
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFabrication of tetraethyl orthosilicate/ethanol–water surfactant-free microemulsions and their applications in self-templating synthesis of monodispersed silica colloidal spheres†
Jiahan Lu,
Longhua Peng,
Ao Zhang,
Jiaqiong Xu,
Min Wu and
Shiyu Ma *
*
Research Center for Water Resources and Interface Science, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, PR China. E-mail: syma@chem.ecnu.edu.cn; Fax: +86-021-54340130; Tel: +86-021-54340130
First published on 11th December 2024
Abstract
Surfactant-free microemulsions (SFMEs) composed of tetraethyl orthosilicate (TEOS), ethanol, and water have been successfully fabricated by visual titration and electrical conductivity methods. Three types of SFMEs, water in TEOS (W/O), bicontinuous (BC) and TEOS in water (O/W), were identified by dynamic light scattering and transmission electron microscopy with negative-staining methods. We demonstrated that there are significant differences in the properties of silica products synthesized with different types of SFMEs, and monodispersed silica colloidal spheres (MSCSs) can only be synthesized in the O/W type SFMEs. Moreover, we found that the particle size of MSCSs is closely related to the size of oil droplets. Cooling the O/W type SFMEs in the early reaction stages, results in the larger MSCSs with different condensation degrees. Furthermore, if the cooling temperature decreased to −20 °C, ring-like spheres could be observed. Based on our results and observations, a self-templating growth mechanism was proposed to explain the formation of silica spheres.
1 Introduction
Conventional surfactant-based microemulsions (SBMEs), composed of water, oil, surfactant and cosurfactant, are generally defined as thermodynamically stable and optically isotropic transparent dispersions.1,2 The SBMEs typically exhibit three different types: O/W, BC and W/O microemulsions,3 which have a broad range of applications across various fields, ranging from nanomaterial synthesis4–7 to life sciences.2,8–10 Among these, microemulsions offer several advantages for the synthesis of nanoparticles, such as precisely controlled size, narrow size distribution and mild reaction conditions, rendering them particularly suitable for some high-quality nanomaterials.11,12 However, SBMEs typically contain a significant quantity of surfactants and co-surfactants, which have a negative impact on the properties of nanoparticles, thereby constraining the development and application of these systems to a certain extent.13In recent years, researchers have discovered that microemulsions can be formed spontaneously in the ternary mixtures of oil, water and an amphiphilic solvent in the absence of conventional surfactants, and such microemulsions are known as surfactant-free microemulsions (SFMEs).14–17 The so-called amphiphilic solvent, which is different from a conventional surfactant, is completely or at least partially miscible with the aqueous and oil phases.18,19 In the late 1970s, Smith et al. reported that stable SFMEs could be fabricated in the n-hexane/i-propanol–water system, which was the first discovered SFMEs.20 Subsequently, Kunz et al. demonstrated the presence of pre-ouzo region in the octanol/ethanol–water system.21,22 The pre-ouzo effect, which primarily occurs in the single-phase region near the multiphase region in a ternary phase diagram, refers to a phenomenon where oil droplets cause a solution to appear milky white due to the refraction and scattering of light.23,24 In 1981, Lara et al. conducted a study in the benzene/iso-propanol/water single-phase system, revealing that three structural types, O/W, BC and W/O, which were similar to those findings in SBMEs.25 Hou et al. investigated the phase behaviour and microenvironment of SFMEs by using electrical conductivity, SAXS, DLS, UV-vis measurements and freeze-fracture and cryogenic TEM observations.26–28 Zhang et al. research results indicated that the phase area and mesostructure of SFMEs of the n-octanol/ethanol–water system could be changed by altering temperature.29
Similar to SBMEs, SFMEs have been used in the synthesis of nanomaterials.30 Chai et al. employed O/W microemulsion in dichloromethane/ethanol–water and ethyl acetate/isopropanol–water systems as a soft template to successfully synthesize monodispersed silica colloidal spheres (MSCSs) and titanium dioxide particles.31,32 El-Hefnawy et al. used an olive oil/n-butanol/water system to synthesize spherical CdS nanoparticles with an average diameter of 45 nm.33 Zhang et al. reported the synthesis of ZnO using a n-hexane/isopropanol/water system. They investigated the effects of temperature, time and varying ammonia concentrations on the morphology and size of ZnO, and further explored its photocatalytic activity and underlying mechanisms.34 Kunz et al. employed SFMEs containing water, alcohols and methyl methacrylate to investigate the synthesis of porous polymers.35,36
Although many studies have explored the use of SFMEs for the synthesis of nanomaterials, there has been limited research on using SFMEs to produce MSCSs. So far, the main methods for the synthesis of MSCSs include the Stöber method,37 the microemulsion method38 and the seed growth method.39 However, they all share the drawback of complicated preparation processes and strict reaction conditions. Therefore, it is of great significance to develop a simple method for preparing silica spheres by SFMEs.
Herein, a new kind of SFMEs composed of tetraethyl orthosilicate (TEOS), ethanol (EtOH) and water (H2O) was fabricated. The mesostructures and properties of the SFMEs were investigated in detail by electrical conductivity, dynamic light scattering (DLS) and transmission electron microscope with negative-staining (negative-staining TEM). The MSCSs were successfully synthesized with the O/W type SFMEs. Furthermore, the larger MSCSs with different condensation degrees could also be synthesized by cooling the O/W type SEMEs in the early reaction stages. The SFMEs provide possibilities for the controllable synthesis of MSCSs. A possible mechanism was proposed to explain the formation of silica spheres.
