
Synthesis of small protein domains by automated flow chemistry†
Kristóf
Ferentzi
 ab,
Dóra
Nagy-Fazekas
ab,
Viktor
Farkas
bc and
András
Perczel
*bc
ab,
Dóra
Nagy-Fazekas
ab,
Viktor
Farkas
bc and
András
Perczel
*bc
aHevesy György PhD School of Chemistry, Institute of Chemistry, Eötvös Loránd University, Pázmány Péter sétány. 1/A, Budapest, H-1117, Hungary
bLaboratory of Structural Chemistry and Biology, Institute of Chemistry, Eötvös Loránd University, Pázmány Péter sétány. 1/A, Budapest, H-1117, Hungary
cHUN-REN-ELTE Protein Modeling Research Group, Institute of Chemistry, Eötvös Loránd University, Pázmány Péter sétány. 1/A, Budapest, H-1117, Hungary. E-mail: perczel.andras@ttk.elte.hu
First published on 20th November 2023
Abstract
The most fundamental topological units of proteins are their autonomously folded domains. The rapid and reliable chemical synthesis of domains in the range of 5–10 kDa in size, remains a challenge. Their bacterial expression is cumbersome, especially when chemical changes, post-translational modifications or the incorporation of non-natural residues are involved. Here, we report an in-house flow-chemistry-based synthetic method that enables one-step, fully automated synthesis of small protein domains without native chemical ligation. Our improved protocol is more efficient, using only 3 equivalent reagents and small amounts of organic solvents (6 ml per cycle) at a scale of 36 μmol. First, we tested the limits of our system with oligotuftins of increasing length, H-(TKPKG)k-NH2 (4 ≤ k ≤ 16), a polypeptide composed of five residue long repeats. Second, four very different small single domain proteins were selected, each representing one specific 3D-fold type. Z amyloid binding affibody 3, Z(Aβ3), is of α-fold, while SRC homology 3 domain (SH3) is a representative of the all β-stand fold. Bovine pancreatic trypsin inhibitor (BPTI) is an example of α + β, while human ubiquitin (UBI) is that of the α/β type domains. Our protocol was developed by optimizing concentration, flow rate, solvent composition, coupling time and reaction temperature allowing the overnight chain assembly (e.g. 7.4–8.6 hours) of a protein such as BPTI. Our smart peptide chemistry in flow (SPF) protocols are versatile and can be successfully applied to produce not only small protein modules, but also their chemical variants such as foldamers, or chimeras in moderate yields providing the synthetic background for current academic and specific pharmaceutical research.
Introduction
In the era of biosimilars, there is a great demand from both academic research and the efficiency-driven pharmaceutical industry for designed and tailored polypeptides and proteins of great diversity. The production of proteins of the most typical size range (200–400 residues in length) has long been successfully carried out by recombinant expression in various bacterial, yeast, insect or even human host cells, but it is far from being a fail-safe method for all. The synthesis of pharmacologically relevant smaller proteins and oligopeptides has remained a challenge, as they are often rapidly digested or silenced by the host when expressed directly. In these cases, chimera or fusion expression systems are used in which the smaller proteins of interest are protected and/or solubilised by another stable partner protein. However, selecting a suitable partner and designing a suitable linker and cleavage site between the two can be difficult. Another common challenge is that pharmaceutical proteins may contain modified or non-natural amino acids, making their expression particularly difficult and uneconomical.1–3 In these cases, chemical synthesis using automated synthetic cycles (involving amino acid activation, coupling and cleavage of the protecting groups) may therefore be useful.Conventional solid-phase peptide synthesis (SPPS) reaches its limits at chain lengths of 40–50 residues, where lower conversion yields per cycle and difficult sequence motifs pose problems. However, larger proteins can be obtained using this method by applying fragment condensation and native chemical ligation (NCL) to join two or more separately synthesised chains.4,5 However, fragment condensation has limitations, for example the individual purification of all fragments and their solubility. Though other ligation methods were also published,6 a typical NCL protocol requires Cys residues. Despite of the advances in the Fmoc synthesis of thioesters, their production still remains a challenge.7–9 Thus, in the case of NCL, using Boc-chemistry would be the first choice, but it is not suitable for flow based automated techniques.10 Synthetic methods that take into account the subsequent purification steps often apply the acetyl capping option after each and every coupling step,11–13 increasing protocol times.
One of the most exciting advancements of peptide chemistry in recent decades has been the rediscovery of flow-based synthesis,14–22 which offers many advantages over conventional, batch reactor-based peptide synthetic techniques. Effective reagent mixing, temperature and pressure control and a high degree of automation with the in-line monitoring options of the reactions are just a few among them23,24 (although – in the field of automation – serious developments were implemented in batchwise synthesis area recently as well).25 In a pioneering paper Hartrampf et al. showed that even the synthesis of a protein (e.g., 164 aa. long) can be carried out in a flow system within a few hours, reducing cycle time to ∼2.5 minutes, although the purity of the published raw materials requires excessive and thorough purification steps.26,27 However, the excess reagent required per coupling cycle varies from 40 to 190 equivalents, which increases production costs, generates a significant amount of waste and represents a serious environmental burden, making this method less economical and certainly not environmentally friendly.
The recent emergence of more efficient and controlled methodologies provides a context for optimising peptide synthesis processes to make them faster, cheaper and more environmentally friendly. With a focus on efficiency, keeping costs as low as possible and making amino acid coupling increasingly environmentally friendly, we are continuously developing both the hardware of the synthesiser and the methods used for SPPS. This complex approach is what we call smart peptide chemistry in flow, or SPF for short. In this sense, SPF is not just an instrument that can be built from relatively simple and easy-to-develop HPLC modules and modified to meet new challenges, nor is it just a synthesis that is under continuous development. It is a combined strategy of both methods and approaches that takes into account the size of the environmental footprint as well as the length of the synthesis time, the amount of reagents and the type of solvent, all of which are optimised at the same time. In this work we aimed to produce representative protein domains within this framework.21
