
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient construction of functionalized pyrroloindolines through cascade radical cyclization/intermolecular coupling†
Yonggang
Jiang
a,
Dongxiang
Liu
a,
Lening
Zhang
a,
Cuirong
Qin
a,
Hui
Li
a,
Haitao
Yang
a,
Patrick J.
Walsh
 *b and
Xiaodong
Yang
*a
*b and
Xiaodong
Yang
*a
aKey Laboratory of Medicinal Chemistry for Natural Resources, Ministry of Education, Yunnan Provincial Center for Research & Development of Natural Products, School of Pharmacy, Yunnan University, Kunming 650091, P. R. China. E-mail: xdyang@ynu.edu.cn
bRoy and Diana Vagelos Laboratories, Penn/Merck Laboratory for High-Throughput Experimentation, Department of Chemistry, University of Pennsylvania, 231 South 34th Street, Philadelphia, Pennsylvania 19104, USA
First published on 4th January 2024
Abstract
Pyrroloindolines are important structural units in nature and the pharmaceutical industry, however, most approaches to such structures involve transition-metal or photoredox catalysts. Herein, we describe the first tandem SET/radical cyclization/intermolecular coupling between 2-azaallyl anions and indole acetamides. This method enables the transition-metal-free synthesis of C3a-substituted pyrroloindolines under mild and convenient conditions. The synthetic utility of this transformation is demonstrated by the construction of an array of C3a-methylamine pyrroloindolines with good functional group tolerance and yields. Gram-scale sequential one-pot synthesis and hydrolysis reactions demonstrate the potential synthetic utility and scalability of this approach.
Introduction
Nitrogen-containing heterocycles are among the most fundamental structural motifs in organic compounds.1–7 Among N-heterocycles, pyrroloindoline alkaloids (also termed hexahydropyrrolo[2,3-b]indoles), particularly C3a-substituted pyrroloindolines, are an important subclass in nature and the pharmaceutical industry (such as alline, physostigmine, flustramide B and F, gllocladin C, folicanthine, Fig. 1).8–12 These molecules have gained interest owing to their extensive biological activities, including antitumor, antimicrobial, antinematodal, vasodilating activities, as well as inhibitory activities against cholinesterases and topoisomerases.13,14 Consequently, the efficient and straightforward construction of functionalized pyrroloindolines remains in demand.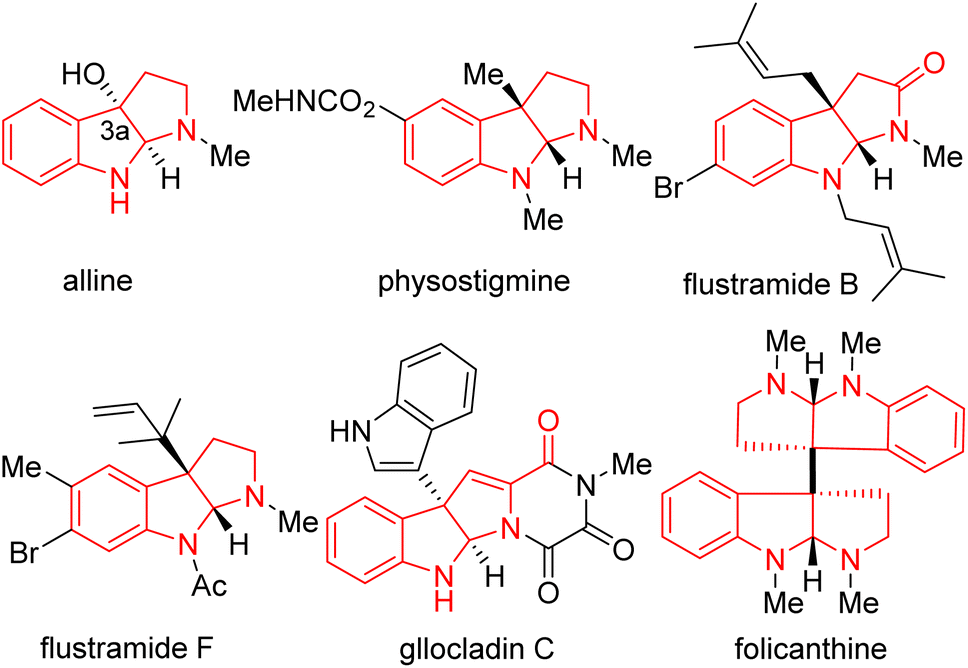 | ||
| Fig. 1 Representative C3a-substituted pyrroloindoline natural products. | ||
The classical approach to C3a-substituted pyrroloindolines involves the transition-metal-catalyzed cyclization (Scheme 1a). For example, MacMillan's group developed the copper-catalyzed arylation/cyclization cascade of indole acetamides with diaryliodonium salts to access enantioenriched C3a-aryl pyrroloindolines,15 while You reported an elegant iridium-catalyzed allylation of tryptamines with Z-cinnamyl acetate to obtain Z-retentive C3a-allyl pyrroloindolines.16 Recently, visible-light- mediated radical cyclizations have emerged as potent approaches for the construction of heterocycles,17–27 as presented in the representative illustrations in Scheme 1b. Knowles developed a Ir(ppy)3 photocatalytic proton-coupled electron transfer reaction for synthesis of C3a-TEMPO-substituted pyrroloindolines,28 and Wang reported a eosin Y visible-light-induced radical cascade reaction of indole acetamides to access C3a-hydroxypyrroloindolines.29 Most of these protocols, however, involve transition-metal catalysts or photoredox catalysts.
