
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe synthesis of bupropion hydrochloride under greener and safer conditions utilizing flow technologies†
Lorinda T.
van Wyk
a,
Nicole C.
Neyt
ab,
Jaimee
Jugmohan
b,
Jenny-Lee
Panayides
 b and
Darren L.
Riley
*a
b and
Darren L.
Riley
*a
aDepartment Chemistry, University of Pretoria, Pretoria, 0028, South Africa. E-mail: Darren.riley@up.ac.za; Tel: +2712 420 3097
bPharmaceutical Technologies, Chemicals cluster, CSIR, Pretoria campus, Pretoria, 0001, South Africa
First published on 14th November 2023
Abstract
Globally, major depressive disorders are a leading cause of disconsolateness affecting more than 300 million individuals. Bupropion is a unique dopamine-norepinephrine reuptake inhibitor (DNRI) commonly utilized in the treatment of depression, smoking cessation, ADHD and other addictions. Herein, we report our attempts to develop a greener, safer and more sustainable process for the preparation of bupropion hydrochloride employing flow chemistry. The use of obnoxious and corrosive liquid bromine was evaded through the employment of polymer-bound pyridinium tribromide and environmentally questionable solvents NMP and DMF were substituted with greener co-solvent systems with appreciable success. The final telescoped flow process afforded bupropion hydrochloride in a 69% overall yield, with improved process mass intensity, productivity and purity, as well as a reduction in reagents/solvents designated as red or amber in terms of H-codes.
Introduction
Major depressive disorder (MDD); also known as clinical depression; is a prevalent chronic, recurring, and enfeebling mood disorder characterised by persistent feelings of sadness and morbidity affecting up to 20% of the population.1–4 According to the World Health Organization, unipolar depressive disorders may become a major leading disease burden by 2030, a quandary which has been intensified in a post-COVID-19 pandemic world where job-losses and economic/emotional hardships continue to be felt in many countries.5–8 Even though depression is amongst one of the most treatable mental disorders, 34–46% of MDD patients are said to have treatment-resistant depression, responding inadequately to the standard antidepressant monotherapies.1,2 Bupropion hydrochloride 1a, was developed and marketed by the Burroughs Wellcome fund in 1985 under trade names, Wellbutrin and Zyban.9,11 It is a unique unicyclic aminoketone drug with an atypical norepinephrine dopamine disinhibitor mode of action which makes it useful for the treatment of depression and as a smoking cessation supplement.12,13 Though effective as an anti-depressant on its own, the drug is commonly prescribed conjointly with Selected Serotonin Reuptake Inhibitors in cases where incomplete response to the first-line antidepressant is seen.3 Additionally, bupropion analogues have recently been found to act as indirect dopamine agonists in the treatment of cocaine and methamphetamine addictions.12In the last decade, the use of flow- and microreactors for the synthesis of organic substrates has increased considerably presenting a means of carrying out reactions in a safer, faster, and more efficient manner. This process technology has gained popularity for its ability to deal with hazardous or noxious materials in a more controllable fashion, and when coupled with solvent recycling, the use of greener solvents, the reuse of reagents and in-line waste treatments it affords a more sustainable means of synthesizing molecules.14,15 The technology is often directly scalable and depending on the process at hand can allow one to telescope multistep processes as continuous, uninterrupted sequences with in-line downstream processing and purification.16
The first total synthesis of bupropion hydrochloride 1a was patented by the Burroughs Wellcome Co. (now GlaxoSmithKline) in 1974 (Scheme 1).9 Since then, several variations have been reported almost exclusively employing the same basic synthetic strategy with variations to the reagents and/or solvents.11,17–19 The general approach involves three stages commencing with an α-bromination of 3′-chloropropiophenone 2 using molecular bromine in dichloromethane (DCM) to afford α-bromoketone 3 which is subsequently treated with excess tert-butylamine in acetonitrile in a nucleophilic displacement reaction to afford the free base 1b. Thereafter, treatment with concentrated hydrogen chloride in diethyl ether affords the commercial salt form of the molecule 1a.9
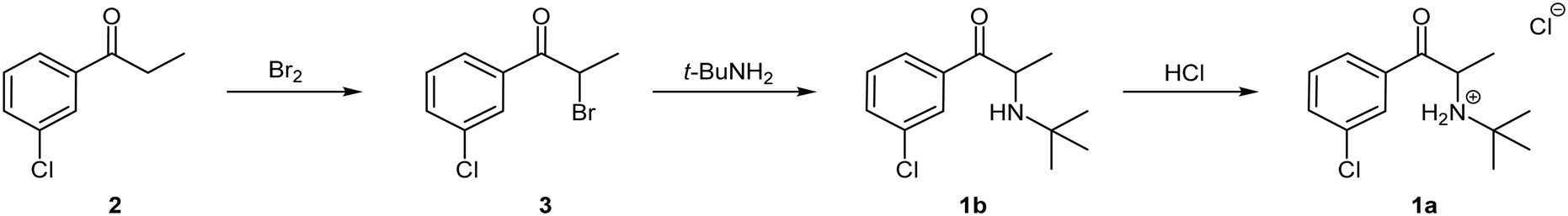 | ||
| Scheme 1 Synthetic route for the preparation of bupropion hydrochloride 1a. | ||
Bromination reactions are pivotal in generating useful intermediary organobromide building blocks, but a notable drawback to their use is that they are almost exclusively prepared using molecular bromine either directly, or indirectly. Molecular bromine poses a significant safety risk as it is toxic, highly corrosive and sublimes at ambient temperature. Furthermore, and of relevance to the synthesis of bupropion hydrochloride 1a, α-brominated species are generally lachrymatory in nature presenting additional safety challenges. A variety of bromination reactions utilizing flow conditions have been reported, often making use of molecular bromine.20–24 Several greener, more sustainable alternatives including photochemical brominations with N-bromosuccinimide, in situ generated metal bromide (FeBr3) and ionic salt bromination sources such as KOBr and NaBr have been reported with enhanced safety while affording comparable yields.21–30 Critically speaking, if reagent lifecycles are considered, the use of molecular bromine, despite its associated hazards, has a cost and sustainability advantage over most “greener” alternatives as they inevitably have their genesis with molecular bromine itself.