2 Experimental section
2.1 Materials and reagents
EtOH and TEOS were purchased from Sinopharm Group Chemical Reagents Co., Ltd (Shanghai, China). Ammonium hydroxide (28–30 wt%) was purchased from Shanghai Aladdin Biochemical Technology Co., Ltd (Shanghai, China). Phosphotungstic acid was purchased from Bide Pharmatech Co., Ltd (Shanghai, China). EtOH, TEOS, phosphotungstic acid, were analytical grade chemicals and ammonium hydroxide was ACS grade chemicals. All chemicals were used without further purification. Ultrapure water with a resistivity of 18.25 MΩ cm was used throughout the experiments.2.2 Apparatus and characterization
An electrical conductivity meter (DDSJ-308F, Shanghai INESA Scientific Instruments Co., Ltd) with a DDSJ-0.1 electrode of cell constant 0.111 cm−1 was used to measure the conductivity of the TEOS/EtOH–H2O ternary system. TEM was carried out on a H-7000 electron microscope (Hitachi, Japan). Scanning electron microscopy (SEM) was carried out on a S-4800 electron microscope (Hitachi, Japan). The oil droplets of O/W type SFMEs were measured at 25.0 ± 0.2 °C using a Zetasizer Nano ZS instrument from Malvern. All the scattering photons were collected at a 173° scattering angle. 29Si MAS NMR measurements were obtained on an Agilent 600 DD2 spectrometer. For 29Si (79.5 MHz), a 4 μs (h = p/2) pulse was used with a repetition time of 360 s. FT-IR spectra of the silica spheres were recorded by Nicolet Fourier transform infrared spectrometer (NEXUS 670) using the KBr technique.2.3 Phase diagram construction
The phase diagram of TEOS/EtOH–H2O ternary systems was constructed by visual titration with H2O for TEOS–EtOH mixtures. The general steps are as follows: A TEOS–EtOH mixture with a desired mass ratio (RT/E) was prepared in a dry test tube. A certain amount of ultrapure water was slowly added to the mixture under magnetic stirring. The phase boundary was determined by observing the phase transition from transparency to turbidity. Repeating this procedure for a series of TEOS–EtOH mixtures with different RT/E allowed the phase diagram to be established. The same procedure for each mixture was repeated thrice for average values. All the experiments were carried out at 25.0 ± 0.2 °C and sealed to prevent evaporation loss.2.4 Electrical conductivity measurements
A TEOS/EtOH–H2O ternay system with a fixed mass ratio (SystemA1) was prepared in a dry test tube. Variations in conductivity were monitored at regular intervals of 5 min, 10 min, 20 min, 30 min, 60 min and 90 min from the initial time of mixing TEOS, EtOH and H2O.An EtOH–H2O mixture with a fixed mass ratio (RW/E) was prepared in a dry test tube. Different amounts of TEOS were added to the test tube and the conductivity was measured at 25.0 ± 0.2 °C. Repeating this procedure for a series of EtOH–H2O mixtures with different RW/E allowed the microregion of single-phase region to be established. All experiments were repeated thrice for average values.
2.5 Negative-staining TEM method
The droplets of the SFMEs were placed onto a copper net (400 mesh) supported by carbon film using a micropipette. After keeping at room temperature for one minute, an absorbent paper was used to absorb the excess solution around the copper net. Then, an aqueous solution of phosphotungstic acid with a mass fraction of 1% was dropped onto the same copper net. Following a one-minute exposure at room temperature, absorbent paper was employed to remove any excess solution again. The samples were maintained at room temperature for 12 h, before the TEM of the SFMEs were observed.2.6 Synthesis of silica products
TEOS, EtOH and H2O were thoroughly mixed and sealed in a dry conical bottle with a desired concentration of TEOS and ethanol–water mass ratio. The mixture was stirred for 20 min and sonicated for 20 min to obtain the SFME. Subsequently, an appropriate amount of ammonium hydroxide was added (the concentration of ammonia, 1.56 mol L−1, obtained based on the total volume of the reaction solution) to start the reaction and reacted for 2 h with a stirring rate of 300 rpm. All the experiments were carried out at 25.0 ± 0.2 °C. The as-synthesized silica samples were directly used to prepare TEM specimens without any further treatment and silica samples for SEM were collected by centrifugation, washed thrice with ultrapure water and ethanol, then dried at 60 °C for 12 h.2.7 Cooling treatment of silica spheres
A pre-reaction/low-temperature strategy: 50 mL of the above SFME was added to a 150 mL stainless steel beaker, and an appropriate amount of ammonium hydroxide was added (the concentration of ammonia, 1.56 mol L−1, obtained based on the total volume of the reaction solution) to start the reaction. Then, the reaction mixture was stirred at room temperature for 1 min (pre-reaction time). Subsequently, the reaction mixtures were placed separately at different cooling temperatures (0 °C, −10 °C, −15 °C and −20 °C) for 4 h. The as-synthesized silica samples were directly used to prepare TEM specimens without any further treatment. Silica samples for the 29Si MAS NMR and IR were collected by centrifugation, washed thrice with ultrapure water and ethanol, and then dried at 60 °C for 12 h.A low-temperature equilibrium/reaction strategy: 50 ml of the above SFME was added in a 150 mL stainless steel beaker and equilibrated at different low temperatures (0 °C, −10 °C, −15 °C, −20 °C) for 30 min, then an appropriate amount of ammonia was added (the concentration of ammonia, 1.56 mol L−1, obtained based on the total volume of the reaction solution) to initiate the reaction, and reaction mixture was stirred at a low temperature for 4 h. The as-synthesized silica samples were directly used to prepare TEM specimens without any further treatment.