The apparatus behind SPF is an HPLC based, easy-to-assemble, modular hardware (a single pump, an auto-injector, a heated column with the resin) with some recent modifications, such as a UV-detector to monitor coupling and Fmoc-cleavage efficiency (Fig. 1a and S1†). We have chosen 4 + 1 proteins for synthesis: Z amyloid binding affibody 3 (Z(Aβ3)) is an α-helical small protein, SRC homology 3 domain (SH3) is of β-strand fold,13,28 bovine pancreatic trypsin inhibitor (BPTI) is an example of the α + β fold-type whereas human ubiquitin (UBI) is an α/β-type single domain protein.29,30 As reference, the completely unstructured oligotuftsin proteins were used. Proteins can be made up of one, sometimes more, and occasionally several domains. However, ∼1/3 of bacterial, and ∼1/4 of eukaryotic proteins consist of a single and relatively small domain, that folds autonomously in water. The four folded proteins we selected for synthesis sample the four basic domain types, those of the α-, β-, α + β- and α/β-types.31
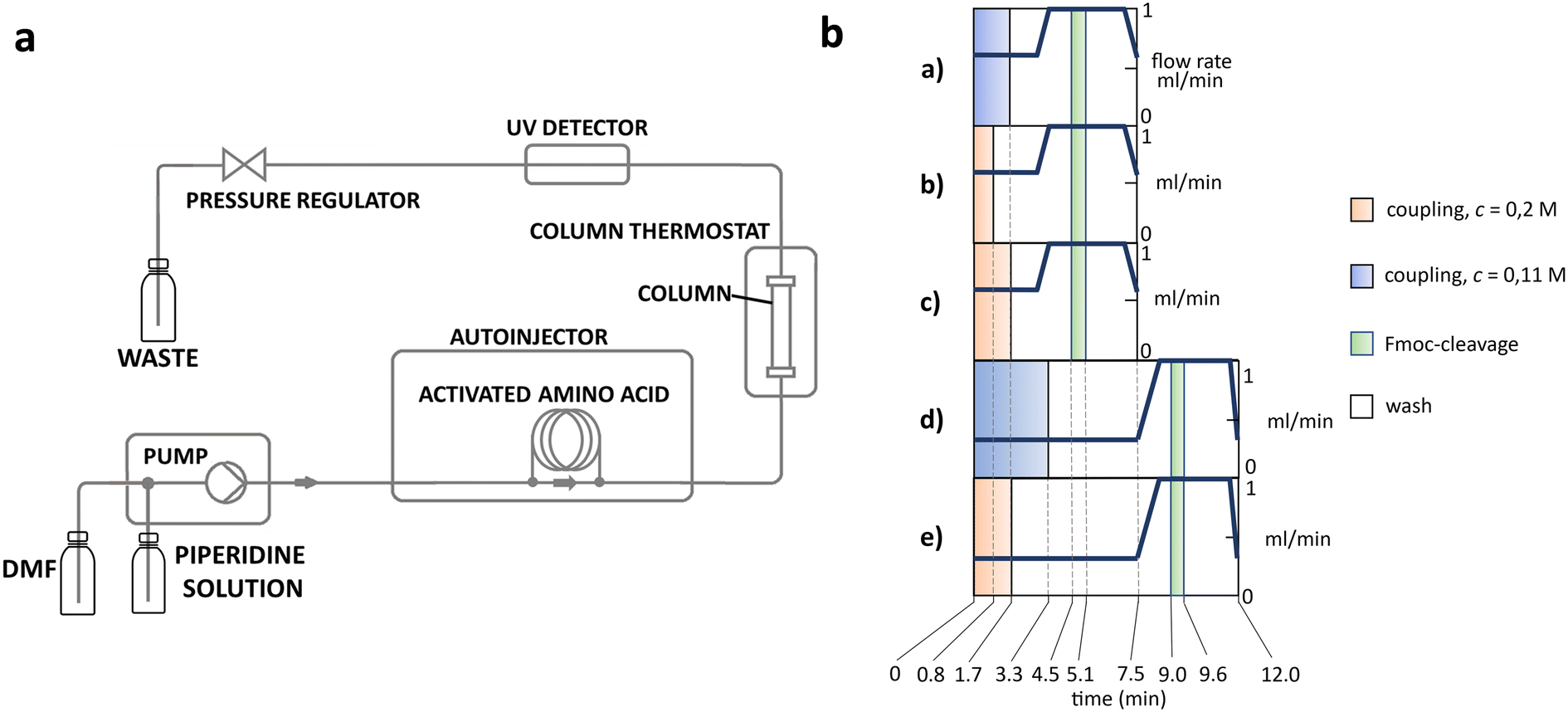 | ||
Fig. 1 a) Layout of the system. A typical SPF synthesis is carried out using the setup shown in panel b): coupling: AA/DIC/oxyma (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2), Fmoc-cleavage: 30% piperidine, T = 80 °C, p: 60–80 bar, TentaGel S RAM resin (0.24 mmol g−1) 130–150 mg. b) Diagrams of the different protocols used for coupling cycles. T = 80 °C, p: 60–80 bar. Protocols a, b, d and e use ∼3 equivalents, while protocol c: ∼6 equivalent of reagents for 150 mg RAM TG resin (0.24 mmol g−1). The five different SPF protocol schemes used, are referred to as protocol a–e. All five protocols consist of the 4 basic elements: coupling, washing, Fmoc-cleavage and washing. While the basic protocols a–c have a total cycle length of 7.5 min, v = 0.6 ml min−1 and c ∼0.11 M, those of protocols d and e are extended by 4.5 min as their flow rate is half of the former ones: v = 0.3 ml min−1. Protocol a and c are identical, but the latter uses higher reagent concentration: c ∼0.2 M. Protocol b uses the same amount of reagent as protocol a, but in a more concentrated form. :1:2), Fmoc-cleavage: 30% piperidine, T = 80 °C, p: 60–80 bar, TentaGel S RAM resin (0.24 mmol g−1) 130–150 mg. b) Diagrams of the different protocols used for coupling cycles. T = 80 °C, p: 60–80 bar. Protocols a, b, d and e use ∼3 equivalents, while protocol c: ∼6 equivalent of reagents for 150 mg RAM TG resin (0.24 mmol g−1). The five different SPF protocol schemes used, are referred to as protocol a–e. All five protocols consist of the 4 basic elements: coupling, washing, Fmoc-cleavage and washing. While the basic protocols a–c have a total cycle length of 7.5 min, v = 0.6 ml min−1 and c ∼0.11 M, those of protocols d and e are extended by 4.5 min as their flow rate is half of the former ones: v = 0.3 ml min−1. Protocol a and c are identical, but the latter uses higher reagent concentration: c ∼0.2 M. Protocol b uses the same amount of reagent as protocol a, but in a more concentrated form. | ||
An important feature of our chosen set of proteins is the diversity of possible dipeptide sequences encountered during their synthesis, providing a versatile testing ground for our methodology (Fig. S2†). The success of any coupling reaction depends on several factors beyond the nature of the two residues to be coupled, such as the side-chain topology of the adjacent and fully protected amino acids, the length of the growing polypeptide chain, the type of resin and the solvent chosen. Self-association, on-resin aggregation, molecular crowding are serious challenges to overcome.32–34 Each characteristic fold type is longer than the typical 40 amino acid length-limit for SPPS (Table 1).
| Name | Z(Aβ3)a | SH3b | BPTIc | UBId | Oligotuftsine |
|---|---|---|---|---|---|
| a Lindgren, J.43 b Mende, F.28 c Ferrer, M.44 d El Oualid45 e Mező46–47 f Based on resin quantity and capacity used. | |||||
| Domain type |
 α α |
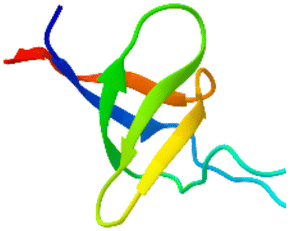 β β |
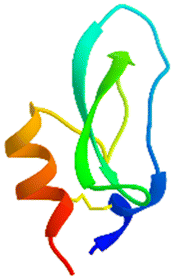 α + β α + β |
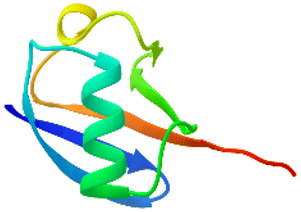 α/β α/β |
 Dynamic reference system Dynamic reference system |
| Total residue number | 58 | 61 | 58 | 76 | 5–40 |
| M av. (Da) | 6307.08 | 7129.83 | 5979.85 | 8433.66 | 2063.52–4107.54 |
| PDB code | 2B89 | 2VKN | 3AUB | 1UBQ | — |
| Total synthesis time as published | 33 h | 81.3 h | 134.3 h | 98 h | 10–70 h |
| HPLC purified yield as publishedf | 8% | Not provided | 2% | 14% | 80–60% |
To fine-tune our protocol, we monitored and analysed the HPLC profile of the raw materials to assess their suitability for conventional HPLC purification. The main source of by-products is ineffective coupling. Several factors exacerbate this problem, such as the amino acid composition of the primary sequence, the hydrophobic residue content of the protein, the distribution and topology of bulky and lipophilic side chain protecting groups, the presence of β-branched residues, local aggregation tendencies, solvent, temperature and resin type.32–35 For example, it was shown, that the crude peptide purity is better if a PEG based resin is used and thus, purity increases with the PEG content of the polymeric support.15 Chain mobility, which facilitates the release of previously aggregated peptide chains, is increased by the higher temperature applied. However, heating can also be a stimulus for side reactions.36,37 The use of chaotropic solvents, polyethylene type sidechain protecting groups, pseudoproline derivatives, depsipeptides and N-alkyl amino acids were shown to improve synthetic results.38–47 As residues are coupled step by step, the properties of the polymer matrix also change, creating the need for residence time control (Fig. S3†). Controlling residence time has been shown to be critical in continuous flow chemistry because – by optimising flow rate and concentration – a higher conversion can be achieved without increasing the reagent excess.
On-resin type aggregation can occur by a number of different mechanisms, hindering coupling and jeopardising success. Intramolecular interactions can cause the polypeptide chain to adopt secondary structures such as backbone folds. Intermolecular interactions can lead to the formation of interconnected, ordered structures that may mask the N-terminal amine of the growing chain, making subsequent coupling difficult. These properties of the resin vary with the nature of the polymeric matrix building up the solid phase. For example, even shorter oligopeptides tend to form aggregates on 100% polystyrene (PS) resin, whereas a 30–40 residue long polypeptide may escape a similar fate if a PEG-grafted PS (TentaGel) resin is used. The use of a 100% PEG based ChemMatrix resin can minimise these unwanted interactions. However, ChemMatrix resins swell to an unfavorable extent, which is a serious obstacle in the use of a fixed volume column. Here we propose a machine and a methodology that, despite the challenges involved, can be successfully applied to synthesise a wide variety of small single domain proteins.
