DOI:
10.1039/D4RA06740A
(Paper)
RSC Adv., 2024,
14, 39061-39068
Highly efficient Ni/Ac–Al2O3 catalysts in the dry reforming of methane: influence of acetic acid treatment and Ni loading†
Received
18th September 2024
, Accepted 6th November 2024
First published on 10th December 2024
Abstract
The presence of abundant hydroxyl groups on the surface of Al2O3 can promote the dispersion of Ni species but produce an inactive NiAl2O4 phase at high temperatures. Moreover, the catalysts prepared by the conventional incipient wetness impregnation method lack the sites for the activation of CO2, which leads to coke deposition and thus affects the catalyst activity. The above restricts the utilization of Ni in conventional Ni/Al2O3 catalysts. In this paper, Al2O3 support was pre-treated by acetic acid to selectively remove hydroxyl groups without affecting the coordination environment of Al. Results revealed that the Al2O3 support after hydroxyl removal not only showed moderate metal–support interaction but also produced more sites for the adsorption and activation of the reactant, which significantly improves the utilization of nickel species and the stability of the catalyst. The conversion of CH4 and CO2 at 700 °C was as high as 88% and 90%, respectively, and has an excellent stability of 50 h. This study provides a feasible strategy for the design of highly active methane dry-reforming catalysts.
Introduction
Syngas plays a significant role in the production of varied chemicals.1–3 Producing syngas by the dry reforming of methane (DRM) with carbon dioxide is a kind of reaction process of natural gas reforming to syngas.4 The basic principle is that methane (CH4) reacts with carbon dioxide (CO2) to produce syngas at high temperatures with the catalyst.5,6 Syngas generation is a highly thermally absorbent reaction, which usually requires temperatures above 700 °C. The H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO ratio of syngas produced by the DRM process is approximately 1,7,8 which is favourable for the synthesis of chemicals.9–11 Compared to the conventional methane steam reforming process to syngas, the DRM method has remarkable advantages, as follows. (1) The process consumes no water but a large amount of carbon dioxide, reducing energy consumption as well as alleviating greenhouse gas emissions;12 (2) the feedstock can be attained by a wide range of sources, such as biogas, coke oven gas, coalbed methane, and natural gas;13,14 (3) two kinds of greenhouse gases are involved in the reaction, which is favourable to the environment; (4) compared to wet reforming and partial oxidation, it reduces methane consumption by 50%.15–17 However, during the DRM reaction process (eqn (1)), the high-temperature environment in the reactor inevitably leads to the occurrence of several parallel reactions such as methane decomposition (eqn (2)), inverse Boudouard reaction (eqn (3)), and reverse water-gas shift (RWGS) (eqn (4)).18–21 These adverse reactions can seriously affect the process efficiency. Coke deposition causes catalyst deactivation and reactor clogging, especially at 550–700 °C, which is a crucial challenge for industrialization. Additionally, the RWGS reaction reduces the H2/CO molar ratio in syngas,22,23 affecting the downstream processing of syngas. Therefore, there is an urgent demand to develop a catalyst with superior catalytic activity, resistance to coke deposition and high temperature for DRM.
:CO ratio of syngas produced by the DRM process is approximately 1,7,8 which is favourable for the synthesis of chemicals.9–11 Compared to the conventional methane steam reforming process to syngas, the DRM method has remarkable advantages, as follows. (1) The process consumes no water but a large amount of carbon dioxide, reducing energy consumption as well as alleviating greenhouse gas emissions;12 (2) the feedstock can be attained by a wide range of sources, such as biogas, coke oven gas, coalbed methane, and natural gas;13,14 (3) two kinds of greenhouse gases are involved in the reaction, which is favourable to the environment; (4) compared to wet reforming and partial oxidation, it reduces methane consumption by 50%.15–17 However, during the DRM reaction process (eqn (1)), the high-temperature environment in the reactor inevitably leads to the occurrence of several parallel reactions such as methane decomposition (eqn (2)), inverse Boudouard reaction (eqn (3)), and reverse water-gas shift (RWGS) (eqn (4)).18–21 These adverse reactions can seriously affect the process efficiency. Coke deposition causes catalyst deactivation and reactor clogging, especially at 550–700 °C, which is a crucial challenge for industrialization. Additionally, the RWGS reaction reduces the H2/CO molar ratio in syngas,22,23 affecting the downstream processing of syngas. Therefore, there is an urgent demand to develop a catalyst with superior catalytic activity, resistance to coke deposition and high temperature for DRM.| | |
CH4 + CO2 → 2CO + 2H2 ΔHθ, 298 K = 247.3 kJ mol−1
| (1) |
| | |
CH4 → C + 2H2 ΔHθ, 298 K = 75.0 kJ mol−1
| (2) |
| | |
2CO → C + CO2 ΔHθ, 298 K = −171.0 kJ mol−1
| (3) |
| | |
CO2 + H2 → CO + H2O ΔHθ, 298 K = 41.0 kJ mol−1
| (4) |
Currently, the focus of increased research into the dry reforming of methane catalysts has been mostly on transition metals (e.g., Ni, Fe, and Co)24,25 and noble metals (e.g., Pd,26 Pt,27,28 Rh,29,30 and Ru31,32) catalysts. Among these, noble metals are widely used in DRM based on the high reactivity, stability and excellent resistance to the deposition of coke. With the same metal size and active component dispersion, the noble metals Ru and Rh exhibited higher activity compared to active components such as Ni, Pd and Pt.33 Zhang et al. modified nickel-based catalysts by introducing a second metal and using a unique support. This provides new ideas for the widespread use of nickel-based catalysts.34–37 However, noble metal-based catalysts are not widely industrialized due to the high cost.
Therefore, attention was gradually focused on transition metals with similar catalytic activities as alternative components. Nickel metal (Ni), usually in the form of nanoparticles, has been widely investigated for the catalysis of DRM because of its high hydrocarbon activation capacity and low cost. For example, Tokunaga et al. investigated the catalytic activity of Ni, Co, and Fe loaded on Al2O3 and found that the catalytic activity of Ni was comparable to that of the noble metal Ru.38,39 However, Ni-based catalysts are vulnerable to catalyst deactivation owing to the deposition of coke and aggregation of Ni particles at high reaction temperature. Currently, two strategies are proposed to solve this problem. On the one hand, the capacity of CO2 adsorption and activation is enhanced by adding alkali metals and defect induction.40,41 On the other hand, the aggregation of Ni particles is inhibited by dispersing Ni on the support with a high specific surface area or tuning the strong interaction between Ni and the support.42,43 It is reported that Al2O3 is widely used as a support for the dry reforming of methane due to its excellent mechanical strength, high thermal stability, tunable surface acidity and alkalinity, and abundant hydroxyl species.44,45 The abundant hydroxyl groups on the Al2O3 surface are favourable for the dispersion of Ni species, but the inactive nickel aluminate phase (NiAl2O4) is readily generated due to high temperature during the preparation of Ni/Al2O3 catalysts and methane dry reforming reaction, which restricts the effective utilization of Ni species and reduces the catalytic activity of methane dry reforming.46,47 Commercial alumina support without any special treatment lacked vacancy defects for the adsorption and activation of CO2.48 Promoting the adsorption and activation of CO2 molecules in the Ni/Al2O3 catalyst for the dry reforming of methane without reducing the utilisation of nickel species is a major challenge in the catalyst design.
