DOI:
10.1039/D3SC05403A
(Edge Article)
Chem. Sci., 2024,
15, 2211-2220
Untangling ancillary ligand donation versus locus of oxidation effects on metal nitride reactivity†
Received
12th October 2023
, Accepted 1st January 2024
First published on 2nd January 2024
Abstract
We detail the relative role of ancillary ligand electron-donating ability in comparison to the locus of oxidation (either metal or ligand) on the electrophilic reactivity of a series of oxidized Mn salen nitride complexes. The electron-donating ability of the ancillary salen ligand was tuned via the para-phenolate substituent (R = CF3, H, tBu, OiPr, NMe2, NEt2) in order to have minimal effect on the geometry at the metal center. Through a suite of experimental (electrochemistry, electron paramagnetic resonance spectroscopy, UV-vis-NIR spectroscopy) and theoretical (density functional theory) techniques, we have demonstrated that metal-based oxidation to [MnVI(SalR)N]+ occurs for R = CF3, H, tBu, OiPr, while ligand radical formation to [MnV(SalR)N]+˙ occurs with the more electron-donating substituents R = NMe2, NEt2. We next investigated the reactivity of the electrophilic nitride with triarylphosphines to form a MnIV phosphoraneiminato adduct and determined that the rate of reaction decreases as the electron-donating ability of the salen para-phenolate substituent is increased. Using a Hammett plot, we find a break in the Hammett relation between R = OiPr and R = NMe2, without a change in mechanism, consistent with the locus of oxidation exhibiting a dominant effect on nitride reactivity, and not the overall donating ability of the ancillary salen ligand. This work differentiates between the subtle and interconnected effects of ancillary ligand electron-donating ability, and locus of oxidation, on electrophilic nitride reactivity.
Introduction
Transition metal complexes featuring terminal nitride (N3−) ligands have attracted significant interest due to their potential role in nitrogen fixation,1–4 stoichiometric nitrene transfer reactions,5–7 catalytic applications,8,9 and their utility in materials chemistry.10–12 In the context of nitrogen fixation, the electronic structure and reactivity of bioinspired Fe nitride complexes have been extensively studied,13–27 along with recent investigations of synthetic FeMo cofactors that have provided insight on the stability and reactivity of activated Fe–N species.28,29 The reactivity of terminal nitride complexes is often rationalized based on the nucleophilic or electrophilic nature of the nitride ligand, which is influenced by the specific metal, oxidation state, and the nature of the ancillary ligands.30–32 Notably, late transition-metal nitrides can exist as isolated or transient intermediates, exhibiting electrophilic reactivity through low energy M![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N π* antibonding orbitals.33–45 Conversely, early transition-metal nitrides are generally more stable46–48 and in some cases demonstrate nucleophilic reactivity via filled MN π orbitals or the nitride lone pair.49–53 Certain metal nitride complexes have been reported to exhibit ambiphilic reactivity, highlighting that subtle modifications to the coordination environment and/or redox changes can enable diverse reactivity.35,54–56 Previous studies by Mayer et al.57,58 and Lau et al.59 have elucidated how changes in ancillary ligands can impact nitride reactivity, work by Holland et al. demonstrated that oxidation of both the metal and ancillary ligand (from amide to nitroxide) led to a change from nucleophilic to electrophilic nitride reactivity,60 and a recent study by Burger and co-workers showed that oxidation of an Ir nitride complex resulted in insertion of the nitride into an aromatic C–C bond of ferrocene.61 Further investigation into the effects of subtle changes in electronic structure on nitride reactivity will yield valuable insight for new reactivity applications.
N π* antibonding orbitals.33–45 Conversely, early transition-metal nitrides are generally more stable46–48 and in some cases demonstrate nucleophilic reactivity via filled MN π orbitals or the nitride lone pair.49–53 Certain metal nitride complexes have been reported to exhibit ambiphilic reactivity, highlighting that subtle modifications to the coordination environment and/or redox changes can enable diverse reactivity.35,54–56 Previous studies by Mayer et al.57,58 and Lau et al.59 have elucidated how changes in ancillary ligands can impact nitride reactivity, work by Holland et al. demonstrated that oxidation of both the metal and ancillary ligand (from amide to nitroxide) led to a change from nucleophilic to electrophilic nitride reactivity,60 and a recent study by Burger and co-workers showed that oxidation of an Ir nitride complex resulted in insertion of the nitride into an aromatic C–C bond of ferrocene.61 Further investigation into the effects of subtle changes in electronic structure on nitride reactivity will yield valuable insight for new reactivity applications.
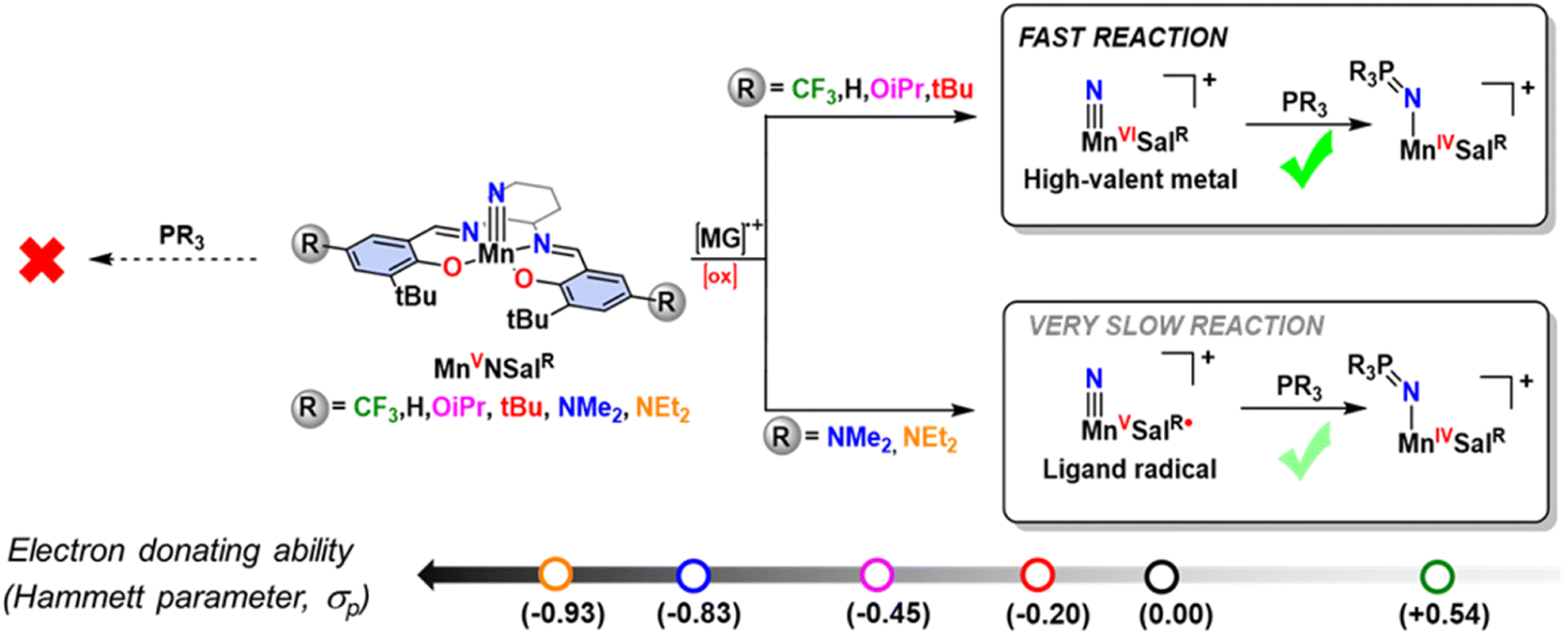
We have shown that the electronic structure of oxidized nitridomanganese(V) salen complexes (where “salen” represents N2O2 bis-phenolate bis-Schiff-base ligands) can be tuned through the adjustment of the electron-donating ability of the para-R phenolate substituents, while preserving the geometry at the metal center.62,63 Salen ligands exhibit redox-active behavior, enabling oxidation or reduction at the ligand in competition with the metal.64–70 Upon oxidation, we and others observed the formation of a MnVI salen nitride, leading to the activation of the MnN bond, and rapid nitride homocoupling to generate N2, even at low temperatures.62,71,72 Intriguingly, the introduction of electron-donating substituents (R = NMe2) at the para position of the salen ligand leads to the formation of a ligand radical, which is stable in solution at 298 K.62 Additionally, a separate study has reported an isolable MnVI nitride complex with a tetraamido macrocyclic ligand (TAML), which exhibits relatively slow nitride homocoupling at 298 K.54 Recent reports have utilized Mn salen nitrides to explore the hydrogen atom bond dissociation free energy of associated imido complexes,73 the generation of ammonia through proton-coupled electron transfer10,74,75 and their application as catalysts for ammonia oxidation.76 Intrigued by the difference in behaviour of the Mn salen nitride complexes upon oxidation, we endeavoured to probe the reactivity difference for an expanded series of oxidized nitridomanganese(V)salen complexes with phosphines and to characterize the resulting phosphoraneiminato adduct. In our previous work with a smaller series of oxidized Cr analogues, we found that while the Cr(VI) nitride complexes reacted with phosphine, a ligand radical derivative did not, and it was unclear whether the locus of oxidation or the electron-donating ability of the salen ligand was primarily responsible for this abrupt reactivity change.77 Factors such as the nature of the ancillary ligands, steric effects, metal oxidation state and overall charge of the complex can influence the analogous oxygen atom transfer reaction efficiency of oxo ligands with triaryl phosphines.78–83 Two-electron nitrogen transfer reactions of metal nitride complexes with phosphines to form the phosphoraneiminato moiety (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PR3) are well known,30 and in the majority of cases the phosphine acts as a nucleophile;25,55,84–86 in some exceptional instances nitride lone pair donation into the phosphine LUMO (generally, a P–C σ*) gives rise to a dual nature transition state.87 Herein, we questioned whether the locus of oxidation, or alternatively the electron-donating ability of the salen ligand, is the major driving factor in the reactivity differences for a series of [Mn(SalR)N]+ complexes with triarylphosphines (Scheme 1), and how the change in locus of oxidation, from metal to ligand, would map against the ligand Hammett parameter, and thus donating ability, of the salen ligand.
