
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAqueous solution of biogenic carboxylic acids as sustainable catalysts and green reaction media for the high-yielding synthesis of Biginelli adducts, Hantzsch esters, and substituted pyridines†
Poornachandra Shamanna Prabhakar‡
a,
Jitendra Sahoo‡a,
Ibrahim A. Alnaserb,
Asiful H. Seikhb,
Mohammad Rezaul Karimb and
Saikat Dutta *a
*a
aDepartment of Chemistry, National Institute of Technology Karnataka (NITK), Surathkal, Mangalore-575025, India. E-mail: sdutta@nitk.edu.in
bCenter of Excellence for Research in Engineering Materials (CEREM), Deanship of Scientific Research, King Saud University, Riyadh 11421, Saudi Arabia
First published on 10th December 2024
Abstract
3,4-Dihydropyrimidin-2(1H)-ones (DHPMs) and 1,4-dihydropyridines (DHPs), prepared by applying the Biginelli and Hantzsch reaction protocols, respectively, are well-documented nitrogen-containing heterocycles with intriguing pharmacological properties. The aqueous solution of biogenic carboxylic acids renewably produced from biomass via catalytic or enzymatic processes can be used as a sustainable catalyst and green reaction media for synthesizing DHPs and DHPMs. This work evaluates the efficacy of various biogenic acids in their aqueous solutions as catalysts for synthesizing DHPs and DHPMs from substituted benzaldehydes. Among the studied biogenic acids, gluconic acid aqueous solution (GAAS) proved to be the most efficient, safe, non-volatile, and recyclable catalyst. The reaction afforded excellent isolated yields (≥85%) of spectroscopically pure DHPs and DHPMs under optimized conditions and employed a straightforward work-up procedure. Aqueous ammonia was successfully employed instead of ammonium salt to improve the atom economy of DHPs. Moreover, substituted pyridines were synthesized from DHPs in a one-pot, two-step process using NaNO2 as an oxidant in the GAAS medium.
Introduction
Synthetic organic chemistry has come a long way over the past two centuries, from disapproving the vitalism theory to creating organic molecules at will with precise control of their structure, properties, and functionality that are indispensable for the comforts of industrialized societies.1 However, the large-scale production of organic compounds in chemical industries generates enormous quantities of toxic wastes as byproducts and side products.2 Green chemistry metrics developed over the past two decades enable the qualitative and quantitative estimation of such wastes and help develop superior synthetic strategies and chemical processes to eliminate or reduce waste at the source.3 One way to achieve source reduction of waste is to minimize the number of synthetic steps for a targeted product since every step requires energy and material input during synthetic transformation and product purification.4 In this regard, multi-component reactions (MCRs) have received much interest over the past decades, allowing three or more types of molecules containing complementary functionalities to react sequentially and afford a product with excellent selectivity and yield. MCRs are convergent and atom-economic, employ readily accessible starting materials, and afford densely functionalized structurally complex molecules in a single-pot reaction.5 Another crucial strategy to minimize waste generation during organic transformation is to use an efficient catalyst and eco-friendly reaction media. A chemical catalyst improves the sustainability of organic synthesis by lowering activation energy and providing a favorable mechanistic pathway. Catalysis leads to a targeted product in excellent selectivity and yield, starting from safe and inexpensive starting materials under energy- and reagent-economic conditions.6,7 However, the preparation, recovery, and recycling of catalysts also produce chemical wastes, which must be accounted for before selecting the appropriate catalyst candidate.8 The reaction medium is pivotal for heat and mass transfer and influences the mechanistic pathway. Traditional organic solvents, which are typically produced from petroleum, are routinely used in organic synthesis in academic and industrial settings. However, they are often expensive, toxic, and volatile and complicate product purification.9 Therefore, coordinated research is needed to identify alternative, sustainable reaction media for organic transformations.10 The use of aqueous media in organic reactions has received particular attention since water is abundant, inexpensive, safe, and conveniently recyclable via distillation.11–13 A major concern of using an aqueous reaction medium is the insolubility of most organic reactants in water.The synthesis of 3,4-dihydropyrimidin-2(1H)-ones (DHPMs) and 1,4-dihydropyridines (DHPs), prepared by following the Biginelli and Hantzsch reaction protocol, respectively, fall under MCRs.14 In the Biginelli reaction, an aldehyde, a β-ketoester, and urea are reacted in the presence of a suitable acid catalyst.15 In the Hantzsch ester synthesis, two equivalents of a β-ketoester react with an aldehyde in the presence of a source