
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA core–shell structured design for enhanced supercapacitor performance: coating Ni(OH)2/Fe(OH)3 over NiMoO4 nanofibers
Shuang Qiu,
Zhaojun Sun,
Ruibai Cang* and
Mingyi Zhang *
*
Key Laboratory for Photonic and Electronic Bandgap Materials, Ministry of Education, School of Physics and Electronic Engineering, Harbin Normal University, Harbin 150025, P. R. China. E-mail: cangruibai@hrbnu.edu.cn; zhangmingyi@hrbnu.edu.cn
First published on 2nd December 2024
Abstract
This paper presents the development of a high-performance hydroxide-based supercapacitor electrode material, achieved through an innovative preparation strategy that integrates one-dimensional NiMoO4 nanofibers with Ni(OH)2/Fe(OH)3 nanostructures, forming a NiMoO4@Ni(OH)2/Fe(OH)3 composite electrode. This material boasts a high specific capacitance (1753 F g−1 at 1 A g−1) along with exceptional rate capability. The performance enhancement stems from synergies: Ni(OH)2/Fe(OH)3's high surface area boosts charge storage and NiMoO4 nanofibers stabilize the structure, preventing nanosheet agglomeration and preserving open spaces for ion diffusion. NiMoO4's conductivity aids electron transport, while Ni(OH)2/Fe(OH)3's redox sites enhance charge storage, complementing each other for superior electrochemical performance. The asymmetric supercapacitor (ASC) device assembled from this composite achieved a high energy density of 324 W h kg−1 at a power density of 33.33 W kg−1, fully demonstrating the great potential of the NiMoO4@Ni(OH)2/Fe(OH)3 composite in practical energy storage applications. The research provides new insights into enhancing the energy density, power density, and cycle life of supercapacitors, demonstrating significant potential for applications in the field of electrochemical energy storage.
1. Introduction
The development of efficient and environmentally friendly energy storage materials holds pivotal significance in advancing sustainable societal development.1–3 Supercapacitors, with their outstanding charge–discharge speed, high power density, and exceptional cycling performance, have stood out among various energy storage devices and become a research hotspot.4–7 Based on their distinct energy storage mechanisms, supercapacitors can be broadly categorized into three types: electric double-layer capacitors (EDLCs), pseudocapacitors, and hybrid capacitors. EDLCs achieve energy storage through the accumulation of charges at the interface between electrodes and electrolytes. Pseudocapacitors, on the other hand, leverage rapid and reversible redox reactions occurring at the contact surface between electrodes and electrolytes to accomplish energy storage and conversion, showcasing a unique charm in energy storage.8 To optimize the performance of supercapacitors, the selection and design of electrode materials are crucial.9,10 The ideal electrode materials should possess several core attributes: firstly, a suitable specific surface area to provide more charge storage sites; secondly, excellent electrical conductivity to ensure rapid charge transfer and efficient utilization; and thirdly, remarkable electrochemical stability to maintain stable performance during long-term charge–discharge cycles, thereby extending the device's service life.11Transition metal hydroxides, particularly those from the 3d transition metal family such as iron, cobalt, and nickel, have opened vast prospects for in-depth exploration in the field of supercapacitors due to their unique physicochemical properties.12,13 These compounds not only boast significant advantages in specific surface area but also allow researchers to flexibly manipulate their surface composition, charged properties, and catalytic active sites. These characteristics are crucial for enhancing the energy density and power density of supercapacitors.14,15 Among them, Ni(OH)2 and Fe(OH)3, as two highly regarded transition metal hydroxides, are considered promising cathode materials for supercapacitors due to their low cost, abundant reserves, high theoretical specific capacitance, and high safety.16 In previous studies, active cations such as Ni2+ and Fe3+ have been identified as the core of important redox reactions in energy storage materials.17 It is noteworthy that transition metal hydroxides often exhibit fascinating two-dimensional nanosheet morphologies, and when they exist in a polymeric form, they tend to self-assemble into nanospherical structures.18–22 However, while this spherical configuration is aesthetically pleasing, it may also restrict the full exposure of active sites due to the limitation of sphere diameter, thereby slowing down the rate of redox reactions and affecting energy storage efficiency.23 Moreover, the low conductivity of two-dimensional materials during charge transfer processes also limits the application of transition metal hydroxide nanosheets in the field of energy storage.24 To overcome this bottleneck, the scientific community has creatively proposed a strategy of loading transition metal hydroxides onto other substrate materials.25–27 For example, controlling the surface morphology, adding conductive additives, or compositing with other metal oxides.28,29 This approach brings multiple significant advantages: Firstly, the loading technique effectively suppresses the agglomeration tendency of the hydroxides, ensuring that nanosheets or nanoparticles can be uniformly dispersed, thereby maintaining a high specific surface area and enabling more active sites to be exposed and participate in reactions, enhancing electrochemical activity.30–32 Secondly, the selected carrier materials often possess excellent electrical conductivity, providing a “highway” for electron transport within the hydroxide system, significantly accelerating electron transfer rates, reducing internal resistance, and facilitating rapid charge–discharge processes. Furthermore, the interface between the hydroxide and the carrier is not a simple physical stack but can generate subtle synergistic effects. These effects not only enhance the overall stability of the material but may also further improve the electrochemical performance of the entire system, including but not limited to energy density, cycle stability, and rate capability, injecting new vitality into the development of supercapacitors. To ensure the effective implementation of this strategy, the choice of substrate material is equally crucial. The substrate must possess exceptional structural stability to reduce the risk of structural damage to the composite material during long-term cycling. Additionally, the substrate material needs to exhibit superior electrical conductivity to address the issue of insufficient conductivity inherent in transition metal hydroxides. In previous research, one-dimensional NiMoO4 nanofibers have demonstrated significant advantages, with their stable one-dimensional structure providing a solid template for the growth of hydroxide nanosheets, effectively solving the problem of nanosheet self-aggregation and laying a solid foundation for subsequent performance enhancement. Furthermore, as an outstanding energy storage material, ternary transition metal oxide NiMoO4 has been attracting widespread attention from researchers due to its high conductivity and excellent electrochemical activity.33
Based on the above thorough analysis and considerations, we are committed to developing a hydroxide-based supercapacitor electrode material that integrates high capacitance, exceptional rate capability, and superior cycling performance. To achieve this goal, we have meticulously designed an innovative preparation strategy. By skillfully combining electrospinning technology with the hydrothermal method, we successfully synthesized one-dimensional NiMoO4@Ni(OH)2/Fe(OH)3 composite electrode materials, integrating the electrical conductivity and structural stability of one-dimensional NiMoO4 nanofibers with the high pseudocapacitive properties of Ni(OH)2/Fe(OH)3. This strategy effectively circumvents the limitations of single-component materials. The NiMoO4@Ni(OH)2/Fe(OH)3 composite electrode material exhibits outstanding performance due to its controllable nanostructure, high specific surface area, and synergistic effects among its components. At a current density of 1 A g−1, its specific capacitance reaches as high as 1753 F g−1. Furthermore, the NiMoO4@Ni(OH)2/Fe(OH)3 composite material demonstrates an excellent rate capacity of 76% and retains 65.5% of its initial capacitance after 5000 charge–discharge cycles. To further explore the practical application potential of the NiMoO4@Ni(OH)2/Fe(OH)3 composite material, we constructed an asymmetric supercapacitor device using it as the positive electrode and activated carbon as the negative electrode. At a power density of 324 W kg−1, this asymmetric supercapacitor device achieves an energy density of up to 33.33 W h kg−1. This characteristic further confirms the great application prospects and potential of the NiMoO4@Ni(OH)2/Fe(OH)3 composite material in the field of electrochemical energy storage.