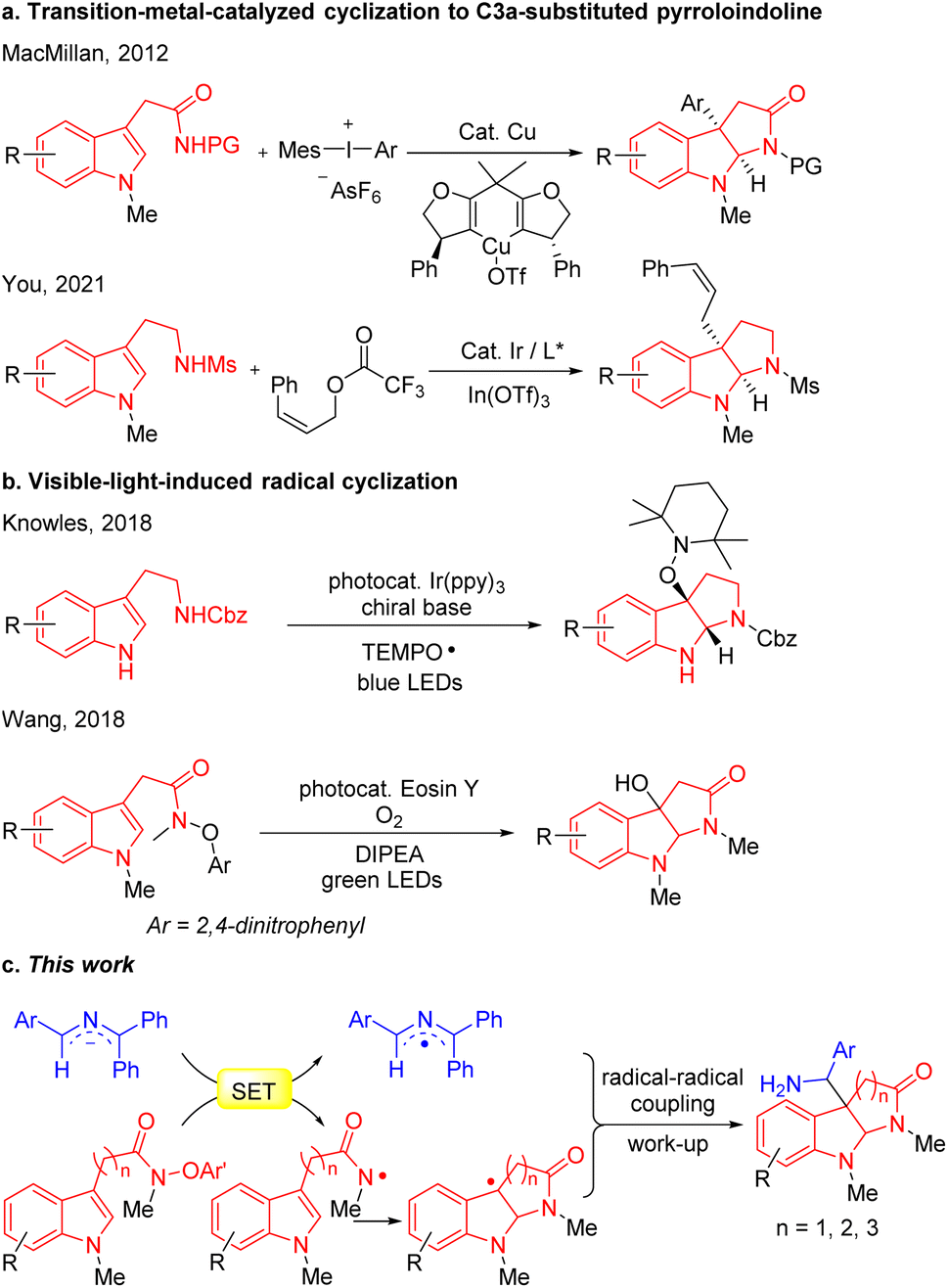 | ||
| Scheme 1 (a) Transition-metal-catalyzed cyclization to C3a-substituted pyrroloindoline. (b) Visible-light-induced radical cyclization to C3a-OH pyrroloindoline. (c) SED 2-azaallyl anions enable synthesis of C3a-substituted pyrroloindolines (this work). | ||
Since the pioneering studies by Murphy,30–32 super electron donors (SEDs), that is neutral and anionic organic compounds that exhibit strong reducing tendencies through single-electron transfer (SET), have emerged as an effective partner in radical–radical couplings to form C–C bonds. In particular, 2-azaallyl anions, which possess the ability to behave as strong single electron reducing agents, have attracted attention in the synthetic community.33–37 Based on the SED properties of 2-azaallyl anions, our team developed a series of tandem cyclization reactions to construct benzofuran, isochromene and isoquinoline derivatives,38–40 among others. We further employed 2-azaallyl anions to develop a series of radical C(sp3)–C(sp2) and C(sp3)–C(sp3) coupling strategies.41–47
In view of the medicinal value of pyrroloindolines, we felt compelled to apply this radical coupling approach to the synthesis of C3a-substituted pyrroloindolines. Based on our prior generation of amidyl radicals,48 we hypothesized that SET between the 2-azaallyl anions and indole N-aryloxy acetamides would generate 2-azaallyl radicals and amidyl radicals, the latter of which would trigger a radical cyclization to furnish C3a-pyrroloindoline radicals. Finally, coupling between 2-azaallyl radicals and pyrroloindoline radicals was expected to afford C3a-methylamine pyrroloindoline derivatives (Scheme 1c).