of ammonia.16 In these reactions, water is formed as the sole innocuous byproduct. These nitrogen-containing heterocyclic compounds (i.e., DHPMs and DHPs) exhibit promising biological activities, including antifungal, antibacterial, antitubercular, and anticancer activity.17–19 DHPs have shown remarkable efficiency as a reagent in various photoredox-catalyzed organic transformations.20 The DHPs can be oxidized selectively to synthesize heavily substituted pyridines, and metal nitrites or nitrates have shown promising activity for this transformation.21–23 Over the years, an exhaustive list of acid catalysts has been explored for both the Biginelli and Hantzsch reactions.24,25 Other reaction parameters, such as the heating method (resistive heating, microwave irradiation) and reaction media (ionic liquid, deep-eutectic solvent, melt), have also been studied.26–29
Carboxylic acids have received much attention as catalysts in MCR because they are metal-free, less corrosive than mineral acids, relatively non-toxic, inexpensive, thermally stable, and available in pure form.30–32 Not surprisingly, the Biginelli and Hantzsch reactions have been performed using various carboxylic acids as catalysts.33,34 The biogenic carboxylic acids that can be produced renewably from biomass by chemical-catalytic or enzymatic routes have additional advantages because they are innocuous and sustainable. Organic acids in their pure form are often corrosive, volatile, thermally unstable, and also solid in some cases, and their use as catalysts requires a secondary reaction medium or solvent. Moreover, the Biginelli and Hantzsch reactions produce water as a byproduct that dilutes the organic acids, and they must be concentrated and dried before subjecting them to the next catalytic cycle. However, the aqueous solution of organic acids is less corrosive, less volatile, and thermally more stable, and could act as both a catalyst and a reaction medium.35 Moreover, the recycling involves evaporation of excess water from the mixture without requiring extensive distillation and drying processes. Gluconic acid aqueous solution (45–50% water) (GAAS) is commercially available and has gained interest as an acid catalyst in various organic transformations.35,36 Gluconic acid (pKa = 3.86) is roughly ten times stronger acid than acetic acid and non-volatile. It is produced by the catalytic or enzymatic oxidation of glucose or directly from cellulose.37,38 Recently, we reported gluconic acid aqueous solution (GAAS) as a sustainable organic acid catalyst and reaction media for synthesizing novel Biginelli and Hantzsch products from biorenewable furfurals.34
This work explores the catalytic efficiency of an aqueous solution of various biogenic acids as a catalyst and reaction medium for synthesizing Biginelli and Hantzsch adducts from substituted benzaldehydes (1a–1j) under conventional heating (Scheme 1). The effects of various reaction parameters, such as reaction temperature, duration, and catalyst loading, were studied for both transformations using GAAS as the catalyst. The products were purified without chromatography, and the catalyst was recycled four times. The Hantzsch products (4a–j) were purified by triturating the crude product in a mixture of n-heptane and ethyl acetate (97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, v/v), while the Biginelli products were purified by recrystallizing from ethanol. Aqueous ammonia instead of ammonium salt (e.g., NH4OAc) was also explored for the atom-economic synthesis of Hantzsch esters. Isolated DHPs were oxidized by NaNO2 in the GAAS medium to form substituted pyridines (7a–7j). Moreover, a one-pot, two-step synthesis of Hantzsch pyridines was attempted without isolating the DHPs.
:3, v/v), while the Biginelli products were purified by recrystallizing from ethanol. Aqueous ammonia instead of ammonium salt (e.g., NH4OAc) was also explored for the atom-economic synthesis of Hantzsch esters. Isolated DHPs were oxidized by NaNO2 in the GAAS medium to form substituted pyridines (7a–7j). Moreover, a one-pot, two-step synthesis of Hantzsch pyridines was attempted without isolating the DHPs.
 | ||
| Scheme 1 Synthesis of Biginelli adducts, Hantzsch esters, and substituted pyridines from substituted benzaldehydes using gluconic acid aqueous solution as a sustainable catalyst. | ||
Experimental section
Materials
Ammonium acetate (96%), oxalic acid (99%), citric acid (99%), succinic acid (99%), and sodium sulfate (99%) were purchased from Loba Chemie Pvt Ltd. Benzaldehyde (98%), 4-phenylbenzaldehyde (98%), ethyl acetoacetate (99.5%), urea (99.5%), formic acid (98%), sodium nitrite (96%) and p-toluenesulfonic acid (98%) were purchased from Spectrochem. 4-Methoxybenzaldehyde (99%), 4-flourobenzaldehyde (98%), 4-nitrobenzaldehyde (99%), 4-bromobenzaldehyde (97%), 4-chlororbenzaldehyde (97%), and gluconic acid aqueous solution (aq. GAAS) (45–50% water) were purchased from TCI Chemicals. p-Tolualdehyde (97%), 3,4,5-trimetoxybenzaldehyde (98%), 4-ethoxybenzaldehyde (99%), phosphotungstic acid (reagent grade), and amberlyst-15(H) were purchased from Sigma-Aldrich. n-Heptane (99%) and glacial acetic acid (99.5%) were purchased from Molychem. Ethanol (99.8%) was purchased from Aqlivia. Ethyl acetate (99%) was purchased from Finar. All the chemicals were used without further purification. Thin-layer chromatography (TLC) plates, silica gel pre-coated on aluminium sheets, were purchased from Merck (TLC Silica Gel 60, F254).Characterization methods