PR3) are well known,30 and in the majority of cases the phosphine acts as a nucleophile;25,55,84–86 in some exceptional instances nitride lone pair donation into the phosphine LUMO (generally, a P–C σ*) gives rise to a dual nature transition state.87 Herein, we questioned whether the locus of oxidation, or alternatively the electron-donating ability of the salen ligand, is the major driving factor in the reactivity differences for a series of [Mn(SalR)N]+ complexes with triarylphosphines (Scheme 1), and how the change in locus of oxidation, from metal to ligand, would map against the ligand Hammett parameter, and thus donating ability, of the salen ligand.
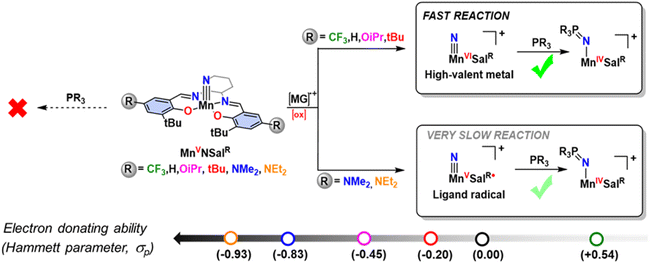 |
| | Scheme 1 Locus of oxidation dependent electrophilic reactivity of a series of oxidized Mn-Salen-Nitrides incorporating different salen ligand para-ring substituents. [MG]+˙ = magic green chemical oxidant. | |
Results and discussion
Synthesis and characterization
To investigate the role of the ligand electronics and locus of oxidation on nitride reactivity, six nitridomagnanese(V) salen complexes were prepared with a range of ancillary ligand electron-donating ability. Mn(SalCF3)N, Mn(SaltBu)N, Mn(SalOiPr)N and Mn(SalNMe2)N have been previously synthesized.62,63 For the two new complexes, unsubstituted Mn(SalH)N was prepared by the reaction of corresponding manganese chloride salt with NH4OH as reported by Carreira and coworkers (Scheme S1 and Fig. S1†).7 On the other hand, Mn(SalNEt2)N was prepared by the photolysis of the precursor manganese–azido complex to avoid a difficult purification procedure (Schemes S2, S3 and Fig. S2†). X-ray quality crystals of Mn(SalH)N were obtained by layering hexanes over a concentrated dichloromethane (CH2Cl2) solution (Fig. S3 and Table S1†). The expected pseudo square pyramidal geometry with a nitride Mn–N bond distance of 1.517 Å was observed, analogous to other reported Mn salen nitrides.62,63
Electrochemistry
The redox properties of the two new complexes were analyzed by cyclic voltammetry (CV) experiments at 298 K under a N2 atmosphere. The voltammogram of Mn(SalH)N displayed a partially reversible redox process with an E1/2 = 0.28 V (vs. Fc+/Fc), attributed to metal-based oxidation (vide infra) (Fig. 1A and S4A†). The partial reversibility at 298 K is indicative of nitride homocoupling observed in analogous Mn(VI) complexes.62,71 In contrast, Mn(SalNEt2)N showed two overlapping quasi-reversible redox processes which were resolved by differential pulse voltammetry (DPV) to afford redox potentials at E1/21 = −0.15 V and E1/22 = −0.01 V respectively (Fig. 2B and S4B†). The electrochemistry data is consistent with two successive oxidations of the phenolate moieties at low potentials for the most electron-donating substituents, as observed for both Mn(SalNMe2)N and Cr(SalNMe2)N.62,77 A small ΔEox (E1/22 − E1/21) value of 140 mV indicates minimal electronic coupling between the redox-active phenolates, supporting a localized ligand radical for [MnV(SalNEt2)N]˙+ upon one-electron oxidation (Table 1).62 We investigated the degree of disproportionation of the mono-oxidized form at 193 K (conditions relevant to the reactivity studies, see ESI† for details), calculating that only ca. 1.5% [MnV(SalNEt2)N]2+ and 1.5% MnV(SalNEt2)N would be present. In addition, the change in E1/2 values for the full series of Mn(SalR)N complexes correlates with the Hammett parameter of the para-ring substituents (Table 1). We note that the Hammett parameter for the R = NEt2 substituent was estimated to be σp = −0.93 on the basis of previous reports directly comparing this substituent to R = NMe2 (ref. 88 and 89) and the consensus value for the latter of σp = −0.83.90 The more negative σp for R = NEt2 in comparison to R = NMe2 is in agreement with our electrochemistry data.
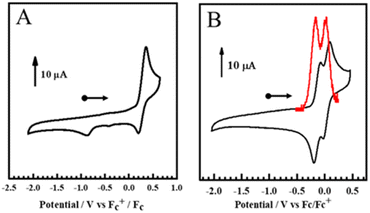 |
| | Fig. 1 (A) Cyclic Voltammogram of Mn(SalH)N and (B) Mn(SalNEt2)N. Conditions: 1.0 mM complex; 0.1 M nBu4NClO4; scan rate: 100 mV s−1; T = 298 K; CH2Cl2. DPV curve in red. | |
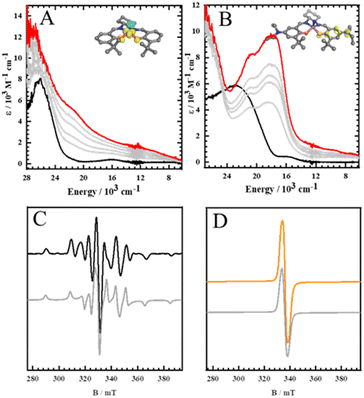 |
| | Fig. 2 Oxidation titration data for (A) Mn(SalH)N and (B) Mn(SalNEt2)N monitored by UV-vis-NIR spectroscopy. Black: neutral; red: oxidized; intermediate grey lines were measured during the oxidation titrations with [N(C6H3Br2)3]+˙[SbF6]−, insets: spin density plot of the oxidized complexes. Conditions: CH2Cl2, 0.1 mM, 193 K (see Experimental section for calculation details) X-band EPR spectra of frozen (C) [Mn(SalH)N]+ and (D) [Mn(SalNEt2)N]+ samples respectively. Simulated spectra are represented by grey lines. Conditions: frequency = 9.38 GHz; power = 2.0 mW; modulation frequency = 100 kHz; modulation amplitude = 0.6 mT; CH2Cl2, T = 20 K. | |
Table 1
E
1/2 of the Mn(SalR)N complexes (vs. Fc+/Fc) and correlation with Hammett parameter (σp). Peak-to-peak separation given in parenthesis [a = ref. 62. b = ref. 63. c = ref. 90]
| Compound |
E
1/2
1 (V) |
E
1/2
2 (V) |
σ
cp
|
|
Mn(SalCF3)N
|
0.42 |
|
0.54 |
|
Mn(SalH)N
|
0.28 (0.16)
|
|
0.00
|
|
Mn(SaltBu)N
|
0.23a |
|
−0.20 |
|
Mn(SalOiPr)N
|
0.21b |
|
−0.45 |
|
Mn(SalNMe2)N
|
−0.10a |
0.05 |
−0.83 |
|
Mn(SalNEt2)N
|
−0.15 (0.14)
|
−0.01 (0.14)
|
−0.93
|
Spectroscopic characterization
The absorption spectra of the neutral Mn(SalR)N complexes are typical of a low spin d2 square pyramidal complex.62,63 Upon one-electron oxidation using an equivalent of the [N(C5H3Br2)3][SbF6] oxidant (Magic Green) at 193 K, a broad low intensity near infrared (NIR) band is observed at 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1 (ε = ∼1200 M−1 cm−1) for [Mn(SalH)N]+, similar to the observed ligand to metal charge transfer (LMCT) transition for [Mn(SaltBu)N]+ and [Mn(SalCF3)N]+, indicating metal-based oxidation (Fig. 2A and S5†).62[Mn(SalH)N]+ was analyzed by electron paramagnetic resonance (EPR) spectroscopy at 20 K, and the EPR spectrum shows a characteristic axial splitting pattern for a d1 metal ion (d1xy) with hyperfine coupling to the 55Mn (I = 5/2) nucleus (gzz = 1.986, gxx = gyy = 1.997; Azz = 528 MHz, Axx = Ayy = 179 MHz), confirming that one-electron oxidation of Mn(SalH)N is metal-based (Fig. 2C). In contrast, for Mn(SalNEt2)N, low temperature oxidation affords a unique visible-NIR feature when compared to [Mn(SalH)N]+, closely resembling that of [Mn(SalNMe2)N]+, indicating ligand-based oxidation (Fig. 2B). The intense envelope of transitions between 15000 cm−1 and 22000 cm−1 are in agreement with ligand radical formation.62,77,91 The EPR spectrum of a frozen solution of [Mn(SalNEt2)N]+ exhibits an isotropic signal centered at 2.003, in agreement with ligand-based oxidation (Fig. 2D). Theoretical calculations on the oxidized complexes match the experimental results, predicting a MnVI (d1xy) ground state for [Mn(SalH)N]+ (Fig. S6B†) and a localized ligand radical for [MnV(SalNEt2)N]˙+ as evident by the spin density plots (Fig. 2A and B insets).