3 Results and discussion
3.1 Phase diagram of ternary systems and mesostructures of the SFMEs
The phase diagram of the TEOS/EtOH–H2O ternary system and microregions of the phase diagram were constructed by visual titration and electrical conductivity measurements, respectively.21,40 The results are shown in Fig. 1. | ||
| Fig. 1 (a) Phase diagram of the TEOS/EtOH–H2O ternary systems at 25 ± 0.2 °C. I, III and II are O/W, BC and W/O microregions respectively. The points A1–A5, B, C and D (SystemA1–SystemA5, SystemB, SystemC, SystemD) were chosen for the synthesis of silica products, respectively. The content of each component is expressed by mass fraction. (b) Electrical conductivity κ as a function of time for the SystemA1. (c) Electrical conductivity κ as a function of fo at different RW/E values. (d) κ as a function of fo with RW/E = 0.33 is zoomed in from (c). | ||
As seen in Fig. 1a, the blank area is the transparent single-phase region, and the gray area is the multiple-phase region. Although the TEOS undergoes gradual hydrolysis and condensation in the EtOH–H2O mixture under neutral conditions, conductivity monitoring results (Fig. 1b) indicated that the rate of TEOS hydrolysis is remarkably slow, which has little effect on the phase boundary within the time required for phase diagram construction (less than 90 min). That is, during the experiment, the SFMEs fabricated could be regarded as an equilibrium system.
The variation curve of electrical conductivity (κ) of a fixed ethanol–water ratio mixture with a mass fraction of TEOS (fo) is used to identify the mesostructure of the SFMEs.41,42 The results are shown in Fig. 1c. It can be seen that, with an increasing fo value, the κ value initially increases and then decreases, at a constant RW/E value. This indicates the fabrication of different mesostructures in the single-phase region of TEOS/EtOH–H2O ternary systems.
For clarity, electrical conductivity κ versus fo curve at the RW/E ratio of 0.33 is also shown in Fig. 1d. In the first stage, the rapid increase of κ with an increase of fo indicates that a gradual formation of O/W microemulsion in the EtOH–H2O mixture. The nonlinear increase of κ with fo in the second stage suggests the transition of microemulsion from O/W to BC microemulsion. The final linear decrease of κ with the increase of fo corresponds to the appearance of W/O microemulsion. A comprehensive analysis of the curve variations allows the single-phase region of the phase diagram to be distinctly classified into three microregions: I (O/W type SFMEs), III (BC type SFMEs) and II (W/O type SFMEs).
The mesostructures of O/W type SFMEs were further verified by DLS, DLS results of oil droplets are shown in Fig. 2 and S1.†
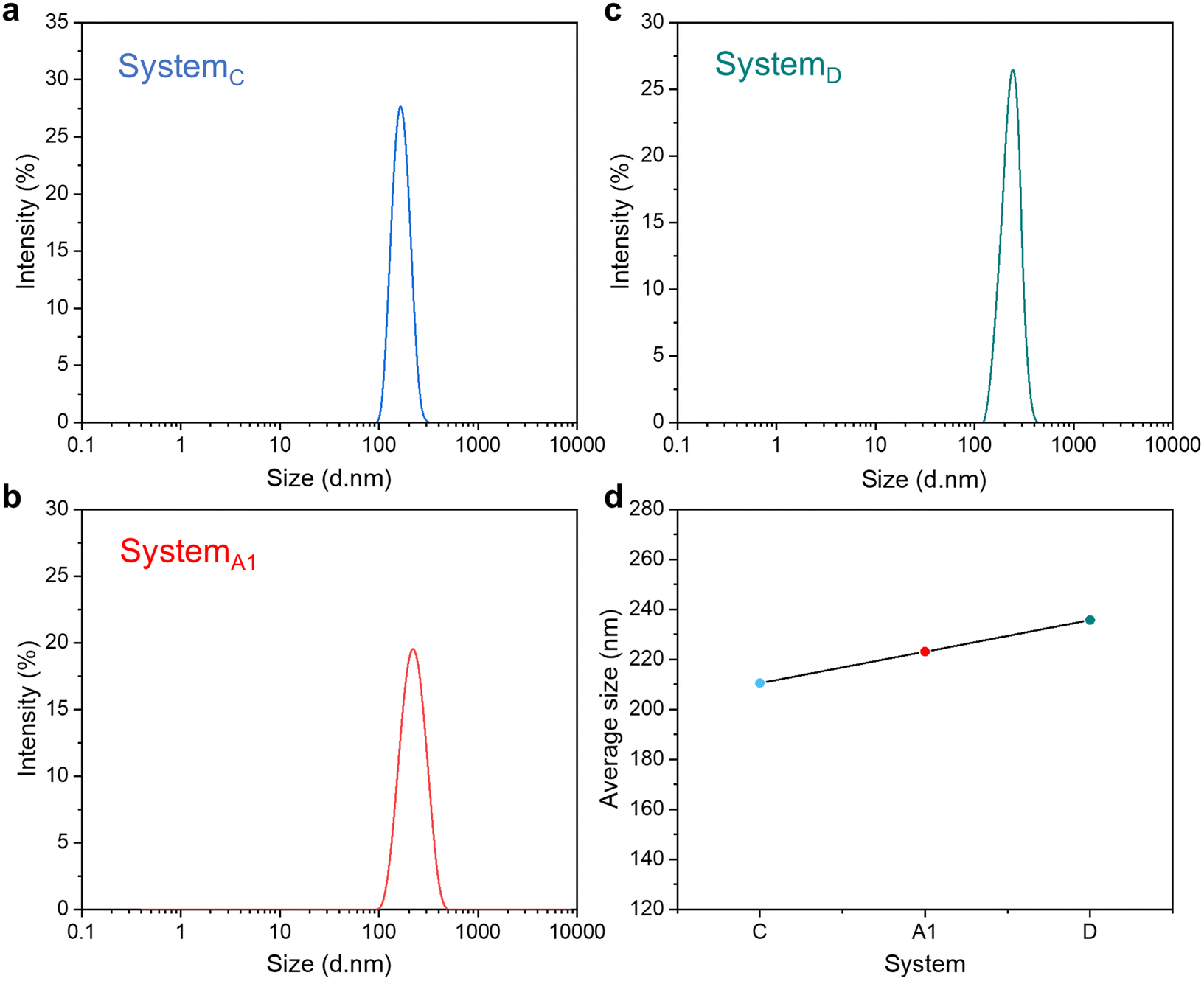 | ||
| Fig. 2 Dynamic light scattering (DLS) results of oil droplets in O/W type SFMEs with different water–ethanol ratios. (a) SystemC, fo = 0.05, RW/E = 0.67, (b) SystemA1, fo = 0.05, RW/E = 0.43, (c) SystemD, fo = 0.05, RW/E = 0.25 and (d) the average size of SystemC, SystemA1 and SystemD. | ||