The synthesized products were characterized by spectroscopic methods and matched with the literature data. Fourier transform infrared (FTIR) spectra were collected using the ATR method in a Bruker Alpha II FTIR instrument equipped with zinc selenide (ZnSe) as the prism material. The compounds were dissolved in dichloromethane, and a thin film was made by evaporating a drop of the solution on the ATR counter. The FTIR spectra were averaged by collecting 24 scans at a scanning speed of 4 scans per second in the 500–4000 cm−1 range. For nuclear magnetic resonance (NMR) spectroscopy, the 1H-NMR spectra were collected using a Bruker NanoBay® NMR instrument at the operating radio frequency of 400 MHz, and the 13C-NMR spectra were recorded using the same instrument at the frequency of 100 MHz (calculated). The melting point of the Biginelli and Hantzsch products was measured using a Stuart SMP3 digital melting point apparatus.Synthetic procedures
Results and discussion
The Biginelli reaction between 1a, EAA (2), and urea (3) was chosen as the model reaction for process optimization. The reaction was carried out by conventional heating under organic solvent-free conditions. Benzaldehyde (1a) was used as the limiting reagent, and the progress of the reaction was monitored using TLC. After the conversion of 1a was complete, the reaction mixture was cooled to room temperature and quenched in crushed ice to obtain a solid product. The solid was filtered through a filter paper (Whatman, grade 5) under vacuum, dried at 60 °C for 12 h in a hot-air oven, and recrystallized using absolute ethanol to get pure ethyl-6-methyl-2-oxo-4-phenyl-1,2,3,4-tetrahydropyrimidine-5-carboxylate (4a). When the reaction was attempted using equimolar amounts of all three reagents without a catalyst, only a trace amount of 4a was observed on TLC, even at 80 °C. However, the reaction progressed much faster even at room temperature using a 25 mol% (based on the starting amount of 1a) GAAS catalyst. However, the reaction was incomplete in 24 h, and only 12% of 4a was obtained. Increasing the reaction temperature to 60 °C afforded a 57% isolated yield of 4a after 24 h because the conversion of 1a was not complete. When the reaction temperature was increased to 80 °C, a complete conversion of 1a was achieved in 6 h, but only 63% of 4a was obtained. This result may be explained by the slower kinetics and the thermal decomposition of urea as a competing reaction. Increasing the temperature to 100 °C afforded a 70% yield of 4a after a 4 h reaction, which increased further to 76% after a 2 h reaction at 120 °C. A further increase in the reaction temperature to 140 °C marginally decreased the yield of 4a to 72% due to the thermal decomposition of the starting material(s) and product (Fig. 1). | ||
| Fig. 1 Effect of reaction temperature on the yield of 4a. Reaction conditions: 1a (0.500 g, 4.71 mmol), EAA (0.613 g, 4.71 mmol), urea (0.282 g, 4.71 mmol), and GAAS (0.231 g, 25 mol%). | ||
After optimizing the temperature, the effect of the molar ratio of the reactants was studied. When a slight excess of EAA and urea was used (1.2 equiv. of 1a), the reaction was completed in 2 h, and an 86% yield of 4a was obtained. Increasing the equivalence of EAA and urea to 1.4 (with respect to 1a) resulted in a 93% yield of 4a. A further increase in the equivalence of EAA and urea showed no noticeable improvement in the yield of 4a. The effect of the loading of the GAAS catalyst was examined next by keeping all the other parameters unaltered. When the catalytic loading was decreased to only 15 mol%, the effect on the yield of 4a was minimal. Interestingly, a 10 mol% loading of GAAS afforded a 96% yield of 4a (120 °C, 2 h) (Fig. 2). A further decrease in catalyst loading to 5 mol% decreased the yield to 90% under similar reaction conditions. Therefore, a 10 mol% GAAS catalyst was selected as the optimized catalyst loading. After optimizing the reaction conditions, the catalytic efficiency of the GAAS catalyst was compared against some frequently used homogeneous and heterogeneous acid catalysts for the Biginelli reaction, including biogenic acids (Table 1). When acetic acid (50% aq., 10 mol%) was used as the catalyst, the reaction was completed within 2 h and afforded a 90% isolated yield of 4a. The use of different biogenic acids, such as oxalic acid, succinic acid, formic acid, and lactic acid (10 mol% of each), resulted in 86%, 89%, 82%, and 83% yields of 4a, respectively, under optimized conditions. Strong inorganic and organic solid acid catalysts, such as Amberlyst-15, pTSA, and PTA, produced 84%, 89%, and 85% of 4a under the reaction conditions, respectively.