Herein, a series of nickel-based alumina catalysts with different Ni loadings and Al2O3 support treated with different concentrations of acetic acid were synthesized. The treatment concentration of acetic acid was modulated to remove some of the hydroxyl groups on the Al2O3 surface, which can improve the utilization of Ni species and the adsorption and activation of CO2 molecules. In this paper, the effect of acetic acid treatment on Ni/Al2O3 catalysts during the DRM reaction is systematically explored by various characterization results. Meanwhile, insights into the acetic acid treatment strategy might be helpful for the design of new catalysts for the DRM reaction.
Experimental
Catalyst preparation
Ni(NO3)2·6H2O (99.0%), commercial γ-Al2O3, and acetic acid (CH3COOH, 99.5%) were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd.
Synthesis of acid-treated Al2O3
2 g of γ-Al2O3 was dispersed in a pre-prepared 0.5/1/2 mol L−1 solution of 80 mL of CH3COOH solution, respectively, and stirred continuously for 2 h at 500 rpm. Then, the mixture was centrifuged and washed at pH 7 several times. All of the obtained catalysts were dried in an oven at 100 °C for 4 h and named as xAc–Al2O3 (x = 0.5, 1 and 2).
Synthesis of 1Ni/xAc–Al2O3 and yNi/Al2O3
1Ni/xAc–Al2O3 and yNi/Al2O3 catalysts were prepared by conventional incipient wetness impregnation method. In a typical procedure, a certain amount of Ni(NO3)2·6H2O was dissolved in deionized water under stirring for 2 h. Then, 2 g Al2O3 or Ac–Al2O3 support was added into the above Ni(NO3)2·6H2O solution. The precipitate was dried at 100 °C for 12 h and then calcined in air from room temperature to 700 °C at a ramp rate of 10 °C per minute for 5 h. The obtained samples were denoted as 1Ni/xAc–Al2O3 and yNi/Al2O3 (x = 0.5, 1, and 2; y = 1, 2, and 5) catalysts, respectively.
Characterization
The detailed methods for X-ray diffraction (XRD), temperature-programmed reduction (H2-TPR), transmission electron microscopy (TEM), Fourier-transform infrared (FT-IR), X-ray photoelectron spectroscopy (XPS), pyridine-dosing FTIR (Py-FTIR), in situ Fourier transform infrared (in situ FTIR), in situ diffuse reflectance Fourier transform infrared spectroscopy (in situ DRIFT), and temperature-programmed desorption of CO2 (CO2-TPD) are given in the ESI.†
Catalytic evaluation
The catalytic activity and stability of the xNi/Ac–Al2O3 and xNi/Al2O3 catalysts in the dry reforming of methane were tested in a fixed-bed flow reactor. 100 mg catalyst (40–60 mesh) was loaded into a quartz reactor and reduced in situ at 750 °C by flowing 10 vol% H2/Ar (30 mL min−1) for 1 h. The reaction gas consisting of CH4, CO2 and N2 (CH4:CO2:N2 = 1:1:1) was fed at a GHSV of 36000 mL h−1 gcat−1. The gas products were analysed by an on-line gas chromatograph (GC).
The CH4/CO2 conversion and H2/CO ratio were calculated as follows.
| |
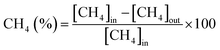 | (5) |
| |
 | (6) |
| |
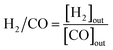 | (7) |
Results and discussion
Catalytic activity measurements
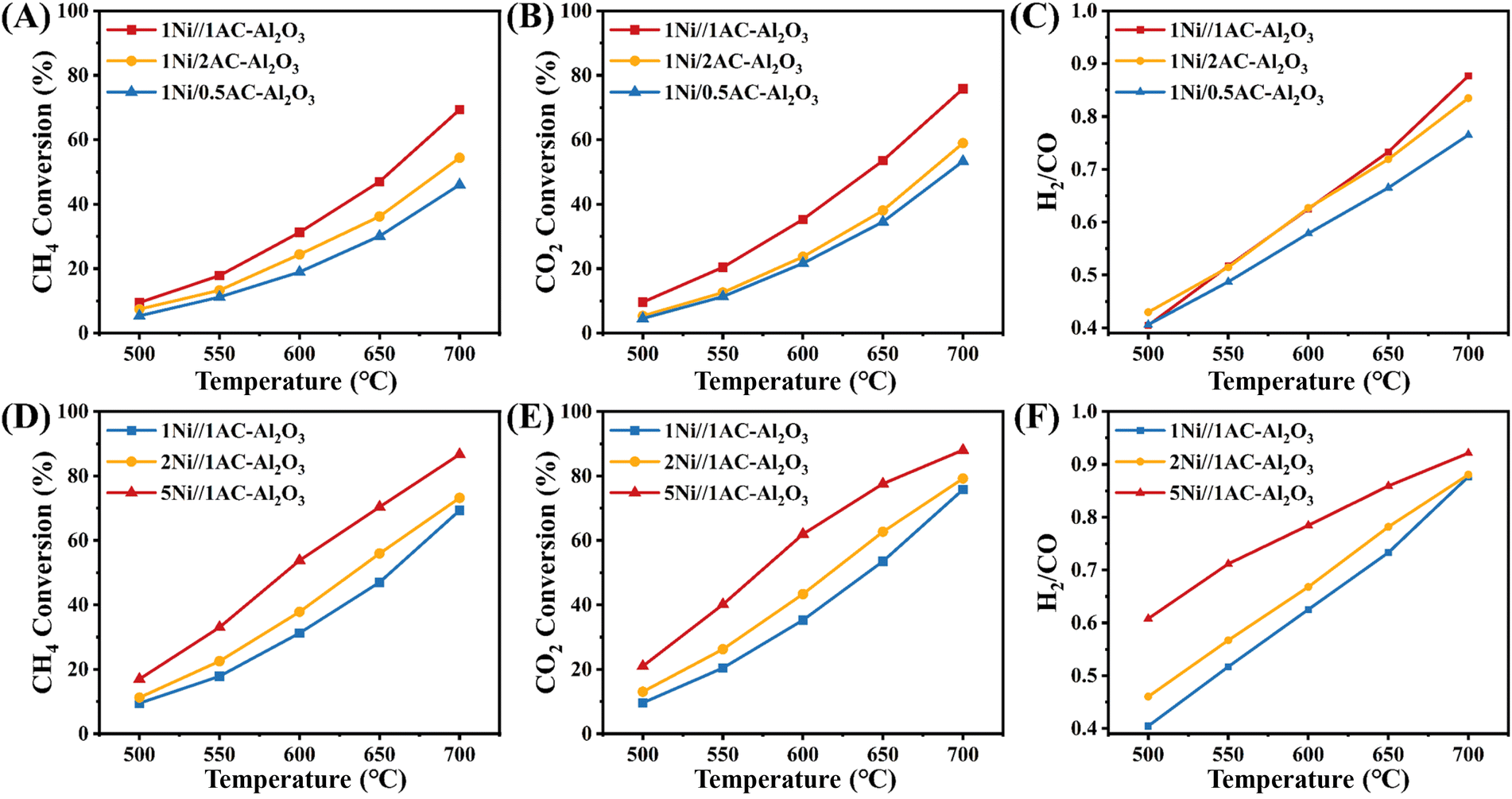