000 cm−1 (ε = ∼1200 M−1 cm−1) for [Mn(SalH)N]+, similar to the observed ligand to metal charge transfer (LMCT) transition for [Mn(SaltBu)N]+ and [Mn(SalCF3)N]+, indicating metal-based oxidation (Fig. 2A and S5†).62[Mn(SalH)N]+ was analyzed by electron paramagnetic resonance (EPR) spectroscopy at 20 K, and the EPR spectrum shows a characteristic axial splitting pattern for a d1 metal ion (d1xy) with hyperfine coupling to the 55Mn (I = 5/2) nucleus (gzz = 1.986, gxx = gyy = 1.997; Azz = 528 MHz, Axx = Ayy = 179 MHz), confirming that one-electron oxidation of Mn(SalH)N is metal-based (Fig. 2C). In contrast, for Mn(SalNEt2)N, low temperature oxidation affords a unique visible-NIR feature when compared to [Mn(SalH)N]+, closely resembling that of [Mn(SalNMe2)N]+, indicating ligand-based oxidation (Fig. 2B). The intense envelope of transitions between 15000 cm−1 and 22000 cm−1 are in agreement with ligand radical formation.62,77,91 The EPR spectrum of a frozen solution of [Mn(SalNEt2)N]+ exhibits an isotropic signal centered at 2.003, in agreement with ligand-based oxidation (Fig. 2D). Theoretical calculations on the oxidized complexes match the experimental results, predicting a MnVI (d1xy) ground state for [Mn(SalH)N]+ (Fig. S6B†) and a localized ligand radical for [MnV(SalNEt2)N]˙+ as evident by the spin density plots (Fig. 2A and B insets).
Electrophilic reactivity
We next investigated the change in electrophilic reactivity of the full [Mn(SalR)N]+ series (R CF3, H, tBu, OiPr, NMe2, NEt2) via reaction with triarylphosphines. Triphenylphosphine (PPh3) was initially chosen to probe potential changes in electrophilic reactivity at the nitride. As expected, the neutral Mn(SalR)N complexes did not react with PPh3, as shown by UV-vis-NIR, ESI-MS, and 31P{H} NMR (Fig. S7–S9†). However, the oxidized [MnVI(SalR)N]+ complexes (RCF3, tBu) react immediately with PPh3 to form a new species as shown in the low temperature UV-vis-NIR spectroscopy. In contrast, when one equiv. of PPh3 was added to the ligand radical species (R = NMe2) at 193 K, a very slow reaction is observed (Fig. S10 and S11†). Unfortunately, the reaction was too fast (completion <1 s) for the MnVI derivatives to obtain reliable reaction rates under our conditions (Fig. S12†). We therefore investigated the less nucleophilic tris-(trifluoromethylphenyl)phosphine (p-CF3Ph)3P to ascertain reactivity differences in this system (Fig. S13†). In addition, we included the full series of Mn salen nitrides with para-ring substituents of varying electron-donating ability to provide a more robust Hammett analysis. As expected, the neutral complexes do not react with (p-CF3Ph)3P with no change observed in the UV-vis-NIR spectra (Fig. S14†) and 31P NMR (Fig. S15†). However, similarly to the reactivity with PPh3, the oxidized [MnVI(SalR)N]+ complexes (RCF3, H, tBu, OiPr) react quickly with (p-CF3Ph)3P to form a new species as monitored by UV-vis-NIR measurements at 193 K. Spectral changes, including isosbestic points, indicate clean conversion to a new species for all MnVI derivatives upon (p-CF3Ph)3P addition (Fig. 3A, B and S16†).
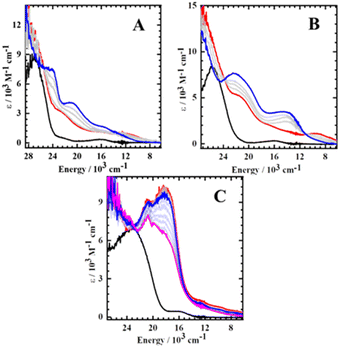 |
| | Fig. 3 UV-vis-NIR spectroscopic monitoring of the reaction of (A) [Mn(SalCF3)N]+; (B) [Mn(SaltBu)N]+; (C) [Mn(SalNMe2)N]+ with (p-CF3Ph)3P. Black: neutral; red: oxidized; blue: one equivalent of (p-CF3Ph)3P added to oxidized complexes. Intermediate grey lines measured during aliquot addition of (p-CF3Ph)3P. For (C) the pink spectrum is 2 h after (p-CF3Ph)3P addition, intermediate opaque blue lines are scans taken within two hours. Conditions: CH2Cl2, 0.1 mM complex, 193 K. See Fig. S16† for other derivatives. | |
The new species (R = tBu) shows minimal decomposition at 193 K over two hours, but decays to a new species upon warming to 298 K (Fig. S17B and S18†). Analysis of the solution at 298 K indicated complete N-atom transfer to form the free iminophosphorane, characterized by the presence of free iminophosphorane [(p-CF3Ph)3PNH] at ∼37 ppm by 31P{H} NMR with no peak corresponding to unreacted phosphine (Fig. S15 and S19A†). The characterization data is in accord with an independently made sample of the iminophosphorane (Fig. S20†). When 50% 15N labelled nitride complex (R = tBu) was used, a small N–P coupling (1JN–P = 2.8 Hz) was observed, with similar 1JN–P constants reported for related compounds (Fig. S20A†).77,92,93 A peak at m/z = 482 in positive mode ESI-MS was observed for all derivatives, corresponding to the protonated iminophosphorane [(p-CF3Ph)3PNH2]+ (Fig. S19B†). Using 50% 15N labelled nitride complex results in a change in the isotopic pattern consistent with incorporation of the nitride nitrogen (Fig. S19C and D†). In contrast, the reaction of the ligand radical derivatives (R = NMe2 and NEt2) with one equivalent of (p-CF3Ph)3P is much slower in comparison to the MnVI analogues (Fig. 3C and S16B†). However, clean conversion to a new species is evident by the presence of the same isosbestic point over a two-hour period. 31P{H} NMR and ESI-MS analysis after warming to 298 K affords the same iminophosphorane product (δ = 37.1 ppm, m/z = 482) (Fig. S21†) as the MnVI analogues, indicating complete N-atom transfer even for the ligand radical derivatives. Similar results were also observed for the reaction with PPh3 (Fig. S22 and S23†). We hypothesize that upon warming the solution to 298 K the rate of adduct formation is increased, and in addition, complete N-atom transfer occurs. While complete N-atom transfer was observed for both PPh3 and (p-CF3Ph)3P, unlike PPh3, the reaction rates with (p-CF3Ph)3P were amenable to kinetic analysis.