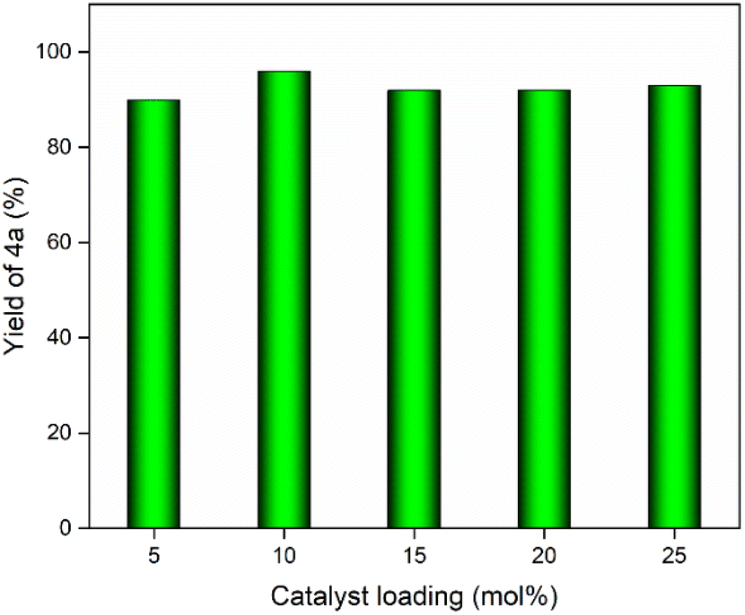 | ||
| Fig. 2 Effect of catalyst loading on the synthesis of 4a. Reaction conditions: 1a (0.500 g, 4.71 mmol), EAA (0.858 g, 6.59 mmol), urea (0.396 g, 6.59 mmol), 120 °C (oil-bath), and 2 h. | ||
| Entry | Catalyst | Reaction time (h) | Yields (%) |
|---|---|---|---|
| a Reaction conditions: 1a (0.500 g, 4.71 mmol), EAA (0.858 g, 6.59 mmol), urea (0.396 g, 6.59 mmol), 120 °C, and catalyst (10 mol%).b Solid heterogeneous acid.c Organic acid other than carboxylic acid.d Homogeneous inorganic acid. | |||
| 1 | Acetic acid | 2 | 90 |
| 2 | Formic acid | 2 | 82 |
| 3 | Succinic acid | 1 | 89 |
| 4 | Lactic acid | 2 | 83 |
| 5 | Citric acid | 2 | 90 |
| 6 | Oxalic acid | 2 | 86 |
| 7 | GAAS | 2 | 96 |
| 8b | Amberlyst-15 | 2 | 84 |
| 9c | pTSA | 2 | 89 |
| 10d | PTA | 2 | 85 |
Finally, the optimized synthesis was applied for the Biginelli reaction of substituted benzaldehydes (1b–j), and the isolated yield of the corresponding DHPM (4b–j) is listed in Table 2. The process worked equally well for substituted benzaldehydes carrying electron-donating or electron-withdrawing functionalities. For example, 4-nitrobenzaldehyde produced a 92% yield of the corresponding DHPM 4c (entry 3), while 4-methylbenzaldehyde also produced the same yield as DHPM 4e (entry 5).
|
||||
|---|---|---|---|---|
| Entry | Substrate | Time (h) | Product | Yields (%) |
| a Reaction conditions: aldehyde (0.500 g), EAA (1.4 eq.), urea (1.4 eq.), GAAS (10 mol%), and 120 °C (oil-bath). | ||||
| 1 |  |
2 |  |
96 |
| 2 |  |
5 | 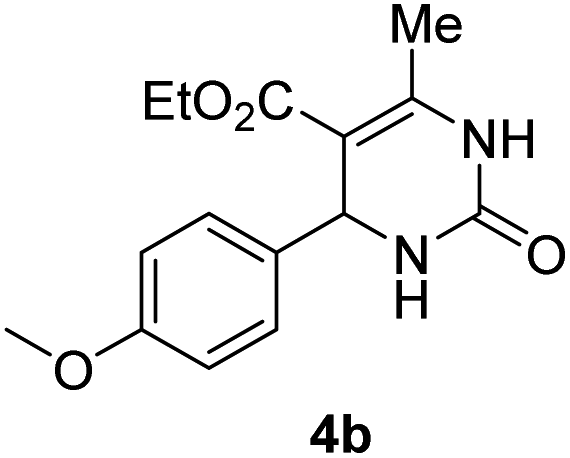 |
88 |
| 3 |  |
2 |  |
92 |
| 4 |  |
2 |  |
85 |
| 5 |  |
3 |  |
92 |
| 6 |  |
2 |  |
85 |
| 7 |  |
3 |  |
86 |
| 8 |  |
2 |  |
94 |
| 9 |  |
2 | 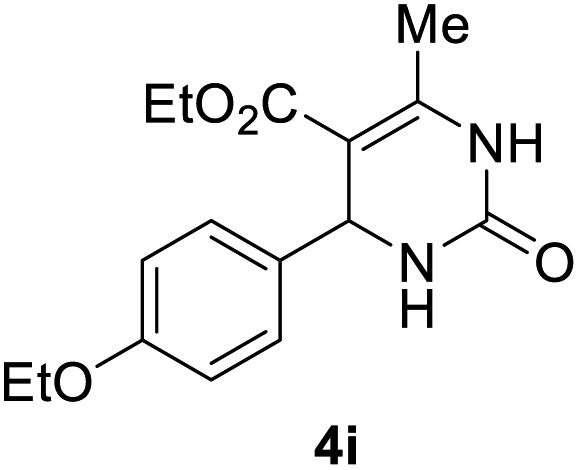 |
92 |
| 10 |  |
2 |  |
85 |

The GAAS catalyst was then used to synthesize DHPs (6a–j) using the Hantzsch reaction protocol. The Hantzsch reaction was optimized using 1a, EAA, and ammonium acetate as model reactants. Initially, the reaction was carried out at room temperature in the presence of GAAS (25 mol%), which remained incomplete even after 24 h, and only a 30% yield of 6a was obtained. The reaction took 8 h for the complete conversion of 1a at 60 °C, and 6a was isolated in only 65% yield. The reaction was completed within 2 h at 80 °C, affording a 92% isolated yield of 6a. Further increasing the temperature to 100 °C led to a marginal decrease in yield of 6a to 90% (Fig. 3).