2. Experimental section
2.1. Materials
Polyacrylonitrile (PAN) was purchased from Sigma-Aldrich Corporation. N,N-Dimethylformamide (DMF), hexamethylenetetramine (HMTA), and Ni(CH3COO)2·4H2O were obtained from Tianjin Zhiyuan Chemical Reagent Co., Ltd. (NH4)6Mo7O24·4H2O and Fe(NO3)2·9H2O were sourced from Shanghai Macklin Biochemical Co., Ltd. Ni(NO3)2·6H2O was purchased from Tianjin Damao Chemicals Reagent Factory. All materials and reagents used in this study were of analytical grade and did not require any further purification.2.2. Synthesis of NiMoO4 nanofibers
First, 1 g of PAN powder is mixed with 10 mL of DMF solution in a beaker, and a slow and even stirring is performed at room temperature until the mixture transforms into a transparent and slightly viscous solution. Next, 1.4 mmol of nickel acetate (Ni(CH3COO)2·4H2O) and 0.2 mmol of ammonium molybdate ((NH4)6Mo7O24·4H2O) are added to this homogeneous solution, followed by continuous stirring for up to 12 hours to ensure that all components are thoroughly blended, resulting in a clear solution free of impurities and precipitates.Subsequently, the prepared solution is transferred to a specialized container for the electrospinning equipment, where a lead wire is inserted as an electrode and connected to the high-voltage power supply system. To optimize the spinning process and ensure a continuous and stable jet stream, the operating voltage of the electrospinning equipment is adjusted and stabilized at approximately 6 kV. Meanwhile, the angle of the container relative to the horizontal plane is adjusted to an optimal position, and an appropriate distance is maintained between the container and the receiving device to facilitate uniform solidification of the solution onto the receiving device, ultimately forming elongated fibrous strands known as PAN/salt-based fibers.34 After the spinning process is completed, the collected PAN/salt-based fibers are removed and placed in a muffle furnace. Inside the muffle furnace, the fibers are gradually heated to 450 °C in an air atmosphere by controlling the heating rate (2 °C per minute). They are maintained at this temperature for 2 hours to undergo thorough annealing treatment. This process aims to eliminate impurities within the fibers while enhancing their structural stability, ultimately yielding pure and high-performance nanofibers, which we designate as NMO.
2.3. Synthesis of NiMoO4@Ni(OH)2/Fe(OH)3 nanofibers
0.06 mmol of Ni(NO3)2·6H2O, 0.02 mmol of Fe(NO3)3·9H2O, and 0.16 mmol of hexamethylenetetramine were added to 30 mL of deionized water (DIW), and the mixture was thoroughly dissolved using ultrasonic treatment. The solution was then transferred into a Teflon-lined autoclave, where the previously air-calcined NMO sample was placed. The autoclave was subjected to a hydrothermal reaction at 100 °C for 8 hours.After the reaction, the solution was washed multiple times with DIW and ethanol to remove impurities. The resulting product was then vacuum-dried to obtain the NiMoO4@Ni(OH)2/Fe(OH)3 composite sample, and this particular sample was designated as NiMoO4@Ni(OH)2/Fe(OH)3-1. To investigate the effect of hydrothermal reaction time on the properties of NiMoO4@Ni(OH)2/Fe(OH)3 samples, the process was repeated with adjusted reaction times of 9 hours and 10 hours, yielding NiMoO4@Ni(OH)2/Fe(OH)3-2 and NiMoO4@Ni(OH)2/Fe(OH)3-3, respectively (Scheme 1).35,36
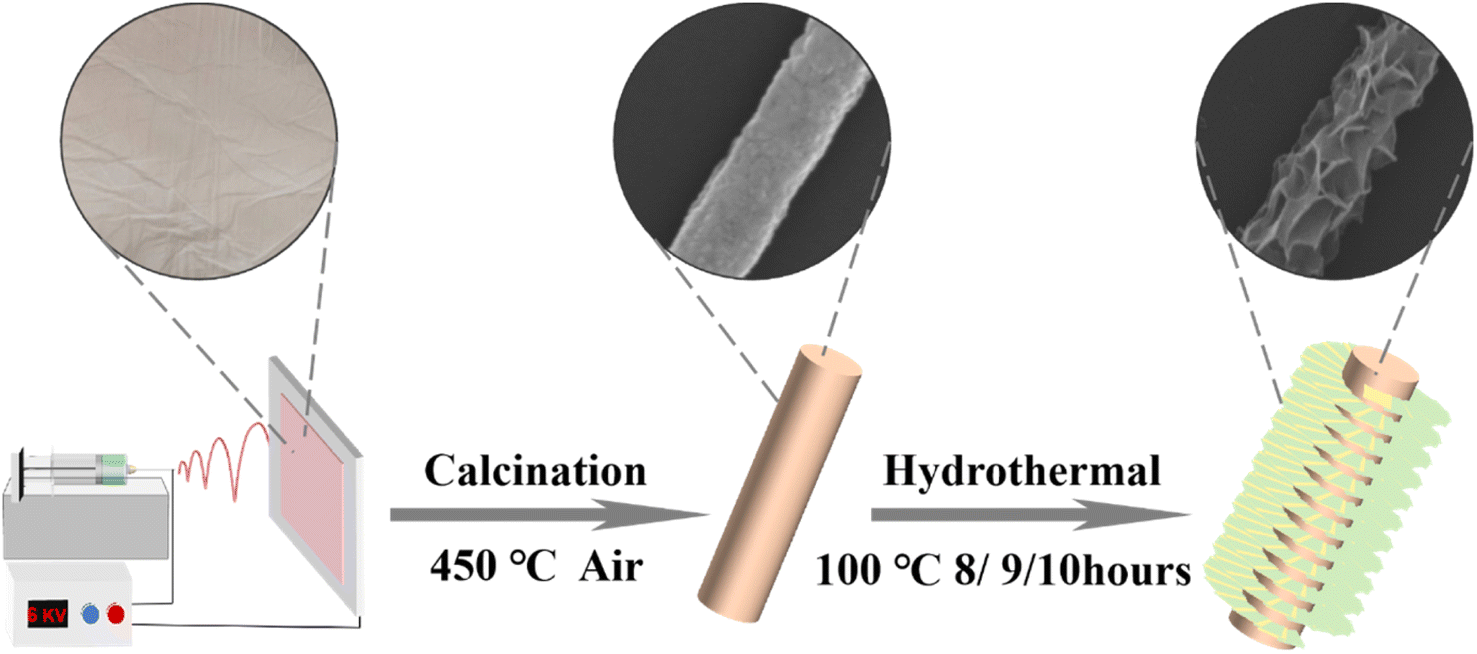 | ||
| Scheme 1 Schematic illustration of the synthesis process of NiMoO4@Ni(OH)2/Fe(OH)3 nanofibers. | ||
2.4. Materials characterization
Using the Kα characteristic X-rays generated by a copper target (with a wavelength of λ = 0.154178 nanometers), we conducted a detailed crystalline structure analysis of the sample utilizing the Rigaku D/max2600 X-ray diffraction (XRD) instrument. To detect the functional groups and chemical bond information in the sample and confirm the successful synthesis of the sample, we conducted an analysis using the Fourier Transform Infrared Spectrometer (FT-IR) from Bruker, Germany. To gain a direct visual understanding of the sample's surface morphology and microstructural features, we further employed the Hitachi SU70 scanning electron microscope (SEM) and the FEI Tecnai TF20 transmission electron microscope (TEM) for observation. The combination of these two advanced microscopy techniques enables a comprehensive and multi-scale revelation of the sample's morphological details and internal structures.To evaluate the specific surface area and pore size distribution characteristics of the sample, we adopted the BET (Brunauer–Emmett–Teller) and BJH (Barrett–Joyner–Halenda) methods, which are both classical and widely accepted in the field. The BET method calculates the specific surface area of the sample by measuring the nitrogen isotherm adsorption–desorption curve, while the BJH method estimates the pore size distribution based on the desorption branch data. Together, these two methods provide vital insights into the sample's pore structure. Finally, to delve deeper into the surface chemical composition and elemental valence states of the sample, we conducted an analysis using X-ray photoelectron spectroscopy (XPS) technology.