As shown in Fig. 2, when the ethanol–water mass ratio was varied from RW/E = 0.67 (SystemC) to RW/E = 0.43 (SystemA1) and RW/E = 0.25 (SystemD), the average sizes of oil droplets were varied from 210.7 nm to 223.1 nm and 235.7 nm. Light scattering may come from the contribution of clearly defined aggregates and critical effects or molecular concentration fluctuations. The strong scattering observed in this study can be attributed to the formation of well-defined aggregates (oil droplets), which provides additional evidence for the existence of mesostructures in the O/W type SFMEs.
The mesostructures of the SFMEs were also determined by negative-staining TEM measurements, and the TEM images were shown in Fig. 3. It can be clearly indicated that the SFMEs fabricated with TEOS, EtOH and H2O have oil droplets (O/W type SFMEs), sponge-like structures (BC type SFMEs) and water droplets (W/O type SFMEs), respectively.43 Therefore, the lower TEOS mass fraction leads to the formation of O/W type SFMEs, while the higher TEOS mass fraction results in the formation of W/O type SFMEs. The BC type SFMEs can be considered as an intermediary state between two structural transitions.31
 | ||
| Fig. 3 TEM images of O/W type SFMEs (SystemA1), BC type SFMEs (SystemA4) and W/O type SFMEs (SystemA5). | ||
3.2 Effect of TEOS contents on the properties of silica products
SystemA1–SystemA5, which contain a constant ethanol–water mass ratio (RW/E = 0.43) and different TEOS mass fractions, were selected as model systems for the synthesis of silica products. The experimental conditions and properties of silica products are listed in Table 1. The TEM and SEM images of silica products are shown in Fig. 4.| System | Type SFMEs | mTEOS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :mEtOH:mH2O (g/g/g) :mEtOH:mH2O (g/g/g) |
TEOS mass fraction | Avg. diameter (nm) | σ (nm) |
|---|---|---|---|---|---|
| A1 | O/W | 10:140:60 |
0.05 | 264 | 12 |
| A2 | O/W | 20:140:60 |
0.09 | 338 | 17 |
| A3 | O/W | 30:140:60 |
0.13 | 427 | 18 |
| A4 | BC | 70:140:60 |
0.26 | — | — |
| A5 | W/O | 130:140:60 |
0.39 | — | — |
 | ||
| Fig. 4 TEM and SEM images of the silica products synthesized with the SFMEs with a constant RW/E = 0.43, same ammonia concentration and different TEOS mass fraction. From (a) to (e), fo = 0.05, 0.09, 0.13, 0.26 and 0.39, respectively. | ||
It can be seen from Table 1 and Fig. 4, the MSCSs can only be synthesized in O/W type SFMEs. When the TEOS mass fraction increased from fo = 0.05 (SystemA1) to fo = 0.13 (SystemA3), the particle size of silica spheres gradually increased from 264 ± 12 nm to 427 ± 18 nm, and the distribution of spheres became wider.44,45 However, as the TEOS fraction further increased to fo = 0.26 (SystemA4) and fo = 0.39 (SystemA5), discrete silica spheres cannot be synthesized (Fig. 4d and e), they displayed strong aggregation behaviour. In addition, similar experimental results could also be obtained for SFMEs fabricated by n-propanol (NPA) or isopropanol (IPA) with TEOS and water, respectively. (Fig. S2 and S3†). As we know that the mesostructure of O/W type SFMEs (SystemA1–SystemA3) is an oil-droplet structure, the mesostrctures of BC type SFMEs (SystemA4) and W/O type SFMEs (SystemA5) are a sponge-like structure and a water-droplet structure, respectively. Therefore, there is a clear correlation between silica morphologies and mesostructures of SFMEs.