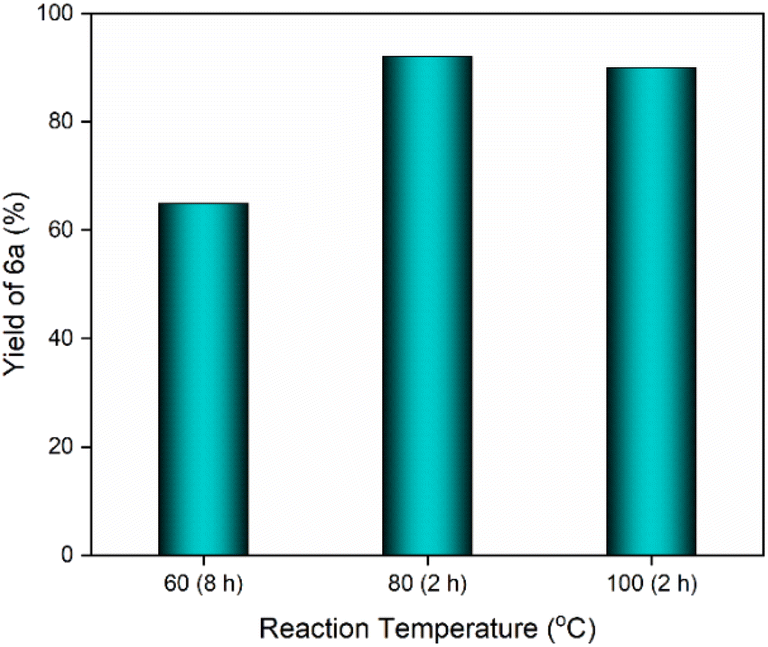 | ||
| Fig. 3 Effect of reaction temperature on the yield of 6a. Reaction conditions: 1a (0.500 g, 4.71 mmol), EAA (1.226 g, 9.42 mmol), NH4OAc (0.544 g, 7.07 mmol), and GAAS (0.231 g, 25 mol%). | ||
After optimizing the temperature, the effect of the catalyst loading was studied. Using 25 mol% of catalyst produced 92% of 6a at 80 °C for 2 h. When the catalyst loading decreased to 15 mol%, it produced an 80% yield after 3 h of reaction time (Fig. 4). The use of 20 mol% of catalyst produced only an 85% yield of 6a. When only 10 mol% of catalyst was used, the reaction required 5 h to complete, and the yield of 6a decreased to 70%. Hence, 25 mol% GAAS was considered the optimum catalyst loading. The lower yields of 6a at lower catalyst loading are attributed to the increased decomposition of EAA and product for the prolonged reaction at increased temperature.
 | ||
| Fig. 4 Effect of catalyst loading for the synthesis of 6a. Reaction conditions: 1a (0.500 g, 4.71 mmol), EAA (1.226 g, 9.42 mmol), NH4OAc (0.544 g, 7.07 mmol), and 80 °C. | ||
Finally, the optimized reaction conditions were applied to synthesize other Hantzsch esters (6b–j), starting from substituted benzaldehydes, and yields are listed in Table 3. All substituted benzaldehydes produced satisfactory yields of DHPs. The electron-donating and electron-withdrawing functionalities attached to the benzaldehyde moiety were equally tolerated.