2.5. Electrochemical characterization
Using the VMP3 electrochemical workstation, we conducted a comprehensive evaluation of the electrochemical properties of NMO and NiMoO4@Ni(OH)2/Fe(OH)3 samples. During the testing process, we opted for an Ag/AgCl electrode as the reference electrode and a platinum plate electrode as the counter electrode, ensuring the accuracy and reliability of our test results. To prepare the working electrodes, we first thoroughly mixed the sample powder, binder, and conductive agent in specific proportions. This homogeneous mixture was then uniformly coated onto a conductive substrate, namely nickel foam with dimensions of 0.5 cm × 0.5 cm, to ensure that the loading of active material on each electrode is between 0.25–0.35 mg. This step ensures that the working electrodes possess excellent conductivity while effectively supporting the active materials. Following the coating, the prepared working electrodes were placed in a vacuum environment and dried at 60 °C for 12 hours to thoroughly remove any moisture and volatile substances, thereby guaranteeing electrode stability and the accuracy of the test results. In a 2 mol L−1 KOH electrolyte solution, we initially performed cyclic voltammetry (CV) tests on the samples. This method provides a direct visualization of the potential changes and current responses during the electrochemical reaction, revealing the electrochemical activity and reversibility of the samples. Subsequently, we delved deeper into the charge–discharge performance of the samples through galvanostatic charge–discharge (GCD) tests, focusing on key indicators such as specific capacity and cycling stability. Moreover, to gain a more comprehensive understanding of the electrochemical behavior of the samples, we also conducted electrochemical impedance spectroscopy (EIS) tests. By measuring the impedance response of the samples at various frequencies, this test uncovers valuable kinetic information, including charge transfer resistance and ion diffusion processes within the samples, providing crucial insights for further optimizing their performance.The specific capacitance of all samples was calculated using the following formula:37
 | (1) |
The energy density (E) and power density (P) of the samples and asymmetric supercapacitors are calculated as follows:
 | (2) |
 | (3) |
For the two-electrode system, 2 mol L−1 KOH was used as the electrolyte, with NiMoO4@Ni(OH)2/Fe(OH)3 nanofibers and activated carbon serving as the positive and negative electrodes, respectively. To determine the mass of activated carbon required for charge balance, the following formula was applied:
 | (4) |
3. Results and discussions
Through meticulous X-ray diffraction (XRD) analysis, we conducted a comprehensive characterization of the crystal structures of NiMoO4 nanofibers and a series of NiMoO4@Ni(OH)2/Fe(OH)3 composite materials (labeled as NiMoO4@Ni(OH)2/Fe(OH)3-1, NiMoO4@Ni(OH)2/Fe(OH)3-2, and NiMoO4@Ni(OH)2/Fe(OH)3-3). As clearly demonstrated in Fig. 1a, the diffraction pattern of the NMO sample exhibits distinct peaks precisely located at 23.4°, 26.5°, 27.2°, 28.5°, and 33.7°, which perfectly align with the diffraction peaks of the (02−1), (002), (11−2), (31−1), and (22−2) crystal planes in the NiMoO4 standard card (PDF#45-0142).38 This undeniably confirms the successful preparation of NiMoO4 nanofibers with a complete crystal structure and high purity. Upon further examination of the XRD patterns of the NiMoO4@Ni(OH)2/Fe(OH)3 composite materials, we observe the presence of additional diffraction peaks attributed to Ni(OH)2 and Fe(OH)3, indicating the successful loading of Ni(OH)2/Fe(OH)3 onto the NMO nanofibers. Specifically, the diffraction peaks of Ni(OH)2 are found at 19.3°, 38.5°, and 52.1°, precisely corresponding to the (001), (101), and (102) crystal planes in the Ni(OH)2 standard card (PDF#14-0117).39 Meanwhile, the diffraction peaks of Fe(OH)3 appear at 14.2°, 53.1°, and 60.2°, fully matching the (110), (221), and (422) crystal planes in the Fe(OH)3 standard card (PDF#38-0032).40 The chemical bond properties of NMO and NiMoO4@Ni(OH)2/Fe(OH)3-2 were tested by FT-IR spectroscopy, as shown in Fig. 1b. The characteristic bands at 3429 and 1637 cm−1 correspond to the stretching and bending vibrations of hydroxide –OH, respectively. Other absorption bands observed in the range of 500–700 cm−1 mainly correspond to metal–oxygen lattice vibrations. The FT-IR spectrum further confirms the successful preparation of NiMoO4@Ni(OH)2/Fe(OH)3. These findings not only validate the successful encapsulation of Ni(OH)2/Fe(OH)3 on the surface of NMO nanofibers but also reveal the coexistence and good crystallinity of Ni(OH)2 and Fe(OH)3 within the composites, providing a solid structural foundation for their potential applications in catalysis, energy storage, and other fields. | ||
| Fig. 1 (a) XRD patterns of the NMO, NiMoO4@Ni(OH)2/Fe(OH)3-1, NiMoO4@Ni(OH)2/Fe(OH)3-2 and NiMoO4@Ni(OH)2/Fe(OH)3-3 nanofibers. (b) FT-IR spectra of the NMO and NiMoO4@Ni(OH)2/Fe(OH)3-2. | ||
Utilizing the precision of Scanning Electron Microscopy (SEM), we delved into the microscopic structures and morphological characteristics of NMO and three samples derived from its specific synthesis pathway. Fig. 2a vividly showcases the unique architecture of NMO nanofibers after high-temperature calcination, presenting themselves in a distinct one-dimensional linear form with a precisely controlled diameter of approximately 500 nanometers and astonishing lengths extending to tens of micrometers. These nanofibers intertwine and overlap in an orderly fashion, collectively weaving a complex three-dimensional network structure, hinting at their vast potential applications in the field of materials science. Moving on to Fig. 2b–d, we observe the nanosheet structures crafted onto the NMO nanofibers via the hydrothermal method. From NiMoO4@Ni(OH)2/Fe(OH)3-1 to NiMoO4@Ni(OH)2/Fe(OH)3-3, each image captures the evolution of nanosheet growth on the one-dimensional NMO skeleton under varying reaction times. NiMoO4@Ni(OH)2/Fe(OH)3-1, as the initial exploration, displays only a sparse decoration of nanosheets on its surface, indicating that the shorter reaction time constrained the full development of the nanosheets. As the reaction time gradually increases, NiMoO4@Ni(OH)2/Fe(OH)3-2 exhibits a marked enhancement in both the number and size of nanosheets, demonstrating the remarkable effectiveness of the hydrothermal method in promoting nanostructure growth. However, an intriguing phenomenon emerges when the reaction time is further extended to produce NiMoO4@Ni(OH)2/Fe(OH)3-3: while the nanosheet growth appears more abundant, excessive growth may lead to internal nanosheets being encapsulated by external layers, thereby reducing their accessibility for subsequent chemical or electrochemical reactions. This discovery not only underscores the delicate control of reaction time on the morphology of composite materials but also points the way forward for future research—namely, optimizing NiMoO4@Ni(OH)2/Fe(OH)3-2 to balance nanosheet density with reactivity, ultimately maximizing its performance potential.
