
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Antimicrobial bianthrones from the crinoid Heterometra sp.†
Vítor F. Freire a,
Lucero Martínez-Fructuosoa,
Rohitesh Kumarb,
Rhone K. Akeeb,
Christopher C. Thornburgb,
Susan Enselbc,
Ekene Okoroaforb,
Jason R. Evansa,
Dongdong Wangd,
Brian D. Peysera,
Tanja Grkovic*ad and
Barry R. O'Keefe*ad
a,
Lucero Martínez-Fructuosoa,
Rohitesh Kumarb,
Rhone K. Akeeb,
Christopher C. Thornburgb,
Susan Enselbc,
Ekene Okoroaforb,
Jason R. Evansa,
Dongdong Wangd,
Brian D. Peysera,
Tanja Grkovic*ad and
Barry R. O'Keefe*ad
aNatural Products Branch, Developmental Therapeutic Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute, Frederick, Maryland 21702-1201, USA
bNatural Products Support Group, Leidos Biomedical Research, Inc., Frederick National Laboratory for Cancer Research, Frederick, Maryland 21702-1201, USA
cDepartment of Chemistry and Physics, Hood College, Frederick, Maryland 21701-8599, USA
dMolecular Targets Program, Center for Cancer Research, National Cancer Institute, Frederick, Maryland 21702-1201, USA
First published on 2nd December 2024
Abstract
Antimicrobial resistance is a global public health problem and identification of new chemical scaffolds is important to overcoming this threat. In a recent high-throughput discovery campaign, fractions derived from the organic extract of crinoid, Heterometra sp. (Echinodermata), showed antibacterial activity. Chemical investigation of this extract led to the isolation of three natural products, namely crinemodin bianthrone (1), the new structure 1′′-dehydrocrinemodin bianthrone (2), and 1′′-hydroxycrinemodin bianthrone (3). Their planar structures were determined through HRESIMS and 1D and 2D NMR analysis while a combination of chemical and chirooptical methods was employed to define their absolute configurations for the first time. Variations in proton chemical shifts as well as instability in DMSO and pyridine were observed for 3. Compounds 1 and 3 showed selective antimicrobial activity against Gram-positive bacterial strains.
1. Introduction
Antimicrobial resistance (AMR) ranks among the top ten global public health issues.1 It is estimated that in 2019, bacteria resistant to antibiotics were associated with 4.95 million deaths, and 1.27 million were directly attributable to this cause worldwide.2 The Centers for Disease Control and Prevention (CDC) in the United States lists 18 antimicrobial-resistant bacteria and fungi, with vancomycin-resistant Enterococci (VRE), multidrug-resistant Pseudomonas aeruginosa, drug-resistant Candida, and methicillin-resistant Staphylococcus aureus (MRSA) classified as serious threats.3 Despite the significant burden of AMR, the development of new molecules for its treatment demonstrates a lack of innovation. From 2017 to 2021 a total of 12 new antibacterial drugs were approved. However, among them, only two—Vabomere® and Lefamulin®—introduced new chemical classes.4To encourage screening of diverse chemical libraries against microbial targets, the National Institute of Allergy and Infectious Diseases (NIAID) and the National Cancer Institute (NCI) partnered to perform a high throughput screen of more than 326![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 natural product fractions against three pathogens, including S. aureus, Escherichia coli (2 strains, including wild type and tolC efflux mutant) and C. albicans. A proof-of-concept study selected 75 active fractions from this discovery campaign for the isolation and identification of the bioactive metabolites. Briefly, HPLC-based subfractions were generated from 1 mg of material, and active subfractions had their chemotype annotated based on NMR, LC-MS and IR data. As previously reported, two selected extracts from this screen led to the isolation of 2-amino imidazole and pyridoacridine alkaloids with potent and selective antifungal and antibacterial activities.5
000 natural product fractions against three pathogens, including S. aureus, Escherichia coli (2 strains, including wild type and tolC efflux mutant) and C. albicans. A proof-of-concept study selected 75 active fractions from this discovery campaign for the isolation and identification of the bioactive metabolites. Briefly, HPLC-based subfractions were generated from 1 mg of material, and active subfractions had their chemotype annotated based on NMR, LC-MS and IR data. As previously reported, two selected extracts from this screen led to the isolation of 2-amino imidazole and pyridoacridine alkaloids with potent and selective antifungal and antibacterial activities.5
As an extension to this proof-of-concept study, herein we describe the scale-up isolation and antibiotic activity of the compounds isolated from Heterometra sp. (Echinodermata). Partially purified subfractions from the organic extract of Heterometra sp. were identified as having selective activity against a Gram-positive bacterium Staphylococcus aureus (Fig. S1†).5 To fully characterize its active constituent(s), the organic extract (NSC number #C4711) was subjected to multiple steps of chromatographic separation, leading to the isolation of three crinemodin bianthrones 1–3.
