) X.-B. Lan, Z. Ye, J. Liu, M. Huang, Y. Shao, X. Cai, Y. Liu and Z. Ke, ChemSusChem, 2020, 13, 2557–2563 CrossRef CAS PubMed;
(c) X.-B. Lan, Z. Ye, M. Huang, J. Liu, Y. Liu and Z. Ke, Org. Lett., 2019, 21, 8065–8070 CrossRef CAS PubMed;
(d) M. Huang, Y. Li, Y. Li, J. Liu, S. Shu, Y. Liu and Z. Ke, Chem. Commun., 2019, 55, 6213–6216 RSC.
-
(a) K. Lindenau, N. Jannsen, M. Rippke, H. Al Hamwi, C. Selle, H.-J. Drexler, A. Spannenberg, M. Sawall, K. Neymeyr, D. Heller, F. Reiß and T. Beweries, Catal. Sci. Technol., 2021, 11, 4034–4050 RSC;
(b) C. Cesari, B. Berti, F. Calcagno, C. Lucarelli, M. Garavelli, R. Mazzoni, I. Rivalta and S. Zacchini, Organometallics, 2021, 40, 2724–2735 CrossRef CAS;
(c) M. Trose, M. Reiß, F. Reiß, F. Anke, A. Spannenberg, S. Boye, A. Lederer, P. Arndt and T. Beweries, Dalton Trans., 2018, 47, 12858–12862 RSC;
(d) T. Miyazaki, Y. Tanabe, M. Yuki, Y. Miyake and Y. Nishibayashi, Organometallics, 2011, 30, 2394–2404 CrossRef CAS.
-
(a) T. Yasue, Y. Kawano and M. Shimoi, Angew. Chem., Int. Ed., 2003, 42, 1727–1730 CrossRef CAS PubMed;
(b) T. Kakizawa, Y. Kawano and M. Shimoi, Organometallics, 2001, 20, 3211–3213 CrossRef CAS.
-
(a) N. V. Belkova, L. M. Epstein, O. A. Filippov and E. S. Shubina, Chem. Rev., 2016, 116, 8545–8587 CrossRef CAS PubMed;
(b) N. V. Belkova, L. M. Epstein and E. S. Shubina, Eur. J. Inorg. Chem., 2010, 2010, 3555–3565 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Complete experimental procedures, details of DFT calculations, original IR and NMR spectra and kinetic profiles for H2 evolution. CCDC 2262301 and 2262302. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc05356c |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b,
Natalia V.
Belkova
*b,
Natalia V.
Belkova
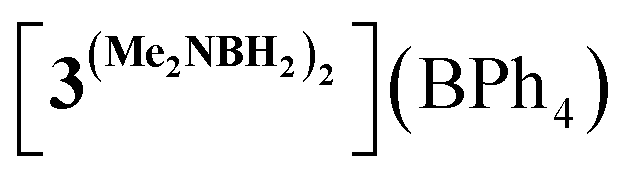 bearing a coordinated [Me2NBH2]2 ligand (Fig. 1(c), see Fig. S17† for νCO band deconvolution). 11B NMR monitoring of the reaction mixture in CD2Cl2 (Fig. S23†) showed the appearance of the characteristic signal of Me2N