Herein, we describe the first tandem SET/radical cyclization/intermolecular coupling between 2-azaallyl anions and indole acetamides, which enables the synthesis of C3a-substituted pyrroloindolines under mild and convenient conditions. The synthetic utility of this transformation is demonstrated by the construction of an array of C3a-methylamine pyrroloindolines with good functional group tolerance and yields (33 examples, up to 88% yield).
Results and discussion
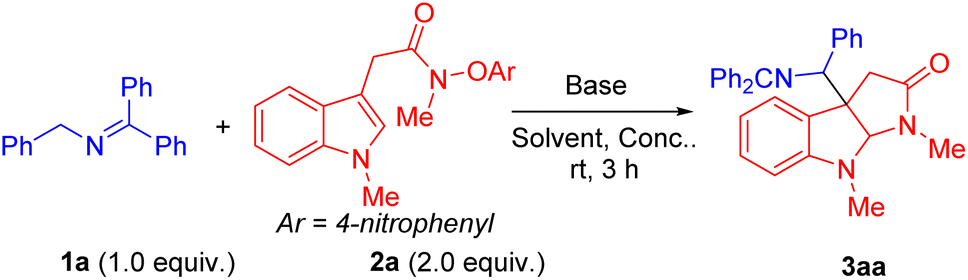
We initiated the reaction optimization by choosing indole N-aryloxy acetamide 2a as the model substrate. Reaction of 2a was performed with N-benzylketimine 1a in the presence of base in DMSO at room temperature for 3 h. Initially, a series of bases including LiOtBu, NaOtBu, KOtBu, LiN(SiMe3)2, NaN(SiMe3)2 and KN(SiMe3)2 were evaluated (Table 1, entries 1–6). Among them, NaN(SiMe3)2 generated the radical cyclization/coupling product 3aa in 89% assay yield (AY, as determined by 1H NMR integration against an internal standard) and 86% isolated yield (dr = 1.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, entry 5), while the other five bases led to product 3aa in 56–65% AY. Next, we then turned our attention to probe the effect of solvent and concentration. Using NaN(SiMe3)2 as base, we examined a range of solvents [THF, DMF, CPME, MTBE (methyl tert-butyl ether) and MeCN]. Surprisingly, no desired products formed in these solvents (entries 7–11). Furthermore, decreasing the concentration to 0.1 M or 0.05 M led to a reduction in the AY to 78% and 68% (entries 12 and 13). When the equiv of base was increased from 1.5 to 2.0 equiv, a decrease in the AY to 72% (entry 14) was observed. Finally, increasing the reaction temperature to 60 °C or decreasing it to 0 °C resulted in a decrease in the AY to 22% or 8%, respectively (entries 15 and 16).
:1, entry 5), while the other five bases led to product 3aa in 56–65% AY. Next, we then turned our attention to probe the effect of solvent and concentration. Using NaN(SiMe3)2 as base, we examined a range of solvents [THF, DMF, CPME, MTBE (methyl tert-butyl ether) and MeCN]. Surprisingly, no desired products formed in these solvents (entries 7–11). Furthermore, decreasing the concentration to 0.1 M or 0.05 M led to a reduction in the AY to 78% and 68% (entries 12 and 13). When the equiv of base was increased from 1.5 to 2.0 equiv, a decrease in the AY to 72% (entry 14) was observed. Finally, increasing the reaction temperature to 60 °C or decreasing it to 0 °C resulted in a decrease in the AY to 22% or 8%, respectively (entries 15 and 16).
|
|
||||
|---|---|---|---|---|
| Entry | Base (equiv.) | Solvent | Conc. [M] | Yield (%)b |
| a Reaction conditions: 1a (0.1 mmol, 1.0 equiv), 2a (0.2 mmol, 2.0 equiv), base, rt., 3 h. b Assay yield (AY) determined by 1H NMR spectroscopy of the crude reaction mixture using C2H2Cl4 as an internal standard. c Isolated yield after chromatographic purification. d 60 °C. e 0 °C. | ||||