To optimize the concentration for acetic acid treatment and Ni loading, a series of Ni-loaded Al2O3 catalysts was prepared by the impregnation method. Among other things, we carried out warming operations during the continuous feeding process. The resultant activity performance of 1Ni/xAc–Al2O3 (A–C) and yNi/1Ac–Al2O3 (D–F) is shown in Fig. 1. As displayed in Fig. 1A–C, for the 1Ni/xAc–Al2O3 catalysts, three catalysts showed significant differences under treatment with different concentrations of acetic acid. Among them, the 1Ni/1Ac–Al2O3 catalyst exhibited the highest conversion of CH4/CO2, while the 1Ni/0.5Ac–Al2O3 catalyst showed the lowest level of conversion. The H2/CO ratio varies between 0.4 and 0.9 throughout the temperature range and increases with increasing temperature. The trend lines of the 1Ni/1Ac–Al2O3 catalyst and 1Ni/2Ac–Al2O3 catalyst largely overlap, and that of the 1Ni/0.5Ac–Al2O3 catalyst remains lower than that of the other two catalysts. This result identified the optimal acetic acid treatment concentration of 1 mol L−1. Moreover, the catalytic performance of yNi/1Ac–Al2O3 catalysts with different Ni loadings in methane dry reforming reaction was also tested, as shown in Fig. 1D–F. From Fig. 1D–F, it is found that the CH4/CO2 conversion and H2/CO ratios of the three catalysts prepared with different Ni loadings are 5Ni/1Ac–Al2O3 > 2Ni/1Ac–Al2O3 > 1Ni/1Ac–Al2O3 in a descending order. Higher CO2 and CH4 conversions were observed over the 5Ni/1Ac–Al2O3 samples in comparison with those of the xNi/1Ac–Al2O3 samples, indicating that the optimal Ni loading for xNi/1Ac–Al2O3 catalyst is 5 wt%.
 |
| | Fig. 1 CH4 and CO2 conversions and H2/CO2 for the dry reforming of methane with 1Ni/xAc–Al2O3 (A–C) and yNi/1Ac–Al2O3 (D–F) (x = 0.5, 1, and 2; y = 1, 2, and 5). | |
The conversions of the yNi/Al2O3 (y = 1, 2, and 5) catalyst and the long-term DRM reaction results of the 5Ni/1Ac–Al2O3 catalyst are shown in Fig. 2 and S1.† It can be seen that the reaction conversions of the 5Ni/Al2O3 catalyst without acetic acid treatment is lower than that of the 5Ni/1Ac–Al2O3 catalyst. However, the catalysts for acetic acid treatment showed obvious advantages in terms of stability, CH4/CO2 conversion rate and H2/CO ratios (Fig. 2B and S1†). In particular, the 5Ni/1Ac–Al2O3 catalyst showed the best performance among all the catalysts. During the 3000 min stability test, it maintained a high conversion rate of 90% and a high H2/CO ratio of 0.97. This indicates that the 5Ni/1Ac–Al2O3 catalyst is extremely stable for the methane dry reforming reaction. It may be a very desirable and reliable choice in production. A comparison of the catalytic performance results revealed that the catalyst treated with acetic acid treatment could maintain the catalytic activity better than the untreated catalyst in the dry reforming of methane. By analysing these data results, it can be concluded that the acetic acid treatment played a positive role in enhancing the catalyst stability and conversion. Further study and exploration of how acetic acid treatment can improve the catalytic performance of the catalyst in the DRM reaction will be very important to further improve the catalyst effect.