|
||||
|---|---|---|---|---|
| Entry | Substrate | Time (h) | Product | Yields (%) |
| a Reaction conditions: aldehyde (0.500 g), EAA (2 equiv.), NH4OAc (1.5 equiv.), GAAS (25 mol%), 80 °C, and 2 h. | ||||
| 1 |  |
2 |  |
92 |
| 2 |  |
5 |  |
90 |
| 3 |  |
2 |  |
88 |
| 4 |  |
2 |  |
88 |
| 5 |  |
3 |  |
89 |
| 6 |  |
2 |  |
91 |
| 7 |  |
3 |  |
84 |
| 8 |  |
2 |  |
89 |
| 9 |  |
5 |  |
88 |
| 10 |  |
2 |  |
82 |

Recyclability of the GAAS catalyst was attempted under optimized reaction conditions to synthesize 4a and 6a (Fig. 5). After the reaction, the mixture was cooled to room temperature and quenched in water. The organic impurities were removed using ethyl acetate, and the aqueous layer was evaporated under reduced pressure to recover gluconic acid. The concentration was then adjusted to 50 wt% by adding deionized water, and the solution was then subjected to the next catalytic cycle. The catalyst showed good activity up to the 4th catalytic cycle.
 | ||
| Fig. 5 Catalyst recyclability for the synthesis of DHPM, 4a (left) and DHP, 6a (right) starting from benzaldehyde, 1a. | ||
The marginal lowering in the product yield in the successive cycles of the catalyst can be attributed to the mass loss of the catalyst after each cycle. Consequently, the reaction takes a longer time to complete. Hence, the product yield was lower due to the incomplete conversion of the starting materials after 2 h of reaction. However, when the reaction was allowed to complete by extending the duration, no apparent dip in the yield of 4a or 6a was observed, demonstrating that the chemical stability and catalytic efficiency of GAAS remained intact.
The use of NH4OAc as the source of ammonia for synthesizing DHPs (6a–j) leads to acetic acid as the byproduct. Therefore, the atom economy of the reaction is lowered, and the acetic acid complicates the purification and recycling of the GAAS catalyst. We argue that aqueous ammonia could be used directly in GAAS. The ammonia reacted with GAAS to form ammonium gluconate, which acted as a reservoir of ammonia. A typical ammonia solution (30%, aq.) was added to GAAS; then, 1a and 2 were added. When a near equivalent amount of ammonia solution was used, the kinetics was relatively slow, and the conversion of 1a was incomplete even after 12 h at 80 °C. This observation can be attributed to the higher thermal stability of ammonium gluconate compared to ammonium acetate. Therefore, excess aq. NH3 had to be used, which acted as the reagent, while ammonium gluconate acted as the catalyst. The reaction afforded a 90% isolated yield of 6a after 4 h at 80 °C. The oxidation of Hantzsch esters is explored next. The isolated esters 6a–j were redissolved in GAAS, and NaNO2 was added as the oxidant. The reaction proceeded rapidly even at RT, and the conversion of the starting material was complete after 30 min, affording excellent isolated yields of the substituted pyridines 7a–j (Table 4). When 6a was used as the substrate, a 98% isolated yield of 7a was obtained (Table 4, entry 1).
|
|||
|---|---|---|---|
| Entry | Substrate | Product | Yields (%) |
| a Reaction conditions: (A) 1a–j (0.500 g), EAA (2 equiv.), aq. NH3, and GAAS (50 mol%), 80 °C, and 2 h; (B) NaNO2 (0.500 g), GAAS (2 mL), RT, and 30 min. | |||
| 1 |  |
 |
98 |
| 2 |  |
 |
97 |
| 3 |  |
 |
97 |
| 4 |  |
 |
98 |
| 5 |  |
 |
96 |
| 6 | 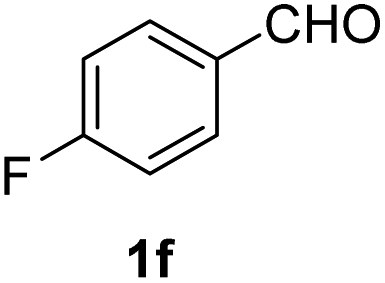 |
 |
97 |
| 7 |  |
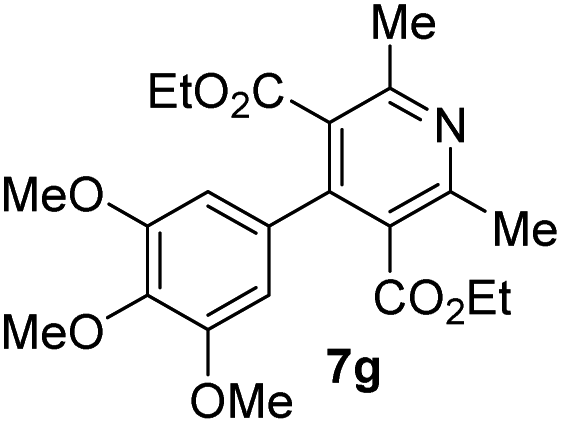 |
94 |
| 8 |  |
 |
95 |
| 9 | 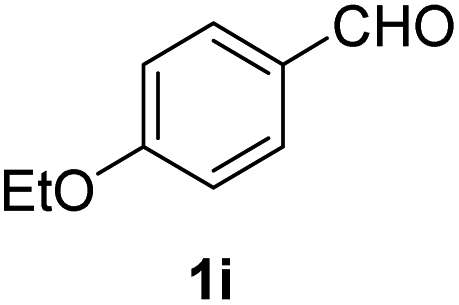 |
 |
96 |
| 10 |  |
 |
95 |

The electron-donating or electron-withdrawing substituents on benzaldehyde had no apparent impact on the reaction kinetics or yield of 7a–7j. Because the Hantzsch ester synthesis and their subsequent oxidation into substituted pyridines were performed in GAAS, a one-pot, two-step synthesis was designed. After the synthesis of 6a was complete (using NH4OAc or aq. NH3 as the reagent), the reaction mixture was cooled to RT. The required amount of NaNO2 and excess GAAS were added to the reaction mixture and magnetically stirred until the disappearance of 6a was complete. The reaction was complete within 30 min and afforded a 90% yield of 7a. In the control reaction, aqueous NaNO2 alone did not afford any oxidation product. Interestingly, the stepwise synthesis of 7a from 1a afforded the same yield as the one-pot, two-step process without isolating 6a. The one-pot, two-step synthesis of 7a was scaled up to 5 g, and the yield marginally improved to 91%.