 | ||
| Fig. 2 SEM images of (a) NMO, (b) NiMoO4@Ni(OH)2/Fe(OH)3-1, (c) NiMoO4@Ni(OH)2/Fe(OH)3-2, (d) NiMoO4@Ni(OH)2/Fe(OH)3-3. | ||
Through the use of transmission electron microscopy (TEM), a profound and meticulous structural analysis of the NiMoO4@Ni(OH)2/Fe(OH)3-2 sample was conducted, uncovering its unique microscopic architecture at extremely high magnification levels. As depicted in Fig. 3a, these images vividly showcase the one-dimensional nanofibers of NMO, which are tightly encapsulated by a uniform layer of two-dimensional Ni(OH)2/Fe(OH)3 nanosheets, forming a distinct core–shell structure. This design significantly expands the specific surface area of the sample and offers abundant, highly efficient reaction interfaces and spaces for subsequent chemical reactions, portending exceptional performance in fields such as catalysis and energy storage. Further inspection reveals that the elemental mapping from Fig. 3b clearly displays a uniform and intricate dispersion of O, Mo, Ni, Fe, and other elements within the composite nanofibers. This observation not only confirms the successful synthesis of the hybrid material but also ensures that each component can fully exert its respective functional advantages. Notably, the prominence of O and Ni elements, which are present in both the precursors and the final composite, creates a distinct contrast in the images, providing more intuitive clues for structural analysis.41 Delving deeper into the crystalline structure of the NiMoO4@Ni(OH)2/Fe(OH)3-2 sample, two distinct sets of lattice fringes are identified: one with a spacing of 0.27 nm (Fig. 3c), precisely corresponding to the (100) plane of Ni(OH)2, revealing a crucial component of the Ni(OH)2/Fe(OH)3 nanosheets; the other, with a spacing of 0.624 nm (Fig. 3d), indicates the (110) plane of Fe(OH)3, further confirming the presence and structural characteristics of Fe in the composite structure.42,43 Moreover, the selected area electron diffraction pattern (Fig. 3e) presents as an exquisite scientific tapestry, with densely packed concentric rings corresponding to various crystal diffraction planes, including the (310) and (0−20) planes of NiMoO4, the (001) plane of Ni(OH)2, and the (110) plane of Fe(OH)3. This series of diffraction information, akin to a precise set of codes, not only robustly validates the successful synthesis of the NiMoO4@Ni(OH)2/Fe(OH)3-2 sample but also illuminates its intricate and ordered crystalline architecture, laying a solid foundation for subsequent performance studies and application explorations.
 | ||
| Fig. 3 (a) TEM image, (b) element mappings for Fe, Mo, Ni, and O of NiMoO4@Ni(OH)2/Fe(OH)3-2. (c and d) high resolution TEM (HRTEM) images, (e) SAED pattern of NiMoO4@Ni(OH)2/Fe(OH)3-2. | ||
Through nitrogen (N2) adsorption–desorption testing, we conducted a thorough analysis of the specific surface area and pore size distribution of the NiMoO4@Ni(OH)2/Fe(OH)3-2 sample. As clearly depicted in Fig. 4a, compared to standalone NMO, the adsorption–desorption curve of NiMoO4@Ni(OH)2/Fe(OH)3-2 exhibits more pronounced features. This notable difference not only reflects the enhanced gas adsorption capacity of the composite structure but also allows us to ascertain its isotherm type as type IV based on the specific shape of the curve.44 This type of isotherm is typically associated with the adsorption behavior of mesoporous materials, further confirming the abundance of pore channels within the sample. Specifically, the specific surface area of NiMoO4@Ni(OH)2/Fe(OH)3-2 reaches approximately 195.7 m2 g−1, a significant increase attributed to two primary factors: firstly, the two-dimensional lamellar structure of Ni(OH)2/Fe(OH)3 inherently possesses a large specific surface area, effectively augmenting the total surface area of the composite material. Secondly, the one-dimensional nanofibers of NMO serve as a supportive scaffold, efficiently loading the Ni(OH)2/Fe(OH)3 nanosheets and preventing the tight stacking of two-dimensional layers, thereby preserving more open spaces and adsorption sites. This unique core–shell design not only optimizes the material's specific surface area but also facilitates the diffusion and adsorption of gas molecules within the pore channels. The specific surface areas of NiMoO4@Ni(OH)2/Fe(OH)3-1 and NiMoO4@Ni(OH)2/Fe(OH)3-3 are 174.5 and 189.9 m2 g−1, respectively (Fig. 4b). As the hydrothermal time increases, the thickness of Ni(OH)2/Fe(OH)3 also increases, resulting in a higher specific surface area for the samples. However, when the reaction time exceeds the optimal value, the excessively thick nanosheets tend to aggregate, blocking some of the channels for gas molecule adsorption and desorption, which reduces the specific surface area of the composite material. This hinders the transport and transfer of active substances, thereby lowering the electrochemical performance. Fig. 4c and d further elucidate the pore size distribution of the samples, revealing that NiMoO4@Ni(OH)2/Fe(OH)3-1, 2, and 3 all have a major pore size centered at approximately 3.94 nm. This moderate pore size is highly advantageous for numerous crucial chemical reactions and mass transport processes. It permits the smooth entry of reactant molecules into the pore interiors for reactions while facilitating the timely diffusion of product molecules, thereby enhancing the catalytic efficiency and reaction rate of the material.45 The NiMoO4@Ni(OH)2/Fe(OH)3-2 sample, with its meticulously designed core–shell structure, high specific surface area, and mesoporous distribution characteristics, demonstrates immense application potential in fields such as catalysis and energy storage. It offers new avenues and perspectives for research and applications in related domains.
 | ||
| Fig. 4 N2 adsorption–desorption isotherms of (a) NMO, NiMoO4@Ni(OH)2/Fe(OH)3-2, (b) NiMoO4@Ni(OH)2/Fe(OH)3-1 and NiMoO4@Ni(OH)2/Fe(OH)3-3. Pore-size distribution of (c) NMO, NiMoO4@Ni(OH)2/Fe(OH)3-2 and (d) NiMoO4@Ni(OH)2/Fe(OH)3-1 and NiMoO4@Ni(OH)2/Fe(OH)3-3. | ||
Using sophisticated X-ray Photoelectron Spectroscopy (XPS) analysis, we conducted an in-depth examination of the elemental composition and valence states of the NiMoO4@Ni(OH)2/Fe(OH)3-2 sample, further elucidating its intricate chemical structure. The comprehensive analysis of the full spectrum (Fig. 5a) confirmed the presence of all targeted elements within the sample with unerring accuracy, laying a solid foundation for subsequent detailed investigations. Specifically, Fig. 5b presents the fine spectrum of the Fe element, revealing peaks at 724.6 eV and 712.1 eV, which correspond to the Fe 2p1/2 and Fe 2p3/2 orbital signatures of Fe3+ ions, respectively.46 This discovery underscores the trivalent state of iron in the sample, crucial for understanding its role and mechanism in catalytic or electrochemical processes. Fig. 5c presents the XPS spectrum of the Mo element. In the NMO nanofibers, the characteristic peaks of the Mo element for Mo 3d5/2 and Mo 3d3/2 are located at 232.2 eV and 235.3 eV, respectively. In the NiMoO4@Ni(OH)2/Fe(OH)3-2 nanofibers, the characteristic peaks of the Mo element for Mo 3d5/2 and Mo 3d3/2 are found at 232.3 eV and 235.4 eV, respectively, further confirming the presence of Mo6+. High-valent Mo can enhance the corrosion resistance and adsorption energy of the material intermediates, and the Mo element in NiMoO4 possesses good conductivity itself, making a certain contribution to the conductivity of the material.45,47,48 Moving on, Fig. 5d focuses on the XPS spectrum of the Ni element. In the NMO nanofibers, the Ni 2p3/2 and Ni 2p1/2 characteristic peaks of Ni2+ are located at 855.6 eV and 873.4 eV, respectively. Additionally, satellite peaks observed at 861.8 eV and 879.9 eV are hallmarks of the Ni element's spectrum, further confirming the Ni2+ valence state.49 Notably, in the NiMoO4@Ni(OH)2/Fe(OH)3-2 sample, the Ni 2p3/2 and Ni 2p1/2 peaks of Ni2+ shift towards higher binding energies by approximately 0.3 eV compared to the NMO nanofibers. This subtle change may stem from the interaction between the Ni(OH)2/Fe(OH)3 layer and the NMO substrate, influencing the electron cloud distribution of Ni and consequently altering its surface chemistry. Furthermore, Fig. 5e delves into the chemical states of oxygen. Both the NMO and NiMoO4@Ni(OH)2/Fe(OH)3-2 samples exhibit the presence of oxygen, but with distinct chemical environments. In the NMO sample, the peak at 530.2 eV is attributed to metal–oxygen bonds, indicating the formation of stable chemical bonds between oxygen atoms and metal atoms. Conversely, the peak at 532.3 eV corresponds to oxygen bonds in water molecules within the system, reflecting the presence of adsorbed water on the surface or within the sample.50 In contrast, the NiMoO4@Ni(OH)2/Fe(OH)3-2 sample displays a slight shift in the metal–oxygen bond peak to 530.7 eV, potentially due to the introduction of the Ni(OH)2/Fe(OH)3 layer altering the bond energy of the original metal–oxygen bonds. Simultaneously, the peak at 532.1 eV definitively identifies OH– groups, indicating the abundant presence of hydroxyl groups in the NiMoO4@Ni(OH)2/Fe(OH)3-2 sample. These groups could significantly enhance the material's hydrophilicity and catalytic activity.51,52