2. Experimental section
2.1. General procedures
Optical rotations were measured on a Rudolph research analytical AUTOPOL IV automatic polarimeter with a 0.25 dm pathlength cell in MeOH at 25 °C. UV spectra were recorded as methanol solutions on a Varian Cary 50-Bio UV/vis spectrophotometer. ECD experiments were recorded as acetonitrile solutions on a J-1500 CD spectropolarimeter using a quartz cell 1 mm path, at room temperature. FTIR spectra were recorded as thin films on a Bruker Alpha II spectrometer. NMR spectra were recorded at 25 °C on either Bruker Avance III HD spectrometer, equipped with a 5 mm TCI Cryo-Probe Prodigy or a Bruker Avance III spectrometer equipped with a 3 mm TCI cryogenic probe, both operating at a frequency of 600 MHz for the 1H nucleus and 151 MHz for the 13C nucleus. For the 3 mm TCI cryogenic probe, all 2D NMR experiments were acquired with non-uniform sampling (NUS) set to 25% using the standard Bruker pulse sequences. For the 5 mm TCI cryogenic probe, all 2D NMR experiments were acquired with non-uniform sampling (NUS) set to 40% for 1H–1H detected experiments or 35% for 1H–13C detected experiments using the standard Bruker pulse sequences. The 1H–13C HMBC experiments were acquired with nJCH = 8.0 Hz. Spectra were calibrated to residual solvent signals at δH 3.31 and δC 49.0 (MeOH-d4). NMR FID processing and data interpretation was done using MestReNova software, version 14.2. High-resolution mass spectra were recorded on an Agilent 1260 Infinity II UHPLC system coupled to an Agilent 6545 QToF equipped with a dual AJS ESI source. Semi-preparative scale HPLC purification was performed with a Gilson HPLC purification system equipped with a GX-281 liquid handler, a 322-binary pump, and a 172-photodiode array detector. All solvents used for chromatography, UV, and MS were HPLC grade, and the H2O was Millipore Milli-Q PF filtered.2.2. Collection, extraction, and isolation
The crinoid Heterometra sp. was collected in Indo-West Pacific (Northwestern Australia) in August 1988 by the Australian Institute of Marine Science under contract to the Natural Products Branch, Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, NCI. The specimen was taxonomically identified by Martin Riddle, and a voucher (Q66C55280) was deposited at the Smithsonian Institution in Suitland, MD. The crinoid (wet weight 316.84 g) was extracted in water, followed by a MeOH/DCM overnight soak according to the Natural Products Branch's standard marine extraction procedure,6 to give 5.84 g of the organic extract (C4711). A portion containing 300 mg of C4711 was prefractionated on a C8 SPE column (2 g), generating seven fractions:7 95:5 H2O:MeOH (C4711_1), 80:20 H2O:MeOH (C4711_2), 60:40 H2O:MeOH (C4711_3), 40:60 H2O:MeOH (C4711_4), 20:80 H2O:MeOH (C4711_5), 0:100 H2O:MeOH (C4711_6), 50:50 MeCN:MeOH (C4711_7).
Fraction C4711_5 (26.5 mg) was submitted for HPLC separation using an Onyx Monolithic C18 column (100 × 10 mm, Phenomenex) and mobile phase of H2O + 0.1% formic acid (solvent A) and MeCN + 0.1% formic acid (solvent B), with the following gradient conditions at 3.8 mL min−1: hold at 70:30 (A:B) for 1.5 min, from 70:30 to 0:100 (A:B) in 7.5 min, hold at 100% (B) for 4.0 min, for a total of 13 min run. Fraction collection was performed in 30 s increments between 1.5 and 12.5 min (22 fractions).8 Fractions C4711_5_8 to 17 were combined (11.3 mg) and further separated using a Kinetex C8 column (150 × 21.2 mm, 5 μm, 100 Å, Phenomenex) and mobile phase of H2O + 0.1% formic acid (solvent A) and MeCN + 0.1% formic acid (solvent B), with the following gradient conditions at 9.0 mL min−1: from 50:50 to 30:70 (A:B) for 32 min, and hold at 30:70 (A:B) for 5.0 min. Fraction collection was performed in 30 s increments, yielding crinemodin bianthrone (1.8 mg, 0.6% of organic extract yield; 1) and 1′′-dehydrocrinemodin bianthrone (1.5 mg, 0.5% of organic extract yield; 2).
Fractions C4711_6 and 7 were combined (90.6 mg) and submitted for HPLC separation using an Onyx monolithic C18 column (100 × 10 mm, Phenomenex) and mobile phase of H2O + 0.1% formic acid (solvent A) and MeCN + 0.1% formic acid (solvent B), with the following gradient conditions at 3.8 mL min−1: hold at 70:30 (A:B) for 1.5 min, from 70:30 to 0:100 (A:B) in 7.5 min, hold at 100% (B) for 4.0 min, for a total of 13 min run. Fraction collection was performed in 30 s increments between 1.5 and 12.5 min (22 fractions).8 Fractions C4711_6_13 and 14 were combined (5.1 mg) and further separated using a Kinetex C8 column (150 × 21.2 mm, 5 μm, 100 Å, Phenomenex) and mobile phase of H2O + 0.1% formic acid (solvent A) and MeCN + 0.1% formic acid (solvent B), with the following gradient conditions at 9.0 mL min−1: from 50:50 to 35:65 (A:B) for 32 min, and hold at 35:65 (A:B) for 5 min, in a total of 37 min run. Fraction collection was performed in 30 s increments, yielding 1′′-hydroxycrinemodin bianthrone (2.5 mg, 0.83% of organic extract yield; 3).