Kinetic analysis
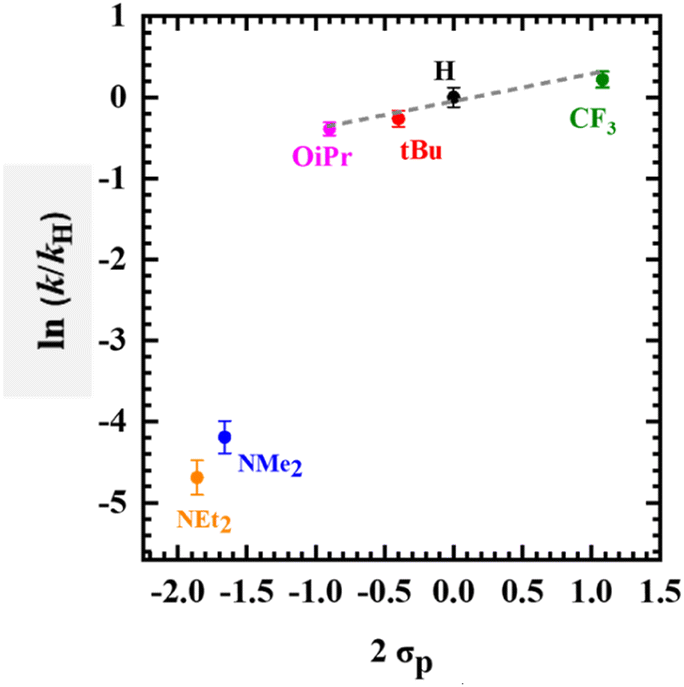
The Hammett parameter (σp) quantifies the electron-donating ability of a para-ring substituent and is commonly compared to a rate constant to gain a mechanistic understanding of a particular reaction.13,87,94 Herein, if the electron-donating capacity of the para-ring substituents in [Mn(SalR)N]+ is the main contributor to the nitride reactivity with (p-CF3Ph)3P, a linear correlation will be expected for all derivatives (RCF3, H, tBu, OiPr, NMe2, NEt2), assuming the reaction mechanism remains unchanged.95,96 However, a substantial deviation from linearity could indicate that the locus of oxidation (metal or ligand), and not the electron-donating ability of a para-ring substituent, plays a more significant role in the reaction, in the absence of a change in reaction mechanism. Kinetic measurements under pseudo-first order conditions of p-(CF3Ph)3P were conducted at 193 K and a substantial difference in reaction rate is observed across all para-ring substituents (Fig. S24–S29†), with the RCF3 substituted complex reacting fastest, and R = NEt2 slowest, in line with the relative electron-donating ability of the para-ring substituents, and thus suggesting that the nitride is acting as an electrophile (Fig. S30†). Interestingly, a clear reaction rate difference was observed for the high-valent MnVI complexes (RCF3, H, tBu, OiPr) in comparison to the ligand radical derivatives (R = NMe2, NEt2), with the MnVI complexes reacting much faster (Fig. S30†). A Hammett plot was obtained by graphing ln(k/kH) versus σp (Fig. 4) where k is the observed pseudo first-order rate constant for each Mn-derivative and kH is the observed pseudo first-order rate constant for [Mn(SalH)N]+.87 A clear break in the plot is observed between the high-valent [MnVI(SalR)N]+ (RCF3, H, tBu, OiPr) complexes and ligand radical [MnV(SalR)N]˙+ (R = NMe2, NEt2) derivatives. For the MnVI derivatives, the reaction rates show a linear correlation (R2 = 0.95) with the Hammett parameter (2σp)87 with a modest reaction constant of r = 0.31, indicating that the [MnVI(SalR)N]+ complexes exhibit electrophilic reactivity, and in addition, that the electron-donating ability of the para-R substituent plays a relatively minor role in influencing the rate of the reaction.87 A linear relationship between phosphine concentration and reaction rate was observed for [MnVI(SaltBu)N]+ (Fig. S31 and 32†), indicating that phosphine is involved in the rate law.
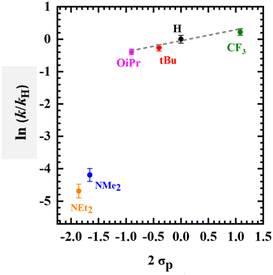 |
| | Fig. 4 Hammett plot using kobs for the reaction between oxidized [Mn(SalR)N]+ and (p-CF3Ph)3P (R = CF3, H, tBu, OiPr, NMe2, NEt2). The x-axis uses 2 σp due to the presence of two R groups in the complex. Note: linear correlation was observed among high-valent MnVI complexes (R2 = 0.95, ρ = 0.31). | |
Although only two ligand radical derivatives are included in the Hammett plot, the general trend in reactivity suggests that the nitride still exhibits electrophilic reactivity, however the rate is significantly reduced (Fig. S30†). Indeed, further analysis of the reaction rate for [Mn(SalNMe2)N]+˙ and (p-RPh)3P, where the donating ability of the phosphine was varied (R = OMe, H, F, and CF3), shows a negative slope consistent with nucleophilic attack of phosphine at the electrophilic nitride (Fig. S33†). The possibility that the reaction of the oxidized MnVI/MnVL˙+ derivatives could occur via outer-sphere single electron transfer (SET) oxidation of PR3 followed by radical recombination was considered. However, the redox potential required for the first oxidation of both the phosphines are significantly higher in comparison to the Mn complexes indicating that initial SET is unlikely (Fig. S34†). Overall, the Hammett plot shows that the locus of oxidation (metal or ligand) is the dominant factor that determines the rate of reaction between the nitride and phosphine, with the electron-donating ability of the salen para-R substituent playing a secondary role.
Adduct characterization
Motivated by stability of the adduct formed between [Mn(SaltBu)N]+ and (p-CF3Ph)3P at low temperature in solution (vide supra), we endeavored to characterize this species by low temperature EPR spectroscopy. A sample of the reaction mixture at 193 K was transferred to a pre-cooled EPR tube and immediately frozen for further analysis. EPR spectra (R = CF3, tBu, OiPr, H, NMe2) of the reaction mixture for each of the oxidized complexes and (p-CF3Ph)3P shows features in the low field region at g ∼5.2 and 3.7 consistent with the expected MnIV phosphoraneiminato adduct (Fig. 5).97,98 For the R = NMe2 derivative, the relatively weak MnIV features in comparison to the strong signal at g = 2.003 is consistent with the slow reactivity for the ligand radical complex as measured by UV-vis-NIR spectroscopy (Fig. 3C). For R = tBu, additional features indicating unreacted oxidized Mn nitride complex (g ∼2.0) and a minor MnIII species at g ∼7.3,97,99 highlight the instability of the adduct, and possible background nitride homocoupling reaction.62
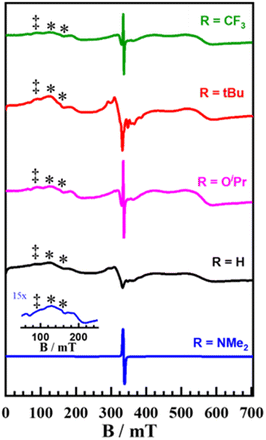 |
| | Fig. 5 X-band EPR spectra of frozen samples of [Mn(SalR)N]+ + (p-CF3Ph)3P. (‡ MnIII feature: g = 7.3; * MnIV features: g = 5.2 and 3.7) Conditions: frequency = 9.38 GHz; power = 2.0 mW; modulation frequency = 100 kHz; modulation amplitude = 0.6 mT; T = 4 K. | |
We suspect these additional species are a result of the EPR sample preparation, and due to the presence of multiple Mn species we did not attempt to simulate the spectra. However, when the MnVI and MnIII EPR spectra were subtracted from the adduct spectrum for the tBu derivative, the resultant spectrum (Fig. S35†) closely resembles EPR data reported by Fujii et al. for MnIV(Sal)–Cl and MnIV(Sal)–OH.97,98 This is in agreement with the UV-vis-NIR data (Fig. 3A and B), with new features for the MnIV phosphoraneiminato adducts consistent with previously reported MnIV complexes.97,98 To further evaluate the decay process of the adduct we analyzed the EPR spectrum of the reaction mixture for [Mn(SaltBu)N]+ and (p-CF3Ph)3P after warming to 298 K. We observe an increase in the MnIII signal at g ∼7.3, and a decrease in the signals associated with the MnIV adduct at ∼5.2 and 3.7 (Fig. S36†). EPR analysis of independently prepared [MnIII(SaltBu)Cl] at 4 K shows a significant feature at g ∼7.3, further corroborating the assignment of a similar species in the adduct and decay spectra. The eventual formation of the MnIII complex is consistent with N-atom transfer from the metal complex to (p-CF3Ph)3P, in accord with the NMR and MS analysis (vide supra), and subsequent reduction of the resulting MnIV complex by either impurities or excess phosphine.77
Theoretical calculations
Theoretical calculations were used to further understand the electronic structure of the oxidized Mn complexes and observed reactivity differences with phosphine. In accord with experimental data, calculations on the one-electron oxidized complexes predict a MnVI electronic ground state for R = CF3, tBu, OiPr, H, while a MnV ligand radical ground state was predicted for R = NMe2, NEt2 (vide supra).62,63 We next evaluated the difference in nitride NPA charge between the neutral and oxidized forms, with the MnVI derivatives exhibiting a much more significant increase in nitride NPA charge and therefore electrophilicity upon oxidation in comparison to the MnV ligand radical derivatives (Fig. 6). In addition, we observe an expected decrease in energy of the MnN π* orbitals upon oxidation. However, the relative decrease in energy for the MnVI derivatives is larger (ca. −68 kcal mol−1) in comparison to the MnV ligand radical analogues (ca. −38 kcal mol−1), implying that the empty MnN π* orbitals for the MnVI derivatives are energetically more accessible to reaction with a nucleophile (Fig. 6).