 |
| | Fig. 2 CH4 and CO2 conversions of 5Ni/Al2O3 (A) and stability test of 5Ni/1Ac–Al2O3 (B). Stability test reaction conditions: Treaction = 700 °C, GHSV = 36000 mL h−1 gcat−1, CH4/CO2/N2 = 1/1/1. | |
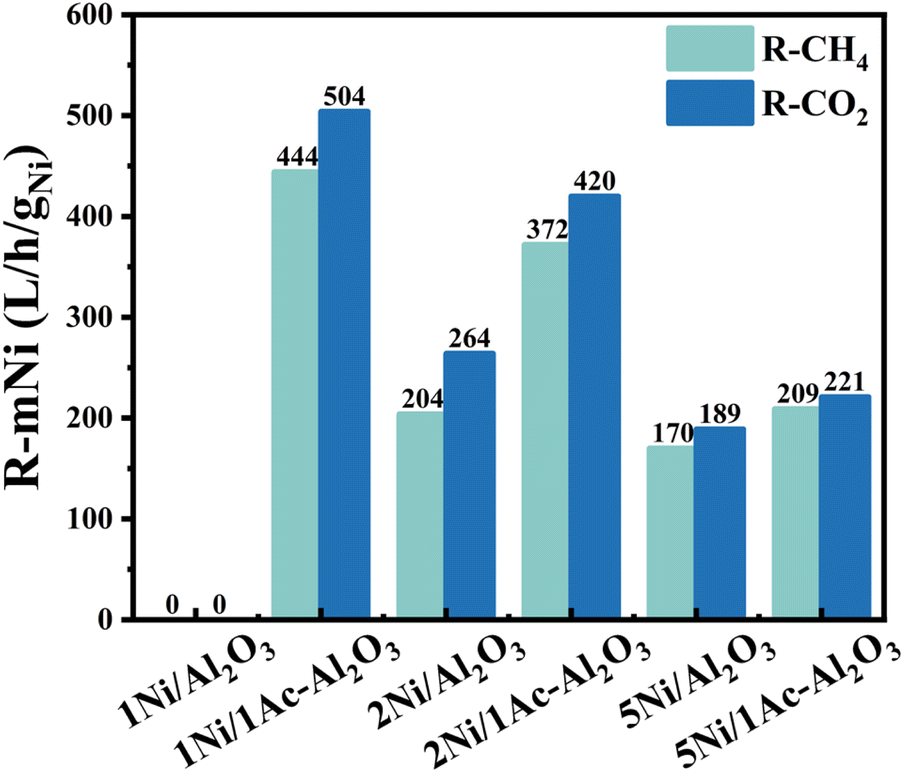
We then compared the reaction rate at 700 °C, and the results are shown in Fig. 3. The rates of conversion of CH4 and CO2 in 1Ni/Al2O3 catalyst are almost zero, which can be attributed to the formation on the catalyst surface of the much inactive NiAl2O4 phase (Fig. S2†). However, the acetic acid-treated catalyst of 1Ni/1Ac–Al2O3 shows high CH4 and CO2 conversion rates and increases from 0 to 444 LCH4 per h per g per Ni and 504 LCO2 per h per g per Ni, respectively. A similar trend is observed in 2Ni/Al2O3, 2Ni/1Ac–Al2O3 and 5Ni/Al2O3, 5Ni/1Ac–Al2O3 catalysts.
 |
| | Fig. 3 The CH4 and CO2 reaction rate during dry reforming of methane over yNi/1Ac–Al2O3 and yNi/Al2O3 (y = 1, 2, and 5) at 700 °C. | |
Apparent activation energy (Ea) experiments were carried out under the conditions of less than 20% conversion. From Fig. 4, the Ea of CH4 (A) and CO2 (B) on 5Ni/1Ac–Al2O3 and 5Ni/Al2O3 catalysts before and after acetic acid treatment was calculated. It can be seen that the Ea of CH4 and CO2 on the 5Ni/Al2O3 catalyst are 57.39 kJ mol−1 and 44.24 kJ mol−1, respectively. However, the Ea values of CH4 and CO2 on the acetic acid-treated catalyst of the 5Ni/1Ac–Al2O3 catalyst are 44.5 kJ mol−1 and 39.6 kJ mol−1, respectively, which are lower than those of the acid-treated 5Ni/Al2O3 catalyst. According to the above results, it can be concluded that the acetic acid-treated catalyst is more effective for both CH4 and CO2 activation, and the specific reasons for promoting the activation are discussed in a later section.
 |
| | Fig. 4 CH4 (A) and CO2 (B) apparent activation energies over 5Ni/Al2O3 and 5Ni/1Ac–Al2O3 catalysts. | |
Effect of acetic acid treatment on the structure of the catalysts
The structure of the unreduced Ac–Al2O3 Ac–Al2O3, Al2O3, yNi/1Ac–Al2O3 and yNi/Al2O3 (y = 1, 2, and 5) samples was determined by XRD and 27Al-NMR characterization. The XRD patterns are displayed in Fig. 5A. As seen, three distinct reflections at 2θ = 38°, 46°, and 66° were detected, which corresponds to the standard for γ-Al2O3 (PDF#10-0425). There were no additional peaks in Fig. 5A after acetic acid treatment, indicating that the structure of these catalysts did not change significantly. Moreover, the diffraction peaks of Ni species were not observed in the XRD results of the yNi/1Ac–Al2O3 and yNi/Al2O3 (y = 1, 2, and 5) catalysts, suggesting that Ni species were well dispersed on the Al2O3 support or formed amorphous phase Ni species.49,50 Additionally, it can be seen from the TEM images in Fig. S3† that the acetic acid treatment has no significant effect on the morphology of Al2O3, which also corresponds to the XRD results before and after the acetic acid treatment in Fig. 5A.
 |
| | Fig. 5 (A) XRD patterns of the yNi/1Ac–Al2O3 and yNi/Al2O3 (y = 1, 2, and 5), (B) 27Al NMR spectra over 1Ac–Al2O3 and Al2O3 samples. | |
The 27Al NMR data are presented in Fig. 5B; it can be observed that there is no significant change in the peak intensity of 27Al NMR before and after acid treatment. This is because 27Al NMR is a holistic detection technique, and changes in the coordination environment of a few aluminum species cannot be detected. Similar 27Al NMR data can only indicate that acid treatment did not damage the overall structure and morphology of the alumina support, which corresponds to the TEM data.
At the same time, the acidic and functional group changes on the catalyst surface after acetic acid treatment were also investigated. As shown in Fig. 6A, the pyridine adsorption peak at 1450 cm−1 is attributed to the Lewis acid sites of xAc–Al2O3 (x = 0.5, 1 and 2) and Al2O3 samples.47 With the increase in the acetic acid concentration, the peak gradually decreased, indicating that the Lewis acid sites of the catalyst are reduced. According to reports, Lewis acid sites can be attributed to hydroxyl species or unsaturated coordination metal species, and nitric acid treatment can remove the hydroxyl group from the surface of Al2O3.47,51
 |
| | Fig. 6 (A) Py-IR spectra and (B) IR of Al2O3, xAc–Al2O3 and Al2O3 (x = 0.5, 1, and 2). | |
The process of hydroxyl removal was further confirmed by IR. The IR spectra of hydroxyl groups before and after acetic acid treatment are compared in Fig. 6B. Three peaks at 3723 cm−1, 3683 cm−1 and 3561 cm−1 were observed, corresponding to the surface terminal hydroxyl, bridged hydroxyl, and tri-bridged hydroxyl groups, respectively.47,52,53 The increase in the acetic acid concentration decreased the intensity of the hydroxyl peaks on the surface of Al2O3, indicating that the surface hydroxyl groups were removed by acetic acid treatment. Additionally, a weak bridged hydroxyl peak was observed on the 2Ac/Al2O3 catalyst surface. This may be attributed to the high concentration of acetic acid treatment, resulting in the exposure of the hydroxyl group of the Al2O3 bulk phase. Therefore, the hydroxyl group on 2Ac/Al2O3 may interact with Ni to form the nickel aluminate phase, which can justify the weaker activity of 1Ni/2Ac–Al2O3 than that of 1Ni/1Ac–Al2O3. According to the results obtained from pyridine-IR and IR analyses, it has been observed that the reduction of Lewis acid sites after acid treatment can be attributed to the elimination of hydroxyl groups. Dehydroxylation can reduce the production of nickel aluminate phases, which helps to form more active sites.