3.3 Effect of ethanol–water ratio on the properties of silica spheres
The O/W type SFMEs, SystemC, SystemA1, and SystemD were selected as model systems to explore the effect of ethanol–water ratios on the properties of silica spheres. The results are shown in Table 2 and Fig. 5.| System | Type SFMEs | mTEOS:mEtOH: mH2O (g/g/g) |
Ethanol–water ratio (RW/E) | Avg. diameter (nm) | σ (nm) |
|---|---|---|---|---|---|
| C | O/W | 10:120:80 |
0.67 | 258 | 8 |
| A1 | O/W | 10:140:60 |
0.43 | 264 | 12 |
| D | O/W | 10:160:40 |
0.25 | 281 | 10 |
 | ||
| Fig. 5 TEM images of the silica spheres synthesized with the O/W type SFMEs with a constant fo = 0.05, same ammonia concentration and different ethanol–water ratio. (a) SystemC, (b) SystemA1, (c) SystemD. | ||
It can be seen from Table 2 and Fig. 5, the MSCSs can be synthesized in SystemA1, SystemC, and SystemD. When the ethanol–water mass ratio was varied from RW/E = 0.67 (SystemC) to RW/E = 0.43 (SystemA1) and RW/E = 0.25 (SystemD), the particle sizes were varied from 258 nm ± 8 nm to 264 nm ± 12 nm and 281 nm ± 10 nm. This result is consistent with the trend of the variation of the size of oil droplets (Fig. 2d). Therefore, the particle size of silica spheres can be further regulated by controlling the size of oil droplets in O/W type SFMEs.
3.4 Effect of cooling temperature on the properties of silica spheres
For the SystemB, two kinds of low-temperature strategies, a pre-reaction/low-temperature strategy and a low-temperature equilibrium/reaction strategy, were employed to assist in understanding of silica sphere formation processes. The TEM images, reaction conditions and properties of silica products are shown in Fig. 6 and Table 3. The 29Si MAS NMR and FT-IR characterization for the silica products synthesized by pre-reaction/low-temperature strategy are shown in Fig. 7 and 8, respectively. | ||
| Fig. 6 (a–d) TEM images of silica products synthesized with the SystemB by the pre-reaction/low-temperature strategy (the pre-reaction time was 1 min, the cooling temperatures were 0 °C, −10 °C, −15 °C and −20 °C, from a to d, respectively). (e–h) TEM images of silica products synthesized by with the SystemB by the low-temperature equilibrium/reaction strategy (the equilibrating time was 30 min, cooling temperatures were 0 °C, −10 °C, −15 °C and −20 °C, from e to h, respectively). | ||
| Strategy | Temperature (°C) | Morphology | Avg. diameter (nm) | σ (nm) |
|---|---|---|---|---|
| a Strategy 1: the pre-reaction/low-temperature strategy. Strategy 2: the low-temperature equilibrium/reaction strategy. | ||||
| 1 | 0 | Sphere | 330 | 13 |
| 1 | −10 | Sphere | 370 | 22 |
| 1 | −15 | Sphere | 460 | 23 |
| 1 | −20 | Ring-like sphere | 170 | 30 |
| 2 | 0 | Sphere | 414 | 30 |
| 2 | −10 | Sphere | 452 | 26 |
| 2 | −15 | Aggregate | — | — |
| 2 | −20 | Aggregate | — | — |
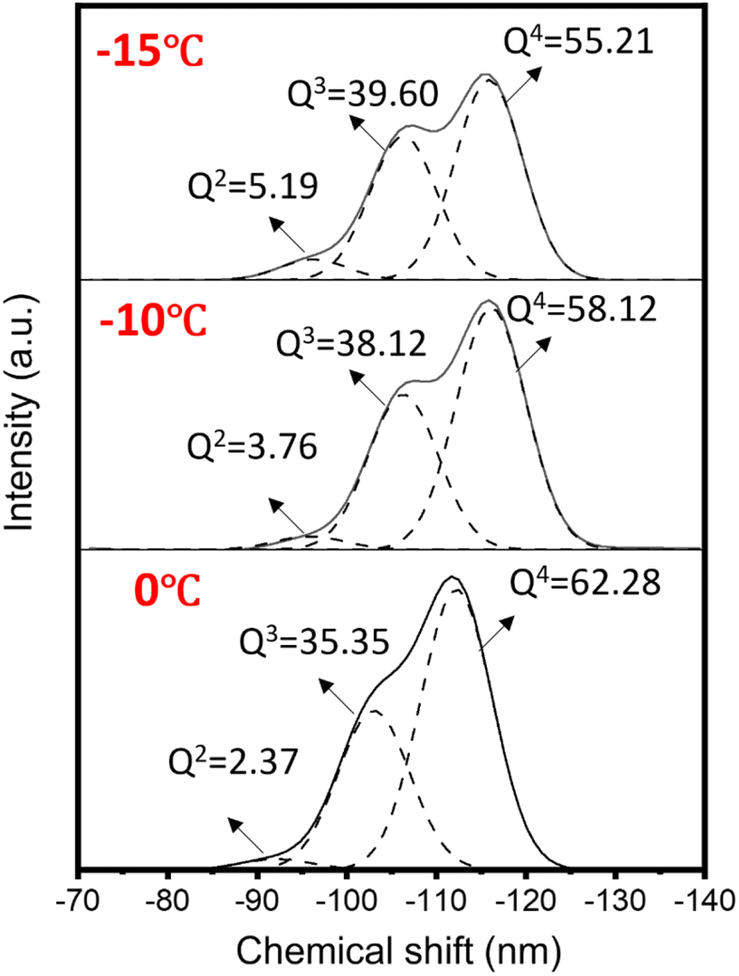 | ||
| Fig. 7 29Si MAS NMR spectra of silica spheres synthesized with the SystemB by the pre-reaction and low-temperature strategy: the pre-reaction time was 1 min, the cooling temperatures were 0 °C, −10 °C and −15 °C, respectively. | ||
 | ||
| Fig. 8 FT-IR diagram of silica spheres synthesized with the SystemB by the pre-reaction and low-temperature strategy: the pre-reaction time was 1 min, the cooling temperatures were 0 °C, −10 °C, −15 °C, and −20 °C, respectively. | ||
As shown in Fig. 6a–d, with the pre-reaction/low-temperature strategy (the pre-reaction time was 1 min), when the cooling temperatures were 0 °C, −10 °C and −15 °C, the main sizes of silica spheres synthesized with the SystemB were 330 nm, 370 nm and 460 nm, respectively (Fig. 6a–c), which are much larger than that of silica spheres synthesized at room temperature (25 °C).46 The yield of silica products synthesized by the pre-reaction/low-temperature strategy was shown in Table S1.† When the cooling temperature was decreased to −20 °C, ring-like spheres (ca. 170 nm in size) were observed in the system (Fig. 6d). This observation aligned with our earlier investigation on the synthesis of hollow spheres via a rapid dilution method.46