Nucleophilic substitution reactions are salient solvent and base dependent transformations. These displacement reactions have been demonstrated under flow conditions with multiple electrophiles, often outperforming their related batch counterparts.31 In a more modernized approach, nucleophilic substitutions have also been performed in ionic-liquids under flow conditions.32 Unfortunately, these reactions are often carried out in unfavourable solvents such as N-methyl-2-pyrrolidone (NMP), N,N-dimethylformamide (DMF) and dimethylsulfoxide (DMSO) which are employed to aid in the solubilization of the reaction matrix but which have associated safety and environmental concerns (NMP and DMF) and are challenging to remove during downstream processing.
Ley and co-workers have previously reported a flow synthesis of the free base form of bupropion 1b while showcasing the use of novel automation technologies. In this instance, the approach outlined in Scheme 1 was adopted and the first two stages were optimized and telescoped under flow conditions to afford the free base 1b. The first stage bromination was performed with molecular bromine affording 3 in 95% yield. The subsequent nucleophilic displacement step was performed in NMP affording the free base 1b in 80% yield. Telescoping of the process; which consisted of four-unit operations; culminated in the production of the free base of bupropion 1b in an overall yield of 80% and production rate of 2.88 g h−1. Although this flow process proved to be efficient in terms of both yield and productivity, we envisioned improving the process further in terms of overall greenness and safety.19
In this paper, we describe our attempts to develop a telescoped synthesis of bupropion hydrochloride 1a under flow conditions with a focus on increasing safety, reducing waste generation, employing the use of greener reagents/solvents, and improving energy efficiency. Critically, hazardous molecular bromine (Stage 1) and N-methyl-2-pyrrolidone (Stage 2) were identified as two key players which required replacement with greener/safer alternatives.
Results and discussion
1st generation – batch processes
The bromination of 3′-chloropropiophenone 2 (Scheme 2) was investigated under batch conditions using several brominating reagents including, molecular bromine, N-bromosuccinimide (NBS), ammonium bromide/oxone and polymer supported pyridinium tribromide (Table 1). Molecular bromine proved to be most favourable when performed in dichloromethane with a short reaction time (40 min) at ambient temperature when employing a modest excess of bromine. Downstream processing only required quenching with aqueous potassium carbonate; followed by extraction to afford 3 in an 87% isolated yield (Table 1, entry 1). Notably, the same approach at reflux with a prolonged reaction time and in the presence of excess bromine resulted in unwanted dibromination affording 4 as a by-product which proved challenging to remove (entry 2). As dichloromethane was undesirable from a sustainability point of view, we elected to screen the process in acetonitrile as a greener alternative. The selection of acetonitrile was driven by the fact that it readily solubilised 2, and concurrent development of the second stage suggested that acetonitrile (in conjunction with a co-solvent) could be utilised for both stages 1 and 2 of the process. Unfortunately, brominating with molecular bromine in acetonitrile under comparable conditions was characterised by the formation of appreciable amounts of the unwanted dibrominated product 4 (entry 3). The employment of a p-TsOH catalysed NBS or ammonium bromide/oxone mediated brominations showed poor conversions and required longer reaction times (entries 5 and 6) and the latter was characterised by significant precipitation which made the translation thereof to flow unappealing. Finally, the use of polymer bound pyridinium tribromide afforded comparable conversions and yields to that achieved with molecular bromine and only required simple extractive downstream processing, albeit at a significantly longer reaction time (12 h vs. 40 min) (entries 7–9).
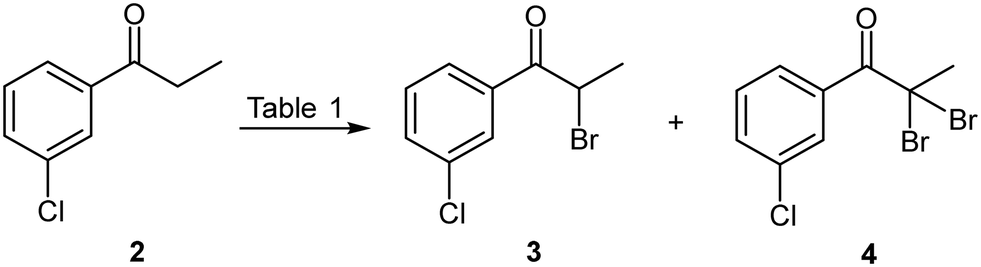 | ||
| Scheme 2 Bromination of 3′-chloropropiophenone 2. | ||
| Entry | Brominating agent | Equivalents | Reaction time | Temperature (°C) | Solvent | Conc. (M) | Conversion%a | ||
|---|---|---|---|---|---|---|---|---|---|
| 2 | 3 | 4 | |||||||
| a Conversions estimated by comparison of integral areas of 2, 3 and 4 in 1H NMR spectra, isolated yields provided in brackets. b Burroughs Wellcome Co. method.9 c Precipitation noted. | |||||||||
| 1b | Bromine | 1.1 | 40 min | rt | DCM | 0.5 | 0 | 100 (87) | 0 |
| 2 | Bromine | 2.0 | 12 h | 40 (1 h) → rt (11 h) | DCM | 0.5 | 0 | 2 | 98 |
| 3 | Bromine | 1.1 | 40 min | rt | ACN | 0.5 | 0 | 76 | 24 |
| 4 | Bromine | 1.1 | 40 min | rt | EtOAc | 0.5 | 0 | 93 | 7 |
| 5 | NBS/p-TsOH | 1.5 | 12 h | Reflux | ACN | 0.5 | 25 | 75 | 0 |
| 6 | NH4Br and oxonec | 1.5 | 48 h | 60 | MeOH | 0.2 | 64 | 36 | 0 |
| 7 | PyBr3 polymer bound | 1.5 | 12 h | 60 | ACN | 0.5 | 0 | 88 | 12 |
| 8 | PyBr3 polymer bound | 1.5 | 12 h | 60 | EtOAc | 0.5 | 0 | 100 | 0 |
| 9 | PyBr3 polymer bound | 1.5 | 12 h | 60 | DCM | 0.5 | 0 | 91 | 9 |
Although the use of molecular bromine utilising flow technology has been well documented,22–24 the employment of polymer bound pyridinium tribromide caught our interest as its use would largely mitigate the toxicity and corrosion risks associated with molecular bromine, thereby improving the safety profile of the process. Critically speaking though, its use would require the employment of packed-bed reactor (PBR) technologies which present engineering challenges, particularly as the reagent is consumed stoichiometrically, and polymer bound reagents also generally have significantly higher costs than analogous non-supported reagents. In this case, however, the polymer can be regenerated; but this then requires treatment with molecular bromine.
Alternatively, the use of an activated alcohol at the alpha position would circumvent the issues of using bromine as previously demonstrated by Coelho and co-workers.33 Unfortunately, in the context of this work the approach was deemed unattractive as it required two additional steps (5 vs. 3) and the triflate group utilised represented twice the atom economy burden of bromine.
 | ||