Inhibition of NiAl2O4 phase formation by acetic acid treatment
Fig. 7A compares the H2-TPR profiles of the 5Ni/1Ac–Al2O3 and 5Ni/Al2O3 catalysts. It can be seen that a significantly reduced peak at 600–800 °C is present, which can be attributed to the reduction of nickel species, and the peak appears at 400–500 °C for 5Ni/1Ac–Al2O3 due to the reduction of NiO with intermediate size.54 The reduction peak shifted to lower temperature after acetic acid treatment, indicating that the interaction between metal Ni and the Al2O3 support was weakened, and the Ni species were more easily reduced. Therefore, the 5Ni/1Ac–Al2O3 catalyst has more active phases, resulting in higher catalytic activity, which is consistent with the activity data.
 |
| | Fig. 7 H2-TPR profiles (A) and XPS spectra (B) over the 5Ni/1Ac–Al2O3 and 5Ni/Al2O3 catalysts. | |
Similar results were confirmed by the XPS characterization. The Ni 2p XPS spectra of 5Ni/1Ac–Al2O3 and 5Ni/Al2O3 are illustrated in Fig. 7B. According to literature, the peaks at 852.5 eV, 855.9 eV, and 857.5 eV are attributed to Ni0, Nioct2+ (Ni2+ in octahedral coordination sites), and Nitet2+ (Ni2+ in tetrahedral coordination sites), respectively.55–58 Nioct2+ and Nitet2+ are Ni species with strong metal–support interaction and difficult to reduce. For the 5Ni/Al2O3 sample, the contents of Ni0, Nioct2+, and Nitet2+ are 2.04%, 43.09%, and 16.11%, respectively. For the Ni/1Ac–Al2O3 sample, the contents of Ni0, Nioct2+, and Nitet2+ are 4.11%, 42.59%, and 14.79%, respectively. Although small Ni0 species may be oxidized due to off-line reduction, it is still observed that after acetic acid treatment, the content of Ni species in the strong metal–support interaction decreases and the content of Ni0 increases. The possible reason is that acetic acid treatment decreases the number of hydroxyl groups on the Al2O3 support, which is conducive to the reduction of Ni species and improves the activity of the catalyst in DRM.
Acetic acid treatment promotes the adsorption and activation of CO2
In situ DRIFTS experiments of reactants were carried out to further identify the intermediate species and compare with the activation abilities of CO2 and CH4 on the 5Ni/Ac–Al2O3 (Fig. 8A–C) and 5Ni/Al2O3 (Fig. 8D–F) catalysts. Both catalysts show peaks located at 3016 cm−1 and 1305 cm−1, which are attributed to the gas phase CH4, as well as another broad one at ∼2360 cm−1 corresponding to the deformation vibration of gas phase CO2 is observed.59–61 Notably, the signals attributed to CH4 and CO2 in the 5Ni/1Ac–Al2O3 catalyst weaken at lower temperatures compared to the 5Ni/Al2O3 catalyst, indicating that the activation capacity for the reactants of the 5Ni/1Ac–Al2O3 catalysts is greater. This is probably because the 5Ni/1Ac–Al2O3 samples have more active sites. The signals corresponding to the reaction intermediates appear from 1200 cm−1 to 1800 cm−1 (Fig. 8C and F). Two signals located at 1442 cm−1 and 1342 cm−1 can be attributed to the deformation vibrations of the  species,62,63 and two other bands at 1655 cm−1 and 1227 cm−1 appear, which are attributed to bicarbonate and carbonate, respectively.64,65 The
species,62,63 and two other bands at 1655 cm−1 and 1227 cm−1 appear, which are attributed to bicarbonate and carbonate, respectively.64,65 The  species are produced by the activation of methane, while the oxygen-containing species (bicarbonate) can be produced by the activation of CO2.59,66 Additionally, it can be observed that the intermediate signals on the 5Ni/1Ac–Al2O3 catalyst is stronger than that on the 5Ni/Al2O3 catalyst. This is because the 5Ni/1Ac–Al2O3 catalyst has more active sites after being treated with acetic acid; thus, its ability to activate the reactants is better than that of the 5Ni/Al2O3 catalyst.
species are produced by the activation of methane, while the oxygen-containing species (bicarbonate) can be produced by the activation of CO2.59,66 Additionally, it can be observed that the intermediate signals on the 5Ni/1Ac–Al2O3 catalyst is stronger than that on the 5Ni/Al2O3 catalyst. This is because the 5Ni/1Ac–Al2O3 catalyst has more active sites after being treated with acetic acid; thus, its ability to activate the reactants is better than that of the 5Ni/Al2O3 catalyst.
 |
| | Fig. 8 In situ DRIFTS spectra of 5Ni/1Ac–Al2O3 (A–C) and 5Ni/Al2O3 (D–F) catalysts. | |
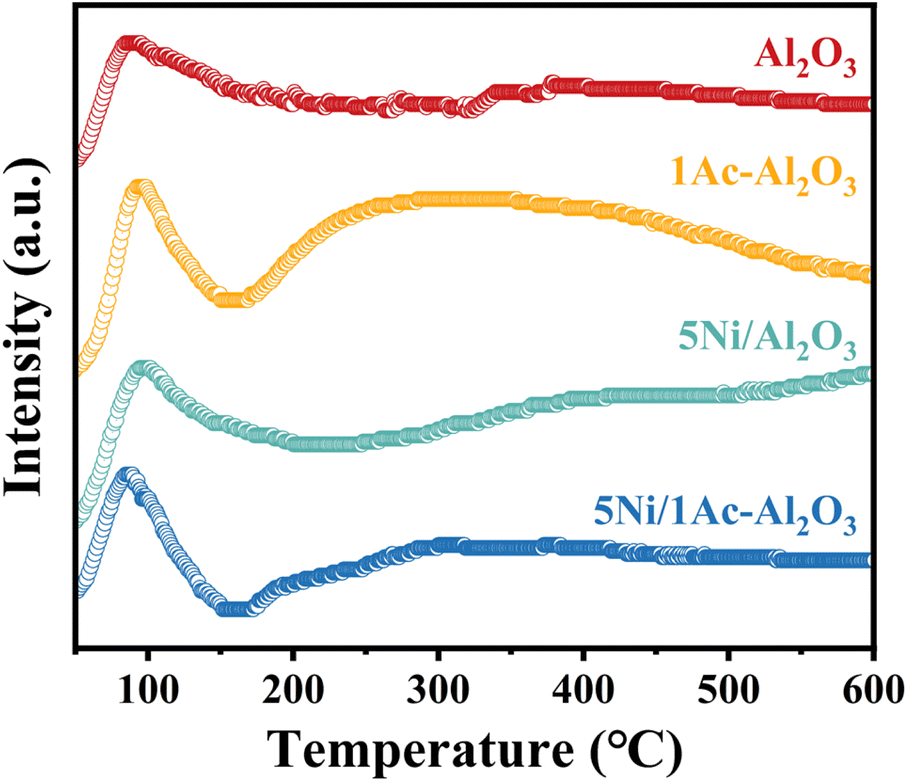
To further prove the CO2 activation capacity over the 5Ni/1Ac–Al2O3 catalyst, CO2-TPD characterization was carried out, as shown in Fig. 9. It can be seen that all the samples show a low temperature adsorption peak at about 100 °C, which could be attributed to the adsorption of CO2 by the supports,67 and the CO2 adsorption signal at 200–500 °C can be attributed to the impregnated Ni species.68 The acetic acid-treated 1Ac/Al2O3, 5Ni/1Ac–Al2O3 exhibits strong CO2 absorption peaks in the 200–500 °C range, whereas no obvious CO2 absorption peaks are observed over the non-acid-treated Al2O3, 5Ni/Al2O3 in the same range. This confirms that the acetic acid-treated catalyst facilitates the adsorption and activation of CO2, corresponding to the in situ DRIFTS results.