bearing a coordinated [Me2NBH2]2 ligand (Fig. 1(c), see Fig. S17† for νCO band deconvolution). 11B NMR monitoring of the reaction mixture in CD2Cl2 (Fig. S23†) showed the appearance of the characteristic signal of Me2N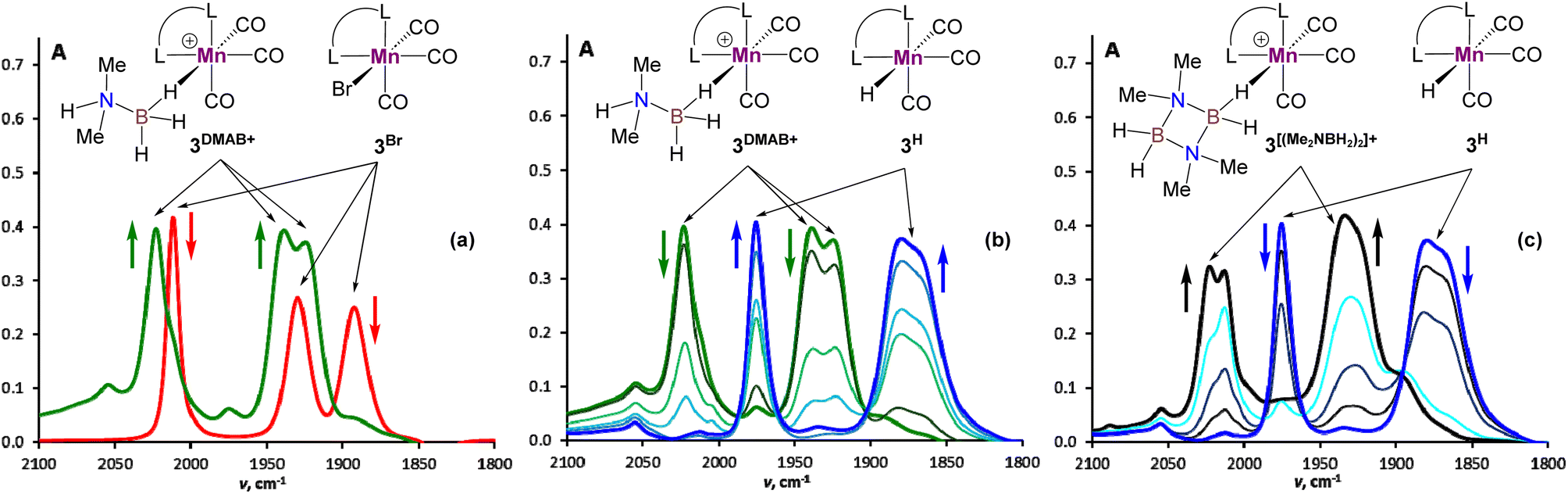
 from Me3NBH3 that is unreactive towards dehydrogenation, 3Br and NaBPh4. Its structure elucidated from X-ray diffraction measurements (Fig. 2) features a η1-BH coordination mode previously observed in half-sandwich Mn(I) analogues.44 The BH3 moiety in the NMR spectra of
from Me3NBH3 that is unreactive towards dehydrogenation, 3Br and NaBPh4. Its structure elucidated from X-ray diffraction measurements (Fig. 2) features a η1-BH coordination mode previously observed in half-sandwich Mn(I) analogues.44 The BH3 moiety in the NMR spectra of  appears as broad signals at δB −11.9 ppm and δH −3.30 ppm. The latter value matches well with that observed during the NMR mechanistic investigation, clearly indicating the participation of such species in the catalytic process. According to IR and NMR data, all transformations occurring in catalytic DMAB dehydrogenation involved only tricarbonyl Mn(I) complexes and we did not find any spectroscopic evidence for the formation of the neutral dicarbonyl hydride species fac-[(CO)2(DMAB)(bis(NHC))MnH] essential for the realization of the route (b), as depicted in Scheme 3.
appears as broad signals at δB −11.9 ppm and δH −3.30 ppm. The latter value matches well with that observed during the NMR mechanistic investigation, clearly indicating the participation of such species in the catalytic process. According to IR and NMR data, all transformations occurring in catalytic DMAB dehydrogenation involved only tricarbonyl Mn(I) complexes and we did not find any spectroscopic evidence for the formation of the neutral dicarbonyl hydride species fac-[(CO)2(DMAB)(bis(NHC))MnH] essential for the realization of the route (b), as depicted in Scheme 3.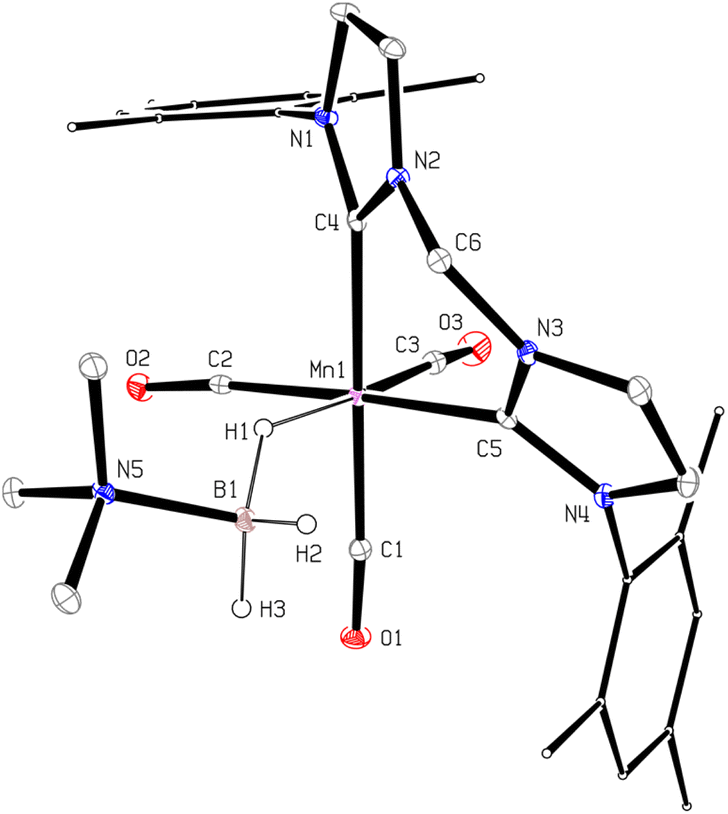
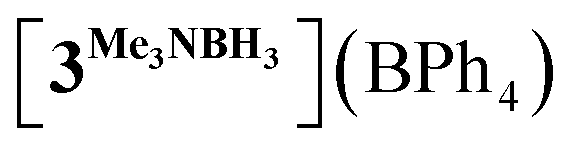 (20% probability ellipsoids, Mes groups represented as a wireframe, most hydrogen atoms, BPh4− anion and CH2Cl2 solvate molecules are not shown).