| Scheme 3 Nucleophilic substitution of 3 with tert-butylamine. | ||
| Entry | Solvent | tert-BuNH4+Br− ppt | Unreacted 3 present | Decomposition observed | Yield% 1b |
|---|---|---|---|---|---|
| a General reaction conditions: 95 °C, 4-hour reaction time, 0.5 M concentration, 3.0 equivalents of tert-butylamine. b Burroughs Wellcome Co. method.9 | |||||
| 1b | ACN | Yes | No | No | 86 |
| 2 | 50% ACN:DCM | No | Yes | No | 24 |
| 3 | 75% ACN:DCM | Yes (minimal) | No | No | 76 |
| 4 | 90% ACN:DCM | Yes | No | No | 78 |
| 5 | 85% ACN:DMSO | No | No | No | 75 |
| 6 | 90% ACN:DMSO | Yes (minimal) | No | No | 77 |
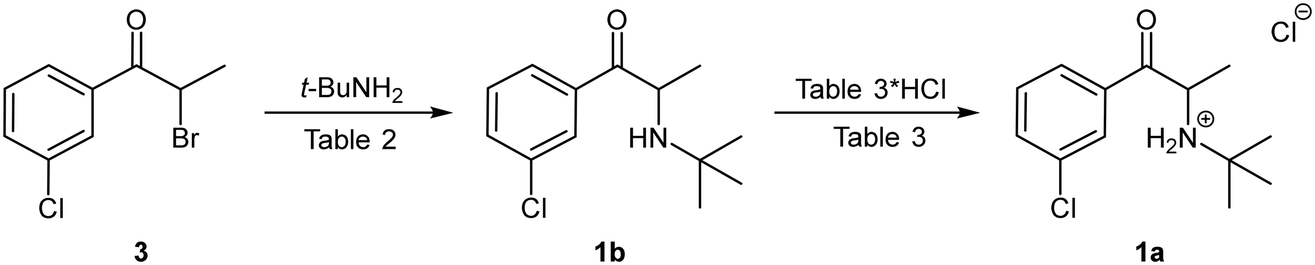
When performed in acetonitrile as described by Burroughs Wellcome Co. using 3.0 equivalents of tert-butylamine at 95 °C for 4 hours an isolated yield of 86% of the free base form 1b was achieved, but the approach was characterised by the formation of a thick precipitate of tert-butylammonium bromide and was not amenable to flow translation (Table 2, entry 1). The addition of a solubilising co-solvent in the form of DCM or DMSO significantly reduced the amount of precipitate observed. In the case of DCM, the yield decreased sharply as the ratio of DCM was increased (entries 2–4). Alternatively, the use of 10–15% DMSO in acetonitrile (entries 5 and 6) largely solubilised the precipitate while affording yields in the range of 75–77%.
| Entry | HCl source | HCl conc. (M) | Solvent | Yield%c1a |
|---|---|---|---|---|
| a General reaction conditions: 0 °C, 2.0 equivalents of HCl, 1.2 gram free base 1b in 100 mL solvent (0.05 M). b 8.0 equivalents of HCl, solvent free. c Isolated yield. d Adaptation of the Burroughs Wellcome Co. method.9 | ||||
| 1d | Et2O*HCl | 2 | Et2O | 95 |
| 2 | Et2O*HCl | 2 | Hexane | 91 |
| 3 | Et2O*HCl | 2 | Cyclohexane | 89 |
| 4 | Et2O*HCl | 2 | EtOAc | 94 |
| 5 | MeOH*HCl | 3 | Et2O | 86 |
| 6 | EtOH*HCl | 1.25 | Et2O | 82 |
| 7 | EtOH*HCl | 1.25 | EtOAc | 30 |
| 8b | HCl (32%) | 8.8 | Neat | 32 |
| 9 | HCl gas | — | EtOAc | 94 |
| 10 | HCl gas | — | Cyclohexane | 89 |
2nd generation – flow translation and optimisation
| Entry | Brominating agent | T R (min) | Temp (°C) | Solvent | Conc. (M) | Conversion%a | ||
|---|---|---|---|---|---|---|---|---|
| 2 | 3 | 4 | ||||||
| a Conversions estimated by comparing the integral areas of 2, 3 and 4 in 1H NMR spectra, isolated yields given in brackets. b General reaction conditions: 0.5 M stock solution of 2, 0.5 M stock solution of elemental bromine. c General reaction conditions: 0.2 M stock solution of 2, crushed ammonium bromide and oxone in a PBR. d General reaction conditions: 1 M stock solution of 2, 1.5 equivalents of pyridinium tribromide polymer housed in a PBR. | ||||||||
| 1 | Bromine liquidb | 10 | 44 | DCM | 0.25 | 0 | 96 (84) | 4 |
| 2 | NH4Br and oxonec | 50 | 130 | MeOH | 0.2 | 50 | 50 | 0 |
| 3 | PyBr3 polymer boundd | 25 | 60 | ACN | 1.0 | 0 | 98 (81) | 2 |
| 4 | PyBr3 polymer boundd | 90 | 40 | EtOAc | 1.0 | 0 | 91 | 9 |
 | ||
| Scheme 4 α-Bromination of 3′-chloropropiophenone 2 under flow conditions. | ||
The spent polymer supported pyridinium tribromide was regenerated by treatment with molecular bromine with a limited loss in activity (regeneration unoptimized). When using the regenerated polymer at the optimised conditions (Table 4, entry 3), 3 was afforded with a conversion of 93% (vs. 98% with new polymer). Comparable yields could be realised by increasing the residence time to 30 minutes and increasing the stoichiometric excess of the supported pyridinium tribromide to 2.0 equivalents.
We were able to demonstrate the displacement under flow conditions (Scheme 5) employing a 95% ACN:DMSO solvent mix without any deleterious effects on the solubility or the yield. Under these conditions, a 1.0 M stock solution of 3 (prepared under batch conditions and purified via column chromatography) in ACN was combined with a 3.0 M stock solution of the tert-butylamine (90% ACN:DMSO) at a T-piece mixer prior to passage through a 25 mL PTFE coil reactor (TR = 20 min, Temp = 90 °C). The resulting solution was then quenched at a second T-piece mixer with water prior to passage through a BPR and collection. The free base bupropion 1b was isolated in a yield of 98% with a production rate of 5.06 g h−1. In a subsequent offline salt formation step the free base 1b was converted into the desired product 1a utilising ethereal hydrogen chloride in cyclohexane in 87% yield.
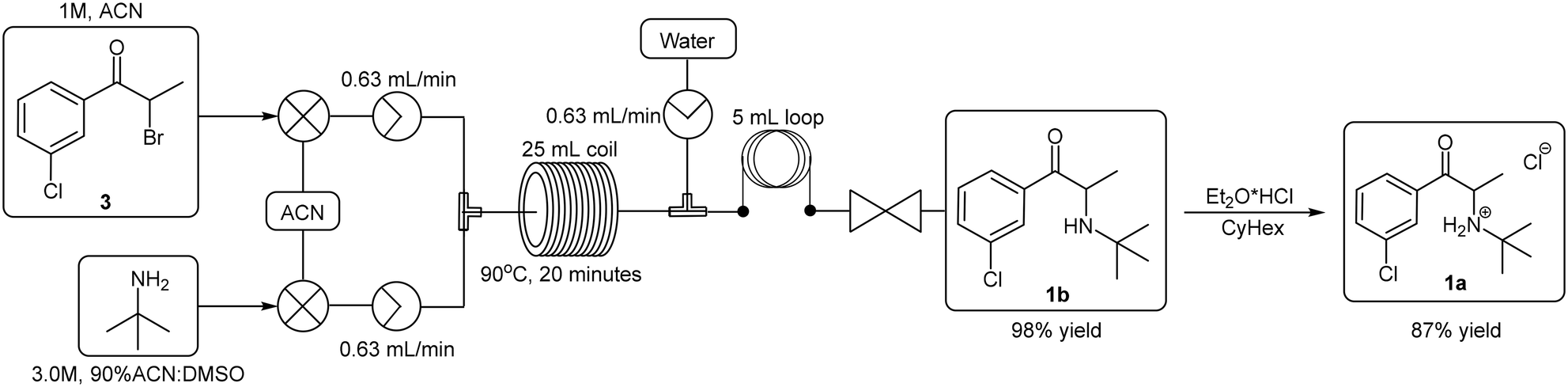 | ||
| Scheme 5 Nucleophilic substitution reaction for the formation of bupropion 1a under flow conditions. | ||
3rd generation – telescoped flow process
We next focused our attention on linking the first two stages to afford a single uninterrupted process. Unfortunately, direct linking of the two stages was characterised by the unexpected in-line precipitation of tert-butylammonium bromide. We suspected that this was arising as bromine and/or hydrogen bromide was leaching into stage 2 and was not being completely neutralised by the potassium carbonate scavenger. To circumvent this issue, we reverted to the use of 85% ACN:DMSO and employed the use of a sonicator at the T-piece mixers to help solubilise the salt. Concurrently we also investigated the use of alternative scavengers including: Amberlite IRA-400, Dowex anion exchange resin and sodium thiosulfate (Table 5).| Entry | Scavenger | Total flow rate (mL Min−1) | Temperature (°C) | Residence time (min) | Yield%b1b | ||
|---|---|---|---|---|---|---|---|
| Column | Coil | Column | Coil | ||||
| a Reactions were performed on a 0.5-gram scale based on 2. b Isolated yield. c Recycling (Scheme 7 – red). | |||||||
| 1 | Amberlite IRA-400 | 0.32 | 60 | 90 | 25 | 78.1 | 51 |
| 2 | Sodium thiosulfate | 0.32 | 60 | 90 | 25 | 78.1 | 51 |
| 3 | Dowex | 0.32 | 60 | 90 | 25 | 78.1 | 55 |
| 4 | Potassium carbonate | 0.32 | 60 | 90 | 25 | 78.1 | 53 |
| 5 | Sodium thiosulfatec | 1.25 | 60 | 90 | 25 | 20 | 63 |
| 6 | Potassium carbonatec | 1.25 | 60 | 90 | 25 | 20 | 77 |