Results and discussion
Previously, as an example, we have successfully synthesized the 51 amino acid long polypeptide that inhibits human myostatin.21,48,49 We optimized the coupling reaction, by modifying flow rate (v), reagent concentration (c) and reagent equivalent. Five different methods were generated to map and provide the necessary alternatives (Fig. 1b).To test the synthetic performance of our SPF protocols, oligotuftsins (OT) of different lengths were used as reference polypeptides. The reference oligopeptides we selected are modular and tunable in length, but lack stable secondary structure, in contrast to folded protein domains.50,51 The canine OT repeat unit, -TKPKG-, contains a Pro residue with structure disrupting properties, making OT less susceptible to the unwanted on-resin aggregation. We will refer to the different oligotuftsin systems as OT(P)x where x is the number of residues making up a given variant. The central residue of the repeat sequence is shown in brackets (Table 2).
| Abbreviation example | Repeating unit | Number of repeating unit | Number of residues |
|---|---|---|---|
| OT(P)20 | -TKPKG- | 4 | 20 |
| OT(V)20 | -TKVKG- | 4 | 20 |
| OT(P)40 | -TKPKG- | 8 | 40 |
The -TKPKG- segment is ideal as a reference unit, as it contains a small (Gly), a branched and bulky (Thr(tBu)), an unbranched and longer (Lys(Boc)) and a secondary amine group comprising the Pro amino acid residue. Furthermore, as the central Pro has a fixed dihedral angle, φPro (∼70°), it partially restrains the internal flexibility of this pentapeptide unit, hindering the peptide to adapt a stable conformation. This feature, repeated at every 5 residues, can suppress both inter- and intramolecular aggregation, even for longer polypeptides, such as OT(P)70. The synthetic performance of the SPF basic protocol a was tested producing OTs of different lengths, in the OT(P)20–OT(P)80 range. Using SPF protocol a (tR = 3.3 min, c = 0.11 M), we were able to efficiently incorporate the first 40–45 amino acids, beyond this size (entering the size-range of our targeted folded domains) the purity of the main product decreased significantly (Fig. 2a and 3 orange line, with respect to the red dashed line). The HPLC chromatograms of the raw OT(P)20, OT(P)30 and OT(P)40 are primarily composed of the main products (Fig. 2a), those of OT(P)50 to OT(P)80 show signs of considerable impurities, although the main product peaks remain sharp and distinguishable (Fig. S5–S11†). As the PEG chains are responsible for preventing the peptide from adopting a stable conformation through solvation, when the length of a nascent polypeptide chain exceeds the length of the PEG chains, the protective power of the PEG drastically decreases. A linear regression (R2 = 0.969) was established between the main product yield and the polypeptide length of OT(P)20–OT(P)50 (Fig. 3 dashed red line), which, extrapolated up to OT(P)80 clearly indicates the decreasing yield. Data collected both during the synthesis and after LC-MS show (see ESI†) that the truncated OTs are Lys-deficient polypeptides. Lys has a long side chain, especially when protected by the bulky and hydrophobic Boc group, which might result slightly lower diffusion rate and slower acylation especially when the peptide chains are longer. This is manifested in small but systematic Lys-loss, especially when the nascent peptide chain grew longer.
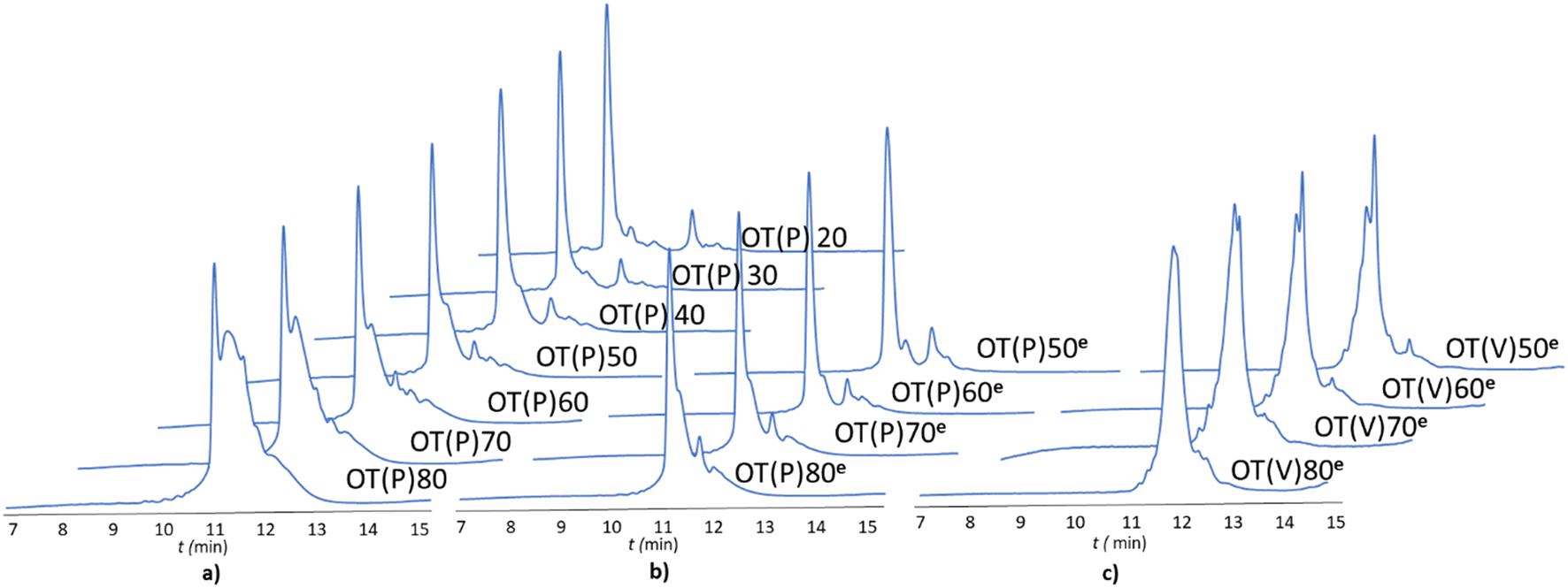 | ||
| Fig. 2 The RP-HPLC chromatograms of the crude oligotuftsin polypeptides, OT(P)x: = [TKPKG]k, where k = 4 for OT20, k = 10 for OT50 and k = 16 for OT80 and x indicates the number of residues. a) SPF protocol a was applied for OT(P)20, …, OT(P)80: c = 0.11 M, v = 0.6 ml min−1, tR = 1.67 min. b) Thr and Gly residues were coupled with protocol a, while Lys and Pro were coupled with protocol e to give OT(P)50e, …, OT(P)80e: c = 0.22 M, v = 0.3 ml min−1, tR = 3.3 min. c) For the synthesis of OT(V)xe polypeptides, [TKVKG]k 10 < k < 16, protocol a was used for all residues, except Lys, for which protocol e was applied. | ||
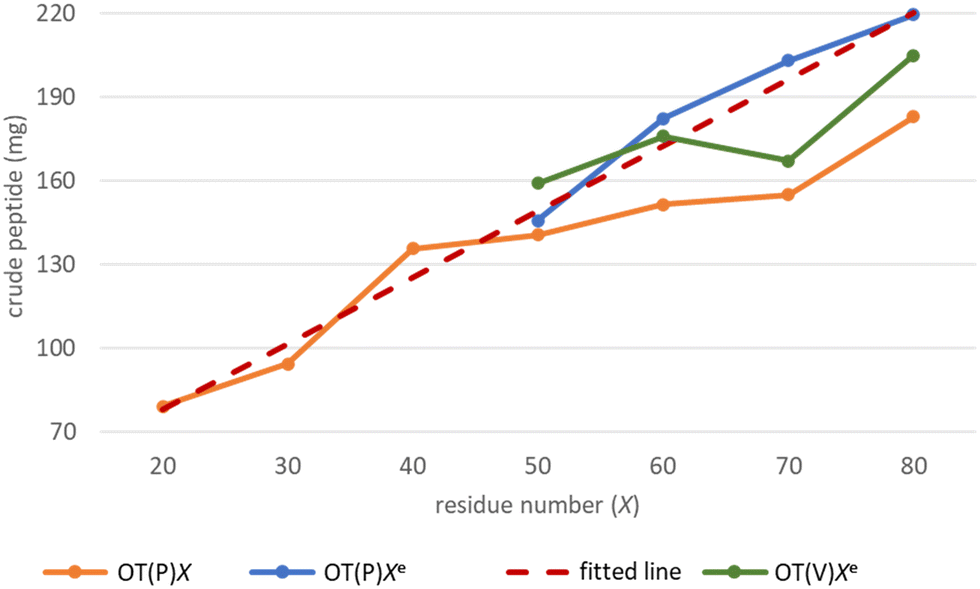 | ||
| Fig. 3 The main product yields of the OT(P)x, -[TKPKG]k-, synthesized by SPF protocol a as a function of the polypeptide length, OT(P)20–OT(P)80: orange line. Red dashed line indicates the established linear correlation based on OT(P)20–OT(P)50. Pearson correlation coefficient, R2 = 0.969. Improved main product yield for OT(P)50e–OT(P)80e (blue line) using protocol e, when coupling both Lys and Pro residues. OT(V)50e–OT(V)80e main product -[TKVKG]10<k<16- yields (green line) using protocol e, when coupling Lys residues. | ||
The above challenges were solved by simultaneously changing both flow rate and concentration, i.e. replacing protocol a with protocol e for coupling Lys and Pro (Fig. 2b). By reducing the flow rate and doubling the reagent concentration, the same residence time on the resin was obtained, but at a higher concentration. The positive effect of protocol e on overall yield and purity is evident (Fig. 2b/blue line). In order to determine the capabilities of protocol e independently of the anti-aggregation protection of the regularly appearing prolines, new OT(V)x was also synthesised placing Val in the center of the repeating unit. A comparison of the crude peptide yields of OT(V)x with those of OT(P)x shows that there is a clear decrease in the purity of the main product (Fig. 2 and 3/green line). Indeed, the profiles of the HPLC chromatograms of the longest OT(V)x (x > 60) show more by-products and predict more difficult purifications (Fig. S16–S19†), confirming the positive role of Pro-like kinks of the backbone for the successful synthesis of longer polypeptide chains by solid-phase peptide synthesis.40 In other words, the most successful synthesis is expected for those protein domains that have more Pro residues. After the first 40 coupling steps, the nascent polypeptide chain gradually becomes more prone to aggregation.