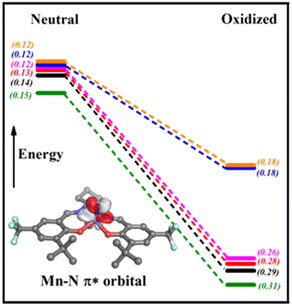 |
| | Fig. 6 Change in predicted MnN π* orbital energies upon oxidation for Mn(SalR)N complexes from natural bond order (NBO) analysis. Green (R = CF3), black (R = H), red (R = tBu), purple (R = OiPr), blue (R = NMe2), orange (R = NEt2). The MnN π* orbital energies are lower by −68.5 kcal mol−1 (CF3, MnVI), by −68.7 kcal mol−1 (H, MnVI), −69.0 kcal mol−1 (tBu, MnVI), −68.0 kcal mol−1 (OiPr, MnVI), −36.8 kcal mol−1 (NMe2, MnVL˙+), −37.0 kcal mol−1 (NEt2, MnVL˙+) in comparison to the neutral analogues. NPA charge on nitride N in brackets, indicating degree of electrophilic character. | |
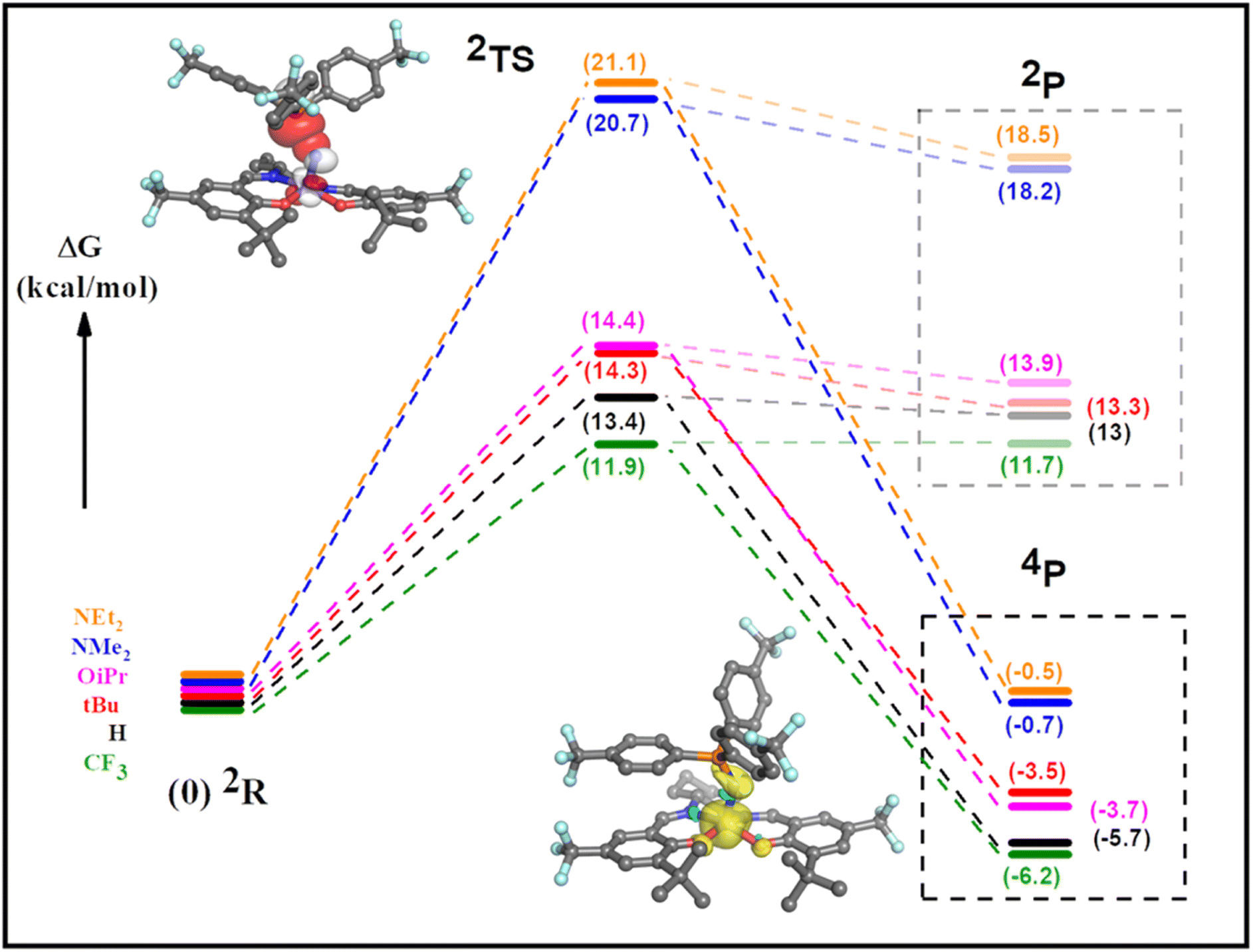
We next investigated the reaction profile of the neutral and oxidized Mn complexes with (p-CF3Ph)3P. As expected, the neutral complexes exhibit high transition state energies for adduct formation and the resulting bent triplet phosphoraneiminato products are de-stabilized by ca. 16–21 kcal mol−1 in comparison to the reactants (Table S2, Fig. S37 and S38†). This data is in agreement with the lack of reaction observed experimentally. In contrast, calculations predict that bent MnIV phosphoraneiminato adducts formed for all six [Mn(SalR)N]+ complexes with (p-CF3Ph)3P are stabilized in comparison to the isolated reactants, with the trend in stability following the electron-donating ability of the para-ring substituents (Fig. 7 and Table S3†). Calculations predict the high spin quartet state to be of lowest energy (Table S3†), with unpaired electrons in dxy, dxz, and dyz orbitals (Fig. S39 and S40†). This is in agreement with the EPR data for the adducts. Analysis of both the doublet and quartet reaction profiles supports that the transition state (TS) is located on the doublet surface with spin-crossover to the quartet state occurring after the TS (Fig. S41†). Indeed, a minimum energy crossing potential (MECP)100–102 calculation for the [Mn(SalCF3)N]+ derivative predicts the spin-crossover to occur at 2.16 Å, with the TS at 2.34 Å, in agreement with the relaxed potential energy surface scan. The calculated TS energies for the [Mn(SalR)N]+ complexes are significantly lower in energy in comparison to the neutral complexes (Fig. 7). However, the high-valent MnVI derivatives exhibit TS of lower energy (11.9–14.4) kcal mol−1 when compared to the MnV ligand radical analogues (20.7–21.2 kcal mol−1) (Fig. 7 and Table S2†). Analysis of the adduct geometry at the TS for all the oxidized derivatives predicts a bent approach of the phosphine nucleophile with a Mn–N–P bond angle of ca. 135° (Fig. S42†). Two different orbital interactions are possible, including phosphorous (P) lone pair (lp) donation into a vacant MnN π* orbital, or alternatively donation by a filled nitride N lp orbital into a P–C σ* orbital (Fig. S43†).87 Further analysis of adduct formation at constrained P–N distances for both [Mn(SaltBu)N]+ and [Mn(SalNMe2)N]˙+ predicts a decrease in P lp occupancy and a concomitant increase in MnN π* occupancy as the reactants approach the TS (Tables S5 and S6†). In contrast, the nitride N lp and P–C σ* orbital occupancies remain unchanged, thereby supporting nucleophilic attack of the phosphine on an electrophilic nitride (Tables S5 and S6†). Additional examination of the adduct formation at fixed P–N distances predicts a rise in the NPA charge on the phosphine coupled with a corresponding reduction in the NPA charge on [Mn(SalR)N]+ (R = CF3 and NMe2) as the distance between the reactants is decreased. This observation is consistent with the empirical findings, which demonstrate a nucleophilic interaction of the phosphine with an electrophilic nitride (Tables S5 and S6†).55 Moreover, a similar trend for both the MnVI and the MnV ligand radical derivatives indicates a similar reaction mechanism. For the high-valent MnVI complexes, the lowest energy transition state is predicted for the least donating para-ring substituent (R = CF3), with a trend in increasing TS energy with increasing electron-donating ability of the para-ring substituent (Fig. 7). The trend in predicted TS energies matches the experimentally observed trend in reaction rates. In addition, the relatively small difference in predicted TS energies for the MnVI complexes, and the significantly larger TS energies for the MnV ligand radical analogues, suggests that the locus of oxidation is the major determinant of the observed reactivity. Theoretical k/kH values were obtained by imputing DFT-calculated activation energies into the Eyring equation (see ESI† for details), and the resulting predicted Hammett plot presents a clear difference in reactivity between the high-valent [MnVI(SalR)N]+ (R = CF3, H, tBu, OiPr) complexes and ligand radical [MnV(SalR)N]˙+ (R = NMe2, NEt2) derivatives. Additionally, among the MnVI derivatives, the reaction rates show a linear correlation (R2 = 0.93) with the Hammett parameter (2σp) as observed experimentally (Fig. S44†).