The telescoping was performed using a 1.0 M 3′-chloropropiophenone 2 stock solution in acetonitrile which was pumped (0.16 mL min−1) through a PBR housing 1.5 equivalents of polymer-bound pyridinium tribromide (TR = 25 min, Temp = 60 °C) followed by a second PBR housing the investigated scavenger (Scheme 6 – black). The resulting mixture was then combined with a 3.0 M stock solution of tert-butylamine in 70% ACN:DMSO (0.16 mL min−1) at a T-piece mixer (sonicated, Temp = 60 °C). The resultant stream was passed through a 25 mL PTFE coil (TR = 78.1 min, Temp = 90 °C) prior to being quenched with water (0.16 mL min−1) at a second T-piece mixer which was submerged in a sonicated bath (Temp = 60 °C) followed by a 5 mL “dissolution” loop and 8 bar BPR. In all instances the scavengers screened performed comparably, but yields were only modest in the range of 51–55% (Table 5, entries 1–4).
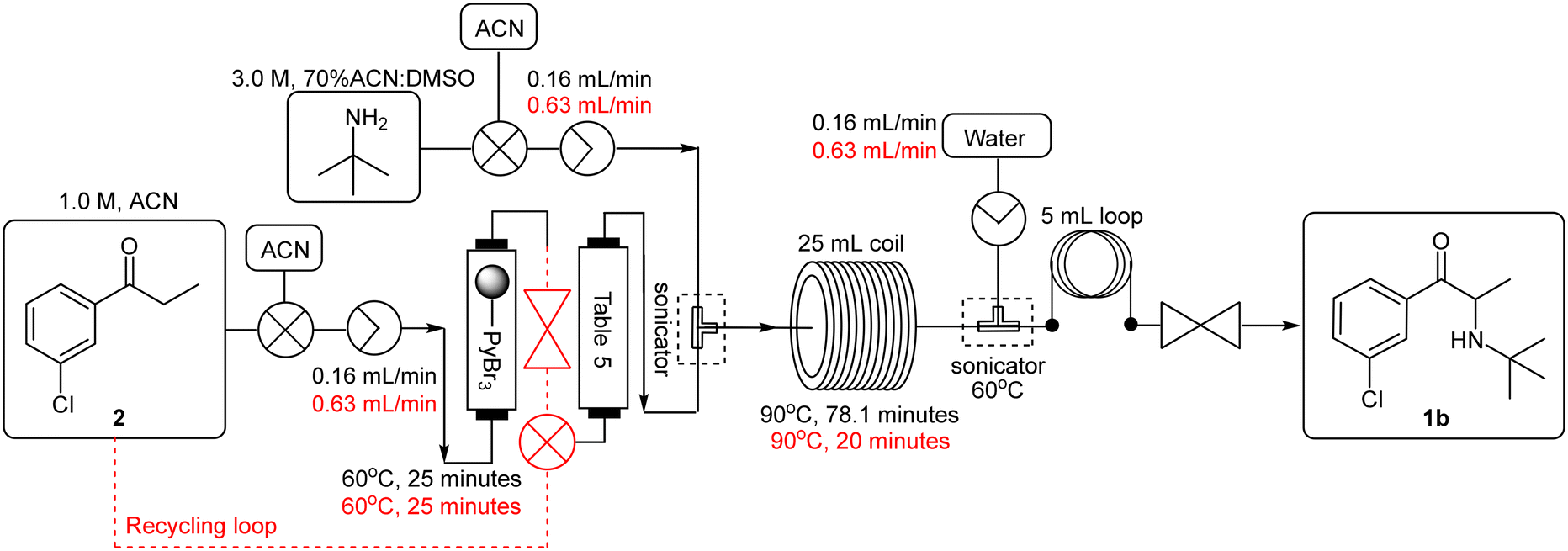 | ||
| Scheme 6 Initial 3rd generation flow synthesis of free base bupropion 1b. | ||
At this stage it was suspected that the drop-off in yield was a result of partial decomposition of the free base product 1b, as a brown discolouration of the reaction matrix was observed. In the set-up employed, the flow rate required for the full conversion of the first stage resulted in a longer than optimal residence time (78.1 minutes) for the second stage in the direct linking. A smaller coil reactor of suitable internal diameter was not accessible at the time of testing, as a result we opted to employ a recycling system for the first stage, affording an optimal flow rate of 1.26 mL min−1 for the second stage (Scheme 6 – red), leading to an improved yield of 77% for the free base 1b when using potassium carbonate as the scavenger (entry 6).
Our ultimate aim was to deliver a fully automated continuous flow process which integrated downstream processing of both the free base 1b and bupropion HCl 1a while minimizing chemical exposure/handling. As such we next shaped our investigation to include a downstream solvent swap to afford the free base 1b in a solvent system that was compatible with the final salt formation step. An in-line solvent swap from acetonitrile to a more appropriate water immiscible extracting solvent was required. The solvent swop also served to i) remove excess tert-butylamine, ii) remove unwanted bromide salts and iii) remove DMSO as part of the aqueous waste fraction. Thereafter, the organic fraction could be triturated with an appropriate hydrogen chloride source to afford the desired salt 1a.
To facilitate the downstream solvent swap and subsequent extraction, the post reactor setup was modified to include an integrated rotary evaporator (Scheme 7, Fig. 1). The rotatory evaporator bleed valve was replaced with a customized screwcap fitted with three 1/16′′ diameter holes through which PTFE tubing could be fed allowing the reaction mixture to collect in the rotatory's evaporation flask. A second line from a peristaltic pump was inserted via the customized cap to facilitate the delivery of extraction solvents and finally, a third line connected to an HPLC pump was inserted to facilitate the removal of material from the evaporation flask.
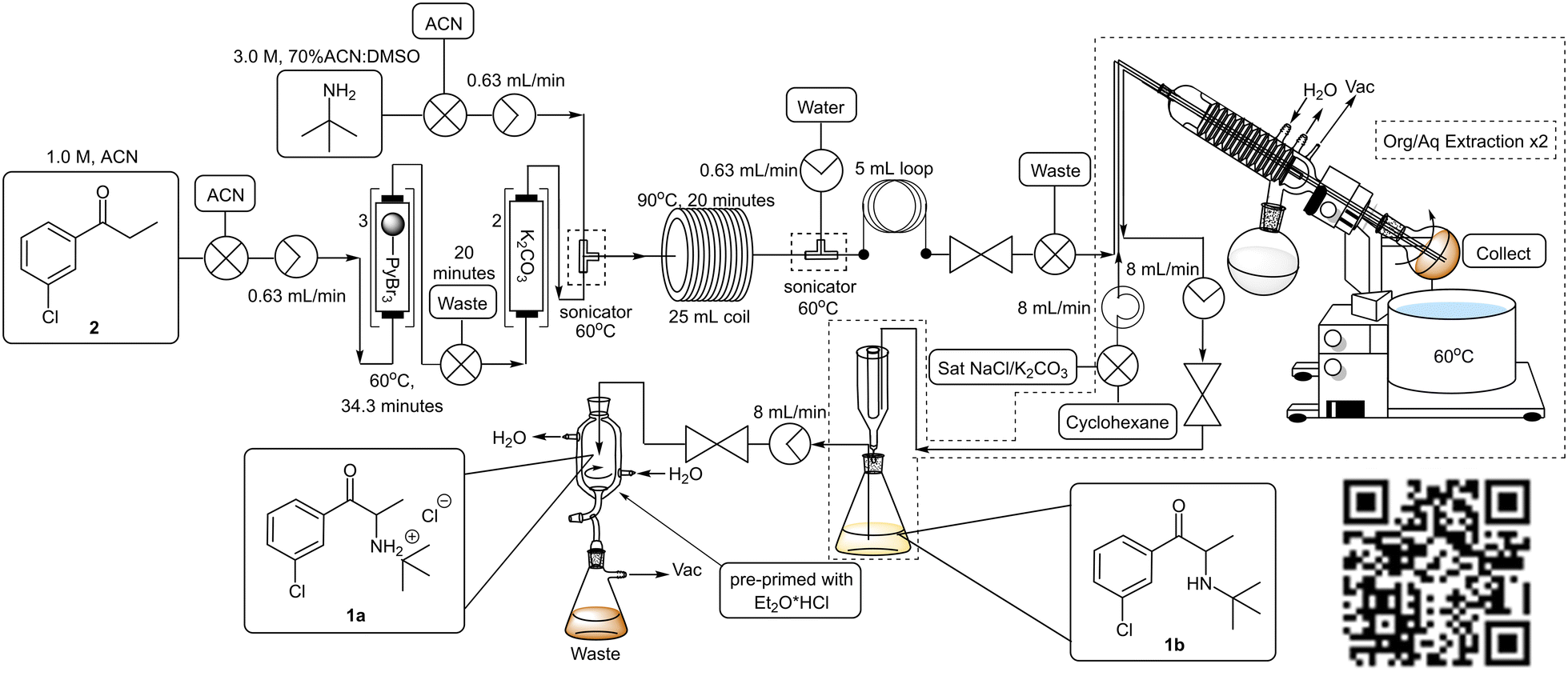 | ||
| Scheme 7 Final 3rd generation staggered flow synthesis of bupropion hydrochloride 1a. | ||
 | ||
| Fig. 1 Final 3rd generation staggered flow synthesis of target bupropion 1a. | ||
Operationally, upon exiting the back-pressure regulator of the main flow reactor, the reaction mixture entered the rotary evaporator. The acetonitrile solvent fraction and unreacted tert-butylamine were evaporated and the rotary was primed with cyclohexane followed by a saturated solution of sodium chloride and/or potassium carbonate (to aid in the removal of the DMSO and ensure complete neutralization) using the second line. Extraction was facilitated by allowing the evaporation flask to rotate, and thereafter the biphasic mixture was allowed to separate, and the organic layer was pumped out of the rotary evaporator using the third line. The resultant mixture was then fed into a Biotage® phase separator allowing the removal of remaining traces of the aqueous phase and subsequent collection of the organic layer. A second extraction was performed in a similar fashion and the organic fractions were combined and pumped directly into a cooled (0 °C) in-line triturator,34 pre-primed with ethereal hydrogen chloride to obtain the desired hydrochloride salt 1a.
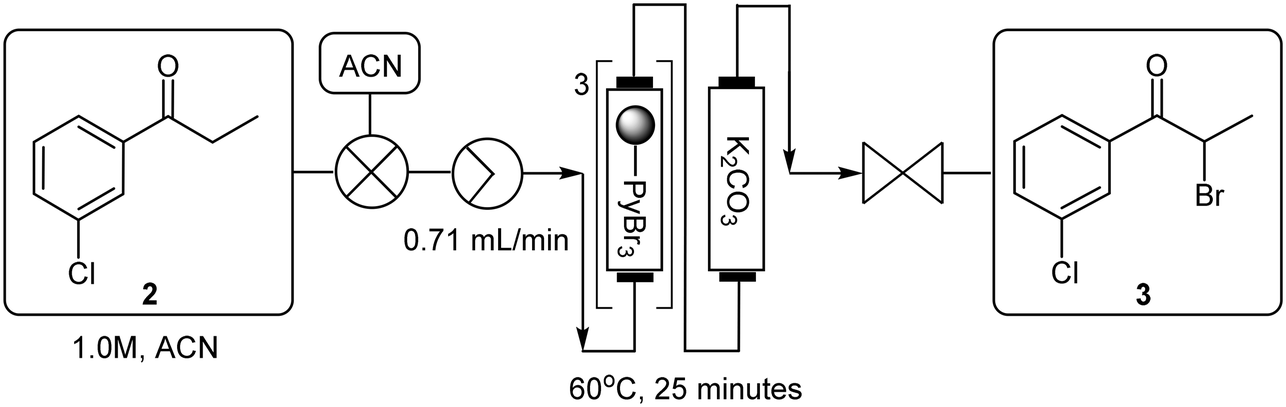
The reaction conditions remained constant as described previously utilizing a 1.0 M 3′-chloropropiophenone 2 stock solution which was pumped through PBR's connected in series housing a combined 1.5 equivalents of pyridinium tribromide (TR = 34.3 min, Temp = 60 °C) which was followed by several PBR's again connected in series housing 14.5 equivalents of ground potassium carbonate. A manual selector valve was included between the final PBR housing the polymer reagent and the first PBR housing the scavenger to divert any front-running leached brominated species (first 20 minutes of reaction) to waste instead of going through and compromising the potassium carbonate scavenger. Upon exiting the final PBR, the reaction mixture was combined with a 3.0 M stock solution of tert-butylamine in 70% ACN:DMSO (17.2 eq.) at a T-piece submerged in a sonicating bath (Temp = 60 °C). The use of excess tert-butylamine was employed to counter the dispersion of 3 as it was pumped through the PBR's. The resulting mixture was subsequently passed through a 25 mL PTFE coil (TR = 20 min, Temp = 90 °C) prior to being quenched with distilled water at a second T-piece mixer (submerged in an ultrasound bath, Temp = 60 °C). The resultant mixture was passed through a 5 mL PTFE “dissolution” loop and ultimately fed through a BPR into the evaporation flask mounted on the rotary evaporator. Upon complete collection, the in-line work-up procedure described previously was performed. The combined organic fractions were subjected to an in-line salt formation affording bupropion hydrochloride 1a in a 62% isolated yield across three stages equating to ∼85% per stage (Scheme 7, Fig. 1, ESI† section 1.6.2). Under these conditions a productivity rate of 1.55 g h−1 for the free base 1b was realised, but disappointingly, the overall purity of the final product 1a decreased significantly from 96.6% (generation 1) and 99.9% (generation 2) to only 86.5%.