ε) 278 (1.72), 362 (1.79) nm; ECD (3.81 × 10−5 M, MeCN) λmax (Δε) 265 (−3.1), 300 (+3.4), 345 (−3.6) and 394 (+3.9) nm; IR (film) νmax 2961, 2933, 2873, 1619, 1602, 1561, 1486, 1377, 1339, 1294, 1249, 1165, 1062, 918, 854, 790, 755, 682, 550 cm−1; 1H and 13C data (MeOH-d4) in Table 1; HRESIMS m/z 567.2015 [M + H]+ (calcd for C34H31O8+ m/z 567.2014, Δ = 0.17 ppm).
| Position | 1 | 2 | ||
|---|---|---|---|---|
| δC, typea | δH, multb (J in Hz) | δC, typea | δH, multb (J in Hz) | |
| a 151 MHz.b 600 MHz. | ||||
| 1 | 102.9, CH | 6.45, br s | 109.8, CH | 6.41, s |
| 2 | 165.5, C | 165.6, C | ||
| 3 | 109.7, CH | 6.28, d (2.2) | 103.0, CH | 6.26, s |
| 4 | 166.5, C | 166.5, C | ||
| 5 | 111.9, C | 111.8, C | ||
| 6 | 191.6, C | 191.4, C | ||
| 7 | 115.1, C | 114.7, C | ||
| 8 | 162.8, C | 163.1, C | ||
| 9 | 117.0, CH | 6.54, s | 114.9, CH | 6.55, s |
| 10 | 151.8, C | 145.2, C | ||
| 11 | 121.6, CH | 5.72, s | 118.1, CH | 5.78, s |
| 12 | 140.7, C | 140.6, C | ||
| 13 | 57.4, CH | 4.37, s | 57.4, CH | 4.27, d (2.9) |
| 14 | 146.9, C | 146.9, C | ||
| 1′ | 102.9, CH | 6.45, br s | 109.7, CH | 6.41, s |
| 2′ | 165.5, C | 165.5, C | ||
| 3′ | 109.7, CH | 6.28, d (2.2) | 102.9, CH | 6.26, s |
| 4′ | 166.5, C | 166.5, C | ||
| 5′ | 111.9, C | 111.8, C | ||
| 6′ | 191.6, C | 191.2, C | ||
| 7′ | 115.1, C | 115.4, C | ||
| 8′ | 162.8, C | 162.8, C | ||
| 9′ | 117.0, CH | 6.54, s | 116.9, CH | 6.50, s |
| 10′ | 151.8, C | 151.7, C | ||
| 11′ | 121.6, CH | 5.72, s | 121.4, CH | 5.65, s |
| 12′ | 140.7, C | 140.6, C | ||
| 13′ | 57.4, CH | 4.37, s | 57.3, CH | 4.17, d (2.9) |
| 14′ | 146.9, C | 146.7, C | ||
| 1′′ | 39.2, CH2 | 2.39, t (7.5) | 131.0, CH | 6.16, d (15.4) |
| 2′′ | 24.4, CH2 | 1.54, m | 131.6, CH2 | 6.13, m |
| 3′′ | 14.3, CH3 | 0.94, t (7.3) | 18.7, CH3 | 1.87, d (5.9) |
| 1′′′ | 39.2, CH2 | 2.39, t (7.5) | 39.2, CH2 | 2.36, t (7.8) |
| 2′′′ | 24.4, CH2 | 1.54, m | 24.4, CH2 | 1.51, m |
| 3′′′ | 14.3, CH3 | 0.94, t (7.3) | 14.3, CH3 | 0.92, t (7.7) |
ε) 282 (2.10), 366 (2.19) nm; ECD (3.94 × 10−5 M, MeCN) λmax (Δε) 273 (−5.2), 303 (+2.5), 342 (−5.3) and 406 (+4.9) nm; IR (film) νmax 2926, 2853, 1617, 1602, 1474, 1376, 1340, 1291, 1251, 1206, 1063, 964, 853, 760 cm−1; 1H and 13C data (MeOH-d4) in Table 1; HRESIMS m/z 565.1860 [M + H]+ (calcd for C34H29O8+ m/z 565.1857, Δ = 0.53 ppm).ε) 277 (4.25), 363 (4.27) nm; ECD (2.75 × 10−5 M, MeCN) λmax (Δε) 272 (−1.3), 306 (+1.8), 348 (−0.8) and 395 (+0.8) nm; IR (film) νmax 2964, 2931, 2874, 1600, 1563, 1483, 1466, 1364, 1337, 1290, 1240, 1163, 1064, 967, 917, 853, 789, 752, 647, 590, 550 cm−1; HRESIMS m/z 605.1785 [M + Na]+ (calcd for C34H30O9Na+ m/z 605.1783, Δ = 0.33 ppm).2.3. Competitive enantioselective conversion (CEC)
The CEC reaction followed by LC-MS analysis was based on previously described procedure by Lee et al.9 Compound 3 (0.25 mg, 0.43 μmol) was transferred to two different vials, and dimethylformamide (90 μL) was added as organic solvent. The catalysts S- and R-HBTM (10.0 μL, 12.9 μmol) were added to their respective vials, along with N,N-diisopropylethylamine (2.2 μL, 12.9 μmol). Propionic anhydride (1.7 μL, 12.9 μmol) was added to initiate the reaction. Aliquots of 2 μL were taken every 5 min and quenched with 100 μL of MeOH prior to LC-MS analysis, for a total reaction time of 60 min.Aliquots (5 μL) of the samples collected at different time intervals were injected on an Agilent 1260 Infinity II HPLC system coupled to an Agilent 6230 ToF equipped with a dual AJS ESI source. A Kinetex C18 column (50.0 × 2.1 mm, 5 μm, Phenomenex) was used. The mobile phase consisted of H2O + 0.1% formic acid (A) and MeCN + 0.1% formic acid (B). The gradient used was maintained at 95:5 (A:B) for 1.0 min, from 95:5 to 0:100 (A:B) for 8.0 min, maintained at 0:100 (A:B) for 1.0 min, from 0:100 to 95:5 (A:B) for 0.5 min and then equilibrated in a post-run at 95:5 (A:B) during 1.5 min. The positive mode ESI conditions were 3.5 V of capillary voltage, 1.5 kV of nozzle voltage, gas temperature at 325 °C and gas flow of 10 L min−1. The MS spectra were acquired using positive mode, mass range of m/z 100–3000 and scan rate of 2 spectra per s. The reaction rates were determined by measuring the peak areas of the fully acylated derivative of compound 3, using the extracted ion chromatogram (EIC) for the sodiated molecule [M + Na]+ at m/z 997.3617 (±20 ppm) on MassHunter Qualitative Analysis 10.0 software.