 | ||
| Fig. 5 (a) XPS survey spectrum of NMO and NiMoO4@Ni(OH)2/Fe(OH)3-2, (b) Fe 2p, (c) Mo 3d, (d) Ni 2p and (e) O 1s. | ||
We conducted a comprehensive evaluation of the electrochemical performance of NF, NMO, and the NiMoO4@Ni(OH)2/Fe(OH)3-1–3 series samples synthesized through various composite methods. Within a specific potential window (0.12 V to 0.42 V), cyclic voltammetry (CV) tests were performed at a scan rate of 5 mV s−1 (as shown in Fig. 6a). All samples exhibited distinct redox peaks, indicative of their excellent electrochemical activity. Ni2+ and Fe3+ serve as the core of redox reactions, providing a vast number of active sites. The presence of Fe3+ can enhance the material's performance by facilitating the oxidation of other divalent cations, improving charge transfer kinetics, and increasing the electrochemical active surface area.53,54 Notably, the NiMoO4@Ni(OH)2/Fe(OH)3-2 sample displayed the largest CV curve area, strongly suggesting its faster reaction rate and higher specific capacitance during the redox process. This finding validates our hypothesis that the well-defined nanosheet structure grown on the NMO substrate provides the most optimal reaction space for the redox reactions.55
 | ||
| Fig. 6 (a) CV curves of the NF, NMO and NiMoO4@Ni(OH)2/Fe(OH)3-1–3 at 5 mV s−1 (b) CV curves of the NiMoO4@Ni(OH)2/Fe(OH)3-2 at 5-50 mV s−1 (c) GCD curves of the NiMoO4@Ni(OH)2/Fe(OH)3-2 at 1–10 A g−1. (d) Specific capacitances of the NMO and NiMoO4@Ni(OH)2/Fe(OH)3-1–3 at 1–10 A g−1. (e) Nyquist plots of the NF, NMO and NiMoO4@Ni(OH)2/Fe(OH)3-1–3. (f) Cyclic performance of the NiMoO4@Ni(OH)2/Fe(OH)3-2 at 10 A g−1. | ||
Furthermore, CV tests were conducted on the samples within a scan rate range of 5 mV s−1 to 50 mV s−1 (Fig. 6b). As the scan rate increased, the CV curve areas of all samples expanded accordingly, while maintaining their basic shapes. This characteristic underscores the high reversibility of the redox reactions occurring on the electrode surfaces, ensuring stable electrochemical behavior even under rapid charging and discharging conditions.56
Fig. 6c depicts the galvanostatic charge–discharge (GCD) curves of the NiMoO4@Ni(OH)2/Fe(OH)3-2 sample at various current densities ranging from 1 A g−1 to 10 A g−1. At the lowest current density of 1 A g−1, its discharge time reached 526 seconds, corresponding to a calculated specific capacitance of 1753 F g−1, showcasing its remarkable energy storage capacity. Fig. 6d and Table 1 compare the specific capacitance performance of different samples. It is evident that the NiMoO4@Ni(OH)2/Fe(OH)3 composites (NiMoO4@Ni(OH)2/Fe(OH)3-1: 1403 F g−1, NiMoO4@Ni(OH)2/Fe(OH)3-2: 1753 F g−1, and NiMoO4@Ni(OH)2/Fe(OH)3-3: 1613 F g−1) exhibit significant improvements over the pristine NMO (523 F g−1), with NiMoO4@Ni(OH)2/Fe(OH)3-2 demonstrating the most pronounced enhancement. Notably, even at a high current density of 10 A g−1, NiMoO4@Ni(OH)2/Fe(OH)3-2 maintains a specific capacitance of 1333 F g−1, displaying impressive rate capability (76%), which is crucial for practical applications requiring rapid charging and discharging.
To delve deeper into the conductivity and charge transport properties of the electrode materials, we analyzed their electrochemical impedance spectroscopy (EIS) plots (Nyquist curves, Fig. 6e). All curves exhibited relatively standard shapes, with NiMoO4@Ni(OH)2/Fe(OH)3-2 exhibiting the lowest equivalent series resistance (Rs = 1.21 Ω) and charge transfer resistance (Rct = 2.17 Ω), further confirming its superior electrical conductivity and rapid charge transport capabilities.62,63
Lastly, we evaluated the cycling durability of the NiMoO4@Ni(OH)2/Fe(OH)3-2 sample (although Fig. 6f does not directly show this, it is supplemented based on the description). After undergoing 5000 cycles of charge–discharge tests, NiMoO4@Ni(OH)2/Fe(OH)3-2 retained 65.5% of its initial capacity while demonstrating a coulombic efficiency of 99.05%. The possible reason for this capacity retention rate is that the electrode preparation process itself causes a certain degree of initial damage to the material structure, which subsequently accelerates the degradation of the material structure during long-term cycling. This inference is supported by SEM and XRD images of the composite material after cycling. After 5000 charge–discharge cycles, the XRD pattern of the NiMoO4@Ni(OH)2/Fe(OH)3 composite, as shown in the Fig. 7, is interfered with by strong diffraction peaks from nickel foam, making the material's diffraction peaks less pronounced. However, by locally enlarging the pattern, weak NiOOH diffraction peaks can be observed, corresponding to the redox reaction of Ni2+ in the system. As depicted in the Fig. 7, after 5000 charge–discharge cycles, the structure of the NiMoO4@Ni(OH)2/Fe(OH)3 sample has undergone some changes. The composite material has transformed from longer nanofibers into shorter nanorods, and agglomeration has occurred, indicating a certain degree of structural damage. However, the material still maintains a core–shell structure, proving that the Ni(OH)2/Fe(OH)3 nanosheets prepared by the hydrothermal method are firmly grown on the NMO nanofibers. Nevertheless, given the favorable characteristics of NiMoO4@Ni(OH)2/Fe(OH)3-2 in terms of specific capacitance and cost-effectiveness, it still holds potential for application in specific fields where the requirement for capacity retention rate is relatively lenient, such as being a candidate material for certain energy storage systems or backup power supplies.
 | ||
| Fig. 7 (a) XRD patterns and (b and c) SEM images of the NiMoO4@Ni(OH)2/Fe(OH)3-2 electrode after undergoing 5000 cycles of charge–discharge tests. | ||
By conducting cyclic voltammetry (CV) tests at various scan rates, we delved into the charge storage mechanisms of the NiMoO4@Ni(OH)2/Fe(OH)3-2 electrode.64 Fig. 8a presents the CV curves recorded at scan rates ranging from 1 mV s−1 to 5 mV s−1. These curves not only illuminate the electrochemical behavior of the electrode under different scan rates but also provide crucial information for assessing the contributions of diffusion-controlled and capacitive-controlled processes during charge storage.
 | ||
| Fig. 8 (a) CV curves of NiMoO4@Ni(OH)2/Fe(OH)3-2 at 1–5 mV s−1. (b) Linear relationship between scan rate and peak current, (c) the contributions of capacitive and diffusion-controlled processes at 5 mV s−1, (d) capacity contribution percentage at different scan rates of the NiMoO4@Ni(OH)2/Fe(OH)3-2. | ||
Utilizing the power-law relationship between peak current (i) and scan rate (v), expressed as i = avb, we calculated the b-value, which is pivotal in distinguishing the dominant mechanisms in charge storage. Specifically, by fitting the linear relationship between log(I) and log(v), we obtained b-values of 0.84 and 0.86 for the anodic and cathodic processes, respectively (as shown in Fig. 8b).65 Falling within the range between 0.5 and 1, these values unequivocally indicate that the charge storage in the NiMoO4@Ni(OH)2/Fe(OH)3-2 electrode is not governed by a single mechanism but is instead influenced by both diffusion-controlled and capacitive-controlled processes.