In line with this, the SPF synthesis of BPTI is expected to be the easiest of those we have selected, as it is rich in prolines after the 40th coupling (P2, P8, P9 and P13) and therefore less prone to aggregation. Therefore, it is safe to assume that if BPTI-SPF synthesis fails for any reason, it will not be due to primary sequence and aggregation problems, but to stepwise coupling inefficiencies. Since our focus in the present SPF synthesis of BPTI was to optimise the main product with respect to all by-products, we synthesised a variant of BPTI containing Cys mutants with only one disulfide bridge (PDB: 3AUB). The performance of SPF is demonstrated by the protocols examined and compared (Tables 3 and S3 and Fig. S20–S26†). As there are 4 prolines in BPTI after the 40th coupling, which are mostly homogeneously distributed, we assumed that faulty sequences were due to ineffective acylation rather aggregation on the resin. The Fmoc cleavage data collected by the in-line UV detector as a function of primary sequence (Fig. 4) show that acylation is typically poorer when coupling with Arg(Pbf), Lys(Boc), Gln(Trt), Pro and Asn(Trt) residues, and does not indicate sequence dependent aggregation. The lower reactivity of Lys(Boc) was previously observed for OTs and we proposed that this was related to the size of the side chain. A similar conclusion can be drawn for Arg, but also for the bulkier Trt-protected Asn, Gln, Cys and His residues. Having successfully overcome this challenge for OTs, we have also overcome this problem for BPTI by fine-tuning both the concentration and the flow rate of the SPF protocols (Table 3). The mainstream approach to solve acylation inefficiency would be to increase the molar equivalent used for coupling,26 which is neither environmentally friendly, nor cost effective. Reducing the flow rate and using a higher reagent concentration in a smaller volume has been successfully tested and now gives a better crude product for BPTI as well. This appears to be an effective compromise between synthesis time and efficacy, as the best and most economical result was obtained with protocol e, which gave a readily purifiable crude product requiring only a small increase in synthesis time (Tables 3 and S3,† compare row 4 to 1 and Fig. 5). It is worth noting that protocol c gives a crude product of similar purity and yield and also reduces the synthesis time for BPTI, but the gain is only about one hour (Fig. S25 and S26†). However, this approach is more costly and less environmentally friendly as the faster route requires the use of 6 equivalent reagents instead of 3.
| # | Protocol | Residence time: tR (min) | Concentration: c (M) | Flow rate: v (ml min−1) | Equiv. | Time (h) | Crude yield (%) |
|---|---|---|---|---|---|---|---|
| 1 | a | 1.67 | 0.11 | 0.6 | 3 | 7.4 | 49% |
| 2 | b | 0.83 | 0.2 | 0.6 | 3 | 7.4 | 59% |
| 3 | c | 1.67 | 0.2 | 0.6 | 6 | 7.4 | 63% |
| 4 | d | 3.34 | 0.11 | 0.3 | 3 | 8.6 | 61% |
| 5 | e | 1.67 | 0.2 | 0.3 | 3 | 8.6 | 62% |
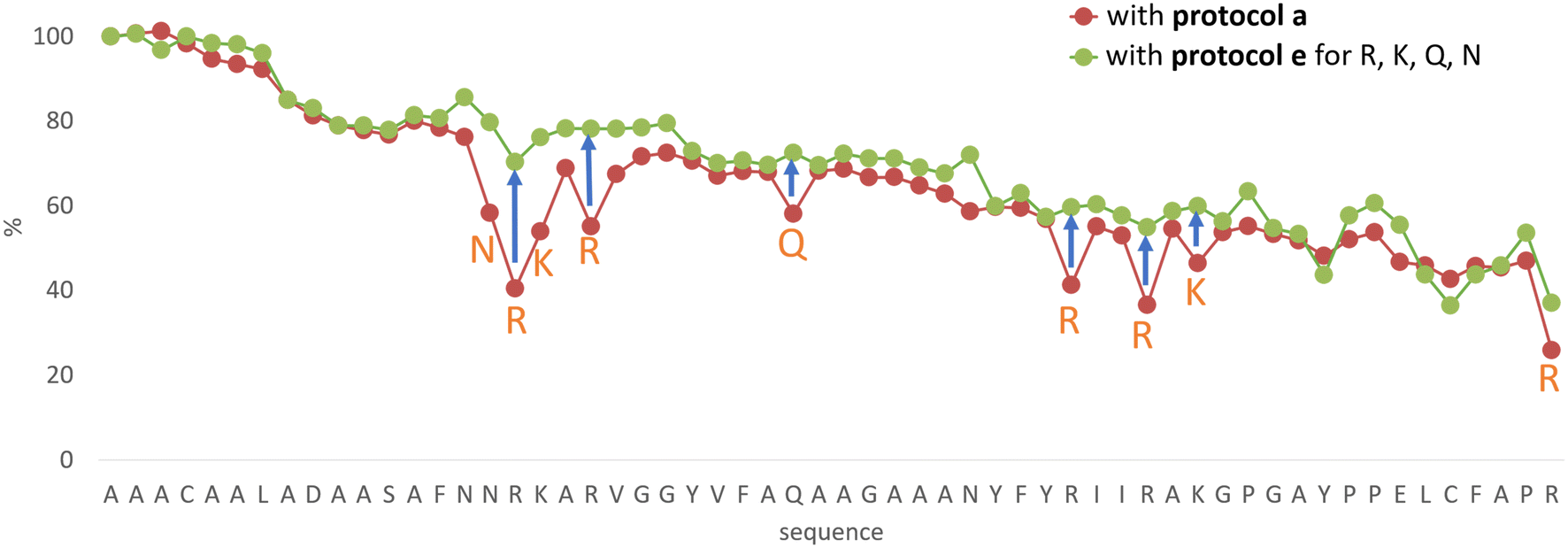 | ||
| Fig. 4 The Fmoc-cleavage area (see text and Fig. S2†) changing as a function of the primary sequence, recorded for the synthesis of BPTI, using protocol a (brown dots). The synthesis of BPTI was repeated using protocol e (green dots) for the highlighted amino acids only. Note that the Fmoc-cleavage area (%) increases when protocol e is used, making the overall trend more “linear”. The systematic decrease of the Fmoc-cleavage area is thought to be the result of resin leakage. | ||
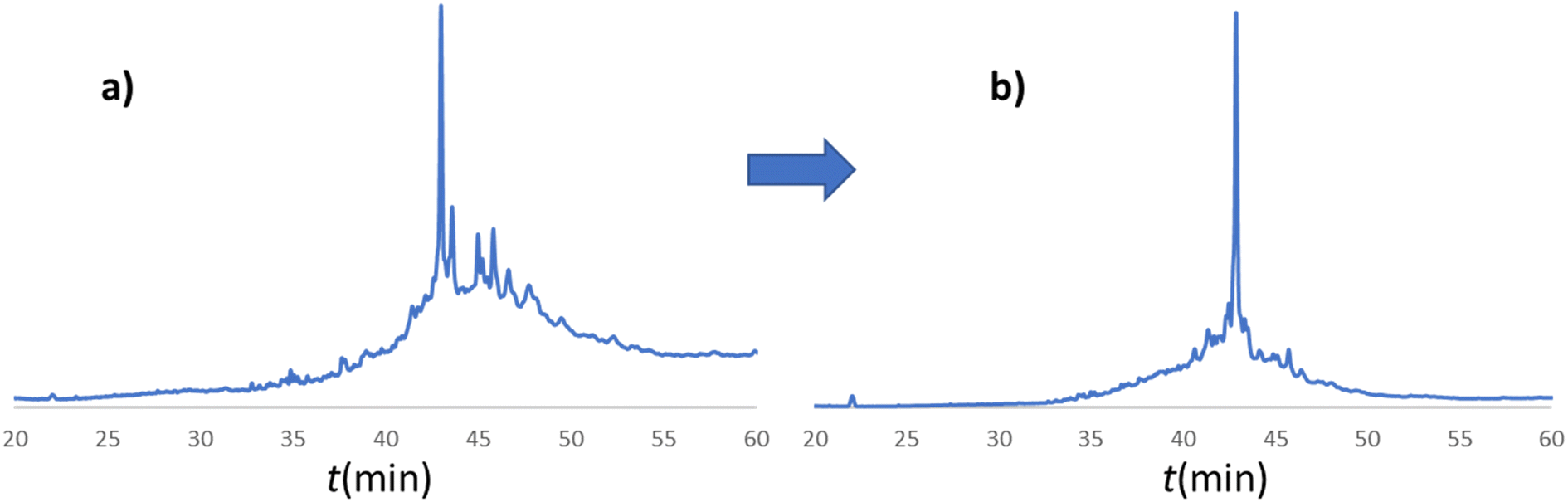 | ||
| Fig. 5 a) RP-HPLC chromatogram of the crude BPTI synthesized with protocol a (Table 3/1): c = 0.11 M, tR = 1.67 min, T = 80 °C, v = 0.6 ml min−1. b) RP-HPLC chromatogram of the crude BPTI synthesized with the optimized protocol e (Table 3/5): c = 0.22 M, tR = 1.67 min, T = 80 °C, v = 0.3 ml min−1. The major peaks correspond to the target molecule. The significant reduction in by-product formation can be attributed to the change in coupling protocol for the specified amino acids (R, K, Q, N, E, P) from protocol a to protocol e. | ||
The optimization of the BPTI synthesis highlighted, that supposedly most of the coupling deficiency likely to be caused by the ineffective acylation, as most of the deletion is amino acid and not sequence specific. Major byproducts are not considered to be responsible for aggregation, as unsatisfactory acylation conversion would be detected regardless of the type of the amino acid and dependent on the primary sequence. In the case of on-resin peptide interaction systematic coupling inefficiencies would appear on the Fmoc-cleavage area plot as consecutive deviation from the trendline (Fig. 4). This highlighted the need for a different coupling protocol for protected amino acids that are larger (protocol e), but this method might not solve the possible acylation inefficiencies caused by on-resin peptide chain interactions. Based on the results and conclusions of the oligotuftsin synthesis series (Fig. 2), the production of the different fold types other than α + β (BPTI) was expected to be more challenging because the selected domains are not proline-rich around the N-terminus (40+ couplings). The less-than ideal number and placement of proline residues in the other protein domains suggests that they are likely to be more prone to aggregation and thus more difficult to produce. This presumption is also supported by Peptide Companion, an empirical software program designed to predict coupling difficulties in a peptide sequence (Fig. S4†).52