| 1 | LiOtBu (1.5) | DMSO | 0.2 | 56 (dr = 1:1) |
| 2 | NaOtBu (1.5) | DMSO | 0.2 | 61 (dr = 1:1) |
| 3 | KOtBu (1.5) | DMSO | 0.2 | 65 (dr = 1.3:1) |
| 4 | LiHMDS (1.5) | DMSO | 0.2 | 59 (dr = 1:1) |
| 5 | NaHMDS (1.5) | DMSO | 0.2 | 89 (86)c (dr = 1.2:1) |
| 6 | KHMDS (1.5) | DMSO | 0.2 | 65 (dr = 1:1) |
| 7 | NaHMDS (1.5) | THF | 0.2 | 0 |
| 8 | NaHMDS (1.5) | DMF | 0.2 | 0 |
| 9 | NaHMDS (1.5) | CPME | 0.2 | 0 |
| 10 | NaHMDS (1.5) | MTBE | 0.2 | 0 |
| 11 | NaHMDS (1.5) | MeCN | 0.2 | 0 |
| 12 | NaHMDS (1.5) | DMSO | 0.1 | 78 (dr = 1:1) |
| 13 | NaHMDS (1.5) | DMSO | 0.05 | 68 (dr = 1:1) |
| 14 | NaHMDS (2.0) | DMSO | 0.2 | 72 (dr = 1:1) |
| 15d | NaHMDS (1.5) | DMSO | 0.1 | 22 (dr = 1:1) |
| 16e | NaHMDS (1.5) | DMSO/THF = 1:1 |
0.1 | 8 (dr = 1:1) |
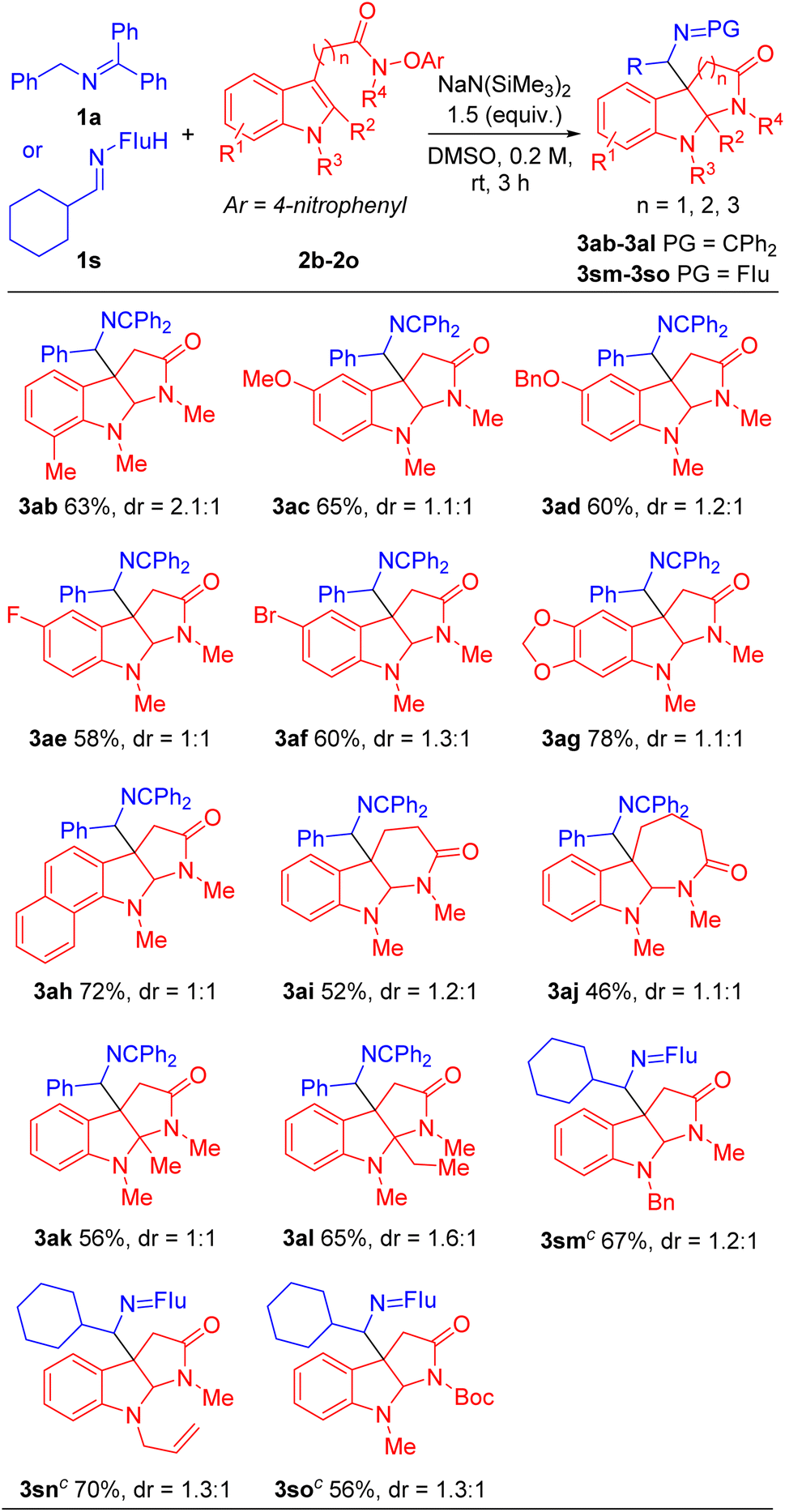
With the optimized conditions in hand (Table 1, entry 5), we next focused our attention on exploring the scope of N-benzyl ketimines. As shown in Table 2, in general, we found that N-benzyl ketimines 1 bearing various substituted N-benzyl or N-alkyl groups provided C3a-substituted pyrroloindolines in moderate to good yields (48–88%) as a mixture of diastereomers. N-Benzyl groups bearing electron-donating substituents 4-OMe (1b) and 3,4-methylenedioxy (1c) generated cyclization products 3ba and 3ca in 56% and 60% yields, respectively. N-Benzyl ketimines decorated with electronegative and electron-withdrawing groups, such as 4-F (1d), 4-Cl (1e), 4-Br (1f), 2,4-di-F (1g) and 4-CF3 (1h) afforded products 3da, 3ea, 3fa, 3ga, 3ha in 80%, 76%, 66%, 63% and 73% yields, respectively. The structures of products 3ga′ and 3ga′′, which were separable by column chromatography and HPLC, were confirmed by X-ray crystallography (CCDC 2293492 and 2293493). The sterically hindered 2-tolyl (1i) and 1-naphthyl (1j) N-benzyl derivatives provided cyclization products 3ia and 3ja in 56% and 68% yields, respectively. Notably, this approach also proved tolerant of medicinally relevant heterocyclic derivatives. N-benzyl groups decorated with 3-pyridyl (1k), 2-furanyl (1l) and 2-thiophenyl (1m) substituents furnished the corresponding products 3ka, 3la and 3ma in 62%, 48% and 56% yields, respectively. Furthermore, switching N-benzyl ketimine with N-(9H-fluoren-9-yl)alkylanimine, we could expand the scope of imine substrates to those with N-alkyl groups. Methyl (1n), i-Pr (1o), isobutyl (1p), cyclobutyl (1q), cyclopentyl (1r) and cyclohexyl (1s) were also suitable substituents, giving the corresponding products (3na-3sa) in 72–88% yields, respectively. Meanwhile, when imines bearing tetraphenyl ketimine and alpha-substituted benzyl amines were employed, the radical cyclization/intermolecular coupling did not take place, likely due to increased steric interactions.