4th generation staggered flow process
We revisited the process to determine whether the loss in yield and purity noted was arising as a result of one or more of the downstream processing operations (see section 1.7.1. of ESI† for additional information). It was determined that i) the absence of an extractive work-up after stage 1 resulted in decreased yields and could be linked to the formation of the thick tert-butylammonium bromide precipitate observed in generation 3 and ii) the use of excess tert-butylamine resulted in a sharp drop-off in yields isolated even when using 3 which had been subjected to an extractive work-up.Practically, both of these issues were overcome through introducing an inline extraction between stages 1 and 2 which allowed us to remove the PBR housing potassium carbonate and replace it with a rotary evaporator and Zaiput™ membrane separator. The flow stream could then be neutralized with 50% aqueous potassium carbonate in the rotary evaporator, at the same time this then allowed us to reduce the tert-butylamine equivalents required for stage 2 from 17.2 to 4.0 equivalents. During this time, we also elected to perform the salt formation step using HCl gas generated in situ to eliminate the use of non-green and hazardous diethyl ether and additional focus was also placed on the minimization of the extraction solvents quantities to reduce waste generation and improve overall greenness (Scheme 8, see ESI† section 1.7.2).
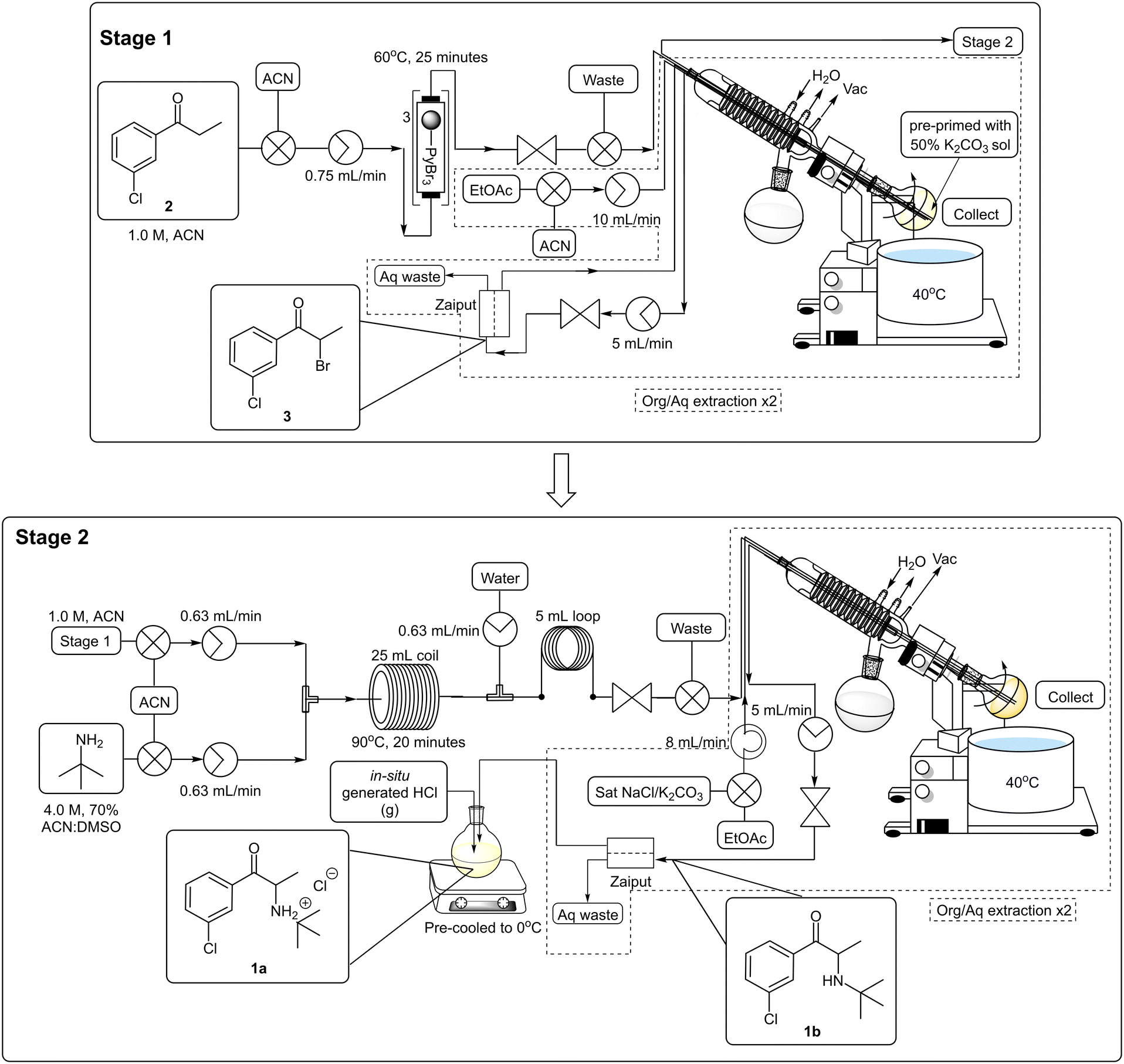 | ||
| Scheme 8 4th generation flow synthesis of bupropion hydrochloride 1a. | ||
The staggered telescoped process was performed as follows: a 1.0 M 3′-chloropropiophenone 2 stock solution was pumped through a series of PBR's housing a total of 1.5 equivalents of pyridinium tribromide (TR = 25 min, Temp = 60 °C). Thereafter, the reaction mixture was passed through a BPR and into the rotatory's evaporation flask which was pre-primed with a 50% potassium carbonate solution. After collection was completed, the acetonitrile solvent was removed in vacuo and ethyl acetate was introduced into the rotary evaporator through a second line. After sufficient rotation, the biphasic mixture was pumped through a Zaiput™ membrane separator (OB-900). The extraction was repeated after which time the combined organic fractions were pumped back into the rotary evaporator fitted with a new evaporation flask. The ethyl acetate solvent (the distillate could be collected and redistilled off-line to recover the ethyl acetate, see ESI† section 1.9.2) was removed in vacuo and the flask was re-primed with acetonitrile to afford a 1.0 M stock solution of 3. The brominated material 3 was then combined with a 4.0 M stock solution of tert-butylamine in 70% ACN:DMSO at a T-piece mixer and the resultant stream passed through a 25 mL PTFE coil reactor (TR = 20 min, Temp = 90 °C). The mixture was then combined with a water quench line at a second T-piece mixer and subsequently passed through a 5 mL “dissolution” loop and BPR prior to collection in the rotary evaporator. The mixture was subjected to a solvent swap and extraction, utilizing ethyl acetate, as described previously. The acetonitrile solvent and unreacted tert-butylamine fraction could be recovered directly from the rotary evaporator's solvent trap if desired (>80% percentage recovery) but not unexpectedly this was contaminated with water (<10%) and unreacted tert-butylamine (∼2%) and complete recycling of the solvent would require additional off-line operations (see ESI† section 1.9.2). The biphasic solution was pumped through the Zaiput™ membrane separator (OB-900) and finally into a pre-cooled flask fitted with a PTFE gas delivery tube. The resultant mixture was treated with HCl gas generated from sulfuric acid and sodium chloride. The final isolation of bupropion hydrochloride 1a was achieved in an overall yield of 69% across three stages equating to ∼88% per stage at a production rate of 1.78 g h−1 for the free base 1b.
The 4th generation staggered telescoped approach outperformed the 3rd generation telescoped flow process in terms of final yield of 1a isolated (69% vs. 62%) and offered several additional advantages such as the reduced need for the off-line distillation to recover unreacted amine due to a 4.3-fold decrease in the amount of tert-butylamine used.
Furthermore, the addition of the work-up step in between the two stages mitigated precipitate formation in the flow system making further scale-up more attractive, it also led to an appreciably increase in the product purity from 86.5 to 97.8% for the final salt 1a while still ensuring a staggered continual process with minimal handling and exposure to chemicals throughout. Lastly, the elimination of the ethereal hydrogen chloride and use of reduced quantities of extraction solvents led to a reduction in associated process costs and the volume of waste produced.
Analysis and comparison of generations 1 to 4
In an effort to better understand and quantify the process improvements in terms of greenness, safety and sustainability we analysed each generation using the CHEM21 toolkit.35 The toolkit was selected as it provides standard quantitative green metrics, and it also holistically assesses non-numerical variables qualitatively through the use of an easy to interpret red, amber and green flag system. In addition, we also performed an analysis of how the mass distribution of reagents and solvents changed over the four generations.Comparison of green metrics
Several sustainability metrics and productivity rates calculated for generations 1–4 are highlighted in Table 6. Following our CHEM21 analysis several observations were noted.| Generation | Yield% | AE% | RME% | OE% | Total PMI | Productivity rate 1b (g h−1) | Space time yield 1b (g h−1 L) | % Purityb |
|---|---|---|---|---|---|---|---|---|