On-resin aggregation can interfere with the free N-terminals, making coupling challenging.32–35 To avoid peptide aggregation in SPPS, the use of chaotropic agents has been suggested in peptide chemistry protocols, namely urea, guanidine hydrochloride, DMSO, n-propanol, SDS, lithium perchlorate.38,39 To verify that aggregation was indeed the cause of the unsatisfactory conversion of the acylation step, each of the 4 selected systems was synthesized in DMF both without and with the addition of denaturing agents. We used the optimized conditions from the synthesis of BPTI: protocol e for R, K, Q, N, E, P and protocol a for the rest of the amino acids. Instead of using solid chaotropic agents (e.g. urea), we decided to use co-solvents. The solid denaturants must be used in high excess, which would increase the viscosity of the solution and result in slower diffusion rates and it has the potential for crystal formation and consequently occlusion of the system which would result high back pressures, thus we avoided their use. We introduced both DMSO and MeCN as chaotropic solvent additives at 25 V/V% amount to DMF.
In the case of BPTI, when chaotropic solvent additives are applied, changes can be observed in the chromatograms of the raw materials compared to those synthesised in DMF alone (Fig. 6), but to a lesser extent compared to the coupling optimization of the bulky amino acids (Fig. 5). This is consistent with our assumption that no systematic aggregation occurs during the synthesis of BPTI. As expected, the crude products of SH3 and Z(Aβ3) show more contamination compared to BPTI, and the effect of DMSO and MeCN is more pronounced. A larger area of contamination peaks disappears from the chromatogram (Fig. 6). While the synthesis of BPTI, Z(Aβ3) and SH3 in the absence of chaotropic agents resulted in raw materials in which the main peak is the target molecule, the synthesis of human ubiquitin did not, as the target molecule could not be characterised by LC-MS analysis. DMSO and MeCN showed a significant positive effect on the purity of the raw material with the appearance of the molecular ion peak, showing that the main cause of the lack of acylation is indeed the formation of aggregates on the resin. This is further supported by the pressure change on the column and the acylation conversion (or change in Fmoc cleavage area) calculated from the UV detector data (Fig. 7) when synthesised without denaturant (as the peptide chain grows, an increase in pressure is expected in a fixed volume reactor such as ours, as there is no room for the resin to swell further). Along with aggregation, a decrease in the pressure rise trend is expected and, of course, a negative change in the Fmoc cleavage area. Our data suggest that aggregation begins around the 66th coupling in the production of ubiquitin, as there is a significant deviation from the trend line for both the pressure change and the change in Fmoc cleavage area (Fig. 7).
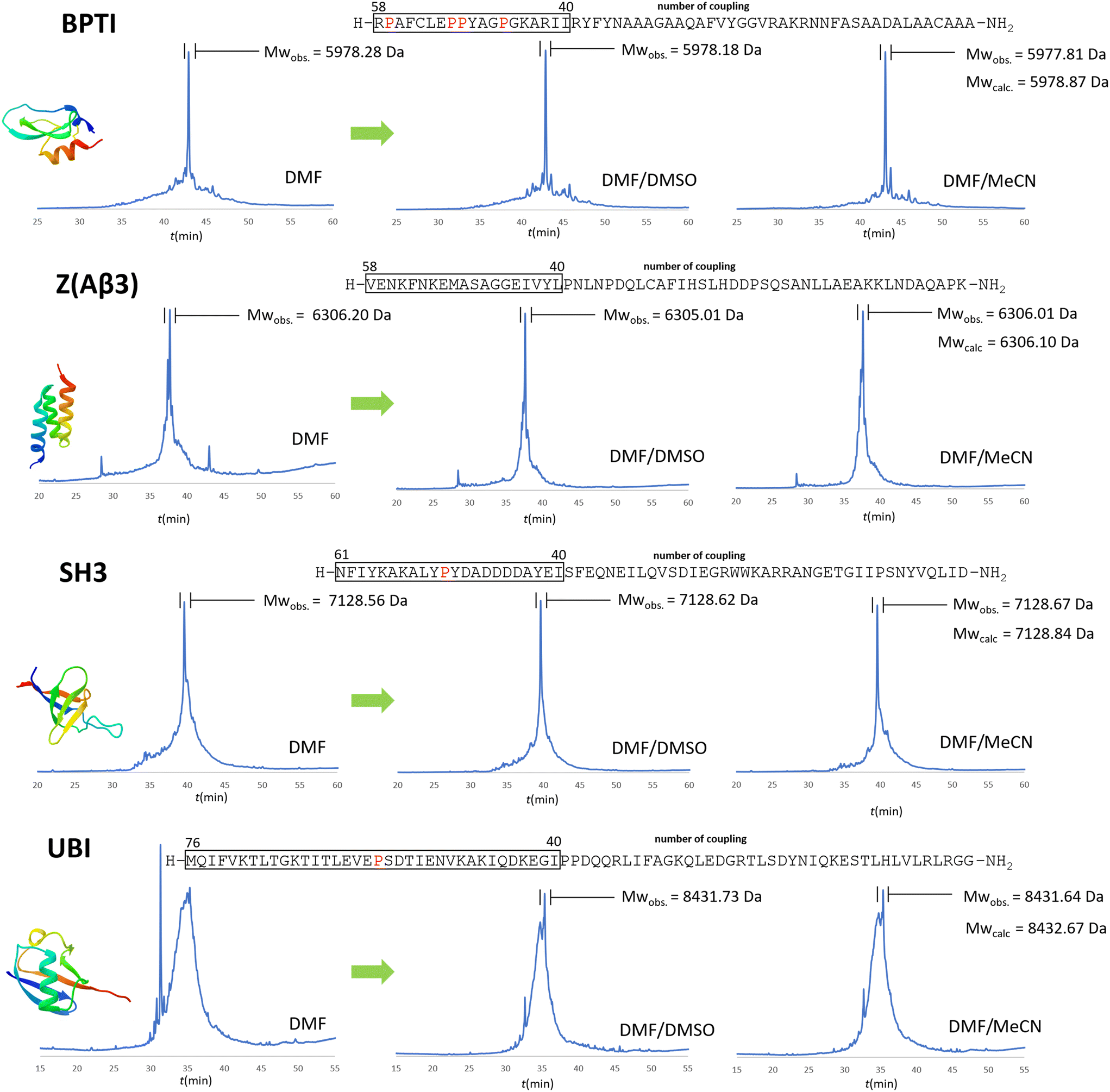 | ||
| Fig. 6 The positive effect of chaotropic additives used in the synthesis of the 4 protein domains. RP-HPLC chromatograms of the crude products using protocols a and e with DMF/DMSO (3:1) or DMF/MeCN (3:1) solvent mixtures versus pure DMF. Protocol e was used only for the following residues: R, K, Q, N, E, P (note that the couplings/amino acids are numbered from right to left according to the SPPS direction as opposed to the conventional notation). As mentioned above, coupling starts to be a challenge after 40+ residues, where the pro-residues could be of help. These “problematic regions” are framed and all prolines within these critical regions are highlighted in red. The MWs of the protein domains are shown in Table 1. | ||
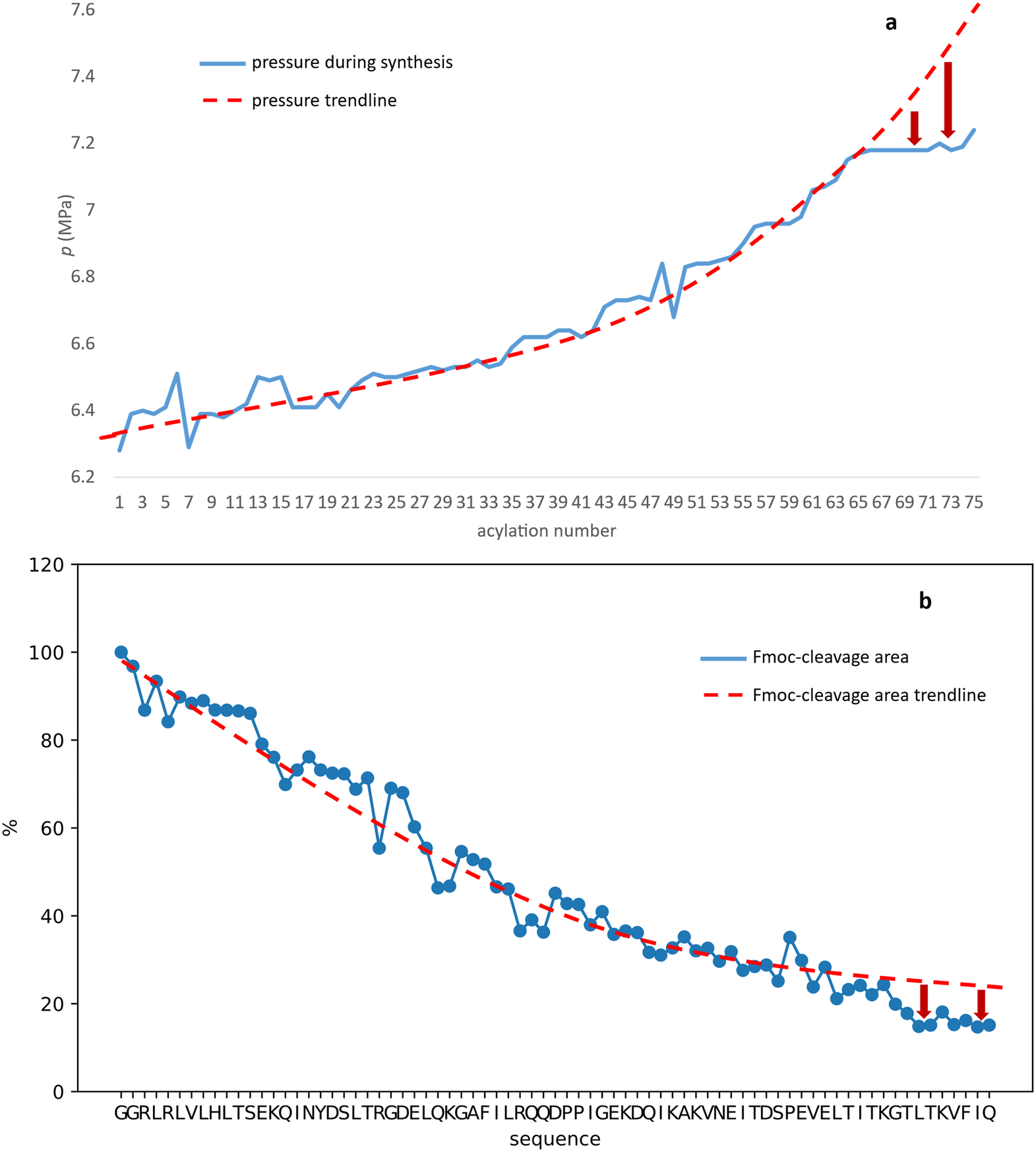 | ||
| Fig. 7 Pressure a) and Fmoc cleavage area b) as a function of primary sequence during the synthesis of human ubiquitin when no chaotropic agents are added to the solvent. Protocol e was used for R, K, Q, N, E, P and protocol a for the other amino acids. | ||