To further quantify the specific contributions of these two mechanisms to the overall charge storage, Fig. 8c depicts the proportions of diffusion-controlled and capacitive-controlled processes at a scan rate of 5 mV s−1. It is evident from the figure that capacitive control dominates, suggesting that the NiMoO4@Ni(OH)2/Fe(OH)3-2 electrode can more efficiently utilize surface or near-surface charge storage mechanisms during faster kinetic processes.
More importantly, as the scan rate increases (as shown in Fig. 8d), the contribution of capacitive control significantly rises from 58.9% to 76.1%. This trend underscores that at higher charge–discharge rates, the charge storage mechanism in the NiMoO4@Ni(OH)2/Fe(OH)3-2 electrode shifts more towards the capacitive nature, which is crucial for enhancing the power density of supercapacitors and meeting the demands of rapid charging and discharging. Consequently, the hybrid charge storage mechanism exhibited by the NiMoO4@Ni(OH)2/Fe(OH)3-2 electrode, particularly its superior capacitive control characteristics, provides a solid theoretical foundation and experimental evidence for its application in high-performance energy storage devices.
To deeply explore the practical application potential of NiMoO4@Ni(OH)2/Fe(OH)3, this study constructed an asymmetric supercapacitor (ASC) device using 2 mol L−1 KOH as the electrolyte, with NiMoO4@Ni(OH)2/Fe(OH)3-2 as the positive electrode and activated carbon (AC) as the negative electrode. Based on the principle of charge balance (eqn (4)), the mass ratio of the positive and negative electrodes was precisely calculated and set to approximately 0.16. Fig. 9a shows that in a three-electrode system, the potential window of NiMoO4@Ni(OH)2/Fe(OH)3-2 is between 0.12 V and 0.42 V, while the potential window of AC covers −1.0 V to 0 V. Therefore, the total potential window of the ASC device composed of NiMoO4@Ni(OH)2/Fe(OH)3-2 and AC can be extended to 1.3 V. When the operating potential exceeds this range, polarization occurs in the ASC device, as shown in Fig. 9b and c. Fig. 9d presents the cyclic voltammetry (CV) curves of the ASC device at scan rates ranging from 5 mV s−1 to 100 mV s−1. Notably, even at a scan rate of up to 100 mV s−1, the shape of the CV curves remains well-preserved, demonstrating the excellent reversibility of the ASC under high scan rates. At current densities of 0.5, 1, 2, 3, 4, and 5 A g−1, the ASC exhibits specific capacitance values of 142, 135, 126, 120, 114, and 112 F g−1, respectively, indicating good rate performance (Fig. 9e and f). The Ragone plot in Fig. 9g visually reveals the relationship between the energy density and power density of the ASC device.65–68 Calculated using eqn (2) and (3), when the energy density of the ASC is 33.33 W h kg−1, the corresponding power density is 324 W kg−1; and when the power density increases to 3251 W kg−1, the energy density can still be maintained at 26.17 W h kg−1. Additionally, as shown in Fig. 9h, after 5000 charge–discharge cycles, the ASC retains 65.64% of its initial capacity, further validating the great potential and application prospects of the NiMoO4@Ni(OH)2/Fe(OH)3 composite as an electrode material for supercapacitors.
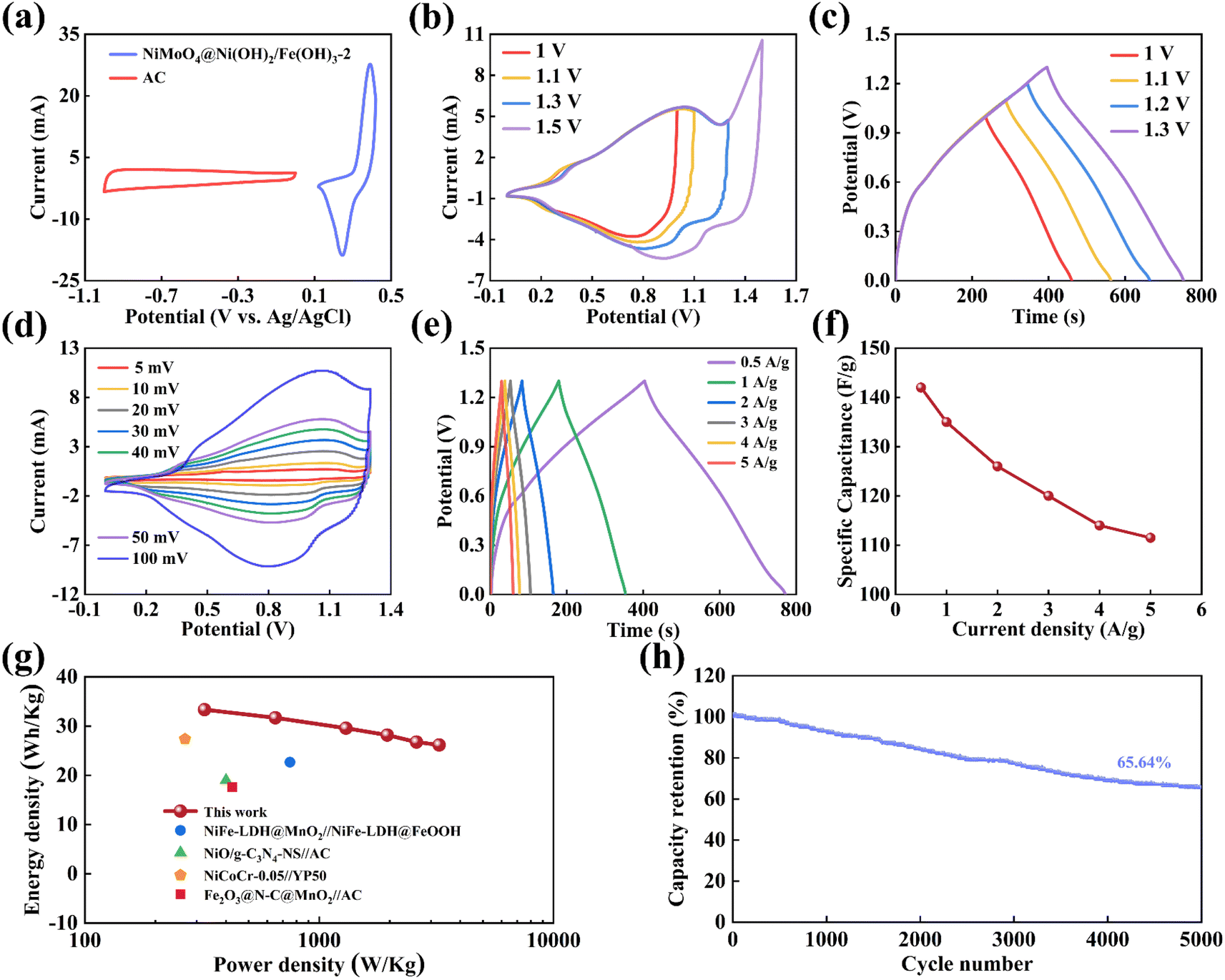 | ||
| Fig. 9 (a) CV curves of the AC and NiMoO4@Ni(OH)2/Fe(OH)3-2 electrodes at 50 mV s−1 (b) CV curves under different potential ranges at 50 mV s−1, (c) GCD curves under different potential ranges at 0.5 A g−1, (d) CV curves at 5–100 mV s−1, (e) GCD curves at 0.5–5 A g−1, (f) specific capacitances at 0.5–5 A g−1, (g) Ragone plots of the power density and energy density, (h) cycling stability after 5000 cycles of the ASC. | ||
4. Conclusion
In this study, an ingenious combination of electrospinning and hydrothermal methods was employed to utilize unique one-dimensional NiMoO4 nanofibers as a substrate, upon which Ni(OH)2 and Fe(OH)3 nanosheets were uniformly grown, thereby constructing a novel hierarchical composite. This composite strategy significantly optimized the electrochemical properties of the material, particularly in terms of a substantial increase in specific capacitance. Specifically, the NiMoO4@Ni(OH)2/Fe(OH)3-2 sample exhibited an excellent specific capacitance of up to 1753 F g−1 at a current density of 1 A g−1. This significant leap in performance can be attributed to the synergistic effect between the one-dimensional NiMoO4 nanofibers as a stable support structure