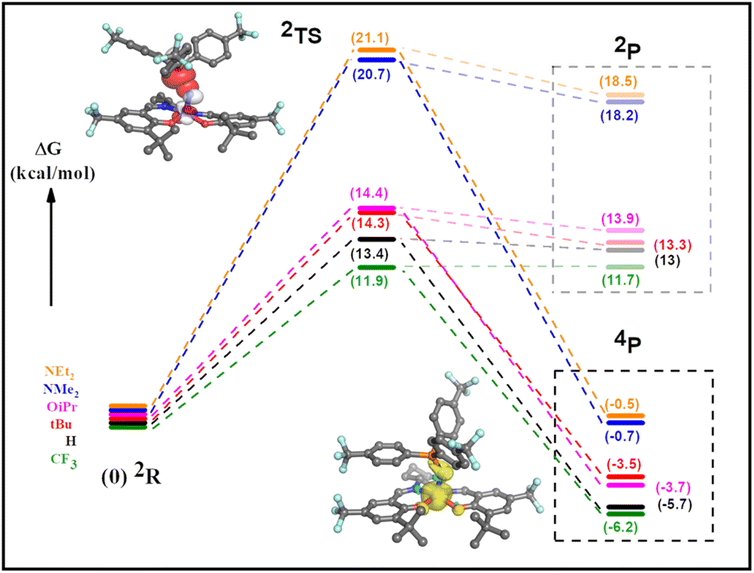 |
| | Fig. 7 Predicted reaction profiles for [Mn(SalR)N]+ and (p-CF3Ph)3P to form a bent MnIV phosphoraneiminato adduct. The initial doublet spin state (2R) proceeds through the transition state (2TS) before crossing to the quartet surface and forming the bent MnIV adduct (4P). Note the predicted product on the doublet surface (2P) is considerably higher in energy. Insets: top left, DFT-computed TS for [Mn(SalCF3)N]+ and (p-CF3Ph)3P, including overlap of MnN π* and P lone pair orbitals; bottom right, bent quartet adduct for [Mn(SalCF3)N]+ and (p-CF3Ph)3P, including spin density. Refer to the experimental section for calculation details. | |
Summary
In this study we investigated how electronic structure influences electrophilic nitride reactivity in a series of oxidized Mn salen nitrides. More specifically, we explored if the locus of oxidation (metal or ligand) was the major factor dictating reactivity at the nitride, or whether this was secondary to the overall electron-donating ability of the ancillary salen ligands. Changing the para-ring substituent of the ancillary salen ligand provided a means to change the electronic structure without significantly altering the geometry at the metal center. In addition to previously synthesized complexes, two new Mn(SalR)N (R = H and NEt2) complexes were synthesized and thoroughly characterized. One-electron oxidation of Mn(SalH)N affords a high-valent MnVI species whereas oxidation of Mn(SalNEt2)N affords a ligand radical, as shown by spectroscopic experiments and supported by DFT calculations. Reactivity of the extended series of neutral Mn(SalR)N and mono-oxidized [Mn(SalR)N]+ (R = CF3, H, tBu, OiPr, NMe2, NEt2) complexes with PPh3 and (p-CF3Ph)3P was then investigated to probe for differences in electrophilic nitride reactivity. While the neutral complexes are unreactive towards PPh3 and (p-CF3Ph)3P, the oxidized complexes react at the nitride to form a bent MnIV phosphoraneiminato adduct. The adduct was identified by low temperature EPR measurements, however, upon warming to 298 K complete N-atom transfer occurs to form the free iminophosphorane and a MnIII complex. Kinetic analysis of the reaction of the full series of [Mn(SalR)N]+ complexes with triarylphosphines proves that the locus of oxidation is the most significant factor in determining the electrophilic reactivity at the nitride, as established by a break in the Hammett plot between high-valent MnVI and MnV ligand radical complexes. Experimental observations are further supported by theoretical calculations in terms of the electronic structure reactivity profile. This work has motivated us to investigate the possibility of C–H bond activation by the more electrophilic nitrides of the [Mn(SalR)N]+ series.
Data availability
The computational details supporting this article have been uploaded as part of the ESI.†
Author contributions
S. M. synthesized and characterized all compounds, performed all CV experiments, collected UV-vis-NIR spectra, acquired kinetic data, and performed computational analysis with T. S. EPR data was collected by G. A. M. and W. V. simulated the data. Crystallographic data was refined by J. M. P. The manuscript was written by S. M. and T. S.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by Natural Sciences and Engineering Research Council (NSERC) Discovery Grants (RGPIN-2019-06749 and RGPAS-2019-00054 to T. S.). The Digital Research Alliance of Canada is thanked for access to computational resources. S. M thanks Mitacs for Globalink Graduate Fellowship (GLF599). W. V. and G. A. M. acknowledge NSERC for postgraduate fellowships. The authors thank Prof. Charles Walsby for access to the EPR instrument. Professor Fabrice Thomas (Grenoble) is thanked for insightful discussions on this work.
Notes and references
- D. V. Yandulov and R. R. Schrock, Science, 2003, 301(5629), 76–78 CrossRef CAS PubMed.
- K. Arashiba, E. Kinoshita, S. Kuriyama, A. Eizawa, K. Nakajima, H. Tanaka, K. Yoshizawa and Y. Nishibayashi, J. Am. Chem. Soc., 2015, 137(17), 5666–5669 CrossRef CAS PubMed.
- J. S. Anderson, J. Rittle and J. C. Peters, Nature, 2013, 501(7465), 84–87 CrossRef CAS PubMed.
- S. J. K. Forrest, B. Schluschaß, E. Y. Yuzik-Klimova and S. Schneider, Chem. Rev., 2021, 121(11), 6522–6587 CrossRef CAS PubMed.
- M. N. Cosio and D. C. Powers, Nat. Rev. Chem., 2023, 7(6), 424–438 CrossRef CAS PubMed.
- J. J. Curley, E. L. Sceats and C. C. Cummins, J. Am. Chem. Soc., 2006, 128(43), 14036–14037 CrossRef CAS PubMed.
- J. Du Bois, C. S. Tomooka, J. Hong and E. M. Carreira, Acc. Chem. Res., 1997, 30(9), 364–372 CrossRef CAS.
- R. L. Gdula and M. J. Johnson, J. Am. Chem. Soc., 2006, 128(30), 9614–9615 CrossRef CAS PubMed.
- M. H. Chisholm, E. E. Delbridge, A. R. Kidwell and K. B. Quinlan, Chem. Commun., 2003, 1, 126–127 RSC.
- S. Wang, H. Ge, S. Sun, J. Zhang, F. Liu, X. Wen, X. Yu, L. Wang, Y. Zhang, H. Xu, J. C. Neuefeind, Z. Qin, C. Chen, C. Jin, Y. Li, D. He and Y. Zhao, J. Am. Chem. Soc., 2015, 137(14), 4815–4822 CrossRef CAS PubMed.
- T.-N. Ye, S.-W. Park, Y. Lu, J. Li, M. Sasase, M. Kitano and H. Hosono, J. Am. Chem. Soc., 2020, 142(33), 14374–14383 CrossRef CAS PubMed.
- R. A. Karaballi, G. Humagain, B. R. A. Fleischman and M. Dasog, Angew. Chem., Int. Ed., 2019, 58(10), 3147–3150 CrossRef CAS PubMed.
- J. J. Scepaniak, C. S. Vogel, M. M. Khusniyarov, F. W. Heinemann, K. Meyer and J. M. Smith, Science, 2011, 331(6020), 1049–1052 CrossRef CAS PubMed.
- C. Vogel, F. W. Heinemann, J. Sutter, C. Anthon and K. Meyer, Angew. Chem., Int. Ed., 2008, 47(14), 2681–2684 CrossRef CAS PubMed.
- J. U. Rohde, T. A. Betley, T. A. Jackson, C. T. Saouma, J. C. Peters and L. Que, Inorg. Chem., 2007, 46(14), 5720–5726 CrossRef CAS PubMed.