The raw materials were purified on a conventional preparative RP-HPLC column. The purifications of 100 mg of raw material yielded good results for BPTI (8 mg before and 4 mg including cyclization), and modest results for Z affibody (5 mg) and SH3 (6 mg), with several byproducts detected by the MS. In conclusion, both DMSO and acetonitrile adjuvants have a positive effect on the total synthesis of protein domains, and this improvement in synthetic efficiency becomes more pronounced as the length of the polypeptide chain increases, especially beyond 40 residues.
Conclusion
The circumstances of SPF synthesis, such as the presence of side-chain protected amino acid residues, aprotic solvent(s), higher temperature (80 °C) and elevated pressure (50–80 bar), differ from physiological conditions. In this altered molecular environment, protein domains are not expected to adopt their native 3D fold. However, even under these altered conditions, the amide bonds of the polypeptide chain interact with each other, forming H-bonds and forming an ensemble of more or less compact nanostructures and/or aggregates, creating undesirable conditions for N-terminal amino group acylation. Our aim was to optimise our SPF system, but with both economic (≤3 reagent equivalents) and environmental (≤6 ml solvent per cycle) aspects in mind.First, we started this work by synthesising oligotuftsin, a polypeptide with a repeating sequence pattern of five amino acids, to see how many amino acids we could comfortably couple if secondary structure-like aggregation was prevented by homogeneously distributed high (secondary structure breaker) Pro residues. We showed that our system could easily build chains of 80 amino acids. We then swapped the prolines for valines to allow a higher degree of aggregation and indeed, a loss of purity was observed. It was concluded that ∼50 amino acid long peptides can be rapidly synthesised even in the absence of Pro residues.
Second, using BPTI as a model system, we investigated the differences in coupling efficiency with respect to different types of residues and found that large and bulky amino acids are the ones that require special conditions during coupling. By controlling the residence time, these conditions could be fine-tuned without increasing the amount of excess reagents beyond 3 equivalents, making our SPF approach more economical and a step closer to being sustainable.
Third, we carried out synthesis of representative small protein domains of α-, β-, α + β- and α/β-fold each of about 65 ± 10 amino acids in length. Our data suggest the number of prolines and their sequential distribution, complemented by the length of the primary sequence are the key determinants of success. We were able to produce raw materials in which the main product was the target molecule, although their contamination profile is not ideal. From the UV and pressure data collected during the synthesis of ubiquitin, we conclude that aggregation does indeed occur during the final acylation steps of synthesis, starting around the 66th amino acid. The use of secondary structure-breaking solvent additives, such as DMSO and MeCN, is beneficial in solubilising the growing polypeptide chain. In this way, ubiquitin was produced overnight in less than 11.5 hours, with modest purity.
We have shown that our SPF system can generate domains up to ∼60 amino acids in length, independent of their native structure, using DMSO and MeCN as chaotropic agents, although there is a much room for improvement. Sequences of up to ∼80 amino acids can be synthesised using our SPF methodology if they are proline rich. However, if the number of Pro residues is low and the primary sequence is too long (longer than 80–90 residues), we are likely to encounter considerable difficulties in completing the synthesis. Although, the purified domains contain truncated sequences, they may be of significant use for example in assay experiments. Of course, these limitations can be overcome using tens to hundreds of times the amount of reagents, but we reject this approach. Instead, we aim to find alternative synthetic solutions. One of which is the use of Thr[ψ(Me,Me)Pro] and Ser[ψ(Me,Me)Pro] residues to mask Thr and Ser respectively.44,47 Similar to native Pro residues, pseudo-prolines also introduce backbone kinks. Recently, also using SPF protocols, we have been able to incorporate pseudoprolines as monomeric building blocks almost as effectively as any other proteinogenic residue. Another one would be the use of a reactor that can change its dimensions during synthesis, allowing the resin to swell, facilitating the diffusion of reagents and allowing the use of resins with greater swelling volumes, such as ChemMatrix.22
As protein- and peptide-based drugs become increasingly important, it is vital that they can be synthesised in an economically and environmentally efficient manner. Our SPF methods appear to provide an ideal background for the design of such synthetic protocols.