and the Ni(OH)2 and Fe(OH)3 with two-dimensional layered structures. Furthermore, the high specific surface area and optimized structural design within the composite provide broader interfaces and pathways for effective charge storage and transport. Additionally, the asymmetric supercapacitor (ASC) device assembled from this composite achieved a high energy density of 324 W h kg−1 at a power density of 33.33 W kg−1, fully demonstrating the great potential of the NiMoO4@Ni(OH)2/Fe(OH)3 composite in practical energy storage applications. With continuous advancements and maturation of related technologies in the future, the NiMoO4@Ni(OH)2/Fe(OH)3 composite is expected to emerge as a promising electrode material with extensive application value and far-reaching significance in the field of energy storage.Data availability
All data included in this study are available upon request by contact with the corresponding author.Author contributions
Shuang Qiu: writing – review & editing, writing – original draft, investigation, formal analysis, data curation, conceptualization. Zhaojun Sun: formal analysis, data curation. Ruibai Cang: writing – review & editing, Mingyi Zhang: writing – review & editing, writing – original draft, funding acquisition, data curation, conceptualization.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (52203257)References
- J. Libich, J. Máca, J. Vondrák, O. Čech and M. Sedlaříková, J. Energy Storage, 2018, 17, 224–227 CrossRef.
- K. Subasinghage and K. Gunawardane, Energies, 2024, 17, 3853 CrossRef CAS.
- F. Ran, M. Hu, S. Deng, K. Wang, W. Sun, H. Peng and J. Liu, RSC Adv., 2024, 14, 11482–11512 RSC.
- H. Zhang, J. Zhang, X. Gao, L. Wen, W. Li and D. Zhao, Ionics, 2021, 28, 515–531 CrossRef.
- V. Augustyn, P. Simon and B. Dunn, Energy Environ. Sci., 2014, 7, 1597 RSC.
- P. Wang, S. Sun, S. Wang, Y. Zhang, G. Zhang, Y. Li, S. Li, C. Zhou and S. Fang, J. Appl. Electrochem., 2017, 47, 1293–1303 CrossRef CAS.
- W. Liu, M. S. Song, B. Kong and Y. Cui, Adv. Mater., 2016, 29, 1603436 CrossRef PubMed.
- S. J. Lee, J. Theerthagiri, P. Nithyadharseni, P. Arunachalam, D. Balaji, A. Madan Kumar, J. Madhavan, V. Mittal and M. Y. Choi, Renew. Sustain. Energy Rev., 2021, 143, 110849 CrossRef CAS.
- Y. Zhang, H.-L. Gao, X.-D. Jia, S.-W. Wang, J. Yan, H.-W. Luo, K.-Z. Gao, H. Fang, A.-Q. Zhang and L.-Z. Wang, J. Renew. Sustain. Energy, 2018, 10, 054101 CrossRef.
- Z.-H. Xu, X.-L. Li, Q.-W. Li, K. Lv, J.-S. Liu, X.-K. Hang and A. Bayaguud, Rare Met., 2024, 43, 4076–4098 CrossRef CAS.
- R. Liu, A. Zhou, X. Zhang, J. Mu, H. Che, Y. Wang, T.-T. Wang, Z. Zhang and Z. Kou, Chem. Eng. J., 2021, 412, 128611 CrossRef CAS.
- H. Jiu, H. Wei, C. Wang, S. Che, Z. Guo, Y. Han, Q. Xu, X. Yu and L. Zhang, Int. J. Hydrogen Energy, 2022, 47, 33719–33727 CrossRef CAS.
- S. D. Dhas, P. N. Thonge, A. C. Mendhe, J. Yun and D. Kim, Renew. Sustain. Energy Rev., 2024, 204, 114819 CrossRef CAS.
- Y. Li, G. Shi, C. Xu, Y. Zhang, B. Liu and P. Huo, J. Power Sources, 2024, 619, 235200 CrossRef CAS.
- M. Guan, Q. Wang, X. Zhang, J. Bao, X. Gong and Y. Liu, Front. Chem., 2020, 8, 390 CrossRef CAS PubMed.
- R. A. Senthil, A. Min, J. Theerthagiri, G.-A. Kim, H. C. Choi and M. Y. Choi, J. Energy Storage, 2023, 72, 108305 CrossRef.
- J. Theerthagiri, K. Karuppasamy, C. Justin Raj, G. Maia, M. L. Aruna Kumari, L. John Kennedy, M. K. R. Souza, E. S. F. Cardoso, S. Kheawhom, H.-S. Kim and M. Y. Choi, Coord. Chem. Rev., 2024, 513, 215880 CrossRef CAS.
- H. Wang, Y. Zhong, J. Ning and Y. Hu, Chin. Chem. Lett., 2021, 32, 3733–3752 CrossRef CAS.
- S. Deka, Dalton Trans., 2023, 52, 839–856 RSC.
- Y. Wang, L. Liu, J. Hong, J. Cao and C. Deng, J. Membr. Sci., 2018, 564, 372–381 CrossRef CAS.
- Y. Ma, M. Yang and X. Jin, Colloids Surf., A, 2020, 588, 124374 CrossRef CAS.
- X. Li, J. Ren, D. Sridhar, B. B. Xu, H. Algadi, Z. M. El-Bahy, Y. Ma, T. Li and Z. Guo, Mater. Chem. Front., 2023, 7, 1520–1561 RSC.
- X. Wang, H. Song, S. Ma, M. Li, G. He, M. Xie and X. Guo, Chem. Eng. J., 2022, 432, 134319 CrossRef CAS.
- S. Panda, K. Deshmukh, S. K. Khadheer Pasha, J. Theerthagiri, S. Manickam and M. Y. Choi, Coord. Chem. Rev., 2022, 462, 214518 CrossRef CAS.
- H. Liang, J. Lin, H. Jia, S. Chen, J. Qi, J. Cao, T. Lin, W. Fei and J. Feng, J. Power Sources, 2018, 378, 248–254 CrossRef CAS.
- F. Zhu, W. Liu, Y. Liu and W. Shi, Chem. Eng. J., 2020, 383, 123150 CrossRef CAS.
- G. Jiang, M. Zhang, X. Li and H. Gao, RSC Adv., 2015, 5, 69365–69370 RSC.