2.4. Antimicrobial assay
Microbes, growth, and testing conditions were identical to those used for the primary screen.52.5. Anti-proliferative assay
Purified compounds were tested for cytotoxicity in the recently described HTS384 NCI60 screen.103. Results
3.1. Structure elucidation
Crinemodin bianthrone (1) has been previously reported as a constituent of the crinoid Lamprometra palmata by Rideout and Sutherland (1985),11 where its structure elucidation was achieved by a combination of chemical degradation, chemical synthesis, and comparison of Rf on TLC, and 1H NMR data. Here, we present full spectrometric and spectroscopic characterization of 1, achieved using HRESIMS, 1D and 2D NMR, and chiroptical methods. HRESIMS analysis showed a protonated molecule [M + H]+ at m/z 567.2015, corresponding to the molecular formula C34H31O8+. The UV spectrum of 1 showed λmax of 278 and 362 nm, consistent with the UV spectra of bianthrones.12,13 Inspection of the 1H NMR spectrum of 1 demonstrated eight proton resonances, while 13C NMR spectrum showed seventeen carbon resonances, indicating symmetry in the molecule (Table 1). The bianthrone core was assigned using 1H–13C HSQC, 1H–13C HMBC correlations and comparison with literature values.14 The propyl group was connected to the bianthrone core through 1H–13C HMBC correlations from H-1′′/1′′′ (δH 2.39) to C-9/9′ (δC 117.0), C-10/10′ (δC 151.8) and C-11/11′ (δC 121.6), completing the planar structure of 1. Due to an inner plane of symmetry in 1, it is possible to categorize the stereoisomers of this homobianthrone by their relative configuration as either meso (cis) or racemic (trans).11,14–16 The chiroptical properties of 1 allowed us to establish its relative configuration as trans, in agreement with that assigned by Rideout and Sutherland.11 Comparison of the experimental ECD spectrum of 1 (Fig. S36†) to reported in silico spectra of two trans-bianthrone series, allianthrones B and C15 and brevianthrones A1 and B1,12 based on the Cotton effects at λmax 265 nm (Δε −3.1), 300 (Δε +3.4) and 345 (Δε −3.6), indicated the absolute configuration of 1 to be 13S,13′S (Fig. 1).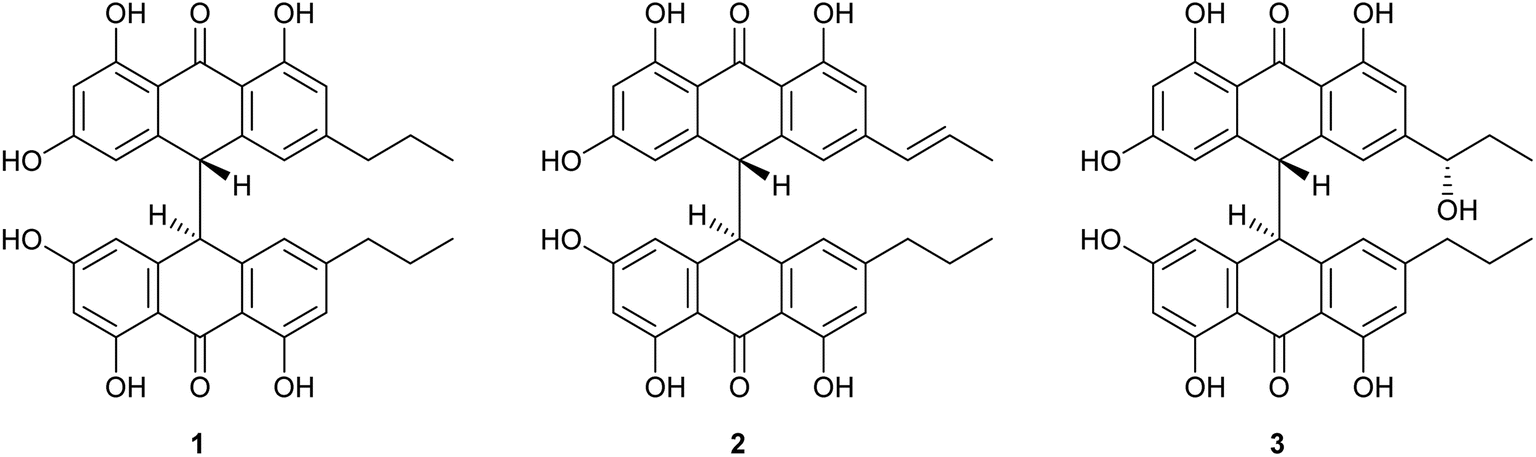 | ||
| Fig. 1 Structures of crinemodin bianthrones (1–3) isolated from Heterometra sp. | ||
1′′-Dehydrocrinemodin bianthrone (2) showed a protonated molecule [M + H]+ at m/z 565.1860 by HRESIMS, corresponding to the molecular formula C34H29O8+. This mass difference and the absence of symmetry observed in the 1H and 13C NMR data, suggested 2 contained an unsaturated alkyl group relative to 1. Analysis of 1D and 2D NMR data revealed that the bianthrone core of the molecule was identical to that of 1. The bianthrone motif was fully characterized with the aid of the 1H–13C LR-HSQMBC17 experiment that showed a 4JCH correlation from H-3/3′ (δH 6.26) to the C-6/6′ (δC 191.4/191.2) carbonyl. Inspection of additional 2D NMR data revealed one side chain composed of a saturated propyl group, H-1′′′ (δH 2.36), H-2′′′ (δH 1.51) and H-3′′′ (δH 0.92), connected to the bianthrone core at