(20% probability ellipsoids, Mes groups represented as a wireframe, most hydrogen atoms, BPh4− anion and CH2Cl2 solvate molecules are not shown).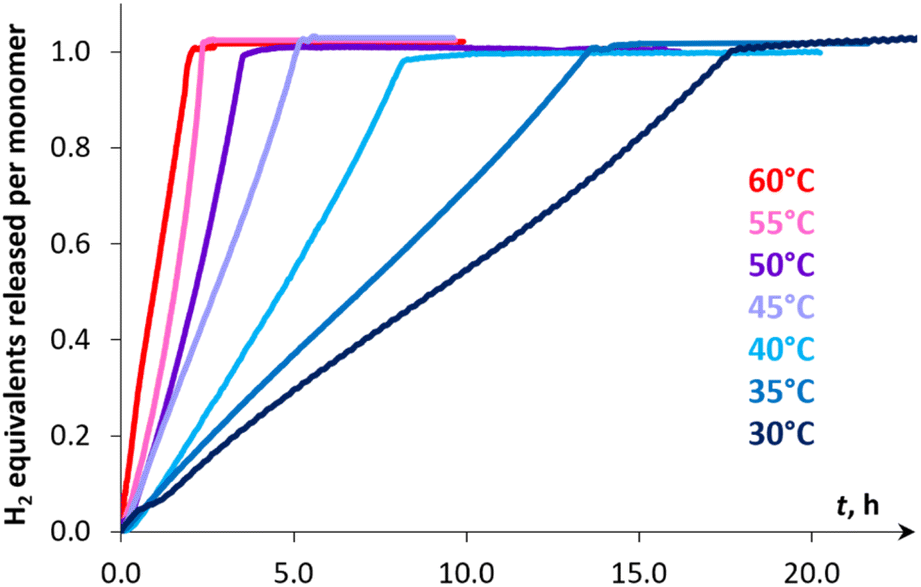
 which is composed of the cationic dihydrogen complex
which is composed of the cationic dihydrogen complex 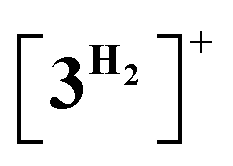 associated with the zwitter-ionic species 5 by hydrogen bonding (2.283 Å), then undergoing dissociation to the individual components. Importantly, the metal-coordinated B–H bond distance in complexes
associated with the zwitter-ionic species 5 by hydrogen bonding (2.283 Å), then undergoing dissociation to the individual components. Importantly, the metal-coordinated B–H bond distance in complexes 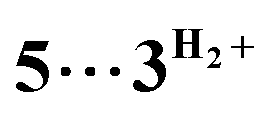 and 5 becomes elongated by 0.093 and 0.234 Å, respectively, relative to that in 4+ with a concomitant shortening of the Mn–H distance by 0.056 and 0.109 Å, respectively. These data are in agreement with the occurrence of a progressive hydride transfer in the sequence of
and 5 becomes elongated by 0.093 and 0.234 Å, respectively, relative to that in 4+ with a concomitant shortening of the Mn–H distance by 0.056 and 0.109 Å, respectively. These data are in agreement with the occurrence of a progressive hydride transfer in the sequence of 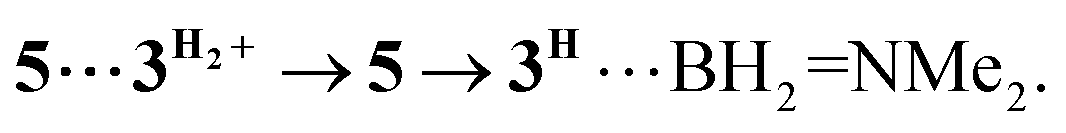 Finally, the essentially barrierless loss of H2 and Me2N
Finally, the essentially barrierless loss of H2 and Me2N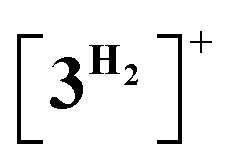 and 3H⋯BH2
and 3H⋯BH2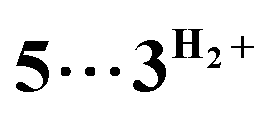 in the DMAB activation process provides additional evidence to consider this reaction mechanism as a viable intermolecular bimetallic cooperation (Scheme 1(d)).
in the DMAB activation process provides additional evidence to consider this reaction mechanism as a viable intermolecular bimetallic cooperation (Scheme 1(d)).