To the best of our knowledge, the TEOS in O/W type SystemB exists in two phases. One is the ethanol aqueous solution (medium), where TEOS exists mainly in a dissolved state. The other is the oil (TEOS) phase, in which TEOS is present in oil-droplet form. And the chemical potential of the TEOS in these two phases is equal.47 When the ammonia catalyst was added to the SystemB at room temperature, the hydrolysis and condensation of the dissolved TEOS could form reactive silica species in the medium, and some of them were captured by oil droplets.48 At the same time, the TEOS in the surface layer of oil droplets could also be hydrolyzed and condensed, and an uneven silica shell was formed in the initial stage of the reaction. When the reaction system was placed at a lower temperature, the chemical potential of the unreacted TEOS in oil droplets would increase accordingly, resulting in the increase of the mass-transfer driving force of the unreacted TEOS. The volume of oil droplets increased with decreasing temperature. When the cooling temperature was above −15 °C, the uneven silica shell could still prevent the migration of a large amount of unreacted TEOS in the oil droplets due to its certain viscoelasticity. If the cooling temperature was lower than −20 °C, the silica shell was no longer able to prevent the expansion caused by the increased driving force, resulting in a significant loss of unreacted TEOS in oil droplets. In addition, the volume shrinkage of ring-like spheres may be caused during specimen preparation for electron microscopy (Fig. 6d).
As it can be seen in Fig. 6e–h, with the low-temperature equilibrium/reaction strategy (the equilibrating time was 30 min), the sphericity of the silica products is lower than that of silica products synthesized by the pre-reaction/low-temperature strategy with the same cooling temperature (Fig. 6e–g). When the cooling temperatures were −15 °C and −20 °C, the silica products exhibited aggregation behaviour (Fig. 6g and h). If the SystemB is placed at a lower temperature, it will tend towards a new equilibrium state under this temperature. And the oil droplets in the SystemB become increasingly unstable. When the cooling temperatures were −15 °C and −20 °C, the transparent SystemB becomes turbid (Fig. S4†), indicating the occurrence of phase separation. Therefore, at lower equilibrium temperatures, it is more difficult to synthesize the MSCSs with the SystemB. The yield of silica products synthesized by the low-temperature equilibrium/reaction strategy was shown in Table S1.†
Fig. 7 shows the 29Si MAS NMR spectra of silica spheres synthesized by the pre-reaction/low-temperature strategy. In Fig. 7, three peaks of Q4, Q3 and Q2 were around −110, −101 and −91 ppm, respectively.49 Moreover, the fraction of those Qn units in the resulting spheres was derived by the deconvolution of the profiles, and the results are summarized in Table S2.† According to previous reports, the Q4 intensity ratio of silica spheres synthesized by the Stöber method was as high as 70%,50 which is significantly higher than our data. This indicates that the condensation degree of silica spheres synthesized at cooling temperatures is not high and the Si–O–Si network structures are not dense. As the cooling temperature decreased from 0 °C to −15 °C, the Q2 and Q3 intensity ratios increased from 2.37% and 35.35% to 5.19% and 39.60%, respectively, while the Q4 intensity ratio decreased from 62.28% to 55.21%. This shows that the lower the cooling temperature, the smaller the Q4 ratio, and the lower the condensation degree (Q2 + Q3/Q2 + Q3 + Q4) of silica spheres (Table S2†).
The FT-IR diagram of silica spheres synthesized by the pre-reaction and low-temperature strategy is shown in Fig. 8. The as-synthesized silica spheres had several IR absorption peaks: a broader one around 3430 cm−1 for υ(O–H) of the Si–OH groups or the adsorbed water, a small but sharp one at 1640 cm−1 for the water molecules, the strongest one at 1111 cm−1 for υ3(Si–O), while the bands at 960 cm−1 and 806 cm−1 were due to the δ(Si–OH) and υ3(Si–O), respectively (Fig. 8). Furthermore, the peaks intensity observed at 960 cm−1, 1640 cm−1 and 3430 cm−1 showed an increasing trend with decreasing cooling temperatures from 0 °C to −20 °C, which aligns with the pattern of condensation degree determined by 29Si MAS NMR measurements.
The results identified by the 29Si MAS NMR and FT-IR measurements indicate that the differences in the condensation degree of silica spheres are mainly caused by the volume increase of silica spheres and the migration of TEOS from oil droplets during the reaction process. Therefore, the low-temperature strategies can be used to control the internal properties of MSCSs.