C-10′ (δC 151.7) (Fig. 2), identical to that of 1. The 1H–1H COSY spectrum of the other side chain revealed a contiguous spin system between H-1′′ (δH 6.16), H-2′′ (δH 6.13) and H-3′′ (δH 1.87), indicative of a propenyl group. The 1H–13C HMBC correlations between H-1′′ and H-2′′ to C-10 (δC 145.2) indicated the connection of the propenyl group to the bianthrone core via C-10. The 1H NMR spectrum showed that H-1′′ (δH 6.16) had a coupling constant of 15.4 Hz, indicating a trans geometry of the double bond. Upon comparison of the specific rotation and ECD spectra of 2 with that of 1 (Fig. S36†), the absolute configuration of 2 was established to be 13S,13′S. The structure of 2 was therefore assigned to be a new natural product, named 1′′-dehydrocrinemodin bianthrone.
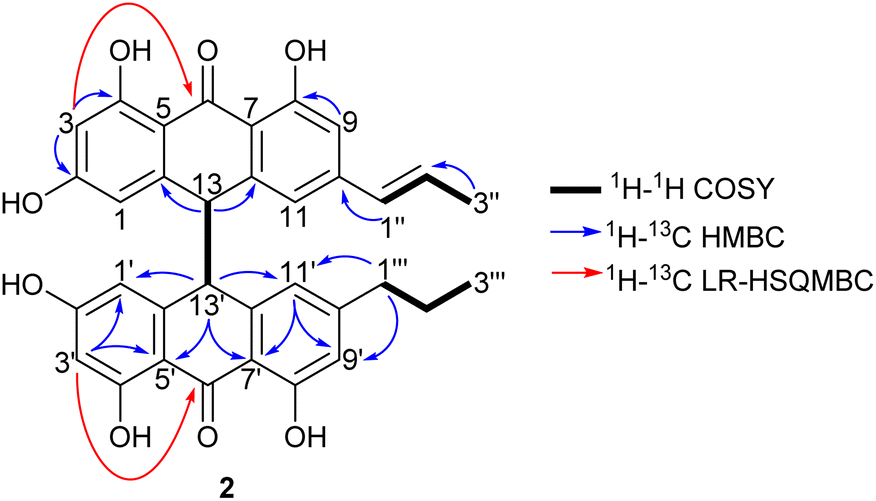 | ||
| Fig. 2 Key 2D NMR correlations used to establish the structure of 1′′-dehydrocrinemodin bianthrone (2). | ||
An additional natural product, 1′′-hydroxycrinemodin bianthrone (3), previously reported from the crinoids Lamprometra palmata11 and Himerometra magnipinna,14 was isolated from the same active fraction. While the planar structure of 3 agreed with that previously described,14 the absolute configuration was never reported for either the bianthrone core or the secondary alcohol on the side chain. Thus, we used the specific rotation and Cotton effects in the ECD spectrum to assign the bianthrone core of 3 as 13S,13′S, identical to that of 1 and 2 (Fig. S36†). Due to the insignificant influence of the secondary alcohol at CH-1′′ in the ECD spectrum, chemical methods were used to establish its configuration. The well-established Mosher's method was not successful due to instability of 3 in pyridine-d5. Alternatively, we applied a competitive enantioselective conversion (CEC) reaction using R- and S-homobenzotetramisole (HBTM) catalysts followed by LC-MS analysis to establish the absolute configuration of the secondary alcohol.9,18,19 This method relies on the kinetic resolution of the acylation of a secondary alcohol by the enantiomeric pair of R- and S-HBTM catalysts. Two parallel reactions were performed, treating compound 3 with the R- or S- catalyst. Aliquots of the reactions were taken every 5 min and quenched with MeOH over a period of 60 min. LC-MS data was acquired and the area under the chromatographic peak was integrated using the extracted ion chromatogram (EIC) of the sodium adduct [M + Na]+ for the fully acylated product of 3. Analytical curves were plotted and the reaction using R-HBTM was established as the fastest one (Fig. 3). The mnemonic used for the determination of the absolute configuration of the secondary alcohol9,19 states that compound 3 should be drawn with the polar (π) group on the left side and the alkyl group on the right side. Since R-HBTM was the faster reaction, the hydroxy group is positioned up and forward allowing us to establish the absolute configuration of C-1′′ to be S for compound 3. Therefore, the absolute configuration of 3 was determined to be 13S,13′S,1′′S.