Conclusion
In conclusion, a series of Biginelli adducts, Hantzsch esters, and substituted pyridines have been synthesized from benzaldehydes using GAAS as a bio-based, eco-friendly, robust, efficient, non-volatile, and recyclable acid catalyst. The promising aspects of the present methodology include simple work-up, high yield, short reaction duration, and use of an innocuous acid catalyst under organic solvent-free conditions. All the products were isolated with good to excellent isolated yields and purified without chromatography. Aqueous ammonia was used instead of ammonium acetate to improve the atom economy of the Hantzsch ester synthesis and simplify the recycling of the GAAS catalyst. Substituted pyridines were synthesized in a one-pot, two-step synthesis by oxidizing the Hantzsch esters using NaNO2 as the oxidant. This work will broaden the scope of GAAS as an eco-friendly catalyst for various organic transformations.Data availability
The original data of the study are included in the article and its ESI material.†Author contributions
Poornachandra Shamanna Prabhakar synthesized the products, analyzed the spectroscopic data, and edited the manuscript. Jitendra Sahoo worked on optimizing the process, purifying the products, and checking the melting points. Ibrahim A. Alnaser, Asiful H. Seikh, and Mohammad Rezaul Karim edited the manuscript and secured research funding. Saikat Dutta ideated the work, supervised the progress, and wrote the original manuscript.Conflicts of interest
The authors declare no competing interest.Acknowledgements
The authors thank the Central Research Facility (CRF), NITK, Surathkal, for assisting in NMR data collection and the Department of Metallurgical and Materials Engineering, NITK Surathkal, for FTIR analysis. The authors would like to acknowledge the Researchers Supporting Project number (RSPD2024R597), King Saud University, Riyadh, Saudi Arabia. SD thanks DST-SERB for the Core Research Grant (File No. CRG/2021/001084 and CRG/2022/009346). PSP thanks NITK, Surathkal, for scholarship support.References
- K. C. Nicolaou, Proc. R. Soc. A, 2014, 470, 20130690 CrossRef CAS PubMed.
- R. Naidu, B. Biswas, I. R. Willett, J. Cribb, B. Kumar Singh, C. Paul Nathanail, F. Coulon, K. T. Semple, K. C. Jones, A. Barclay and R. J. Aitken, Environ. Int., 2021, 156, 106616 CrossRef CAS PubMed.
- R. A. Sheldon, M. L. Bode and S. G. Akakios, Curr. Opin. Green Sustainable Chem., 2022, 33, 100569 CrossRef CAS.
- J. Schwan and M. Christmann, Chem. Soc. Rev., 2018, 47, 7985–7995 RSC.
- S. E. John, S. Gulati and N. Shankaraiah, Org. Chem. Front., 2021, 8, 4237–4287 RSC.
- R. A. Sheldon, Pure Appl. Chem., 2000, 72, 1233–1246 CrossRef CAS.
- V. K. Tiwari, A. Kumar, S. Rajkhowa, G. Tripathi and A. K. Singh, Green Chemistry, Springer Nature Singapore, Singapore, 2022, pp. 207–260 Search PubMed.
- M. Marafi and A. Stanislaus, Resour., Conserv. Recycl., 2008, 52, 859–873 CrossRef.
- M. Ikeda, Toxicol. Lett., 1992, 64–65, 191–201 CrossRef CAS PubMed.
- R. A. Sheldon and C. Opin, Green Sustainable Chem., 2019, 18, 13–19 CrossRef.
- M. Cortes-Clerget, J. Yu, J. R. A. Kincaid, P. Walde, F. Gallou and B. H. Lipshutz, Chem. Sci., 2021, 12, 4237–4266 RSC.