3.5 Possible mechanism of silica spheres formation
When the NH3 is added to the O/W type SFMEs, the hydrolysis-condensation of TEOS, both in the medium and at the interface of oil droplets, occur immediately. According to the thermodynamic metastable state and new phase formation theory,48 nucleation and growth occur preferentially on the surface of oil droplets, resulting in the formation of shell of primary silica spheres. As the reaction progresses, the shell will become thicker, and the TEOS in the oil droplets will decrease. In addition, the reactive silica species generated in the medium are preferentially captured by the interfaces already existed rather than the accumulation of a high concentration to reduce the Gibbs free energy of the system.48 Therefore, the MSCSs can be prepared in the O/W type SFMEs (Scheme 1a). | ||
| Scheme 1 Possible mechanism of MSCSs (a) and ring-like silica (b) formation in the O/W type SFMEs. | ||
For the ring-like spheres, the formation process is illustrated in Scheme 1b. When the NH3 is added to the O/W type SFMEs, the silica spheres with different shell thickness are formed quickly. If the pre-reaction/low-temperature strategy was introduced to the systems in certain stages of reaction, the volume of oil droplets would expand due to the increase of the chemical potential of the unreacted TEOS, resulting in larger silica spheres. When the cooling temperature was below −20 °C, the silica shell was no longer able to prevent the expansion, and the ring-like spheres were formed.
4. Conclusions
In summary, three types of SFMEs (O/W, BC and W/O) composed of TEOS, EtOH and H2O were successfully fabricated. The mesostructures of SFMEs, oil droplets, sponge-like structure and water droplets, were confirmed by DLS and negative-staining TEM methods. The MSCSs can only be synthesized with the O/W type SFMEs, and there is a strong relationship between the particle size of silica spheres and the size of oil droplets. Larger MSCSs spheres could also be synthesized by cooling the O/W type SEMEs in the early reaction stages. Combining the thermodynamic metastable state and new phase formation theory, we proposed a self-templating growth mechanism to explain the formation of MSCSs. The research is very helpful for both expanding the application of the SFMEs and the controllable synthesis of MSCSs.Data availability
The date that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
Jiahan Lu: methodology, software, investigation, experiment, writing. Longhua Peng: validation, supervision. Ao Zhang: supervision. Jiaqiong Xu: supervision. Min Wu: supervision. Shiyu Ma: methodology, experiment, writing – review & editing.Conflicts of interest
No conflict of interest exits in the submission of this manuscript, and manuscript is approved by all authors for publication. I would like to declare on behalf of my co-authors that the work described was original research that has not been published previously, and not under consideration for publication elsewhere, in whole or in part. All the authors listed have approved the manuscript that is enclosed.Acknowledgements
We are very grateful to Prof. Chengzhong Yu and Kun Zhang from East China Normal University for their guidance and help in this work.Notes and references
- Y. Zhang, Y. Zhang, C. Wang, X. Liu, Y. Fang and Y. Feng, Green Chem., 2016, 18, 392–396 RSC.
- A. W. Du and M. H. Stenzel, Biomacromolecules, 2014, 15, 1097–1114 CrossRef CAS.
- S. P. Moulik and B. K. Paul, Adv. Colloid Interface Sci., 1998, 78, 99–195 CrossRef CAS.
- Q. Wang and D. O'Hare, Chem. Rev., 2012, 112, 4124–4155 CrossRef CAS PubMed.
- J. Xu, L. Zhang, D. Li, J. Zhao and W. Hou, Colloid Polym. Sci., 2013, 291, 2515–2521 CrossRef CAS.
- J. Xu, Y. Cui, R. Wang, Z. Shi, C. Wu and D. Li, Colloid Surf. A-Physicochem. Eng. Asp., 2021, 624, 126689 CrossRef CAS.
- M. Jalali-Jivan, F. Garavand and S. M. Jafari, Adv. Colloid Interface Sci., 2020, 283, 102227 CrossRef CAS PubMed.
- T. Nhujak, W. Saisuwan, M. Srisa-art and A. Petsom, J. Sep. Sci., 2006, 29, 666–676 CrossRef CAS PubMed.
- G. Mojeiko, M. de Brito, G. C. Salata and L. B. Lopes, Int. J. Pharm., 2019, 560, 365–376 CrossRef CAS PubMed.
- M. Zoumpanioti, H. Stamatis, V. Papadimitriou and A. Xenakis, Colloids Surf. B Biointerfaces, 2006, 47, 1–9 CrossRef CAS.
- R. A. Martınez-Rodrıguez, F. J. Vidal-Iglesias, J. Solla-Gullón, C. R. Cabrera and J. M. Feliu, J. Am. Chem. Soc., 2014, 136, 1280–1283 CrossRef.
- K. Min and H. Gao, J. Am. Chem. Soc., 2012, 134, 15680–15683 CrossRef CAS PubMed.
- W. Hou and J. Xu, Curr. Opin. Colloid Interface Sci., 2016, 25, 67–74 CrossRef CAS.
- T. N. Zemb, M. Klossek, T. Lopian, J. Marcus, S. Schöettl, D. Horinek, S. F. Prevost, D. Touraud, O. Diat, S. Marčelja and W. Kunz, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 4260–4265 CrossRef CAS.
- N. F. Borys, S. L. Holt and R. E. Barden, J. Colloid Interface Sci., 1979, 71, 526–532 CrossRef CAS.
- R. F. Hankel, P. E. Rojas, M. Cano-Sarabia, S. Sala, J. Veciana, A. Braeuer and N. Ventosa, Chem. Commun., 2014, 50, 8215–8218 RSC.
- Y. L. Khmelnitsky, A. Van Hoek, C. Veeger and A. J. W. G. Visser, J. Phys. Chem., 1989, 93, 872–878 CrossRef CAS.