 | ||
| Fig. 3 Competitive enantioselective conversion (CEC) reaction with R- and S-homobenzotetramisole (HBTM) coupled to LC-MS. (A) Reaction rates of 3 treated with propionic anhydride using R- and S-HBTM catalysts. (B) Adapted mnemonic from Lee et al.9 used to predict the configuration of secondary alcohols in CEC reaction. (C) Proposed transition state of 3 in CEC reaction. | ||
3.2. Solution behavior of bianthrones
During isolation and NMR data acquisition, instability of bianthrones 1–3 was observed when exposed to DMSO, DMSO-d6 and pyridine-d5. Previous reports indicated that the bianthrones can suffer polymerization15 or cleavage to its monomers,20 which can be triggered by changes in pH or pressure, and exposure to UV or organic solvents.15,20 However, while unstable in some organic solvents, compounds 1–3 showed stability in methanol, water, and acetonitrile which allowed the full assignment of their structure, and revealed unexpected solution behavior as outlined below. Two different fractions containing compound 3 with consistent retention time, MS1 spectra, UV profile, [α]D value, and ECD spectra, showed significant differences in δH chemical shifts, especially for two methine doublets at δH 4.2–4.6 (H-13/13′) and the two singlets at δH 6.0–5.8 (H-11/11′). Subsequently, the 1H NMR spectrum of the same sample acquired several days apart showed variation of the proton chemical shifts (Fig. 4). The relative level of water in the MeOH-d4 solution in the NMR tube was speculated to be responsible for the observed behavior. To test this hypothesis, we performed a titration by incrementally adding D2O to a solution of 3 in MeOH-d4. First, 3 was dried under high vacuum overnight, and then solubilized using MeOH-d4 (99.9%, ampule). A 1H NMR spectrum was initially acquired with neat MeOH-d4, followed by titration with D2O (1–20 μL) directly into the NMR tube and subsequent acquisition of additional 1H NMR spectra to determine any measurable changes. The addition of D2O to the sample caused the proton chemical shifts of 3 to change, especially for H-13/13′ and H-11/11′ (Fig. S37†). We hypothesize that a shielding effect at positions H-13/13′ and H-11/11′ is taking place, due to the higher population of crossed A/A′ (I) gauche conformers of 3 when solvated by D2O in solution, compared to the higher population of crossed B/B′ (II) conformers when D2O is not present. Although further studies may be required to understand the dynamics of these bianthrones in solution, researchers should be cautious on relying exclusively on 1H NMR chemical shifts for configurational assignments of this group of natural products. | ||
| Fig. 4 1H NMR spectra comparison of 3 acquired in MeOH-d4 on different days showing the variation of proton chemical shifts. | ||
3.3. Biological activity
The crinemodin bianthrones (1–3) were tested against a range of pathogenic bacterial and fungal strains (Table 2), which included E. coli wild-type (BW25113) and E. coli efflux-(JW5503-1), P. aeruginosa (PAM12626), S. aureus (ATCC29213), E. faecalis wild-type (ATCC29212), vancomycin-resistant E. faecium (VRE, ATCC 700221), C. albicans (ATCC90028), and A. fumigatus (ATCC MYA-3626), as well as a range of human tumor cell lines in the NCI-60 cell lines screen. Compound 1 was the most active, with MIC values of 5.0, 2.5 and 2.5 mg L−1 against the Gram-positive strains S. aureus, E. faecalis and E. faecium, respectively. Compound 3 showed MIC values of 10.0 and 20.0 mg L−1 against S. aureus and E. faecalis, while 2 did not show activity against any of the tested strains. These results suggest that the side chains of the bianthrones are important features for activity against Gram-positive bacteria, with the saturated aliphatic chain exhibiting the highest activity. Notably, no antimicrobial activity was observed for compounds 1–3 against any of the Gram-negative bacteria, the fungal strains tested, or any of the NCI-60 cell lines, demonstrating selectivity of this chemotype for Gram-positive bacterial strains.| E. coli WT | E. coli efflux | P. aeruginosa efflux | S. aureus WT | E. faecalis WT | E. faecium VRE | C. albicans WT | A. fumigatus WT | ||