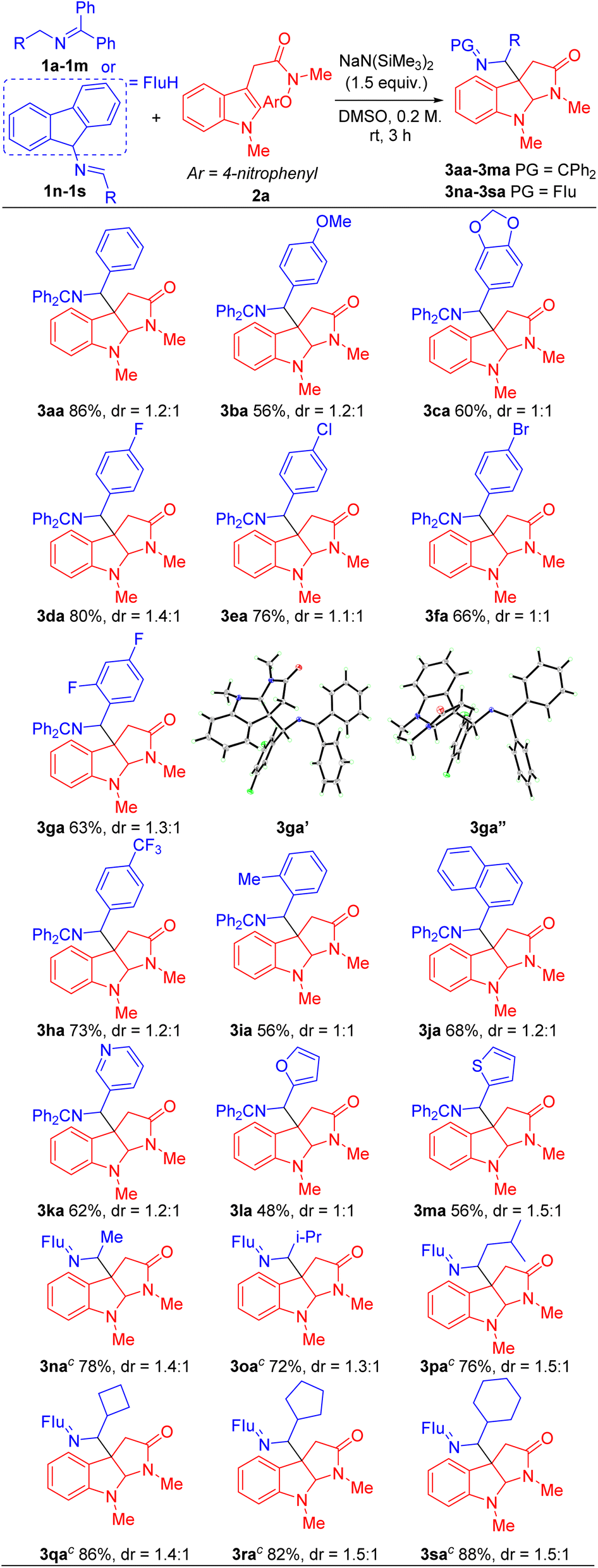
Next, we evaluated the scope of the indole acetamides 2, which were easily synthesized using the method of Wang29,49 (see ESI for details†). A wide range of indole N-aryloxy acetamides bearing various groups were all compatible with our method, generating the pyrroloindoline products in moderate to good yields (46–78%, Table 3). For instance, indole derivatives with electron-donating substituents, such as 7-Me (2b), 5-OMe (2c) and 5-OBn (2d), afforded cyclization products 3ab, 3ac and 3ad in 63%, 65% and 60% yields, respectively. Indole acetamides with electronegative substituents, such as 5-F (2e) and 5-Br (2f), provided the corresponding products 3ae and 3af in 58% and 60% yields. It is noteworthy that indole derivatives with a heterocyclic piperonyl group (2g) and sterically hindered naphthyl group (2h) led to coupling products 3ag and 3ah in 78% and 72% yields, respectively. In addition, we explored a range of indole N-aryloxy acetamide substrates. Gratifyingly, when we extended the alkyl chain of indole acetamides to two or three methylenes, the corresponding six- and seven-member ring products 3ai and 3aj were obtained in 52% and 46% yields, respectively. Furthermore, we introduced steric hindrance at the C2 position of indole substrates [2-methyl (2k) and 2-ethyl (2l)], leading to cyclization products 3ak and 3al in 56% and 65% yields. Next, we use indole derivatives bearing benzyl (2m), allyl (2n) and Boc (2o) substituents, which afforded cyclization products 3sm, 3sn and 3so in 67, 70 and 56% yields, respectively.
To demonstrate the utility and scalability of our cascade radical cyclization/intermolecular coupling reaction, a gram-scale sequential one-pot synthesis and product hydrolysis were conducted. A telescoped gram-scale experiment was performed by employing benzylamine and diphenyl methyl imine in THF at 50 °C for 12 h, followed by solvent removal to afford imine 1a. The unpurified 1a was coupled with indole N-aryloxy acetamide 2a under the standard reaction conditions. The product 3aa was obtained in 74% yield (1.40 g, Scheme 2a). Subsequently, imine hydrolysis of the cyclization product 3aa under mildly acidic conditions furnished the free C3a-methylamine pyrroloindoline derivative 4aa in excellent yield (92%, Scheme 2b).