- P. Ni and W.-G. Hou, Chin. J. Chem., 2008, 26, 1985–1990 CrossRef CAS.
- P. Ni and W.-G. Hou, Chin. J. Chem., 2008, 26, 1335–1338 CrossRef CAS.
- G. D. Smith, C. E. Donelan and R. E. Barden, J. Colloid Interface Sci., 1977, 60, 488–496 CrossRef CAS.
- S. Schöttl, J. Marcus, O. Diat, D. Touraud, W. Kunz, T. Zemb and D. Horinek, Chem. Sci., 2014, 5, 2949–2954 RSC.
- M. L. Klossek, D. Touraud, T. Zemb and W. Kunz, Chemphyschem, 2012, 13, 4116–4119 CAS.
- M. L. Klossek, D. Touraud and W. Kunz, Phys. Chem. Chem. Phys., 2013, 15, 10971–10977 RSC.
- O. Diat, M. L. Klossek, D. Touraud, B. Deme, I. Grillo, W. Kunz and T. Zemb, J. Appl. Crystallogr., 2013, 46, 1665–1669 CrossRef CAS.
- J. Lara, G. Perron and J. E. Desnoyers, J. Phys. Chem., 1981, 85, 1600–1605 CrossRef CAS.
- J. Xu, A. Yin, J. Zhao, D. Li and W. Hou, J. Phys. Chem. B, 2013, 117, 450–456 CAS.
- N. I. Peng and W. G. Hou, Chin. J. Chem., 2010, 26, 1335–1338 Search PubMed.
- Y. Liu, J. Xu, H. Deng, J. Song and W. Hou, RSC Adv., 2018, 8, 1371–1377 RSC.
- Y. Zhang, X. Chen and X. Liu, Langmuir, 2019, 35, 14358–14363 CrossRef CAS PubMed.
- X. Ma, Y. Chen, J. Qian, Y. Yuan and C. Liu, Mater. Chem. Phys., 2016, 183, 220–229 CrossRef CAS.
- B. Sun, J. Chai, Z. Chai, X. Zhang, X. Cui and J. Lu, J. Colloid Interface Sci., 2018, 526, 9–17 CrossRef CAS PubMed.
- X. Cui, J. Wang, X. Zhang, Q. Wang, M. Song and J. Chai, Langmuir, 2019, 35, 9255–9263 CrossRef CAS PubMed.
- M. El-Hefnawy, Mod. Appl. Sci., 2012, 6, 101–105 CAS.
- X. Zhang, Y. Han, W. Liu, N. Pan, D. Li and J. Chai, J. Ind. Eng. Chem., 2021, 97, 326–336 CrossRef CAS.
- J. Blahnik, J. Schuster, R. Müller, E. Müller and W. Kunz, J. Colloid Interface Sci., 2024, 655, 371–382 CrossRef CAS.
- J. Blahnik, S. Krickl, K. Schmid, E. Müller, J. Lupton and W. Kunz, J. Colloid Interface Sci., 2023, 648, 755–767 CrossRef CAS PubMed.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- A. Guerrero-Martinez, J. Perez-Juste and L. M. Liz-Marzan, Adv. Mater., 2010, 22, 1182–1195 CrossRef CAS PubMed.
- X. Meng, Z. Wang, Y. Qiao and D. Qiu, Chem. Eng. J., 2023, 456 Search PubMed.
- J. Drapeau, M. Verdier, D. Touraud, U. Kroeckel, M. Geier, A. Rose and W. Kunz, Chem. Biodivers., 2009, 6, 934–947 CrossRef CAS PubMed.
- A. Wang, L. Chen, D. Jiang, H. Zeng and Z. Yan, Ind. Crop. Prod., 2014, 62, 515–521 CrossRef CAS.
- R. Pramanik, S. Sarkar, C. Ghatak, V. G. Rao, P. Setua and N. Sarkar, J. Phys. Chem. B, 2010, 114, 7579–7586 CrossRef CAS.
- M. Song, W. Liu, Q. Wang, J. Wang and J. Chai, J. Ind. Eng. Chem., 2020, 83, 81–89 CAS.
- G. H. Bogush, M. A. Tracy and C. F. Zukoski, J. Non-Cryst. Solids, 1988, 104, 95–106 CrossRef CAS.
- M. T. Harris, R. R. Brunson and C. H. Byers, J. Non-Cryst. Solids, 1990, 121, 397–403 CrossRef CAS.
- Z. Chen, B. Peng, J.-Q. Xu, X.-C. Xiang, D.-F. Ren, T.-Q. Yang, S.-Y. Ma, K. Zhang and Q.-M. Chen, Nanoscale, 2020, 12, 3657–3662 CAS.
- S. Trasatti, P. Atkins and J. De Paula, Atkins’ Physical Chemistry, Oxford University Press Oxford, 8th edn, 2007, vol. 52, pp. 2729–2730 Search PubMed.
- P. C. Hiemenz and R. Rajagopalan, Principles of Coslloid and Surface Chemistry, Marcel Dekker, 3rd edn, 2016. Search PubMed.
- G. Engelhardt and D. Michel, High-Resolution Solid-State NMR of Silicates and Zeolites, John Wiley & Sons, New York NY, 1987 Search PubMed.
- S. Chen, A. Osaka, S. Hayakawa, K. Tsuru, E. Fujii and K. Kawabata, J. Sol-Gel Sci. Technol., 2008, 48, 322–335 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra07679f |
| This journal is © The Royal Society of Chemistry 2024 |