Abbreviations
| c | Concentration |
| v | Flow rate |
| CF-SPPS | Continuous flow solid phase peptide synthesis |
| DIC | N,N′-Diizopropylcarbodiimide |
| DMF | Dimethylformamide |
| DMSO | Dimethyl sulfoxide |
| EDT | Ethanedithiol |
| MeCN | Acetonitrile |
| n | Amount of substance |
| NCL | Native chemical ligation |
| NMM | N-Methylmorpholine |
| NMP | N-Methylpirrolidone |
| OT(P)5 | -(TKPKG)- oligotuftsin pentapeptide |
| OT(V)5 | -(TKVKG)- oligotuftsin P3V pentapeptide |
| PEG | Polyethyleneglycol |
| PS | Polystyrene |
| RP-HPLC | Reversed-phase high-performance liquid chromatography |
| SDS | Sodiumdodecylsulphate |
| SPF | Smart peptide chemistry in flow |
| TFA | Trifluoracetic acid |
| TG | TentaGel |
| TIS | Triizopropylsilane |
| t R | Residence time |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was completed in the ELTE Thematic Excellence Programme supported by the Hungarian Ministry for Innovation and Technology (SzintPlus). Project no. 2018-1.2.1-NKP-2018-00005 has been implemented with the support provided from the National Research, Development, and Innovation Fund of Hungary, financed under the 2018-1.2-1-NKP funding scheme (HunProtExc). Project number RRF-2.3.1-21-2022-00015 is implemented with the support of the European Union's Recovery and Resilience Instrument (PharmaLab). Servier Beregi Scholarship founded by Servier Research Institute of Medicinal Chemistry.References
- D. de la Torre and J. W. Chin, Reprogramming the Genetic Code, Nat. Rev. Genet., 2021, 22(3), 169–184, DOI:10.1038/s41576-020-00307-7.
- E. D. Hankore, L. Zhang, Y. Chen, K. Liu, W. Niu and J. Guo, Genetic Incorporation of Noncanonical Amino Acids Using Two Mutually Orthogonal Quadruplet Codons, ACS Synth. Biol., 2019, 8(5), 1168–1174, DOI:10.1021/acssynbio.9b00051.
- T. Hohsaka and M. Sisido, Incorporation of Non-Natural Amino Acids into Proteins, Curr. Opin. Chem. Biol., 2002, 6(6), 809–815, DOI:10.1016/S1367-5931(02)00376-9.
- P. Dawson, T. Muir, I. Clark-Lewis and S. Kent, Synthesis of Proteins by Native Chemical Ligation, Science, 1994, 266(5186), 776–779, DOI:10.1126/science.7973629.
- P. Lloyd-Williams, M. Gairí, F. Albericio and E. Giralt, Convergent Solid-Phase Peptide Synthesis. X. Synthesis and Purification of Protected Peptide Fragments Using the Photolabile Nbb-Resin, Tetrahedron, 1991, 47(47), 9867–9880, DOI:10.1016/S0040-4020(01)80724-9.
- C. L. Lee, H. Liu, C. T. T. Wong, H. Y. Chow and X. Li, Enabling N-to-C Ser/Thr Ligation for Convergent Protein Synthesis via Combining Chemical Ligation Approaches, J. Am. Chem. Soc., 2016, 138(33), 10477–10484, DOI:10.1021/jacs.6b04238.
- L. Raibaut, N. Ollivier and O. Melnyk, Sequential Native Peptide Ligation Strategies for Total Chemical Protein Synthesis, Chem. Soc. Rev., 2012, 41(21), 7001, 10.1039/c2cs35147a.
- J.-C. M. Monbaliu and A. R. Katritzky, Recent Trends in Cys- and Ser/Thr-Based Synthetic Strategies for the Elaboration of Peptide Constructs, Chem. Commun., 2012, 48(95), 11601, 10.1039/c2cc34434c.
- G.-M. Fang, Y.-M. Li, F. Shen, Y.-C. Huang, J.-B. Li, Y. Lin, H.-K. Cui and L. Liu, Protein Chemical Synthesis by Ligation of Peptide Hydrazides, Angew. Chem., Int. Ed., 2011, 50(33), 7645–7649, DOI:10.1002/anie.201100996.
- T. J. Lukas, M. B. Prystowsky and B. W. Erickson, Solid-Phase Peptide Synthesis under Continuous-Flow Conditions, Proc. Natl. Acad. Sci. U. S. A., 1981, 78(5), 2791–2795, DOI:10.1073/pnas.78.5.2791.
- S. F. Loibl, Z. Harpaz, R. Zitterbart and O. Seitz, Total Chemical Synthesis of Proteins without HPLC Purification, Chem. Sci., 2016, 7(11), 6753–6759, 10.1039/C6SC01883A.
- R. Zitterbart, N. Berger, O. Reimann, G. T. Noble, S. Lüdtke, D. Sarma and O. Seitz, Traceless Parallel Peptide Purification by a First-in-Class Reductively Cleavable Linker System Featuring a Safety-Release, Chem. Sci., 2021, 12(7), 2389–2396, 10.1039/D0SC06285E.
- F. Mende, M. Beisswenger and O. Seitz, Automated Fmoc-Based Solid-Phase Synthesis of Peptide Thioesters with Self-Purification Effect and Application in the Construction of Immobilized SH3 Domains, J. Am. Chem. Soc., 2010, 132(32), 11110–11118, DOI:10.1021/ja101732a.
- C. P. Gordon, The Renascence of Continuous-Flow Peptide Synthesis – an Abridged Account of Solid and Solution-Based Approaches, Org. Biomol. Chem., 2018, 16(2), 180–196, 10.1039/C7OB02759A.
- B. G. de la Torre, A. Jakab and D. Andreu, Polyethyleneglycol-Based Resins as Solid Supports for the Synthesis of Difficult or Long Peptides, Int. J. Pept. Res. Ther., 2007, 13(1–2), 265–270, DOI:10.1007/s10989-006-9077-5.
- I. M. Mándity, B. Olasz, S. B. Ötvös and F. Fülöp, Continuous-Flow Solid-Phase Peptide Synthesis: A Revolutionary Reduction of the Amino Acid Excess, ChemSusChem, 2014, 7(11), 3172–3176, DOI:10.1002/cssc.201402436.
- L. K. Spare, M. Menti, D. G. Harman, J. R. Aldrich-Wright and C. P. Gordon, A Continuous Flow Protocol to Generate, Regenerate, Load, and Recycle Chlorotrityl Functionalised Resins, React. Chem. Eng., 2019, 4(7), 1309–1317, 10.1039/C8RE00318A.
- L. K. Spare, V. Laude, D. G. Harman, J. R. Aldrich-Wright and C. P. Gordon, An Optimised Approach for Continuous-Flow Solid-Phase Peptide Synthesis Utilising a Rudimentary Flow Reactor, React. Chem. Eng., 2018, 3(6), 875–882, 10.1039/C8RE00190A.
- M. D. Simon, P. L. Heider, A. Adamo, A. A. Vinogradov, S. K. Mong, X. Li, T. Berger, R. L. Policarpo, C. Zhang, Y. Zou, X. Liao, A. M. Spokoyny, K. F. Jensen and B. L. Pentelute, Rapid Flow-Based Peptide Synthesis, ChemBioChem, 2014, 15(5), 713–720, DOI:10.1002/cbic.201300796.
- A. J. Mijalis, D. A. Thomas, M. D. Simon, A. Adamo, R. Beaumont, K. F. Jensen and B. L. Pentelute, A Fully Automated Flow-Based Approach for Accelerated Peptide Synthesis, Nat. Chem. Biol., 2017, 13(5), 464–466, DOI:10.1038/nchembio.2318.
- V. Farkas, K. Ferentzi, K. Horváti and A. Perczel, Cost-Effective Flow Peptide Synthesis: Metamorphosis of HPLC, Org. Process Res. Dev., 2021, 25(2), 182–191, DOI:10.1021/acs.oprd.0c00178.
- E. T. Sletten, M. Nuño, D. Guthrie and P. H. Seeberger, Real-Time Monitoring of Solid-Phase Peptide Synthesis Using a Variable Bed Flow Reactor, Chem. Commun., 2019, 55(97), 14598–14601, 10.1039/C9CC08421E.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, The Hitchhiker's Guide to Flow Chemistry, Chem. Rev., 2017, 117(18), 11796–11893, DOI:10.1021/acs.chemrev.7b00183.
- A. Adamo, R. L. Beingessner, M. Behnam, J. Chen, T. F. Jamison, K. F. Jensen, J.-C. M. Monbaliu, A. S. Myerson, E. M. Revalor, D. R. Snead, T. Stelzer, N. Weeranoppanant, S. Y. Wong and P. Zhang, On-Demand Continuous-Flow Production of Pharmaceuticals in a Compact, Reconfigurable System, Science, 2016, 352(6281), 61–67, DOI:10.1126/science.aaf1337.
- K. Kiss, S. Ránky, Z. Gyulai and L. Molnár, Development of a Novel, Automated, Robotic System for Rapid, High-Throughput, Parallel, Solid-Phase Peptide Synthesis, SLAS Technol., 2023, 28(2), 89–97, DOI:10.1016/j.slast.2023.01.002.
- N. Hartrampf, A. Saebi, M. Poskus, Z. P. Gates, A. J. Callahan, A. E. Cowfer, S. Hanna, S. Antilla, C. K. Schissel, A. J. Quartararo, X. Ye, A. J. Mijalis, M. D. Simon, A. Loas, S. Liu, C. Jessen, T. E. Nielsen and B. L. Pentelute, Synthesis of Proteins by Automated Flow Chemistry, Science, 2020, 368(6494), 980–987, DOI:10.1126/science.abb2491.