 |
| | Fig. 9 CO2-TPD patterns over Al2O3, 1Ac–Al2O3, 5Ni/Al2O3 and 5Ni/1Ac–Al2O3. | |
Conclusions
In summary, a strategy to directly remove hydroxyl groups from the surface of alumina support has been developed. The hydroxyl species can be removed by simple acetic acid treatment, inhibiting the formation of NiAl2O4 and significantly improving the utilization efficiency of the nickel species, thereby enhancing the catalytic activity. Additionally, the catalyst after acetic acid treatment promoted the activation of CO2. This study provides a feasible approach for nickel-based alumina catalysts to undergo the methane dry-reforming methane reaction through simple acetic acid treatment.
Data availability
Data will be made available on request to the corresponding author.
Author contributions
Han Xiao: methodology, writing – original draft. Jiaming Dong: writing – original draft. Yimin Zhang: conceptualization. Xiaohua Cao: writing-review & editing. Yanhong Li: formal analysis. Dedong He: conceptualization, investigation. Yongming Luo: methodology, investigation. Pingyan Wang: supervision, investigation. Hao Wang: supervision, writing-review & editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge the financial support from the National Natural Science Foundation of China (42030712, 22306081), Excellent Youth Project of Natural Science Foundation of Yunnan Province (202201AW070007), Young Academic and Technical Leader Raising Foundation of Yunnan Province (202205AC160011). Thanks for the support by Key Laboratory of Yunnan Province for synthesizing sulfur. Containing Fine Chemicals/The Innovation Team for Volatile Organic Compounds Pollutants Control and Resource Utilization of Yunnan Province/The Higher Educational Key Laboratory for Odorous Volatile Organic Compounds Pollutants Control of Yunnan Province, Kunming 650500, P. R. China.
Notes and references
- A. Kurlov, E. B. Deeva, P. M. Abdala, D. Lebedev, A. Tsoukalou, A. Comas-Vives, A. Fedorov and C. R. Müller, Nat. Commun., 2020, 11, 4920 CrossRef CAS.
- H. K. Min, S. Kweon, Y. W. Kim, H. An, D. Jo, E. D. Park, C. H. Shin and M. B. Park, Appl. Catal., B, 2021, 298, 120627 CrossRef CAS.
- Y. Song, E. Ozdemir, S. Ramesh, A. Adishev, S. Subramanian, A. Harale, M. Albuali, B. A. Fadhel, A. Jamal, D. Moon, S. H. Choi and C. T. Yavuz, Science, 2020, 367, 777–781 CrossRef CAS.
- H. Qu, H. Yang, L. B. Han, S. H. He, J. D. Liu, R. J. Hu, H. Q. Su and Y. Su, Chem. Eng. J., 2023, 453, 139694 CrossRef CAS.
- K. H. Han, S. Wang, N. Hu, W. D. Shi and F. G. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 23487–23495 CrossRef CAS PubMed.
- L. Wang and F. G. Wang, Energy Fuels, 2022, 36, 5594–5621 CrossRef CAS.
- Y. Tang, Y. C. Wei, Z. Y. Wang, S. R. Zhang, Y. T. Li, L. Nguyen, Y. X. Li, Y. Zhou, W. J. Shen, F. F. Tao and P. J. Hu, J. Am. Chem. Soc., 2019, 141, 7283–7293 CrossRef CAS PubMed.
- A. S. Al-Fatesh, R. Kumar, S. O. Kasim, A. A. Ibrahim, A. H. Fakeeha, A. E. Abasaeed, R. Alrasheedu, A. Bagabas, M. L. Chaudhary, F. Frusteri and B. Chowdhury, Catal. Today, 2020, 348, 236–242 CrossRef CAS.
- S. Dama, S. R. Ghodke, R. Bobade, H. R. Gurav and S. Chilukuri, Appl. Catal., B, 2018, 224, 146–158 CrossRef CAS.
- A. T. Ashcroft, A. K. Cheetham, M. L. H. Green and P. D. F. Vernon, Nature, 1991, 352, 225–226 CAS.
- S. T. Oyama, P. Hacarlioglu, Y. F. Gu and D. Lee, Int. J. Hydrogen Energy, 2012, 37, 10444–10450 CrossRef CAS.
- X. Y. Gao, J. Ashok, S. Widjaja, K. Hidajat and S. Kawi, Appl. Catal., A, 2015, 503, 34–42 CrossRef CAS.
- W. H. Chen, C. L. Hsu and S. W. Du, Energy, 2015, 86, 758–771 CrossRef CAS.
- Q. Yi, G. S. Wu, M. H. Gong, Y. Huang, J. Feng, Y. H. Hao and W. Y. Li, Appl. Energy, 2017, 193, 149–161 CrossRef CAS.
- X. Y. Gao, Z. T. Lin, T. T. Li, L. T. Huang, J. M. Zhang, S. Askari, N. Dewangan, A. Jangam and S. Kawi, Catalysts, 2021, 11, 455 CrossRef CAS.
- L. C. Buelens, V. V. Galvita, H. Poelman, C. Detavernier and G. B. Marin, Science, 2016, 354, 449–452 CrossRef CAS PubMed.