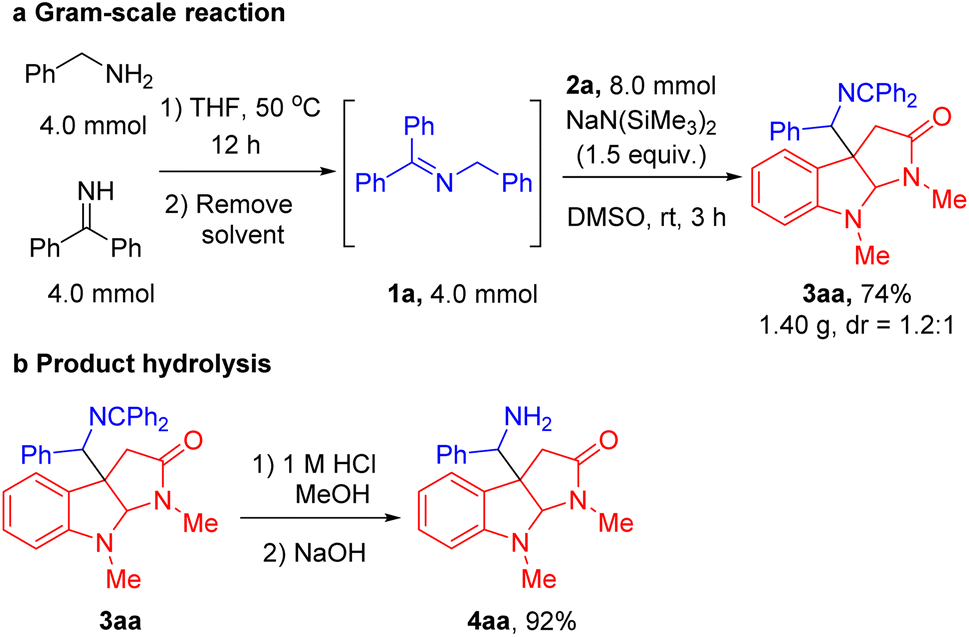 | ||
| Scheme 2 (a) Gram-scale sequential one-pot synthesis. (b) Hydrolysis of the product imine. | ||
Finally, to gain some information on the reaction mechanism, we carried out control experiments. First, the experiment with the addition of 2.0 equiv of radical scavenger 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) was conducted under the standard conditions. However, no desired product 3aa was detected, only affording TEMPO trapping compounds 5aa and 6aa in 70% and 10% yields (Scheme 3a). A control experiment with 2.0 equiv of TEMPO in the absence of N-benzyl ketimine 1a was carried out under the standard conditions, and radical coupling product 5aa was not observed (Scheme 3b). The lack of product formation indicates that NaN(SiMe3)2 is not the active reductant in this chemistry. Together, these results suggest that the reaction proceeds via a radical pathway, supporting the key SET/radical cyclization/coupling pathway proposed in Scheme 1c.
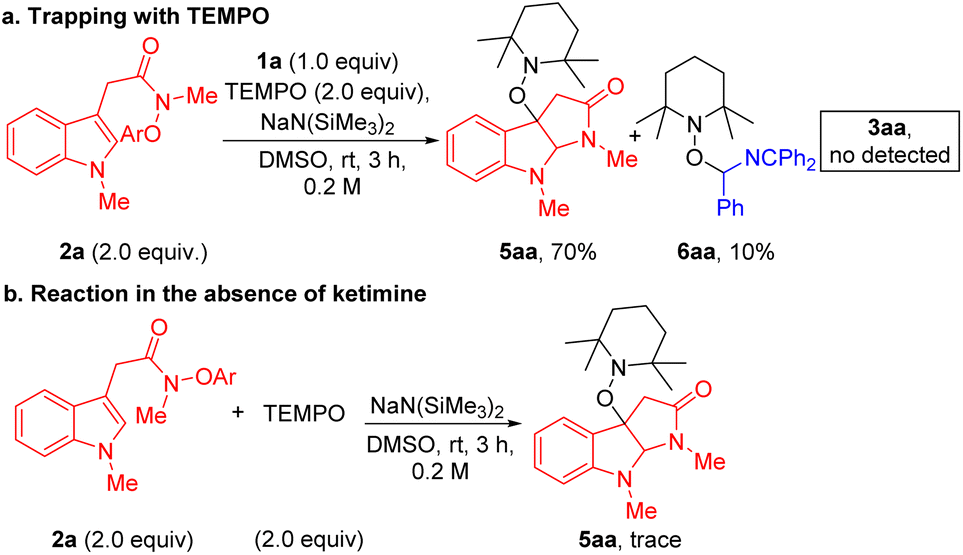 | ||
| Scheme 3 Control experiments. (a) Radical trapping experiment. (b) Reaction in the absence of ketimine. | ||
A plausible mechanism for the reaction is outlined in Scheme 4. Ketimine 1a is deprotonated by the NaN(SiMe3)2 to afford the 2-azaallyl anion 7. Next, SED 7 undergoes an SET process with acetamides 2a to form azaallyl radical 8 and N-centered radical 9. The amidyl radical 9 initiates a radical cyclization to generate C3a-pyrroloindoline radical 10. Finally, pyrroloindoline radical 10 couples with 2-azaallyl radical 8 to obtain coupling product 3aa.