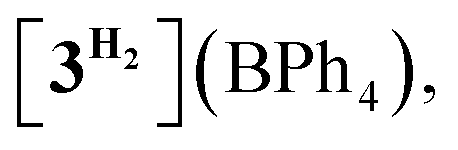 which rapidly exchanges the H2 ligand for DMAB and thus restores the initial [3DMAB](BPh4). The first-order kinetics on the metal, spectroscopic detection of 3H as a resting state and KIE values are consistent with the low steady-state concentration of the cation [3DMAB](BPh4) in the reaction mixture and its deprotonation by 3Hvia the formation of [4](BPh4) as the rate-determining step. Thus, the overall reaction rate is dependent on variable 3DMAB+ concentration, while 3H concentration remains constant and gives the effective reaction rate law: r3 = keff[3DMAB+] (see Scheme S2 and the associated text in the ESI† for a complete kinetic model). For this reason, despite the simultaneous presence of two metal complexes (3H and 3DMAB+) in the reaction mixture, first reaction order in overall metal concentration was actually observed. In this context, the beneficial effect of NHC on the series of Mn(I) complexes with bidentate ligands (Scheme 2) can be rationalized due to the synergy between a better stabilization of the cationic species and a higher basicity of the corresponding hydrides. Importantly, complex 3H may be observed in the reaction mixture uniquely in the presence of an amine-borane substrate, whereas isolated hydride decomposes rapidly in PhCl at 50–60 °C to form mainly chloride derivative 3Cl. We suppose that such counter-intuitive survival of 3H in a variety of chlorinated solvents (nBuCl, PhCl, CH2Cl2) may be explained by the faster dehydrogenation kinetics vs. chlorination and efficient regeneration of the cationic complex [3DMAB]+ from 3Cl by the excess of halide abstractor (Scheme 4), thus explaining the observed trend in the catalytic activity in the order of PhCl ≫ nBuCl > CH2Cl2 and the overall robustness of our catalytic system that results in high TON values.
which rapidly exchanges the H2 ligand for DMAB and thus restores the initial [3DMAB](BPh4). The first-order kinetics on the metal, spectroscopic detection of 3H as a resting state and KIE values are consistent with the low steady-state concentration of the cation [3DMAB](BPh4) in the reaction mixture and its deprotonation by 3Hvia the formation of [4](BPh4) as the rate-determining step. Thus, the overall reaction rate is dependent on variable 3DMAB+ concentration, while 3H concentration remains constant and gives the effective reaction rate law: r3 = keff[3DMAB+] (see Scheme S2 and the associated text in the ESI† for a complete kinetic model). For this reason, despite the simultaneous presence of two metal complexes (3H and 3DMAB+) in the reaction mixture, first reaction order in overall metal concentration was actually observed. In this context, the beneficial effect of NHC on the series of Mn(I) complexes with bidentate ligands (Scheme 2) can be rationalized due to the synergy between a better stabilization of the cationic species and a higher basicity of the corresponding hydrides. Importantly, complex 3H may be observed in the reaction mixture uniquely in the presence of an amine-borane substrate, whereas isolated hydride decomposes rapidly in PhCl at 50–60 °C to form mainly chloride derivative 3Cl. We suppose that such counter-intuitive survival of 3H in a variety of chlorinated solvents (nBuCl, PhCl, CH2Cl2) may be explained by the faster dehydrogenation kinetics vs. chlorination and efficient regeneration of the cationic complex [3DMAB]+ from 3Cl by the excess of halide abstractor (Scheme 4), thus explaining the observed trend in the catalytic activity in the order of PhCl ≫ nBuCl > CH2Cl2 and the overall robustness of our catalytic system that results in high TON values.
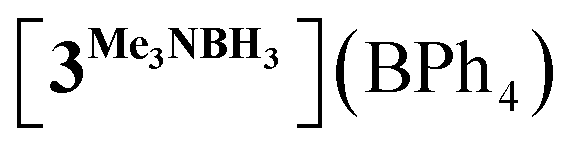 ) contain the supplementary crystallographic data for this paper.
) contain the supplementary crystallographic data for this paper.