- I. V. Yentekakis, P. Panagiotopoulou and G. Artemakis, Appl. Catal., B, 2021, 296, 120210 CrossRef CAS.
- K. K. Bu, J. Deng, X. Y. Zhang, S. Kuboon, T. T. Yan, H. R. Li, L. Y. Shi and D. S. Zhang, Appl. Catal., B, 2020, 267, 118692 CrossRef CAS.
- J. Martin-del-Campo, M. Uceda, S. Coulombe and J. Kopyscinski, J. CO2 Util., 2021, 46, 101474 CrossRef CAS.
- B. Yang, J. Deng, H. R. Li, T. T. Yan, J. P. Zhang and D. S. Zhang, Iscience, 2021, 24, 102747 CrossRef CAS.
- Y. Wang, L. Yao, S. H. Wang, D. H. Mao and C. W. Hu, Fuel Process. Technol., 2018, 169, 199–206 CrossRef CAS.
- G. J. Zhang, J. W. Liu, Y. Xu and Y. H. Sun, Int. J. Hydrogen Energy, 2018, 43, 15030–15054 CrossRef CAS.
- M. M. Barroso-Quiroga and A. E. Castro-Luna, Int. J. Hydrogen Energy, 2010, 35, 6052–6056 CrossRef CAS.
- A. M. O'Connor, Y. Schuurman, J. R. H. Ross and C. Mirodatos, Catal. Today, 2006, 115, 191–198 CrossRef.
- S. Therdthianwong, A. Therdthianwong, C. SiangChin and S. Yonprapat, Int. J. Hydrogen Energy, 2008, 33, 991–999 CrossRef CAS.
- S. Damyanova, B. Pawelec, K. Arishtirova, J. L. G. Fierro, C. Sener and T. Dogu, Appl. Catal., B, 2009, 92, 250–261 CrossRef CAS.
- P. G. Schulz, M. G. Gonzalez, C. E. Quincoces and C. E. Gigola, Ind. Eng. Chem. Res., 2005, 44, 9020–9029 CrossRef CAS.
- H. Y. Wang and E. Ruckenstein, Appl. Catal., A, 2000, 204, 143–152 CrossRef CAS.
- R. Wang, H. Y. Xu, X. B. Liu, Q. J. Ge and W. Z. Li, Appl. Catal., A, 2006, 305, 204–210 CrossRef CAS.
- C. Carrara, J. Múnera, E. A. Lombardo and L. M. Cornaglia, Top. Catal., 2008, 51, 98–106 CrossRef CAS.
- J. X. Chen, C. C. Yao, Y. Q. Zhao and P. H. Jia, Int. J. Hydrogen Energy, 2010, 35, 1630–1642 CrossRef CAS.
- D. L. Li, R. L. Li, M. M. Lu, X. Y. Lin, Y. Y. Zhan and L. L. Jiang, Appl. Catal., B, 2017, 200, 566–577 CrossRef CAS.
- D. Pakhare, C. Shaw, D. Haynes, D. Shekhawat and J. Spivey, J. CO2 Util., 2013, 1, 37–42 CrossRef CAS.
- J. J. Zheng, S. Impeng, J. Liu, J. Deng and D. S. Zhang, Appl. Catal., B, 2024, 342, 123369 CrossRef CAS.
- X. Y. Zhang, J. Deng, T. W. Lan, Y. J. Shen, Q. D. Zhong, W. Ren and D. S. Zhang, ACS Catal., 2022, 12, 14152–14161 CrossRef CAS.
- X. Y. Zhang, J. Deng, M. Pupucevski, S. Impeng, B. Yang, G. R. Chen, S. Kuboon, Q. D. Zhong, K. Faungnawakij, L. R. Zheng, G. Wu and D. S. Zhang, ACS Catal., 2021, 11, 12087–12095 CrossRef CAS.
- J. Deng, M. Gao, J.-y. Hasegawa, X. Zhang, A. Wang, A. Chen and D. Zhang, CCS Chem., 2023, 5, 2111–2124 CrossRef CAS.
- O. Tokunaga, Y. Osada and S. Ogasawara, Fuel, 1989, 68, 990–994 CrossRef CAS.
- O. Tokunaga and S. Ogasawara, React. Kinet. Catal. Lett., 1989, 39, 69–74 CrossRef CAS.
- C. Crisafulli, S. Scirè, S. Minicò and L. Solarino, Appl. Catal., A, 2002, 225, 1–9 CrossRef CAS.
- N. Oyama, Y. Iwami, T. Yamamoto, S. Machida, T. Higuchi, H. Sato, M. Sato, K. Takeda, Y. Watanabe, M. Shimizu and K. Nishioka, ISIJ Int., 2011, 51, 913–921 CrossRef CAS.
- T. Huang, W. Huang, J. Huang and P. Ji, Fuel Process. Technol., 2011, 92, 1868–1875 CrossRef CAS.
- E. García-Bordejé, A. B. Dongil, J. M. Conesa, A. Guerrero-Ruiz and I. Rodríguez-Ramos, Chem. Eng. J., 2023, 472, 144953 CrossRef.
- Z. A. Xie, Z. Li, P. Tang, Y. Y. Song, Z. Zhao, L. A. Kong, X. Q. Fan and X. Xiao, J. Catal., 2021, 397, 172–182 Search PubMed.
- T. Margossian, K. Larmier, S. M. Kim, F. Krumeich, A. Fedorov, P. Chen, C. R. Müller and C. Copéret, J. Am. Chem. Soc., 2017, 139, 6919–6927 Search PubMed.
- S. S. Zhang, M. Ying, J. Yu, W. C. Zhan, L. Wang, Y. Guo and Y. L. Guo, Appl. Catal., B, 2021, 291, 120074 CAS.
- Y. M. Zhang, Y. Zu, D. D. He, J. Liang, L. H. Zhu, Y. Mei and Y. M. Luo, Appl. Catal., B, 2022, 315, 121539 CAS.
- K. Li, C. L. Pei, X. Y. Li, S. Chen, X. H. Zhang, R. Liu and J. L. Gong, Appl. Catal., B, 2020, 264, 118448 Search PubMed.
- Z. J. Huang, D. D. He, W. H. Deng, G. W. Jin, K. Li and Y. M. Luo, Nat. Commun., 2023, 14, 100 Search PubMed.
- L. Zhang, S. J. Wu, Y. Zhang, T. H. Ai, D. K. Chen, Y. M. Luo and D. D. He, Chem. Eng. J., 2024, 486, 150337 CrossRef CAS.