| a Calculations were performed with the aid of the CHEM21 green metrics toolkit, see ESI† section 1.9 for description of metrics and summary of Green Metrics analysis. b Determined by ECIC qNMR method.10 c The HCl (g) was generated chemically and we were unable to quantify the amount introduced into the reactor, as such we assumed the use of 2.0 equivalents HCl which proved to be sufficient in generations 2 and 3 when calculating the green metrics. d H&S: Br2 (red: H330, H400), tert-butylamine (amber: H331), Et2O*HCl (amber: H331, H224), Et2O (H224) & DCM (H351). e H&S: tert-butylamine (amber: H331), Et2O*HCl (amber: H331, H224) & cyclohexane (red: H400, H410). f H&S: tert-butylamine (amber: H331), Et2O*HCl (amber: H331, H224) & cyclohexane (red: H400, H410). g H&S: tert-butylamine (amber: H331). | ||||||||
| 1d | 71.1 | 63.1 | 32.9 | 52.1 | 135.3 | 1.1 | 10.1 | 96.6 |
| 2e | 69.2 | 63.1 | 29.7 | 47.0 | 155.1 | 1.5 | 24.6 | 99.9 |
| 3f | 62.1 | 63.1 | 9.9 | 15.7 | 203.0 | 1.6 | 18.3 | 86.5 |
| 4c,g | 69.4 | 63.1 | 24.5 | 38.9 | 114.7 | 1.8 | 29.6 | 97.8 |
i) The calculated reaction mass efficiencies RME (and related operational efficiencies OE) suggest that the batch approach and standalone flow approach (generations 1 and 2) are superior, affording more product per unit mass of starting reactants. This was not unexpected, as when moving to generation 3 the excess of tert-butylamine employed was increased from 3.0 to 17.2 equivalents before being reduced back to 4.0 equivalents in generation 4.
ii) The atom economy is modest, but the shortfall is solely resultant from the need to install and subsequently displace a bromine at the alpha-position of 2. Unfortunately, few viable alternative synthetic approaches exist to install the required tert-butylamine.
iii) The process mass intensity (PMI), which is arguably the more important green metric, suggests that generation 4 is more attractive. The initial flow translation and telescoping (generations 2 and 3) performed worse than generation 1, this was also not unexpected as larger unoptimized work-up solvent volumes were utilised; these were subsequently reduced during the optimisation of generation 4.
iv) The productivity of the flow-based approaches was superior with generation 4 affording a 2.6-fold increase in space–time yield over that of generation 1.
v) Finally, in terms of safety we reduced the number of reagents, reactants, and solvents with red or amber health and safety (H&S) codes. The final generation only has an amber flag for the use of tert-butylamine (H331) which cannot be avoided as it is structurally required in the final molecule.
Comparison of raw materials mass distribution
An analysis of the mass of raw materials distribution for the reaction was performed, revealing in the case of both generations 1 and 2 that the organic solvents contributed between 70 and 77% of the total reaction mass suggesting a high waste and cost burden (Fig. 2, see ESI† section 1.8 for detailed breakdown). Translation to generation 3 resulted in a substantial improvement with the organic solvent contribution reducing to 42% of the total mass. This change arose as the extractive work up after stage 1 was avoided, but as noted previously, the overall yield and purity for generation 3 was inferior to that of generations 1 and 2. To circumvent these shortfalls we elected to include an extractive work-up between the first two stages in generation 4. Fortunately, the resulting mass penalty was largely negated through i) the reduction of the work-up solvent masses and ii) by swopping to the use of gaseous hydrogen chloride for the final salt formation.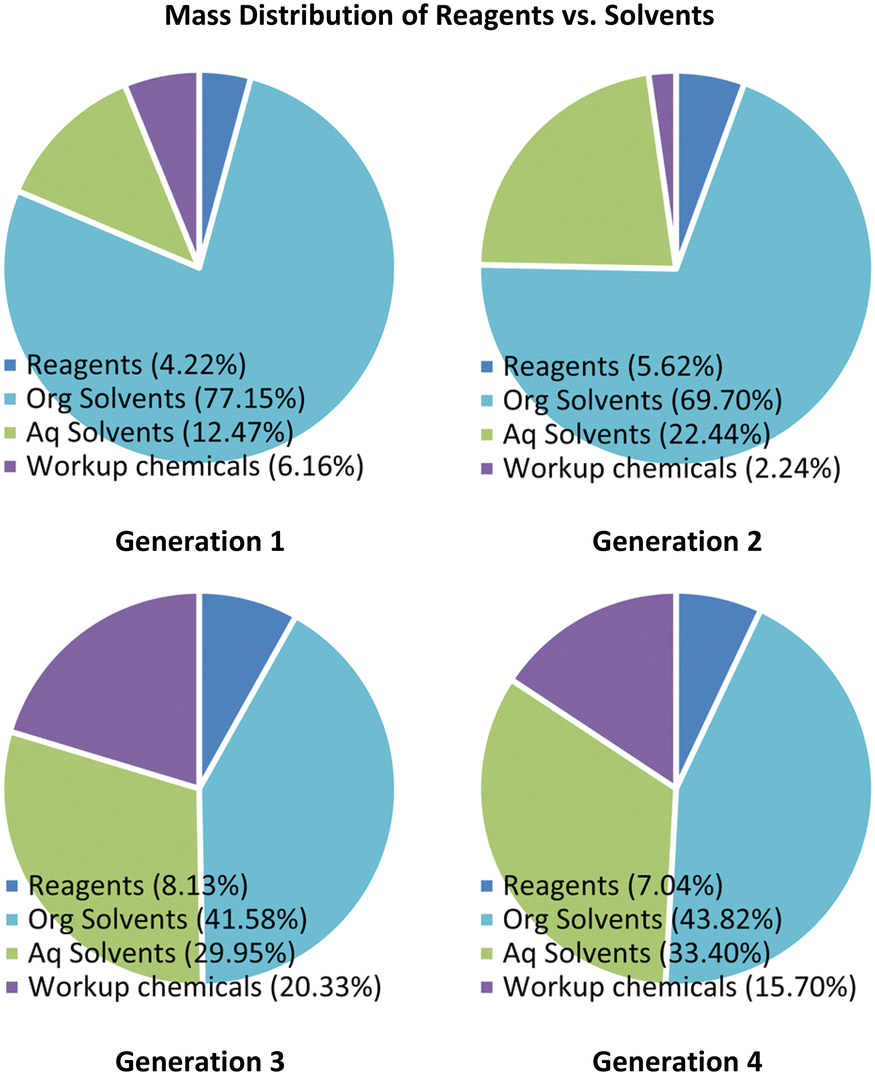 | ||
| Fig. 2 Mass distribution of reagent vs. solvents. | ||
Overall, we believe that generation 4 represents an efficient synthetic process with tangible improvements linked to safety and greenness which are well aligned with eight of the twelve principles of green chemistry (Fig. 3).
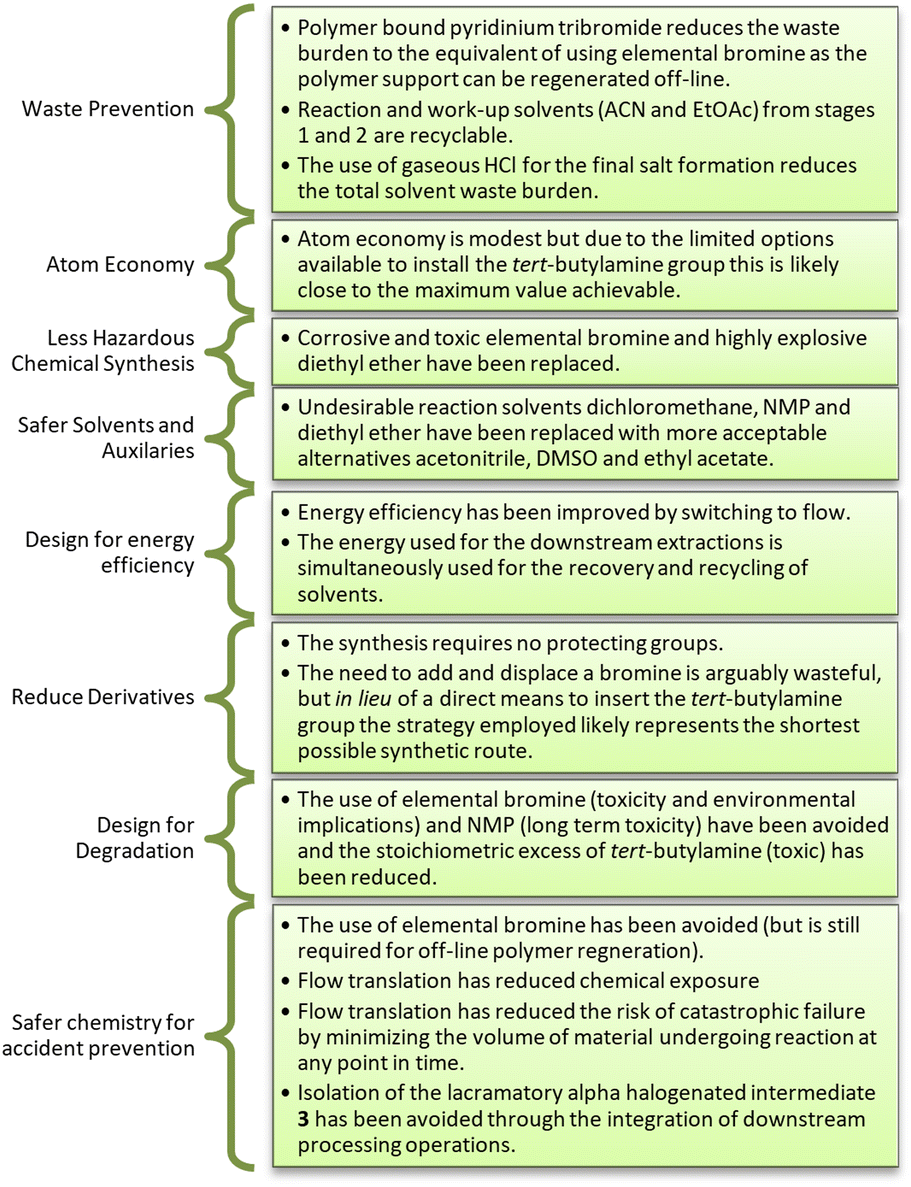 | ||
| Fig. 3 Alignment of generation 4 with eight of the twelve principles of green chemistry. | ||
Comparison of generation 4 with previously reported processes
Finally, the CHEM21 toolkit35 was employed to benchmark generation 4 against several previously reported syntheses of bupropion 1b and bupropion hydrochloride 1a (Table 7, see section 1.9.1 of ESI† for a detailed comparison of the previously reported approaches).| Metric | Burroughs Wellcome9,b | Perrine11 | Ley19,c | Z. A. J. Pharmad | Hurst and Sherwood36 | Rileye,f |
|---|---|---|---|---|---|---|
| a Calculations were performed with the aid of the Chem21 green metrics toolkit, in the case of qualitative variables red = undesirable, amber = acceptable and green = desirable. b Based on in-house validation of the Burroughs Wellcome Co. approach (generation 1), original patent does not have sufficient information for full analysis. c Analysis for the preparation of the free base 1b, preparation of the salt form 1a not reported. d Values in brackets represent those obtained during in-house validation of the process. e Analysis of generation 4, values in brackets represent a scenario including an 80% recovery of ethyl acetate and acetonitrile and use of 2.0 equivalents of HCl (g). f Analysis performed using Br2 as an input for Polymer bound PyBr3 as the spent polymer is not consumed in the reaction and is readily regenerated with Br2. g Offline work-up solvents used as reported for the single flow reactions in the supplementary information of the manuscript. h Combined residence time. | ||||||
| Yield | 71 | 80 | 80g | 78 (21) | 68 | 69 |
| Purity | 96.6 | 98 | NR | ≥99.9 (ND) | NR | 97.8 |
| AE% | 63.1 | 63.1 | 59.7 | 63.1 | 60.5 | 63.1 |
| PMITotal | 135.3 | 174.2 | 566.1g | 17.4 (72.6) | 167.7 | 114.7 (72.9) |
| PMISolv | 132.1 | 170.0 | 563.3g | 13.8 (50.9) | 160.6 | 110.3 (68.5) |
| PMIRRC | 3.2 | 4.2 | 2.9 | 3.6 (21.8) | 7.1 | 4.1 |
| RME | 32.9 | 24.1 | 35.0 | 28.9 (4.7) | 14.2 | 24.5 |
| Reaction time | >4 h | >20 min | 29.7 minh | >8.5 h | 90 min | 45 minh |
| Solvents – stage 1 |