- N. Aliaga-Alcalde, S. DeBeer George, B. Mienert, E. Bill, K. Wieghardt and F. Neese, Angew. Chem., 2005, 117(19), 2968–2972 CrossRef.
- K. Meyer, J. Bendix, N. Metzler-Nolte, T. Weyhermüller and K. Wieghardt, J. Am. Chem. Soc., 1998, 120(29), 7260–7270 CrossRef CAS.
- J. F. Berry, E. Bill, E. Bothe, S. D. George, B. Mienert, F. Neese and K. Wieghardt, Science, 2006, 312(5782), 1937–1941 CrossRef CAS PubMed.
- J. J. Scepaniak, R. P. Bontchev, D. L. Johnson and J. M. Smith, Angew. Chem., Int. Ed., 2011, 50(29), 6630–6633 CrossRef CAS PubMed.
- S. B. Munoz, W. T. Lee, D. A. Dickie, J. J. Scepaniak, D. Subedi, M. Pink, M. D. Johnson and J. M. Smith, Angew. Chem., Int. Ed., 2015, 54(36), 10600–10603 CrossRef CAS PubMed.
- S. Aghazada, M. Miehlich, J. Messelberger, F. W. Heinemann, D. Munz and K. Meyer, Angew. Chem., Int. Ed., 2019, 58(51), 18547–18551 CrossRef CAS PubMed.
- N. B. Thompson, M. T. Green and J. C. Peters, Nitrogen Fixation via a Terminal Fe(IV) Nitride, J. Am. Chem. Soc., 2017, 139(43), 15312–15315 CrossRef CAS PubMed.
- A. K. Maity, J. Murillo, A. J. Metta-Magaña, B. Pinter and S. Fortier, J. Am. Chem. Soc., 2017, 139, 15691–15700 CrossRef CAS PubMed.
- T. J. Del Castillo, N. B. Thompson and J. C. Peters, J. Am. Chem. Soc., 2016, 138(16), 5341–5350 CrossRef CAS PubMed.
- J. J. Scepaniak, M. D. Fulton, R. P. Bontchev, E. N. Duesler, M. L. Kirk and J. M. Smith, J. Am. Chem. Soc., 2008, 130(32), 10515–10517 CrossRef CAS PubMed.
- T. A. Betley and J. C. Peters, J. Am. Chem. Soc., 2004, 126, 6252–6254 CrossRef CAS PubMed.
- M. Keilwerth, L. Grunwald, W. Mao, F. W. Heinemann, J. Sutter, E. Bill and K. Meyer, J. Am. Chem. Soc., 2021, 143(3), 1458–1465 CrossRef CAS PubMed.
- A. McSkimming and D. L. M. Suess, Nat. Chem., 2021, 13(7), 666–670 CrossRef CAS PubMed.
- A. Sridharan, A. C. Brown and D. L. M. Suess, Angew. Chem., Int. Ed., 2021, 60(23), 12802–12806 CrossRef CAS PubMed.
- J. M. Smith, Prog. Inorg. Chem., 2014, 58, 417–470 CAS.
- R. A. Eikey and M. M. Abu-Omar, Coord. Chem. Rev., 2003, 243(1–2), 83–124 CrossRef CAS.
- J. F. Berry, Comments Inorg. Chem., 2009, 30(1–2), 28–66 CrossRef CAS.
- A. G. Maestri, K. S. Cherry, J. J. Toboni and S. N. Brown, J. Am. Chem. Soc., 2001, 123(30), 7459–7460 CrossRef CAS PubMed.
- T. J. Crevier, S. Lovell, J. M. Mayer, A. L. Rheingold and I. A. Guzei, J. Am. Chem. Soc., 1998, 120(26), 6607–6608 CrossRef CAS.
- T. J. Crevier, B. K. Bennett, J. D. Soper, J. A. Bowman, A. Dehestani, D. A. Hrovat, S. Lovell, W. Kaminsky and J. M. Mayer, J. Am. Chem. Soc., 2001, 123, 1059–1071 CrossRef CAS PubMed.
- T. J. Meyer and M. H. V. Huynh, Inorg. Chem., 2003, 42(25), 8140–8160 CrossRef CAS PubMed.
- W.-L. Man, W. W. Y. Lam, H.-K. Kwong, S.-M. Yiu and T.-C. Lau, Angew. Chem., Int. Ed., 2012, 51, 9101–9104 CrossRef CAS PubMed.
- W.-L. Man, T.-M. Tang, T.-W. Wong, T.-C. Lau, S.-M. Peng and W.-T. Wong, J. Am. Chem. Soc., 2004, 126(2), 478–479 CrossRef CAS PubMed.
- W. L. Man, W. W. Y. Lam, S. M. Yiu, T. C. Lau and S. M. Peng, J. Am. Chem. Soc., 2004, 126, 15336–15337 CrossRef CAS PubMed.
- E. M. Zolnhofer, M. Käβ, M. M. Khusniyarov, F. W. Heinemann, L. Maron, M. van Gastel, E. Bill and K. Meyer, J. Am. Chem. Soc., 2014, 136, 15072–15078 CrossRef CAS PubMed.
- M. G. Scheibel, Y. L. Wu, A. C. Stuckl, L. Krause, E. Carl, D. Stalke, B. de Bruin and S. Schneider, J. Am. Chem. Soc., 2013, 135(47), 17719–17722 CrossRef CAS PubMed.
- J. Schoffel, A. Y. Rogachev, S. DeBeer George and P. Burger, Angew. Chem., 2009, 48(26), 4734–4738 CrossRef PubMed.
- T. Schmidt-Räntsch, H. Verplancke, J. N. Lienert, S. Demeschko, M. Otte, G. P. Van Trieste III, K. A. Reid, J. H. Reibenspies, D. C. Powers, M. C. Holthausen and S. Schneider, Angew. Chem., Int. Ed., 2022, 61, e2021156 CrossRef PubMed.
- V. Vreeken, M. A. Siegler, B. de Bruin, J. N. Reek, M. Lutz and J. I. van der Vlugt, Angew. Chem., 2015, 54(24), 7055–7059 CrossRef CAS PubMed.
- M. G. Scheibel, B. Askevold, F. W. Heinemann, E. J. Reijerse, B. de Bruin and S. Schneider, Nat. Chem., 2012, 4(7), 552–558 CrossRef CAS PubMed.
- C. E. Laplaza and C. C. Cummins, Science, 1995, 268(5212), 861–863 CrossRef CAS PubMed.
- C. E. Laplaza, M. J. A. Johnson, J. C. Peters, A. L. Odom, E. Kim, C. C. Cummins, G. N. George and I. J. Pickering, J. Am. Chem. Soc., 1996, 118(36), 8623–8638 CrossRef CAS.
- J. J. Curley, T. R. Cook, S. Y. Reece, P. Muller and C. C. Cummins, J. Am. Chem. Soc., 2008, 130(29), 9394–9405 CrossRef CAS PubMed.
- R. Thompson, B. L. Tran, S. Ghosh, C. H. Chen, M. Pink, X. Gao, P. J. Carroll, M. H. Baik and D. Mindiola, J. Inorg. Chem., 2015, 54(6), 3068–3077 CrossRef CAS PubMed.
- L. N. Grant, M. Bhunia, B. Pinter, C. Rebreyend, M. E. Carroll, P. J. Carroll, B. de Bruin and D. Mindiola, J. Inorg. Chem., 2021, 60(8), 5635–5646 CrossRef CAS PubMed.
- L. N. Grant, B. Pinter, T. Kurogi, M. E. Carroll, G. Wu, B. C. Manor, P. J. Carroll and D. J. Mindiola, Chem. Sci., 2017, 8(2), 1209–1224 RSC.
- L. N. Grant, B. Pinter, J. Gu and D. J. Mindiola, J. Am. Chem. Soc., 2018, 140(50), 17399–17403 CrossRef CAS PubMed.
- T. Kurogi, P. J. Carroll and D. J. Mindiola, J. Am. Chem. Soc., 2016, 138(13), 4306–4309 CrossRef CAS PubMed.
- H. Shi, H. K. Lee, Y. Pan, K.-C. Lau, S.-M. Yiu, W. W. Y. Lam, W.-L. Man and T.-C. Lau, J. Am. Chem. Soc., 2021, 143(38), 15863–15872 CrossRef CAS PubMed.
- F. S. Schendzielorz, M. Finger, C. Volkmann, C. Würtele and S. Schneider, Angew. Chem., Int. Ed., 2016, 55(38), 11417–11420 CrossRef CAS PubMed.
- D. Sieh and P. Burger, J. Am. Chem. Soc., 2013, 135(10), 3971–3982 CrossRef CAS PubMed.
- T. J. Crevier and J. M. Mayer, J. Am. Chem. Soc., 1998, 120, 5595–5596 CrossRef CAS.
- A. Dehestani, W. Kaminsky and J. M. Mayer, Inorg. Chem., 2003, 42(2), 605–611 CrossRef CAS PubMed.