- K. Hadjiivanov, in Advances in Catalysis, ed. F. C. Jentoft, Academic Press, 2014, vol. 57, pp. 99–318 Search PubMed.
- T. Cao, R. You, X. Y. Zhang, S. L. Chen, D. Li, Z. H. Zhang and W. X. Huang, Phys. Chem. Chem. Phys., 2018, 20, 9659–9670 RSC.
- J. J. Dai and H. B. Zhang, J. Catal., 2022, 410, 266–279 CrossRef CAS.
- Y. Bai, D. Y. Shen, G. W. Yu, J. Wang, S. A. Lyu, Y. H. Zhang, G. H. Wang, J. L. Li and L. Li, New J. Chem., 2023, 47, 17186–17193 RSC.
- S. Q. Li, Y. Fu, W. B. Kong, B. R. Pan, C. K. Yuan, F. F. Cai, H. Zhu, J. Zhang and Y. H. Sun, Appl. Catal., B, 2020, 277, 118921 CrossRef CAS.
- P. Yan, S. Xi, H. Peng, D. R. G. Mitchell, L. Harvey, M. Drewery, E. M. Kennedy, Z. Zhu, G. Sankar and M. Stockenhuber, J. Am. Chem. Soc., 2023, 145, 9718–9728 CrossRef CAS PubMed.
- P. H. Yan, X. X. Tian, E. M. Kennedy and M. Stockenhuber, Catal. Sci. Technol., 2022, 12, 2184–2196 RSC.
- Y. Zhang, R. Zeng, Y. Zu, L. Zhu, Y. Mei, Y. Luo and D. He, Chin. J. Chem. Eng., 2022, 48, 76–90 CrossRef CAS.
- H. Wang, G. Q. Cui, H. Lu, Z. Y. Li, L. Wang, H. Meng, J. Li, H. Yan, Y. S. Yang and M. Wei, Nat. Commun., 2024, 15, 3765 CrossRef CAS.
- P. Yan, H. Peng, X. Zhang, H. Rabiee, M. Ahmed, Y. Weng, A. Rozhkovskaya, J. Vogrin, M. Konarova and Z. Zhu, Fuel, 2024, 374, 132373 CrossRef CAS.
- D. He, J. Yan, K. Chen, L. Zhang, J. Lu, J. Liu and Y. Luo, ACS Catal., 2023, 13, 12114–12124 CrossRef CAS.
- Y. Wang, L. Li, G. Y. Li, Q. Zhao, X. S. Wu, Y. N. Wang, Y. F. Sun and C. W. Hu, ACS Catal., 2023, 13, 6486–6496 CrossRef CAS.
- G. X. Deng, G. F. Zhang, X. Zhu, Q. J. Guo, X. B. Liao, X. Chen and K. Z. Li, Appl. Catal., B, 2021, 289, 120033 CrossRef CAS.
- Q. S. Pan, J. X. Peng, S. Wang and S. D. Wang, Catal. Sci. Technol., 2014, 4, 502–509 RSC.
- L. Azancot, L. F. Bobadilla, M. A. Centeno and J. A. Odriozola, Appl. Catal., B, 2021, 285, 119822 CrossRef CAS.
- D. D. He, S. J. Wu, X. H. Cao, D. K. Chen, L. Zhang, Y. Zhang and Y. M. Luo, Applied Catalysis B: Environment and Energy, 2024, 346, 123728 CrossRef CAS.
- P. Yan, H. Peng, X. Wu, H. Rabiee, Y. Weng, M. Konarova, J. Vogrin, A. Rozhkovskaya and Z. Zhu, J. Catal., 2024, 432, 115439 CrossRef CAS.
- C. Sun, P. Beaunier and P. Da Costa, Catal. Sci. Technol., 2020, 10, 6330–6341 RSC.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Jiaming Dongab,
Yimin Zhanga,
Xiaohua Caoab,
Yanhong Li
ab,
Jiaming Dongab,
Yimin Zhanga,
Xiaohua Caoab,
Yanhong Li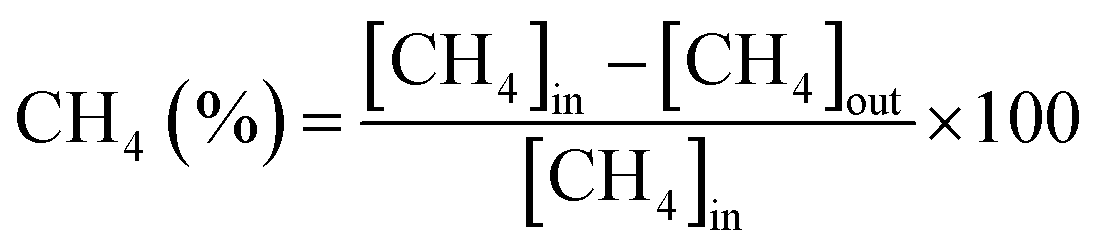

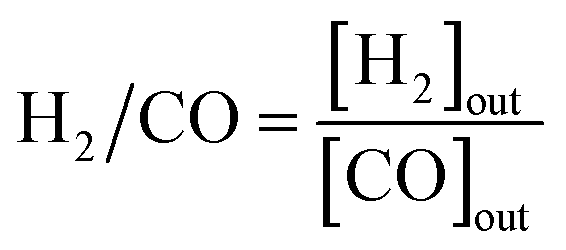





 species,62,63 and two other bands at 1655 cm−1 and 1227 cm−1 appear, which are attributed to bicarbonate and carbonate, respectively.64,65 The
species,62,63 and two other bands at 1655 cm−1 and 1227 cm−1 appear, which are attributed to bicarbonate and carbonate, respectively.64,65 The  species are produced by the activation of methane, while the oxygen-containing species (bicarbonate) can be produced by the activation of CO2.59,66 Additionally, it can be observed that the intermediate signals on the 5Ni/1Ac–Al2O3 catalyst is stronger than that on the 5Ni/Al2O3 catalyst. This is because the 5Ni/1Ac–Al2O3 catalyst has more active sites after being treated with acetic acid; thus, its ability to activate the reactants is better than that of the 5Ni/Al2O3 catalyst.
species are produced by the activation of methane, while the oxygen-containing species (bicarbonate) can be produced by the activation of CO2.59,66 Additionally, it can be observed that the intermediate signals on the 5Ni/1Ac–Al2O3 catalyst is stronger than that on the 5Ni/Al2O3 catalyst. This is because the 5Ni/1Ac–Al2O3 catalyst has more active sites after being treated with acetic acid; thus, its ability to activate the reactants is better than that of the 5Ni/Al2O3 catalyst.