|

|

|

|

|

|
| Solvents – stage 2 |

|

|

|

|

|

|
| Solvents – stage 3 |

|

|
N/A |

|

|

|
| Solvents – work-up |

|

|

|

|

|

|
| Stoichiometric excess |

|

|

|

|

|

|
| Critical elements |

|

|

|

|

|

|
| Energy |

|

|

|

|

|

|
| Batch/flow |

|

|

|

|

|

|
| Work up |

|

|

|

|

|

|
| Health and safety |

|

|

|

|

|

|
In contrast to the approaches by Burrough's Welcome Co., Perrine and Ley the use of molecular bromine and problematic dichloromethane, NMP and diethyl ether were avoided through replacement with more appropriate solvents: acetonitrile (amber), DMSO (green) and ethyl acetate (green). The process still required the use of excess tert-butylamine (an issue experienced in all previously reported approaches), but we were able to restrict this to 4.0 equivalents without any deleterious effects. We also avoided the use of sulphur containing reagents commonly employed to quench brominations (e.g. sodium thiosulfate and sodium metabisulfite) as sulphur is amber flagged in terms of critical elements with an estimate global supply of 50–500 years remaining. The overall PMI of 114.7 is lower than previously reported approaches with the exception of that patented by Z.A.J. Pharma. The value is approaching the desirable value of 100 which is often targeted for pharmaceutical preparations, and if recycling of solvents (acetonitrile and ethyl acetate) is considered this could readily be reduced to a value close to 70. In the case of Z.A.J. Pharma a solvent free approach with a total PMI value of only 17.4 is claimed, however, the approach employs extensive heating for prolonged periods (>8.5 h). This heating was at odds with our observations linked to unwanted dibromination in stage 1 and free base decomposition in stage 2. We elected to validate the approach in-house and were only able to isolate 1a in an overall yield of 21%, notably the first stage showed ∼20% dibromination (see sections 1.4.6 and 1.9.1 of ESI† for additional information).
In terms of safety the staggered continuous flow approach reduced chemical exposure, limited the volume of material reacting at a given time and allowed on-the-fly processing of the lachrymatory intermediate 3. Finally, from an energy perspective the translation to flow is predicted to reduce the overall energy burden relative to the analogous batch processes, and the design also allowed us to utilise the energy required during the solvent swops to recover partially purified ethyl acetate and acetonitrile for recycling.
Conclusion
The synthesis of bupropion hydrochloride 1a has been reimagined and revised in an effort to improve greenness, sustainability and safety. The approach adopted avoided the use of toxic and corrosive molecular bromine and undesirable solvents like dichloromethane, NMP and diethyl ether commonly utilised in previously reported approaches.In summary, the process is comprised of a three-stage staggered flow synthesis affording bupropion hydrochloride 1a in an overall yield of 69% (97.8% purity). The approach afforded an appreciable decrease in process mass intensity relative to previous reports. The approach is well suited for solvent recycling (acetonitrile and ethyl acetate) and the use of integrated downstream processing limits chemical exposure to the preparation of stock solutions and recovery of the final salt 1a.
Critically speaking, it is important to consider that i) the offline regeneration of the polymer supported pyridinium tribromide requires the use of molecular bromine, despite this we feel its use is warranted as it reduces the risk in the primary process as well as the additional waste burden associated with the use of other unsupported alternatives to molecular bromine, and ii) the use of excess tert-butylamine is not desirable from a health and safety point-of-view but due to the mechanistic limitations of the reaction it cannot be reduced significantly while maintaining adequate process performance.
Author contributions
The manuscript was compiled with contributions from all authors. This final version of the manuscript was approved by all authors.Conflicts of interest
The authors declare no competing conflicts.Acknowledgements
This work was supported by the National Research Foundation of South Africa (grant number SRUG190329425006), the University of Pretoria and the Council for Scientific and Industrial Research (CSIR). Opinions expressed in this publication and the conclusions arrived at are those of the authors, and are not necessarily attributed to the NRF or CSIR. The authors would like to gratefully acknowledge Mamoalosi Selepe for NMR spectroscopy services, Madelien Wooding for mass spectroscopy services, Frikkie Malan for SC-XRD services, Solly Motaung for PXRD services, Ncholu Manyala and Vusani Maphiri for raman spectroscopy, Erna van Wilpe for SEM assistance at the University of Pretoria's microscopy laboratory, Uniqsis Ltd. and Vapourtec Ltd. for flow equipment.References
- M. Bares, T. Novak, M. Kopecek, P. Stopkova, J. Cermak, J. Kozeny and C. Höschl, Antidepressant monotherapy compared with combinations of antidepressants in the treatment of resistant depressive patients: A randomized, open-label study, Int. J. Psychiatry Clin. Pract., 2013, 17(1), 35–43 CrossRef CAS PubMed.
- Z. Zuilhof, S. Norris, C. Blondeau, P. Tessier and P. Blier, Optimized regimens of combined medications for the treatment of major depressive disorder: a double-blind, randomized-controlled trial, Neuropsychiatr. Dis. Treat., 2018, 14, 3209–3218, DOI:10.2147/NDT.S175203.
- K. Patel, S. Allen, M. N. Haque, I. Angelescu, D. Baumeister and D. K. Tracy, Bupropion: a systematic review and meta-analysis of effectiveness as an antidepressant, Ther. Adv. Psychopharmacol., 2016, 6(2), 99–144 CrossRef CAS PubMed.
- S. Bai, Q. Hu, Z. Chen, Z. Liang, W. Wang, P. Shen, T. Wang, H. Wang and P. Xie, Brain region-specific metabolite networks regulate antidepressant effects of venlafaxine, RSC Adv., 2017, 7(73), 46358–46369, 10.1039/c7ra08726h.
- H. C. Lee, H. K. Ko, B. E. Huang, Y. H. Chu and S. Y. Huang, Antidepressant-like effects of Perilla frutescens seed oil during a forced swimming test, Food Funct., 2014, 5(5), 990–996, 10.1039/c3fo60717h.
- D. Witteveen and E. Velthorst, Economic hardship and mental health complaints during COVID-19, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(44), 27277–27284, DOI:10.1073/pnas.2009609117.