- D. K. Romney, F. H. Arnold, B. H. Lipshutz and C.-J. Li, J. Org. Chem., 2018, 83, 7319–7322 CrossRef CAS PubMed.
- B. H. Lipshutz and S. Ghorai, Green Chem., 2014, 16, 3660–3679 CAS.
- J. E. Biggs-Houck, A. Younai and J. T. Shaw, Curr. Opin. Chem. Biol., 2010, 14, 371–382 CrossRef CAS PubMed.
- M. Puripat, R. Ramozzi, M. Hatanaka, W. Parasuk, V. Parasuk and K. Morokuma, J. Org. Chem., 2015, 80, 6959–6967 CrossRef CAS PubMed.
- L. Shen, S. Cao, J. Wu, J. Zhang, H. Li, N. Liu and X. Qian, Green Chem., 2009, 11, 1414 CAS.
- H. Nagarajaiah, A. Mukhopadhyay and J. N. Moorthy, Tetrahedron Lett., 2016, 57, 5135–5149 CrossRef CAS.
- G. Liu, R. Pan, Y. Wei and L. Tao, Macromol. Rapid Commun., 2021, 42, 2000459 CrossRef CAS PubMed.
- A. M. Vijesh, A. M. Isloor, S. K. Peethambar, K. N. Shivananda, T. Arulmoli and N. A. Isloor, Eur. J. Med. Chem., 2011, 46, 5591–5597 CrossRef CAS PubMed.
- P.-Z. Wang, J.-R. Chen and W.-J. Xiao, Org. Biomol. Chem., 2019, 17, 6936–6951 RSC.
- S. H. Mashraqui and M. A. Karnik, Synthesis, 1998, 713–714 CrossRef CAS.
- B.-H. Lou, S.-B. Chen, J. Wang, Y. Chen and J.-H. Li, J. Chem. Res., 2013, 37, 409–412 CrossRef CAS.
- M. A. Zolfigol, F. Shirini, A. G. Choghamarani and I. Mohammadpoor-Baltork, Green Chem., 2002, 4, 562–564 RSC.
- L. V. Chopda and P. N. Dave, ChemistrySelect, 2020, 5, 5552–5572 CrossRef CAS.
- I. S. S. Anantha, N. Kerru, S. Maddila and S. B. Jonnalagadda, Front. Chem., 2021, 9, 800236 CrossRef CAS.
- F. Felluga, F. Benedetti, F. Berti, S. Drioli and G. Regini, Synlett, 2018, 29, 1047–1054 CrossRef CAS.
- M. S. Khan, M. A. Nawaz, S. Jalil, F. Rashid, A. Hameed, A. Asari, H. Mohamad, A. U. Rehman, M. Iftikhar, J. Iqbal and M. al-Rashida, Bioorg. Chem., 2022, 118, 105457 CrossRef PubMed.
- L. Ming, G. Wei-Si, W. Li-Rong, L. Ya-Feng and Y. Hua-Zheng, J. Mol. Catal. A: Chem., 2006, 258, 133–138 CrossRef.
- M. Leonardi, M. Villacampa and J. C. Menéndez, Chem. Sci., 2018, 9, 2042–2064 RSC.
- M. Kangani, N. Hazeri, A. Yazdani-Elah-Abadi and M.-T. Maghsoodlou, Polycyclic Aromat. Compd., 2018, 38, 322–328 CrossRef CAS.
- M. Saroha and J. M. Khurana, New J. Chem., 2019, 43, 8644–8650 RSC.
- Z. Madanifar, M.-T. Maghsoodlou, M. Kangani and N. Hazeri, Res. Chem. Intermed., 2015, 41, 9863–9869 CrossRef CAS.
- A. S. Suresh, D. Kumar and J. S. Sandhu, Green Chem. Lett. Rev., 2009, 2, 29–33 CrossRef.
- H. N. Anchan, P. Naik C, N. S. Bhat, M. Kumari and S. Dutta, ACS Omega, 2023, 8, 34077–34083 CrossRef CAS.
- H. Y. Lim and A. V. Dolzhenko, Sustainable Chem. Pharm., 2021, 21, 100443 CrossRef CAS.
- B. Zhou, J. Yang, M. Li and Y. Gu, Green Chem., 2011, 13, 2204–2211 RSC.
- H. Zhang, N. Li, X. Pan, S. Wu and J. Xie, ACS Sustainable Chem. Eng., 2017, 5, 4066–4072 CrossRef CAS.
- J. F. Kornecki, D. Carballares, P. W. Tardioli, R. C. Rodrigues, Á. Berenguer-Murcia, A. R. Alcántara and R. Fernandez-Lafuente, Catal. Sci. Technol., 2020, 10, 5740–5771 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Spectroscopic characterization (FTIR, 1H-NMR, and 13C-NMR) data, melting point values, and spectra of all synthesized compounds. The compounds are known in the literature and these references for the synthesized compounds are cited in the ESI file. See DOI: https://doi.org/10.1039/d4ra07772e |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2024 |