|---|---|---|---|---|---|---|---|---|---|
| 1 |  |
>20 | >20 | >20 | 5 | 2.5 | 2.5 | >10 | >10 |
| 2 |  |
>20 | >20 | >20 | >20 | >20 | >20 | >10 | >10 |
| 3 | 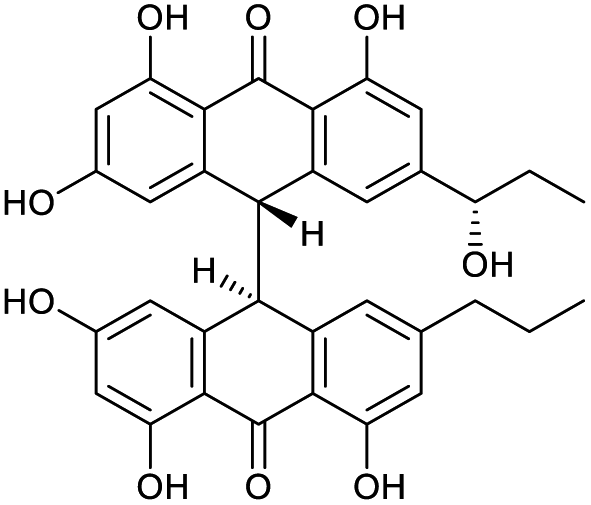 |
>20 | >20 | >20 | 10 | 20 | >20 | >10 | >10 |
4. Conclusions
Bianthrones 1–3 were isolated from the organic extract of Heterometra sp., with 1′′-dehydrocrinemodin bianthrone (2) being a new natural product. For the first time the absolute configuration of crinemodin bianthrones were established by a combination of modern chemical and spectroscopic approaches, enabling the complete configurational assignment of the bianthrone core and side chains. The competitive enantioselective conversion method proved to be a good alternative to solve the absolute configuration of the secondary alcohol in 3. The bianthrones showed interesting solution behavior, which should be taken in consideration when working with this chemotype. The isolated compounds presented selective activity against Gram-positive strains, with 1 showing greater activity against E. faecalis wild-type and vancomycin resistant E. faecium strains.Data availability
The data that support the findings of the present study are available in ESI† of this article. In addition, the raw HRMS and NMR data for the natural products 1–3 has been deposited in the Harvard Dataverse (https://dataverse.harvard.edu/) and can be found at https://doi.org/10.7910/DVN/RJOMB5.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors thank Dr Ryan Arends, JMI Laboratories, for facilitating the antimicrobial testing. We also thank Drs Erin Zeituni and Joseph Campbell from the National Institute of Allergy and Infectious Diseases for helpful discussions, the Molecular Pharmacology Branch, DTP, DCTD, NCI for performing the NCI 60-cell cytotoxicity assays in support of this study, and Dr Sergey G. Tarasov and Ms Marzena Dyba of the Biophysics Resource in the Center for Structural Biology, NCI-Frederick for technical support and help with the ECD measurements. This project has been funded in whole or in part with Federal funds from the Division of Microbiology and Infectious Diseases, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract No. 75N93019D00011/75N93019F00210. This research was supported [in part] by the Intramural Research Program of the NIH, National Cancer Institute, under contract HHSN261200800001E and by the National Cancer Institute's Cancer MoonshotSM NCI Program for Natural Products Discovery – Cure (ZIA BC 011854). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.References
- T. R. Walsh, A. C. Gales, R. Laxminarayan and P. C. Dodd, PLoS Med., 2023, 20, e1004264 CrossRef PubMed
.
- C. J. L. Murray, K. S. Ikuta, F. Sharara, L. Swetschinski, G. R. Aguilar, A. Gray, C. Han, C. Bisignano, P. Rao, E. Wool, S. C. Johnson, A. J. Browne, M. G. Chipeta, F. Fell, S. Hackett, G. Haines-Woodhouse, B. H. K. Hamadani, E. A. P. Kumaran, B. McManigal, S. Achalapong, R. Agarwal, S. Akech, S. Albertson, J. Amuasi, J. Andrews, A. Aravkin, E. Ashley, F.-X. Babin, F. Bailey, S. Baker, B. Basnyat, A. Bekker, R. Bender, J. A. Berkley, A. Bethou, J. Bielicki, S. Boonkasidecha, J. Bukosia, C. Carvalheiro, C. Castañeda-Orjuela, V. Chansamouth, S. Chaurasia, S. Chiurchiù, F. Chowdhury, R. C. Donatien, A. J. Cook, B. Cooper, T. R. Cressey, E. Criollo-Mora, M. Cunningham, S. Darboe, N. P. J. Day, M. De