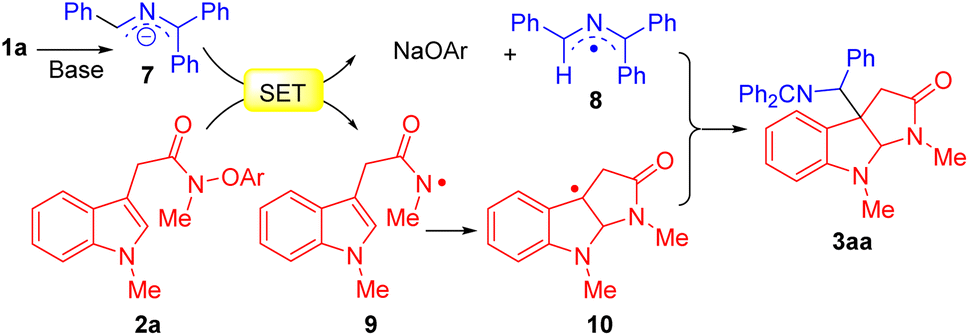 | ||
| Scheme 4 Proposed reaction pathway. | ||
Conclusions
In summary, we have developed a unique strategy for constructing functionalized pyrroloindolines in a single synthetic step. Unlike many previous reports, which generally involve transition-metal catalysts or photoredox catalysts, this chemistry utilizes readily generated SED, 2-azaallyl anions. In this transformation, the tandem SET/radical cyclization/intermolecular coupling between 2-azaallyl anions and indole N-aryloxy acetamides provides the functionalized pyrroloindolines related to biologically active compounds by simple combination of base and DMSO at room temperature. A gram-scale sequential one-pot synthesis and hydrolysis reaction demonstrate the potential synthetic utility and scalability of this approach. It is noteworthy that this method includes a multistep tandem reaction with a rapid increase in molecular complexity. The sustainability of this method enhances its potential utility in the pharmaceutical industry.50Data availability
All experimental data, procedures for data analysis, and pertinent data sets are provided in the ESI.†Author contributions
X. Y. and Y. J. conceived of the project. X. Y. and P. J. W. designed the experiments. Y. J., D. L., L. Z., C. Q., H. L. and H. Y. performed the research. X. Y. and P. J. W. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the National Key R&D Program of China (2019YFE0109200), NSFC (22361050), NSF of Yunnan (202207AA110007, 202207AB110002), Ling-Jun Scholars Yunnan Province (202005AB160003), Program for Xingdian Talents (Yun-Ling Scholars), IRTSTYN and Yunnan University Professional Degree Graduate Student Practice and Innovation Fund (H. L.). P. J. W. thanks the US National Science Foundation (CHE-2154593) for financial support.Notes and references
- M. E. Welsch, S. A. Snyder and B. R. Stockwell, Curr. Opin. Cell Biol., 2010, 14, 347–361 CrossRef CAS PubMed.
- C. Lamberth, Pest Manage. Sci., 2013, 69, 1106–1114 CrossRef CAS PubMed.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- R. V. Patel, Y.-S. Keum and S. W. Park, Eur. J. Med. Chem., 2015, 97, 649–663 CrossRef CAS PubMed.
- K. Chaudhari, S. Surana, P. Jain and H. M. Patel, Eur. J. Med. Chem., 2016, 124, 160–185 CrossRef CAS PubMed.
- R. Kaur, L. Dahiya and M. Kumar, Eur. J. Med. Chem., 2017, 141, 473–505 CrossRef CAS PubMed.
- J. Davies, T. D. Svejstrup, D. F. Reina, N. S. Sheikhb and D. Leonori, J. Am. Chem. Soc., 2016, 138, 8092–8095 CrossRef CAS PubMed.
- A. Huang, J. J. Kodanko and L. E. Overman, J. Am. Chem. Soc., 2004, 126, 14043–14053 CrossRef CAS PubMed.
- S. Tadano, Y. Mukaeda and H. Ishikawa, Angew. Chem., Int. Ed., 2013, 52, 7990–7994 CrossRef CAS PubMed.
- B. D. Joshi and J. D. Chisholm, Tetrahedron Lett., 2021, 77, 153256 CrossRef CAS PubMed.
- J. J. Kodanko and L. E. Overman, Angew. Chem., Int. Ed., 2003, 42, 2528–2531 CrossRef CAS PubMed.
- A. Steven and L. E. Overman, Angew. Chem., Int. Ed., 2007, 46, 5488–5508 CrossRef CAS PubMed.
- P. Ruiz-Sanchis, S. A. Savina, F. Albericio and M. Alvarez, Chem.–Euro. J., 2011, 17, 1388–1408 CrossRef CAS PubMed.
- C. Sun, W. Tian, Z. Lin and X. Qu, Nat. Prod. Rep., 2022, 39, 1721–1765 RSC.
- S. Zhu and D. W. MacMillan, J. Am. Chem. Soc., 2012, 134, 10815–10818 CrossRef CAS PubMed.
- R. Jiang, L. Ding, C. Zheng and S.-L. You, Science, 2021, 371, 380–386 CrossRef CAS PubMed.
- J. R. Chen, X. Q. Hu, L. Q. Lu and W. J. Xiao, Chem. Soc. Rev., 2016, 45, 2044–2056 RSC.
- S. Fukuzumi and K. Ohkubo, Org. Biomol. Chem., 2014, 12, 6059–6071 RSC.
- L. Furst, J. M. Narayanam and C. R. Stephenson, Angew. Chem., Int. Ed., 2011, 50, 9655–9659 CrossRef CAS PubMed.
- M. N. Hopkinson, B. Sahoo, J. L. Li and F. Glorius, Chem.–Euro. J., 2014, 20, 3874–3886 CrossRef CAS PubMed.