- D. Mojtahedi, N. Dagnall, A. Denovan, P. Clough, S. Hull, D. Canning, C. Lilley and K. A. Papageorgiou, The Relationship Between Mental Toughness, Job Loss, and Mental Health Issues During the COVID-19 Pandemic, Front. Psychiatry, 2020, 11, 607246, DOI:10.3389/fpsyt.2020.607246.
- D. Posel, A. Oyenubi and U. Kollamparambil, Job loss and mental health during the COVID-19 lockdown: Evidence from South Africa, PLoS One, 2021, 16(3), e0249352, DOI:10.1371/journal.pone.0249352.
- N. B. Mehta, Meta chloro substituted-alpha-butylamino-propiophenones, US Pat., US3819706A, 1974 Search PubMed.
- G. F. Pauli, S.-N. Chen, C. Simmler, D. C. Lankin, T. Gödecke, B. U. Jaki, J. B. Friesen, J. B. McAlpine and J. G. Napolitano, Importance of Purity Evaluation and the Potential of Quantitative 1H NMR as a Purity Assay, J. Med. Chem., 2014, 57(22), 9220–9231, DOI:10.1021/jm500734a.
- D. M. Perrine, J. T. Ross, S. J. Nervi and R. H. Zimmerman, A short, one-pot synthesis of bupropion (Zyban, Wellbutrin), J. Chem. Educ., 2000, 77(11), 1479 CrossRef CAS.
- F. I. Carroll, B. E. Blough, P. Abraham, A. C. Mills, J. A. Holleman, S. A. Wolckenhauer, A. M. Decker, A. Landavazo, K. T. McElroy and H. A. Navarro, Synthesis and biological evaluation of bupropion analogues as potential pharmacotherapies for cocaine addiction, J. Med. Chem., 2009, 52(21), 6768–6781 CrossRef CAS PubMed.
- F. I. Carroll, B. E. Blough, S. W. Mascarella, H. A. Navarro, J. B. Eaton, R. J. Lukas and M. I. Damaj, Synthesis and biological evaluation of bupropion analogues as potential pharmacotherapies for smoking cessation, J. Med. Chem., 2010, 53(5), 2204–2214 CrossRef CAS PubMed.
- L. Vaccaro, D. Lanari, A. Marrocchi and G. Strappaveccia, Flow approaches towards sustainability, Green Chem., 2014, 16(8), 3680–3704, 10.1039/C4GC00410H.
- C. J. Clarke, W.-C. Tu, O. Levers, A. Bröhl and J. P. Hallett, Green and Sustainable Solvents in Chemical Processes, Chem. Rev., 2018, 118(2), 747–800, DOI:10.1021/acs.chemrev.7b00571.
- S. V. Ley and I. R. Baxendale, New tools and concepts for modern organic synthesis, Nat. Rev. Drug Discovery, 2002, 1(8), 573–586, DOI:10.1038/nrd871.
- K. Debak, H. Keskin, A. Yurdakul and N. Ridvanoglu, Process for the preparation of bupropion hydrochloride, PCT, WO2004024674A1, 2004 Search PubMed.
- C. Wu, H. Xiang, X. Yu, C. He, F. Li, X. Shi, C. Lu, G. Chen and Y. Ge, Process for preparing bupropion hydrochloride, US Pat., US7737302B2, 2010 Search PubMed.
- D. E. Fitzpatrick, T. Maujean, A. C. Evans and S. V. Ley, Across-the-World Automated Optimization and Continuous-Flow Synthesis of Pharmaceutical Agents Operating Through a Cloud-Based Server, Angewandte Chemie, 2018, 130(46), 15348–15352 CrossRef.
- D. Cantillo and C. O. Kappe, Halogenation of organic compounds using continuous flow and microreactor technology, React. Chem. Eng., 2017, 2(1), 7–19, 10.1039/c6re00186f.
- Y. Manabe, Y. Kitawaki, M. Nagasaki, K. Fukase, H. Matsubara, Y. Hino, T. Fukuyama and I. Ryu, Revisiting the Bromination of C–H Bonds with Molecular Bromine by Using a Photo-Microflow System, Chem. – Eur. J., 2014, 20(40), 12750–12753, DOI:10.1002/chem.201402303.
- P. Löb, V. Hessel, H. Klefenz, H. Löwe and K. Mazanek, Bromination of Thiophene in Micro Reactors, Lett. Org. Chem., 2005, 2(8), 767–779 CrossRef.
- Q. Deng, R. Shen, R. Ding and L. Zhang, Bromination of Aromatic Compounds using Bromine in a Microreactor, Chem. Eng. Technol., 2016, 39(8), 1445–1450, DOI:10.1002/ceat.201400723.
- F. E. A. Van Waes, S. Seghers, W. Dermaut, B. Cappuyns and C. V. Stevens, Efficient Continuous-Flow Bromination of Methylsulfones and Methanesulfonates and Continuous Synthesis of Hypobromite, J. Flow Chem., 2014, 4(3), 118–124, DOI:10.1556/jfc-d-14-00006.
- R. Becker, S. A. M. W. van den Broek, P. J. Nieuwland, K. Koch and F. P. J. T. Rutjes, Optimisation and Scale-up of α-Bromination of Acetophenone in a Continuous Flow Microreactor, J. Flow Chem., 2012, 2(3), 87–91, DOI:10.1556/jfc-d-12-00007.
- T. Fukuyama, M. T. Rahman, N. Kamata, M. Tokizane, Y. Fukuda and I. Ryu, Continuous Microflow Bromination of Alkenes Combined with a Circulatory Recycling of a Fluorous Polyether as a Bromine Support, J. Flow Chem., 2013, 3(1), 4–6, DOI:10.1556/jfc-d-12-00023.
- D. Cantillo, O. de Frutos, J. A. Rincon, C. Mateos and C. O. Kappe, A scalable procedure for light-induced benzylic brominations in continuous flow, J. Org. Chem., 2014, 79(1), 223–229, DOI:10.1021/jo402409k.
- D. Cantillo, B. Gutmann and C. Oliver Kappe, Safe generation and use of bromine azide under continuous flow conditions--selective 1,2-bromoazidation of olefins, Org. Biomol. Chem., 2016, 14(3), 853–857, 10.1039/c5ob02425k.
- D. Šterk, M. Jukič and Z. Časar, Application of Flow Photochemical Bromination in the Synthesis of a 5-Bromomethylpyrimidine Precursor of Rosuvastatin: Improvement of Productivity and Product Purity, Org. Process Res. Dev., 2013, 17(1), 145–151, DOI:10.1021/op300248y.
- M. Vögtle and B. Leforestier, Safe Generation and Direct Use of Chlorine Azide in Flow Chemistry: 1,2-Azidochlorination of Olefins and Access to Triazoles, Synlett, 2016, 27(13), 1957–1962, DOI:10.1055/s-0035-1561659.
- A. Nagaki, K. Imai, S. Ishiuchi and J. Yoshida, Reactions of difunctional electrophiles with functionalized aryllithium compounds: remarkable chemoselectivity by flash chemistry, Angew. Chem., Int. Ed., 2015, 54(6), 1914–1918, DOI:10.1002/anie.201410717.
- Z. Wang, Z. Li and W. Bao, Nucleophilic Substitution Reactions in Ionic Liquid: Towards a Real Continuous-Flow Process for Synthesis of Alkyl Bromides and Cyanides, Lett. Org. Chem., 2007, 4, 72–74 CrossRef CAS.
- G. W. Amarante, M. Cavallaro and F. Coelho, Hyphenating the curtius rearrangement with Morita-Baylis-Hillman adducts: synthesis of biologically active acyloins and vicinal aminoalcohols, J. Braz. Chem. Soc., 2011, 22, 1568–1584 CrossRef CAS.
- N. C. Neyt and D. L. Riley, Batch–flow hybrid synthesis of the antipsychotic clozapine, React. Chem. Eng., 2018, 3(1), 17–24, 10.1039/c7re00146k.
- C. R. McElroy, A. Constantinou, L. C. Jones, L. Summerton and J. H. Clark, Towards a holistic approach to metrics for the 21st century pharmaceutical industry, Green Chem., 2015, 17(5), 3111–3121, 10.1039/c5gc00340g.
- O. B. Andrew, J. Sherwood and G. A. Hurst, A Greener Synthesis of the Antidepressant Bupropion Hydrochloride, J. Chem. Educ., 2022, 99(9), 3277–3282, DOI:10.1021/acs.jchemed.2c00581.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3re00443k |
| This journal is © The Royal Society of Chemistry 2024 |