- A. Saebi, J. S. Brown, V. M. Marando, N. Hartrampf, N. M. Chumbler, S. Hanna, M. Poskus, A. Loas, L. L. Kiessling, D. T. Hung and B. L. Pentelute, Rapid Single-Shot Synthesis of the 214 Amino Acid-Long N-Terminal Domain of Pyocin S2, ACS Chem. Biol., 2023, 18(3), 518–527, DOI:10.1021/acschembio.2c00862.
- J. Lindgren, A. Wahlström, J. Danielsson, N. Markova, C. Ekblad, A. Gräslund, L. Abrahmsén, A. E. Karlström and S. K. T. S. Wärmländer, N-Terminal Engineering of Amyloid-β-Binding Affibody Molecules Yields Improved Chemical Synthesis and Higher Binding Affinity: Improved Aβ-Binding Affibody Molecules, Protein Sci., 2010, 19(12), 2319–2329, DOI:10.1002/pro.511.
- M. Ferrer, C. Woodward and G. Barany, Solid-Phase Synthesis of Bovine Pancreatic Trypsin Inhibitor (BPTI) and Two Analogues. A Chemical Approach for Evaluating the Role of Disulfide Bridges in Protein Folding and Stability, Int. J. Pept. Protein Res., 1992, 40(3–4), 194–207 CrossRef CAS.
- F. El Oualid, R. Merkx, R. Ekkebus, D. S. Hameed, J. J. Smit, A. de Jong, H. Hilkmann, T. K. Sixma and H. Ovaa, Chemical Synthesis of Ubiquitin, Ubiquitin-Based Probes, and Diubiquitin, Angew. Chem., Int. Ed., 2010, 49(52), 10149–10153, DOI:10.1002/anie.201005995.
- J. Hou, G. E. Sims, C. Zhang and S.-H. Kim, A Global Representation of the Protein Fold Space, Proc. Natl. Acad. Sci. U. S. A., 2003, 100(5), 2386–2390, DOI:10.1073/pnas.2628030100.
- M. Narita, S. Honda, H. Umeyama and T. Ogura, Infrared Spectroscopic Conformational Analysis of Polystyrene Resin-Bound Human Proinsulin C-Peptide Fragments. β-Sheet Aggregation of Peptide Chains during Solid-Phase Peptide Synthesis, Bull. Chem. Soc. Jpn., 1988, 61(4), 1201–1206, DOI:10.1246/bcsj.61.1201.
- B. D. Larsen, D. H. Christensen, A. Holm, R. Zillmer and O. F. Nielsen, The Merrifield Peptide Synthesis Studied by Near-Infrared Fourier-Transform Raman Spectroscopy, J. Am. Chem. Soc., 1993, 115(14), 6247–6253, DOI:10.1021/ja00067a044.
- S. Wang and Y. Ishii, Revealing Protein Structures in Solid-Phase Peptide Synthesis by 13 C Solid-State NMR: Evidence of Excessive Misfolding for Alzheimer's β, J. Am. Chem. Soc., 2012, 134(6), 2848–2851, DOI:10.1021/ja212190z.
- C. Dhalluin, C. Boutillon, A. Tartar and G. Lippens, Magic Angle Spinning Nuclear Magnetic Resonance in Solid-Phase Peptide Synthesis, J. Am. Chem. Soc., 1997, 119(43), 10494–10500, DOI:10.1021/ja971795l.
- B. Bacsa, K. Horváti, S. Bõsze, F. Andreae and C. O. Kappe, Solid-Phase Synthesis of Difficult Peptide Sequences at Elevated Temperatures: A Critical Comparison of Microwave and Conventional Heating Technologies, J. Org. Chem., 2008, 73(19), 7532–7542, DOI:10.1021/jo8013897.
- S. L. Pedersen, A. P. Tofteng, L. Malik and K. J. Jensen, Microwave Heating in Solid-Phase Peptide Synthesis, Chem. Soc. Rev., 2012, 41(5), 1826–1844, 10.1039/C1CS15214A.
- C. Hyde, T. Johnson and R. C. Sheppard, Internal Aggregation during Solid Phase Peptide Synthesis. Dimethyl Sulfoxide as a Powerful Dissociating Solvent, J. Chem. Soc., Chem. Commun., 1992,(21), 1573, 10.1039/c39920001573.
- J. W. Choi, H. Y. Kim, M. Jeon, D. J. Kim and Y. Kim, Efficient Access to Highly Pure β-Amyloid Peptide by Optimized Solid-Phase Synthesis, Amyloid, 2012, 19(3), 133–137, DOI:10.3109/13506129.2012.700287.
- L. Kocsis, T. Bruckdorfer and G. Orosz, The Concept of Internal Solubilization in Peptide Synthesis: Ethylene Glycol-Based Protecting Groups, Tetrahedron Lett., 2008, 49(49), 7015–7017, DOI:10.1016/j.tetlet.2008.09.126.
- T. Haack and M. Mutter, Serine Derived Oxazolidines as Secondary Structure Disrupting, Solubilizing Building Blocks in Peptide Synthesis, Tetrahedron Lett., 1992, 33(12), 1589–1592, DOI:10.1016/S0040-4039(00)91681-2.
- P. M. Fischer and D. I. Zheleva, Liquid-Phase Peptide Synthesis on Polyethylene Glycol (PEG) Supports Using Strategies Based on the 9-Fluorenylmethoxycarbonyl Amino Protecting Group: Application of PEGylated Peptides in Biochemical Assays, J. Pept. Sci., 2002, 8(9), 529–542, DOI:10.1002/psc.413.
- M. Mutter, H. Oppliger and A. Zier, Makromol. Chem., Rapid Commun., 1992, 13(3), 151–157, DOI:10.1002/marc.1992.030130303.
- D. A. Senko, N. D. Timofeev, I. E. Kasheverov and I. A. Ivanov, Scope and Limitations of Pseudoprolines as Individual Amino Acids in Peptide Synthesis, Amino Acids, 2021, 53(5), 665–671, DOI:10.1007/s00726-021-02973-1.
- T. Johnson, M. Quibell and R. C. Sheppard, N,O-bisFmoc Derivatives ofN-(2-Hydroxy-4-Methoxybenzyl)-Amino Acids: Useful Intermediates in Peptide Synthesis, J. Pept. Sci., 1995, 1(1), 11–25, DOI:10.1002/psc.310010104.
- L. A. Carpino, E. Krause, C. D. Sferdean, M. Schümann, H. Fabian, M. Bienert and M. Beyermann, Synthesis of ‘Difficult’ Peptide Sequences: Application of a Depsipeptide Technique to the Jung–Redemann 10- and 26-Mers and the Amyloid Peptide Aβ(1–42), Tetrahedron Lett., 2004, 45(40), 7519–7523, DOI:10.1016/j.tetlet.2004.07.162.
- S. Szaniszló, K. Ferentzi, A. Perczel and V. Farkas, Improved Acylation of Pseudoproline: Masked Threonine in Flow Peptide Chemistry, Org. Process Res. Dev., 2023, 27(6), 1053–1060, DOI:10.1021/acs.oprd.3c00029.
- T. R. Cotton, G. Fischer, X. Wang, J. C. McCoy, M. Czepnik, T. B. Thompson and M. Hyvönen, Structure of the Human Myostatin Precursor and Determinants of Growth Factor Latency, EMBO J., 2018, 37(3), 367–383, DOI:10.15252/embj.201797883.
- D. Nagy-Fazekas, Z. Fazekas, N. Taricska, P. Stráner, D. Karancsiné Menyhárd and A. Perczel, Inhibitor Design Strategy for Myostatin: Dynamics and Interaction Networks Define the Affinity and Release Mechanisms of the Inhibited Complexes, Molecules, 2023, 28(15), 5655, DOI:10.3390/molecules28155655.
- G. Mezö, A. Kalászi, J. Reményi, Z. Majer, Á. Hilbert, O. Láng, L. Köhidai, K. Barna, D. Gaál and F. Hudecz, Synthesis, Conformation, and Immunoreactivity of New Carrier Molecules Based on Repeated Tuftsin-like Sequence: New Carrier Molecules, Biopolymers, 2004, 73(6), 645–656, DOI:10.1002/bip.20024.
- M. Manea, F. Hudecz, M. Przybylski and G. Mezõ, Synthesis, Solution Conformation, and Antibody Recognition of Oligotuftsin-Based Conjugates Containing a β-Amyloid(4−10) Plaque-Specific Epitope, Bioconjugate Chem., 2005, 16(4), 921–928, DOI:10.1021/bc0500037.
- V. Krchňák, Z. Flegelová and J. Vágner, Aggregation of Resin-Bound Peptides during Solid-Phase Peptide Synthesis: Prediction of Difficult Sequences, Int. J. Pept. Protein Res., 2009, 42(5), 450–454, DOI:10.1111/j.1399-3011.1993.tb00153.x.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3re00324h |
| This journal is © The Royal Society of Chemistry 2024 |