- M. D. Karkas, J. A. Porco, Jr. and C. R. Stephenson, Chem. Rev., 2016, 116, 9683–9747 CrossRef CAS PubMed.
- D. A. Nicewicz and T. M. Nguyen, ACS Catal., 2013, 4, 355–360 CrossRef.
- C. K. Prier, D. A. Rankic and D. W. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- J. Xuan and W. J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef CAS PubMed.
- T. P. Yoon, ACS Catal., 2013, 3, 895–902 CrossRef CAS PubMed.
- D. P. Hari and B. Konig, Chem. Commun., 2014, 50, 6688–6699 RSC.
- E. C. Gentry, L. J. Rono, M. E. Hale, R. Matsuura and R. R. Knowles, J. Am. Chem. Soc., 2018, 140, 3394–3402 CrossRef CAS PubMed.
- K. Wu, Y. Du, Z. Wei and T. Wang, Chem. Commun., 2018, 54, 7443–7446 RSC.
- J. P. Barham, G. Coulthard, K. J. Emery, E. Doni, F. Cumine, G. Nocera, M. P. John, E. Leonard, A. Berlouis, T. McGuire, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2016, 138, 7402–7410 CrossRef CAS PubMed.
- J. P. Barham, S. E. Dalton, M. Allison, G. Nocera, A. Young, M. P. John, T. McGuire, S. Campos, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2018, 140, 11510–11518 CrossRef CAS PubMed.
- J. A. Murphy, J. Org. Chem., 2014, 79, 3731–3746 CrossRef CAS PubMed.
- Y. Lei, J. Yang, R. Qi, S. Wang, R. Wang and Z. Xu, Chem. Commun., 2018, 54, 11881–11884 RSC.
- S. Tang, X. Zhang, J. Sun, D. Niu and J. J. Chruma, Chem. Rev., 2018, 118, 10393–10457 CrossRef CAS PubMed.
- Q. Wang, M. Poznik, M. Li, P. J. Walsh and J. J. Chruma, Adv. Synth. Catal., 2018, 360, 2854–2868 CrossRef CAS.
- B. Wang, M. Li, G. Gao, A. Sanz-Vidal, B. Zheng and P. J. Walsh, J. Org. Chem., 2022, 87, 8099–8103 CrossRef CAS PubMed.
- G. B. Panetti, P. J. Carroll, M. R. Gau, B. C. Manor, E. J. Schelter and P. J. Walsh, Chem. Sci., 2021, 12, 4405–4410 RSC.
- G. Deng, M. Li, K. Yu, C. Liu, Z. Liu, S. Duan, W. Chen, X. Yang, H. Zhang and P. J. Walsh, Angew. Chem., Int. Ed., 2019, 58, 2826–2830 CrossRef CAS PubMed.
- K. Yu, M. Li, G. Deng, C. Liu, J. Wang, Z. Liu, H. Zhang, X. Yang and P. J. Walsh, Adv. Synth. Catal., 2019, 361, 4354–4359 CrossRef CAS.
- Q. Zi, M. Li, J. Cong, G. Deng, S. Duan, M. Yin, W. Chen, H. Jing, X. Yang and P. J. Walsh, Org. Lett., 2022, 24, 1786–1790 CrossRef CAS PubMed.
- M. Li, S. Berritt, L. Matuszewski, G. Deng, A. Pascual-Escudero, G. B. Panetti, M. Poznik, X. Yang, J. J. Chruma and P. J. Walsh, J. Am. Chem. Soc., 2017, 139, 16327–16333 CrossRef CAS PubMed.
- M. Li, O. Gutierrez, S. Berritt, A. Pascual-Escudero, A. Yesilcimen, X. Yang, J. Adrio, G. Huang, E. Nakamaru-Ogiso, M. C. Kozlowski and P. J. Walsh, Nat. Chem., 2017, 9, 997–1004 CrossRef CAS PubMed.
- Z. Liu, M. Li, G. Deng, W. Wei, P. Feng, Q. Zi, T. Li, H. Zhang, X. Yang and P. J. Walsh, Chem. Sci., 2020, 11, 7619–7625 RSC.
- G. Deng, S. Duan, J. Wang, Z. Chen, T. Liu, W. Chen, H. Zhang, X. Yang and P. J. Walsh, Nat. Commun., 2021, 12, 3860 CrossRef CAS PubMed.
- S. Duan, G. Deng, Y. Zi, X. Wu, X. Tian, Z. Liu, M. Li, H. Zhang, X. Yang and P. J. Walsh, Chem. Sci., 2021, 12, 6406–6412 RSC.
- S. Duan, Y. Zi, L. Wang, J. Cong, W. Chen, M. Li, H. Zhang, X. Yang and P. J. Walsh, Chem. Sci., 2022, 13, 3740–3747 RSC.
- L. Zhang, Z. Liu, X. Tian, Y. Zi, S. Duan, Y. Fang, W. Chen, H. Jing, L. Yang and X. Yang, Org. Lett., 2021, 23, 1714–1719 CrossRef CAS PubMed.
- Y. Jiang, D. Liu, M. E. Rotella, G. Deng, Z. Liu, W. Chen, H. Zhang, M. C. Kozlowski, P. J. Walsh and X. Yang, J. Am. Chem. Soc., 2023, 145, 16045–16057 CrossRef CAS PubMed.
- K. Wu, Y. Du and T. Wang, Org. Lett., 2017, 19, 5669–5672 CrossRef CAS PubMed.
- C. E. Garrett and K. Prasad, Adv. Synth. Catal., 2004, 346, 889–900 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2293492 and 2293493. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc05210a |
| This journal is © The Royal Society of Chemistry 2024 |