- T.-W. Wong, T.-C. Lau and W.-T. O. Wong, Inorg. Chem., 1999, 38(26), 6181–6186 CrossRef CAS PubMed.
- G. P. Connor, B. Q. Mercado, H. M. C. Lant, J. M. Mayer and P. L. Holland, Inorg. Chem., 2019, 58(16), 10791–10801 CrossRef CAS PubMed.
- C. Schiller, D. Sieh, N. Lindenmaier, M. Stephan, N. Junker, E. Reijerse, A. A. Granovsky and P. Burger, J. Am. Chem. Soc., 2023, 145(20), 11392–11401 CrossRef CAS PubMed.
- R. M. Clarke and T. Storr, J. Am. Chem. Soc., 2016, 138(47), 15299–15302 CrossRef CAS PubMed.
- N. M. Hein, G. A. MacNeil and T. Storr, Inorg. Chem., 2021, 60(22), 16895–16905 CrossRef CAS PubMed.
- Y. Shimazaki, F. Tani, K. Fukui, Y. Naruta and O. Yamauchi, J. Am. Chem. Soc., 2003, 125, 10512–10513 CrossRef CAS PubMed.
- T. Kurahashi and H. Fujii, J. Am. Chem. Soc., 2011, 133(21), 8307–8316, DOI:10.1021/ja2016813.
- L. Chiang, K. Herasymchuk, F. Thomas and T. Storr, Inorg. Chem., 2015, 54(12), 5970–5980 CrossRef CAS PubMed.
- R. M. Clarke, K. Herasymchuk and T. Storr, Coord. Chem. Rev., 2017, 352, 67–82 CrossRef CAS.
- J. Andrez, V. Guidal, R. Scopelliti, J. Pécaut, S. Gambarelli and M. Mazzanti, J. Am. Chem. Soc., 2017, 139, 8628–8638 CrossRef CAS PubMed.
- T. Storr, E. C. Wasinger, R. C. Pratt and T. D. P. Stack, Angew. Chem., Int. Ed., 2007, 46, 5198–5201 CrossRef CAS PubMed.
- M. Orio, O. Jarjayes, H. Kanso, C. Philouze, F. Neese and F. Thomas, Angew. Chem., Int. Ed., 2010, 49, 4989–4992 CrossRef CAS PubMed.
- M. Keener, M. Peterson, R. Hernández Sánchez, V. F. Oswald, G. Wu and G. Ménard, Chem.–Eur. J., 2017, 23(48), 11479–11484 CrossRef CAS PubMed.
- T. Chantarojsiri, A. H. Reath and J. Y. Yang, Angew. Chem., Int. Ed., 2018, 57(43), 14037–14042 CrossRef CAS PubMed.
- N. G. Léonard, T. Chantarojsiri, J. W. Ziller and J. Y. Yang, J. Am. Chem. Soc., 2022, 144(4), 1503–1508 CrossRef PubMed.
- S. Kim, H. Y. Zhong, Y. Park, F. Loose and P. J. Chirik, J. Am. Chem. Soc., 2020, 142(20), 9518–9524 CrossRef CAS PubMed.
- F. Loose, D. A. Wang, L. Tian, G. D. Scholes, R. R. Knowles and P. J. Chirik, Chem. Commun., 2019, 55(39), 5595–5598 RSC.
- H. Toda, K. Kuroki, R. Kanega, S. Kuriyama, K. Nakajima, Y. Himeda, K. Sakata and Y. Nishibayashi, Chempluschem, 2021, 86(11), 1511–1516 CrossRef CAS PubMed.
- D. Martelino, S. Mahato, W. VandeVen, N. M. Hein, R. M. Clarke, G. A. MacNeil, F. Thomas and T. Storr, J. Am. Chem. Soc., 2022, 144(26), 11594–11607 CrossRef CAS PubMed.
- D. D. Dumez and J. M. Mayer, Inorg. Chem., 1995, 34(25), 6396–6401 CrossRef CAS.
- D. D. DuMez and J. M. Mayer, Inorg. Chem., 1998, 37(3), 445–453 CrossRef CAS PubMed.
- S. B. Seymore and S. N. Brown, Inorg. Chem., 2000, 39, 325–332 CrossRef CAS PubMed.
- S. N. Brown and J. M. Mayer, J. Am. Chem. Soc., 1996, 118(48), 12119–12133 CrossRef CAS.
- R. H. Holm, Chem. Rev., 1987, 87(6), 1401–1449 CrossRef CAS.
- Y. M. Lee, M. Yoo, H. Yoon, X. X. Li, W. Nam and S. Fukuzumi, Chem. Commun., 2017, 53(67), 9352–9355 RSC.
- B. K. Bennett, E. Saganic, S. Lovell, W. Kaminsky, A. Samuel and J. M. Mayer, Inorg. Chem., 2003, 42, 4127–4134 CrossRef CAS PubMed.
- X. Y. Yi, T. C. H. Lam, Y.-K. Sau, Q.-F. Zhang, I. D. Williams and W.-H. Leung, Inorg. Chem., 2007, 46, 7193–7198 CrossRef CAS PubMed.
- C. Besson, Y. V. Geletii, F. Villain, R. Villanneau, C. L. Hill and A. Proust, Inorg. Chem., 2009, 48, 9436–9443 CrossRef CAS PubMed.
- J. J. Scepaniak, C. G. Margarit, J. N. Harvey and J. M. Smith, Inorg. Chem., 2011, 50(19), 9508–9517 CrossRef CAS PubMed.
- C. C. Price and W. J. Belanger, J. Am. Chem. Soc., 1954, 76(10), 2682–2684 CrossRef CAS.
- W. G. Herkstroeter, J. Am. Chem. Soc., 1973, 95(26), 8686–8691 CrossRef CAS.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91(2), 165–195 CrossRef CAS.
- K. Herasymchuk, L. Chiang, C. E. Hayes, M. L. Brown, J. S. Ovens, B. O. Patrick, D. B. Leznoff and T. Storr, Dalton Trans., 2016, 45(31), 12576–12586 RSC.
- G. Zhang, T. Liu, J. Song, Y. Quan, L. Jin, M. Si and Q. Liao, J. Am. Chem. Soc., 2022, 144, 2444–2449 CrossRef CAS PubMed.
- C. Lopez-Leonardo, M. Alajarin, P. Llamas-Lorente, D. Bautista, M. L. Jimeno, I. Alkorta and J. Elguero, Struct. Chem., 2003, 14(4), 391–397 CrossRef CAS.
- B. E. Nadeau, D. D. Beattie, E. K. J. Lui, M. Tewkesbury, J. A. Love and L. L. Schafer, Organometallics, 2023, 42(17), 2326–2334 CrossRef CAS.
- M. J. Zdilla, J. L. Dexheimer and M. M. Abu-Omar, J. Am. Chem. Soc., 2007, 129(37), 11505–11511 CrossRef CAS PubMed.
- H. M. Neu, T. Yang, R. A. Baglia, T. H. Yosca, M. T. Green, M. G. Quesne, S. P. de Visser and D. P. Goldberg, J. Am. Chem. Soc., 2014, 136(39), 13845–13852 CrossRef CAS PubMed.
- T. Kurahashi, A. Kikuchi, T. Tosha, Y. Shiro, T. Kitagawa and H. Fujii, Inorg. Chem., 2008, 47(5), 1674–1686 CrossRef CAS PubMed.
- T. Kurahashi, A. Kikuchi, Y. Shiro, M. Hada and H. Fujii, Inorg. Chem., 2010, 49(14), 6664–6672 CrossRef CAS PubMed.
- K. A. Campbell, M. R. Lashley, J. K. Wyatt, M. H. Nantz and R. D. Britt, J. Am. Chem. Soc., 2001, 123(24), 5710–5719 CrossRef CAS PubMed.
- P. L. Holland, Acc. Chem. Res., 2015, 48, 1696 CrossRef CAS PubMed.
- T. Yang, M. G. Quesne, H. M. Neu, F. G. CantúReinhard, D. P. Goldberg and S. P. de Visser, J. Am. Chem. Soc., 2016, 138, 12375 CrossRef CAS PubMed.
- R. Poli and J. N. Harvey, Chem. Soc. Rev., 2003, 32, 1 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details, X-ray structures, CV data, UV-vis-NIR data, kinetics, EPR analysis, 1H NMR, 31P NMR and ESI-MS, reactivity profiles, and DFT calculation data including molecular orbitals, spin densities, NBO analysis, NPA analysis, transition state geometries, calculations and free energy profiles; theoretical eyring plot, crystallographic data for Mn(SalH)N, DFT metrical parameters. CCDC 2298460. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc05403a |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Warren
VandeVen
,
Gregory A.
MacNeil
,
Warren
VandeVen
,
Gregory A.
MacNeil