Luca, K. Dokova, A. Dramowski, S. J. Dunachie, T. D. Bich, T. Eckmanns, D. Eibach, A. Emami, N. Feasey, N. Fisher-Pearson, K. Forrest, C. Garcia, D. Garrett, P. Gastmeier, A. Z. Giref, R. C. Greer, V. Gupta, S. Haller, A. Haselbeck, S. I. Hay, M. Holm, S. Hopkins, Y. Hsia, K. C. Iregbu, J. Jacobs, D. Jarovsky, F. Javanmardi, A. W. J. Jenney, M. Khorana, S. Khusuwan, N. Kissoon, E. Kobeissi, T. Kostyanev, F. Krapp, R. Krumkamp, A. Kumar, H. H. Kyu, C. Lim, K. Lim, D. Limmathurotsakul, M. J. Loftus, M. Lunn, J. Ma, A. Manoharan, F. Marks, J. May, M. Mayxay, N. Mturi, T. Munera-Huertas, P. Musicha, L. A. Musila, M. M. Mussi-Pinhata, R. N. Naidu, T. Nakamura, R. Nanavati, S. Nangia, P. Newton, C. Ngoun, A. Novotney, D. Nwakanma, C. W. Obiero, T. J. Ochoa, A. Olivas-Martinez, P. Olliaro, E. Ooko, E. Ortiz-Brizuela, P. Ounchanum, G. D. Pak, J. L. Paredes, A. Y. Peleg, C. Perrone, T. Phe, K. Phommasone, N. Plakkal, A. Ponce-de-Leon, M. Raad, T. Ramdin, S. Rattanavong, A. Riddell, T. Roberts, J. V. Robotham, A. Roca, V. D. Rosenthal, K. E. Rudd, N. Russell, H. S. Sader, W. Saengchan, J. Schnall, J. A. G. Scott, S. Seekaew, M. Sharland, M. Shivamallappa, J. Sifuentes-Osornio, A. J. Simpson, N. Steenkeste, A. J. Stewardson, T. Stoeva, N. Tasak, A. Thaiprakong, G. Thwaites, C. Tigoi, C. Turner, P. Turner, H. R. van Doorn, S. Velaphi, A. Vongpradith, M. Vongsouvath, H. Vu, T. Walsh, J. L. Walson, S. Waner, T. Wangrangsimakul, P. Wannapinij, T. Wozniak, T. E. M. W. Y. Sharma, K. C. Yu, P. Zheng, B. Sartorius, A. D. Lopez, A. Stergachis, C. Moore, C. Dolecek and M. Naghavi, Lancet, 2022, 399, 629–655 CrossRef CAS PubMed
- CDC, Antibiotic Resistance Threats in the United States, U.S. Department of Health and Human Services, CDC, Atlanta, GA, 2019 Search PubMed
- World Health Organization, 2021 Antibacterial Agents in Clinical and Preclinical Development, 2022 Search PubMed
- L. Martinez-Fructuoso, S. J. R. Arends, V. F. Freire, J. R. Evans, S. DeVries, B. D. Peyser, R. K. Akee, C. C. Thornburg, R. Kumar, S. Ensel, G. M. Morgan, G. D. McConachie, N. Veeder, L. R. Duncan, T. Grkovic and B. R. O'Keefe, ACS Infect. Dis., 2023, 9, 1245–1256 CrossRef CAS PubMed
- T. G. McCloud, Molecules, 2010, 15, 4526–4563 CrossRef CAS PubMed
- C. C. Thornburg, J. R. Britt, J. R. Evans, R. K. Akee, J. A. Whitt, S. K. Trinh, M. J. Harris, J. R. Thompson, T. L. Ewing, S. M. Shipley, P. G. Grothaus, D. J. Newman, J. P. Schneider, T. Grkovic and B. R. O'Keefe, ACS Chem. Biol., 2018, 13, 2484–2497 CrossRef CAS PubMed
- T. Grkovic, R. K. Akee, C. C. Thornburg, S. K. Trinh, J. R. Britt, M. J. Harris, J. R. Evans, U. Kang, S. Ensel, C. J. Henrich, K. R. Gustafson, J. P. Schneider and B. R. O'Keefe, ACS Chem. Biol., 2020, 15, 1104–1114 CrossRef CAS PubMed
- S. R. Lee, H. B. Park and K. H. Kim, Anal. Chem., 2018, 90, 13212–13216 CrossRef CAS PubMed
- M. W. Kunkel, N. P. Coussens, J. Morris, R. C. Taylor, T. S. Dexheimer, E. M. Jones, J. H. Doroshow and B. A. Teicher, Cancer Res., 2024, 84(15), 2403–2416 CrossRef CAS PubMed
- J. A. R. M. S. Sutherland, Aust. J. Chem., 1985, 38, 793–808 CrossRef
- M. Lin, D. A. Adpressa, M. Feng, Y. Lu and B. R. Clark, Phytochemistry, 2021, 188, 112792 CrossRef CAS PubMed
- L. Du, T. Zhu, H. Liu, Y. Fang, W. Zhu and Q. Gu, J. Nat. Prod., 2008, 71, 1837–1842 CrossRef CAS PubMed
- N. Shao, G. Yao and L. C. Chang, J. Nat. Prod., 2007, 70, 869–871 CrossRef CAS PubMed
- P. E. Mandelare, D. A. Adpressa, E. N. Kaweesa, L. N. Zakharov and S. Loesgen, J. Nat. Prod., 2018, 81, 1014–1022 CrossRef CAS PubMed
- N.-Y. Ji, X.-R. Liang, R.-R. Sun and F.-P. Miao, RSC Adv., 2014, 4, 7710 RSC
- R. T. Williamson, A. V. Buevich, G. E. Martin and T. Parella, J. Org. Chem., 2014, 79, 3887–3894 CrossRef CAS PubMed
- A. J. Wagner and S. D. Rychnovsky, J. Org. Chem., 2013, 78, 4594–4598 CrossRef CAS PubMed
- A. J. Wagner, J. G. David and S. D. Rychnovsky, Org. Lett., 2011, 13, 4470–4473 CrossRef CAS PubMed
- A. Lagarde, L. Mambu, P. Y. Mai, Y. Champavier, J. L. Stigliani, M. A. Beniddir and M. Millot, Fitoterapia, 2021, 149, 104811 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra05594b |
| This journal is © The Royal Society of Chemistry 2024 |