- J. Theerthagiri, R. A. Senthil, P. Nithyadharseni, S. J. Lee, G. Durai, P. Kuppusami, J. Madhavan and M. Y. Choi, Ceram. Int., 2020, 46, 14317–14345 CrossRef CAS.
- J. Chen, Z. Yan, G. Wang, Y. Ding, M. Xiang and Z. Xu, J. Alloys Compd., 2023, 968, 171841 CrossRef CAS.
- M. Cui and X. Meng, Nanoscale Adv., 2020, 2, 5516–5528 RSC.
- X. Zhang and Y. Xu, Inorg. Chem. Commun., 2018, 87, 8–11 CrossRef CAS.
- S. Chen, M. Zhang, G. Jiang, Z. Zhang and X. Zhou, J. Alloys Compd., 2020, 814, 152253 CrossRef CAS.
- K. Narthana, G. Durai, P. Kuppusami, J. Theerthagiri, S. Sujatha, S. J. Lee and M. Y. Choi, Int. J. Energy Res., 2021, 45, 9983–9998 CrossRef CAS.
- R. K. Mishra, P. Mishra, K. Verma, A. Mondal, R. G. Chaudhary, M. M. Abolhasani and S. Loganathan, Environ. Chem. Lett., 2018, 17, 767–800 CrossRef.
- M. Zhang, Z. Dong, L. Fang, C. Zhi, H. Dong, Z. Ke, J. Liu and L. Yu, Diamond Relat. Mater., 2023, 139, 110395 CrossRef CAS.
- S. Jemai, A. Hajjaji, F. Baig, I. Harabi, B. M. Soucase and B. Bessais, Mater. Charact., 2021, 175, 111036 CrossRef CAS.
- R. Darabi, N. Zare, H. Karimi-Maleh and F. Karimi, Adv. Compos. Hybrid Mater., 2024, 7, 184 CrossRef CAS.
- D. Amiri, E. Kamali Heidari, A. Kamyabi-Gol, S. A. Sajjadi and M. Hoor, J. Energy Storage, 2022, 55, 105490 CrossRef.
- Q. Li, Y. Liao, Y. J. Liu, Q. Y. Mu and Y. D. Wang, Mater. Technol., 2014, 27, 104–106 CrossRef.
- T.-T. Sun, Z.-F. Zhao, Z.-K. Xu and S.-Y. Lin, Rare Met., 2023, 42, 1453–1459 CrossRef CAS.
- J. Yao, D. Xu, X. Ma, J. Xiao, M. Zhang and H. Gao, J. Power Sources, 2022, 524, 231068 CrossRef CAS.
- C. Guo, Y. H. Tang, E. L. Zhang, X. C. Li and J. L. Li, J. Mater. Sci.: Mater. Electron., 2008, 20, 1118–1122 CrossRef.
- X. Xu, X. Liu, J. Zhao, D. Wu, Y. Du, T. Yan, N. Zhang, X. Ren and Q. Wei, J. Colloid Interface Sci., 2022, 606, 1374–1379 CrossRef CAS PubMed.
- J. Wei, D. Qiu, M. Li, Z. Xie, A. Gao, H. Liu, S. Yin, D. Yang and R. Yang, RSC Adv., 2019, 9, 10237–10244 RSC.
- S. Xiong, S. Weng, Y. Tang, L. Qian, Y. Xu, X. Li, H. Lin, Y. Xu, Y. Jiao and J. Chen, J. Colloid Interface Sci., 2021, 602, 355–366 CrossRef CAS PubMed.
- L.-M. Cao, J.-W. Wang, D.-C. Zhong and T.-B. Lu, J. Mater. Chem. A, 2018, 6, 3224–3230 RSC.
- W. Kang, Y. Sun, B. Xu, J. Li, X. Kong, D. Huang, X. Zhang, H. Yang and B. Lin, Electrochim. Acta, 2019, 323, 134819 CrossRef CAS.
- N. Padmanathan, H. Shao and K. M. Razeeb, Int. J. Hydrogen Energy, 2020, 45, 30911–30923 CrossRef CAS.
- B. S. Singu, U. Male, S. E. Hong and K. R. Yoon, Ionics, 2016, 22, 1485–1491 CrossRef CAS.
- L. Zheng, Y. Zhao, P. Xu, Y. Pan, P. Yang, H.-E. Wang, N. Li, X. Shi and H. Zheng, J. Alloys Compd., 2022, 911, 165115 CrossRef CAS.
- Y. Sun, K. Lai, N. Li, Y. Gao and L. Ge, Appl. Catal., B, 2024, 357, 124302 CrossRef CAS.
- F. Zhao, D. Zheng, Y. Liu, F. Pan, Q. Deng, C. Qin, Y. Li and Z. Wang, Chem. Eng. J., 2021, 415, 128871 CrossRef CAS.
- T. I. Singh, G. Rajeshkhanna, U. N. Pan, T. Kshetri, H. Lin, N. H. Kim and J. H. Lee, Small, 2021, 17, 2101312 CrossRef CAS PubMed.
- Y. Lu, B. Jiang, L. Fang, F. Ling, F. Wu, B. Hu, F. Meng, K. Niu, F. Lin and H. Zheng, J. Alloys Compd., 2017, 714, 63–70 CrossRef CAS.
- B. Xing, J. Zhao, S. Jiang, M. Pang, Q. Pan, Y. Geng, G. Ma, Z. Li and P. Han, Synth. Met., 2021, 271, 116638 CrossRef CAS.
- W. Jiao and L. Zhang, Curr. Appl. Phys., 2016, 16, 115–119 CrossRef.
- X. Li, J. Zai, Y. Liu, X. He, S. Xiang, Z. Ma and X. Qian, J. Power Sources, 2016, 325, 675–681 CrossRef CAS.
- W. Zheng, S. Sun, Y. Xu, R. Yu and H. Li, J. Alloys Compd., 2018, 768, 240–248 CrossRef CAS.
- X. Xu, S. Wu, Y. Liu, C. Liu, X. Sun, S. Tian, L. Wu, Y. Sun, Z. Wang and Q. Yang, J. Energy Storage, 2023, 62, 106869 CrossRef.
- I. K. Durga, D. K. Kulurumotlakatla, T. Ramachandran, Y. A. Kumar, D. A. Reddy, K. V. G. Raghavendra, A. A. Alothman and S. S. Rao, J. Phys. Chem. Solids, 2024, 186, 111811 CrossRef CAS.
- L. Li, J. Chen, H. Gao, F. Liu, Z. Li, Q. Li and J. Zhang, J. Electroanal. Chem., 2022, 907, 116065 CrossRef CAS.
- T. Feng, J. Ding, H. Li, W. Wang, B. Dong and L. Cao, ChemSusChem, 2021, 14, 3382–3390 CrossRef CAS PubMed.
- S. Hao, Y. Wei, X. Chen, M. Cong, X. Ding and Y. Gao, Int. J. Hydrogen Energy, 2022, 47, 17263–17270 CrossRef CAS.
- Y. Jiang, R. Xu, B. Cai, H. Gu, L. Zhang, X. Yang, H. Shen and G. Liu, Electrochim. Acta, 2024, 474, 143542 CrossRef CAS.
- Y. Tan, Y. Zhang and Y. Ren, Mater. Chem. Phys., 2024, 320, 129456 CrossRef CAS.
- Q. Sun, K. Yao and Y. Zhang, Chin. Chem. Lett., 2020, 31, 2343–2346 CrossRef CAS.
- R. S. Santos, R. Suresh Babu, T. S. Lessa, L. M. Samyn, R. Vinodh, R. Vivekananth and A. L. F. de Barros, J. Ind. Eng. Chem., 2024, 133, 410–418 CrossRef CAS.
- L. Liu, C. Lu, Y. Ma, Y. Yang, S. Li and M. Zhu, J. Electron. Mater., 2023, 52, 4988–4999 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |