
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBent naphthodithiophenes: synthesis and characterization of isomeric fluorophores†
Emmanuel B. A. Adusei a,
Vincent T. Casettia,
Calvin D. Goldsmitha,
Madison Caswella,
Drecila Alinja,
Jimin Parka,
Matthias Zellerb,
Alexander A. Rusakova and
Zacharias J. Kinney*a
a,
Vincent T. Casettia,
Calvin D. Goldsmitha,
Madison Caswella,
Drecila Alinja,
Jimin Parka,
Matthias Zellerb,
Alexander A. Rusakova and
Zacharias J. Kinney*a
aDepartment of Chemistry, Oakland University, Rochester, Michigan, USA. E-mail: kinney@oakland.edu; Tel: +1-248-370-2347
bDepartment of Chemistry, Purdue University, West Lafayette, Indiana, USA
First published on 12th August 2024
Abstract
Thiophene-containing heteroarenes are one of the most well-known classes of π-conjugated building blocks for photoactive molecules. Isomeric naphthodithiophenes (NDTs) are at the forefront of this research area due to their straightforward synthesis and derivatization. Notably, NDT geometries that are bent – such as naphtho[2,1-b:3,4-b′]dithiophene (α-NDT) and naphtho[1,2-b:4,3-b′]dithiophene (β-NDT) – are seldom employed as photoactive small molecules. This report investigates how remote substituents impact the photophysical properties of isomeric α- and β-NDTs. The orientation of the thiophene units plays a critical role in the emission: in the α(OHex)R2 series conjugation from the end-caps to the NDT core is apparent, while in the β(Oi-Pent)R2 series minimal change is observed unless strong electron acceptors, such as β(Oi-Pent)(PhCF3)2, are employed. This push–pull acceptor–donor–acceptor (A–D–A) fluorophore exhibits positive fluorosolvatochromism that correlates with increasing solvent polarity parameter, ET(30). In total, these results highlight how remote substituents are able to modulate the emission of isomeric bent NDTs.
1 Introduction
Heteroarenes, polyaromatic hydrocarbons (PAHs) bearing main group elements, are well established as building blocks for π-conjugated functional materials.1–4 Thiophene-containing heteroarenes with benzene, naphthalene, or anthracene cores have been thoroughly investigated as electron rich moieties within both small molecule and polymer systems with applications in organic electronics.1,5,6 Of the thiophene-based heteroarenes, naphthodithiophenes (NDTs) are of particular interest due to the plethora of isomers available and their straightforward derivatization.7,8 Linear and angular NDTs shown in Fig. 1a represent two classes of isomeric NDTs that have found applications throughout organic electronics, most notably as organic field-effect transistors (OFETs),9–11 organic light-emitting diodes (OLEDs),12 organic13,14 and dye-sensitized15 solar cells, and more recently as hole-transporting materials (HTMs).12,16,17 Due to the orientation of the residual α-positions (2- and 7-positions, labelled in Fig. 1a), NDT-1 and NDT-2 are considered to be linear rods, while NDT-3 and NDT-4 are described as angular.7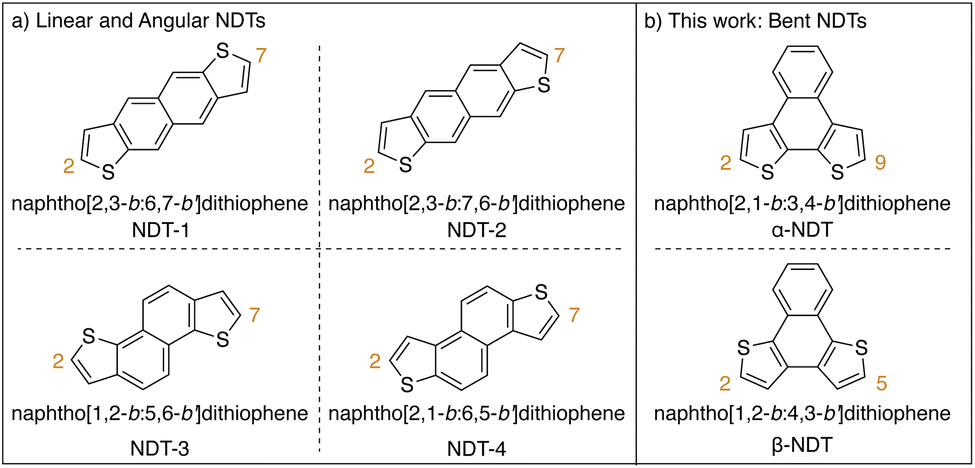 | ||
| Fig. 1 Classes of naphthodithiophenes. (a) Isomeric linear and angular naphthodithiophenes. (b) This work: isomeric bent naphthodithiophenes. | ||
Alternatively bent NDTs – such as naphtho[2,1-b:3,4-b′]dithiophene (α-NDT) and naphtho[1,2-b:4,3-b′]dithiophene (β-NDT) shown in Fig. 1b – are known building blocks in polymeric materials,18,19 but are underrepresented as luminescent small molecules.16,20 At their core, unfunctionalized α- and β-NDTs are isoelectronic to triphenylene, but can be easily fused to other motifs to yield electron deficient cores (e.g. phenazines21 or imides19,22). The α-positions of α- and β-NDTs, labelled in Fig. 1b, offer a facile means to modulate the emissive properties of the core via peripheral derivatization (i.e. V-shaped23 acceptor–donor–acceptor systems). These bent push–pull fluorophores24 are reminiscent of Höger and co-workers isomeric dithienylphenazines (termed α- and β-DTPs), whose systems have inverted electronics (i.e. the electronics are aligned to be donor–acceptor–donor systems). Their series of α- and β-DTPs were shown to have variable conjugation paths based on the isomer employed, ultimately yielding photoactive molecules that are fluorescent (α-DTP) or dual emitters (β-DTP).25,26 With this in mind, probing how the peripheral functionalization of α- and β-NDTs impacts the photoactive cores – and ultimately their application as push–pull fluorophores – is of interest.
Herein we investigate the structure–property relationships in a series of isomeric bent NDTs – naphtho[2,1-b:3,4-b′]dithiophene (α-NDT) and naphtho[1,2-b:4,3-b′]dithiophene (β-NDT) – with variable end-caps. The initial premise of this research effort was to establish if the peripheral R-groups are (1) in communication with the NDT core and (2) if there are distinct differences between the isomers. NMR spectroscopy can afford preliminary evidence of communication to the core in the ground state via modulation of interior resonances far removed from the R-groups; while emission spectra are informative of excited state communication. Ultimately, there are clear differences in the photophysical properties between the isomers, with α-NDT being more receptive to all types of peripheral substituents and β-NDT only being responsive to electron-deficient end-caps.
2 Results and discussion
2.1 α(OR′)R2 series: synthesis and structural analysis
With the goal of having both solution- and solid-state metrics for the α(OR′)R2 series two monomers with differing solubility are needed. For the solution-state analysis, hexyloxy groups were used to ensure that all α-NDT derivatives are soluble across a range of concentrations, particularly with aggregation being a concern for similar PAH systems.27 To investigate solid-state metrics methoxy groups were used to maximize the chance of growing single crystals for X-ray diffraction. Both α(OHex)R2 and α(OMe)R2 can be synthesized via a streamlined bottom-up approach. Scheme 1 details the synthetic path to the α(OHex)R2 series beginning with the alkylation of 4,5-dibromocatechol28 with iodohexane to yield solubilized building block 1(OHex). Installation of the thiophene units is accomplished via palladium-mediated cross-coupling to afford 2(OHex) in modest yield. Planarization of 2(OHex) under typical Scholl oxidation conditions29 affords α(OHex) as an off-white crystalline solid after column purification. Bromination of the open α-positions with NBS yields α(OHex)Br2, a versatile core molecule for attaching end-caps with variable electronic properties. Commercially available phenyl boronic acid (R = Ph, H σp = 0.00) and para-functionalized phenyl boronic acid derivatives p-tolyl boronic acid (R = Tol, Me σp = −0.17), p-methoxyphenyl boronic acid (R = PhOMe, OMe σp = −0.27), and p-(trifluoromethyl)phenyl boronic acid (R = PhCF3, CF3 σp = +0.54) were chosen as end-caps due to their variable electronic properties that are poised to be in resonance with the NDT cores. Due to this arrangement for the R-groups Hammett parameters30 for σp, which include resonance effects, are more informative than electronically isolated σm. Suzuki cross-coupling of the boronic acids proved to be efficient across all derivatives, with modest isolated yields for the α(OHex)R2 series (Scheme 1, inset).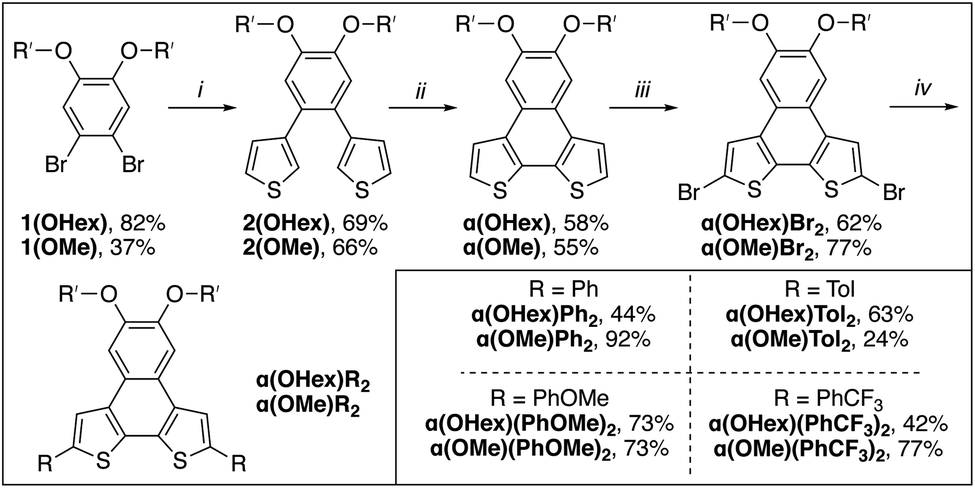 | ||
| Scheme 1 Synthesis of α(OR′)R2 series. (i) 3-thiophene boronic acid, Pd(PPh3)2Cl2, 2 M K2CO3, THF, 80 °C, overnight; (ii) FeCl3, MeNO2, DCM, 0 °C to RT, 1 h; (iii) NBS, CHCl3, RT, 2 d; (iv) R boronic acid, Pd(PPh3)2Cl2, 2 M K2CO3, THF, 80 °C, overnight. (Inset): Isolated yields for α(OHex)R2 and α(OMe)R2 series. | ||
The α(OMe)R2 series follows an identical synthetic path as α(OHex)R2 (shown in Scheme 1), with the exception of starting from 4,5-dibromoveratrole.31 The solubility of the α(OMe)R2 series is demonstrably worse than the hexyloxy series, which proved challenging for purification and analysis (see ESI†). Due to this poor solubility the α(OMe)R2 series was used almost explicitly for crystallography. Single crystals of sufficient quality for X-ray diffraction were grown for each derivative via vapor diffusion of hexanes into saturated solutions of α(OMe)R2 in THF (see ESI†). Of particular interest are the inter-ring bond lengths, which can detail the level of communication the remote substituents have with the α-NDT core, and the supramolecular organization of the α(OMe)R2 series. The C–C linkage between the α-NDT core and the appended R-groups are expected to display minor elongation due to torsional strain between the ring systems (C–C ≈ 1.45 Å),32 with measurements under this value being indicative of increased conjugation with the α-NDT core. Each α(OMe)R2 derivative has an inter-ring bond length of ≈1.46 Å, revealing negligible bond elongation with no dependence on the R-group. The herringbone arrays formed in the crystallographic packing of the α(OMe)R2 series reveal that the planar cores of the α-NDTs are arranged into slipped-stacks with arene–arene distances of ≈3.5 Å (see ESI† for full details).
With these solid-state metrics established, we turned our attention to the solubilized α(OHex)R2 derivatives for solution-state structural analysis. Specifically of interest are resonances far removed from the R-groups that are part of the NDT core, as these signals are expected to shift if there is communication from the end-caps to the core. The NDT cores themselves are electron-rich, therefore R-groups that are electron withdrawing are expected to function as intramolecular acceptor–donor–acceptor (A–D–A) systems, while electron-rich groups are expected to yield highly polarized all donor systems. Intramolecular A–D–A species are posited to increase the contribution of the quinoidal resonance, a highly desirable characteristic in photoactive molecules.23 Key proton resonances 2b and 3b are highlighted in Fig. 2 and are matched between each derivative of the α(OHex)R2 series to ease comparison.
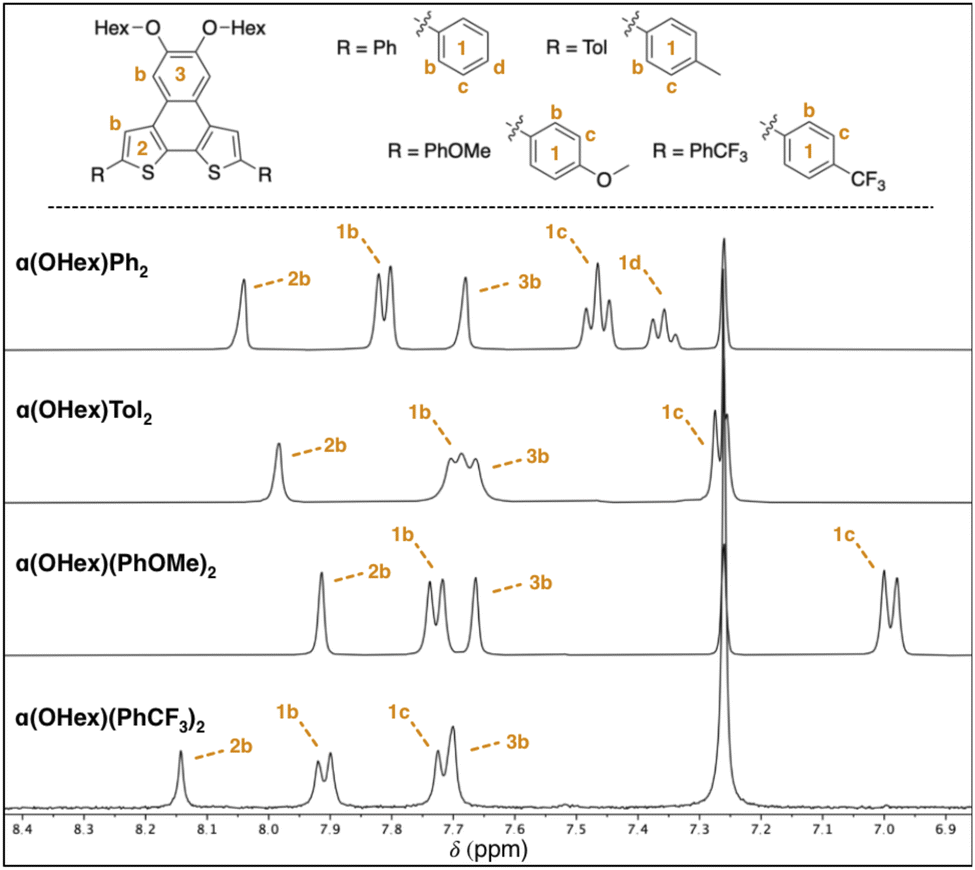 | ||
| Fig. 2 Partial 1H NMR spectra for the α(OHex)R2 series with assignment of aromatic resonances (400 MHz, CDCl3). See ESI† for full spectra and assignments. | ||
Due to the planar nature of the NDT cores aggregation is likely, thus comparison of NMR spectra at similar dilute concentrations is required. Inspection of the aromatic region of dilute (≤10 mM) solutions of each α(OHex)R2 species reveals slight perturbations of the interior 2b proton (Δδ ≈ 0.20 ppm, Fig. 2) and carbon (Δδ ≈ 3 ppm, see ESI†) resonances. Modulation of the 2b resonances is quite unique: these signals are far removed from the functional group and indicate that there is remote communication to the NDT core upon end-capping. Relative to parent α(OHex)Ph2 the electron donating groups shift the 2b resonance upfield with α(OHex)Tol2 (Δδ −0.06 ppm) and α(OHex)(PhOMe)2 (Δδ −0.13 ppm) both being above the threshold of significance (Δδ ≥ 0.05 ppm). α(OHex)(PhCF3)2 is shifted downfield in comparison (Δδ +0.10 ppm), indicative of a shift towards a higher energy state (with respect to parent α(OHex)Ph2). Overall, the shift differences observed for the 2b proton resonances are quite small (Δδ < 0.25 ppm); thus, inspection of the 13C signals for the 2b resonances could be informative. Indeed, the 13C resonances follow the same trend, wherein α(OHex)Tol2 (Δδ −0.6 ppm) and α(OHex)(PhOMe)2 (Δδ −1.1 ppm) are shifted upfield relative to α(OHex)Ph2 and α(OHex)(PhCF3)2 is shifted downfield (Δδ +1.6 ppm). This trend fits well with σp for the R-groups for both the observed 1H and 13C resonances of 2b, while σm yields poor fits (see ESI†). To ensure these observations are not artefacts of aggregation, a series of variable concentration solutions were probed for the α(OHex)R2 series (see ESI,† 50–1 mM in CDCl3). These data for α(OHex)Tol2 and α(OHex)(PhOMe)2 suggest that there is minimal propensity for aggregation in CDCl3 (Δδ ≤ 0.12 ppm for all resonances), with measured Kd of ≈3.2 × 10−4 M and ≈1.2 M for these two derivatives respectively.33,34
To verify the attribution of the small perturbations observed via NMR to the remote R-groups, we modelled the NMR spectra of the α(OHex)R2 series computationally. To simplify simulations and clarify the assignment of NMR features, we replaced the hexyloxy groups with methoxy. To differentiate from the synthesized α(OMe)R2 series the model series is represented as α(OMe)R2′. In our 1H and 13C calculated NMR spectra, we rely on the widely adopted non-relativistic density functional theory (DFT) protocol with gauge including atomic orbitals (GIAO)35,36 as implemented in the Gaussian 16 (ref. 37) programme suite. The justification of the non-relativistic approach comes from the smallness of the heavy atom effect in compounds containing sulfur and lighter elements only.38–42 In all DFT calculations, we use a popular global hybrid exchange–correlation functional (XCF) approximation B3LYP43,44 widely regarded in the literature as a reliable choice45–60 even in light of recent advances.47,51,53,61–64 Finally, we choose to work with polarization-consistent basis sets developed by Jensen for rational and systematic reduction of basis-set error specifically in DFT methods.65–67 In particular, we use pc-2 (ref. 68) for geometry optimizations and vibrational frequency calculations, and aug-pc-Sseg-2 (ref. 69) for modelling NMR spectra. The latter set belongs to the pc-Sseg-n family of basis sets augmented with additional tight exponents for the description of the electronic density near atomic nuclei and optimised for nuclear magnetic shielding. These sets have been known to markedly outperform other available choices for core-related properties.70,71 Due to the poor solubility of α(OHex)(PhCF3)2 – and the entire α(OMe)R2 series – only experimental resonances extracted from the 1H–13C HSQC spectra were used for comparison to calculated parameters. Comparison of the 1H and 13C isotropic shieldings for the model α(OMe)R2′ series to the synthesized α(OHex)R2 (shown in Fig. 3) and α(OMe)R2 (see ESI†) series yield quality fits (r2 > 0.9), further indicating that the modulation of the interior chemical shifts is directly related to the remote R-groups.
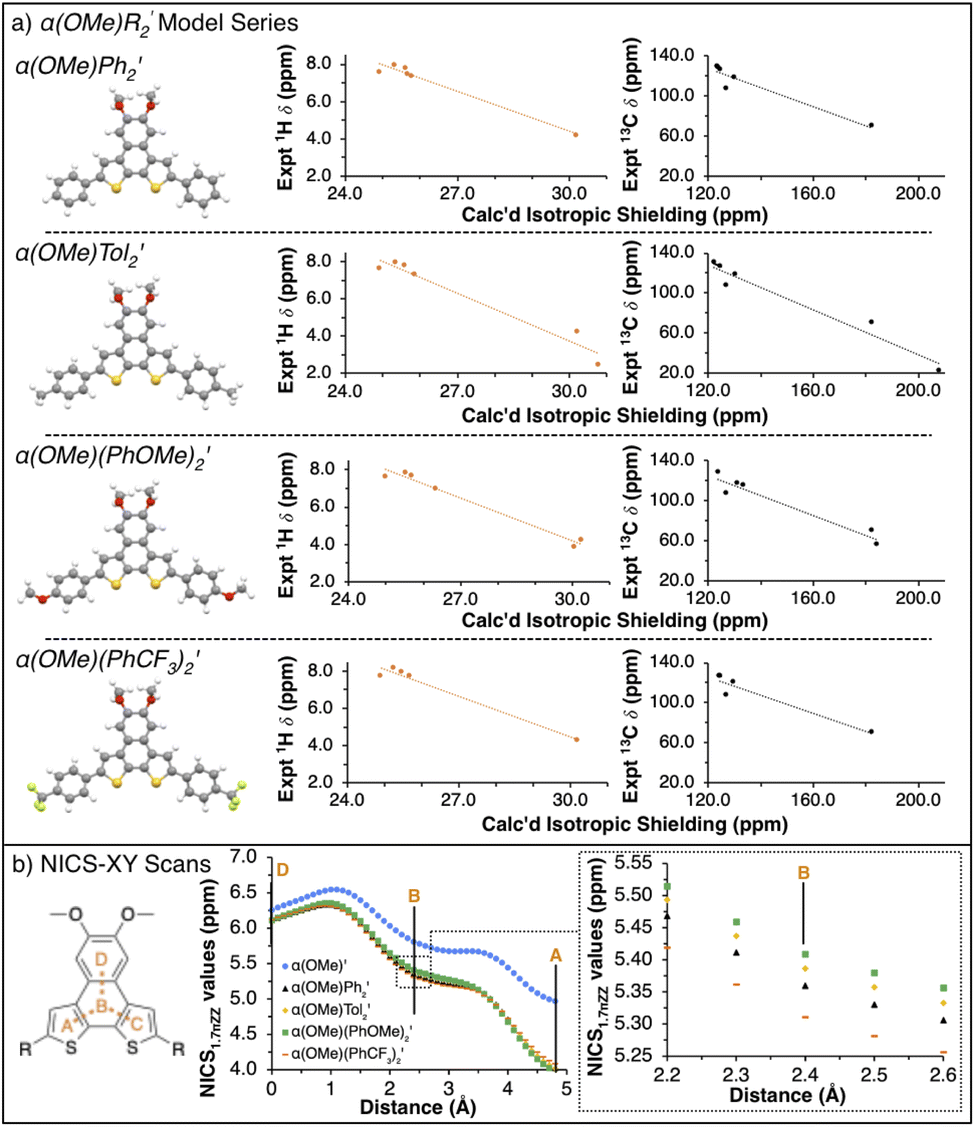 | ||
| Fig. 3 Computational analysis of α(Me)R2′ series. (a) Ball-and-stick representations of model compounds α(OMe)R2′ and their respective experimental vs. calculated NMR plots (1H is shown in orange, 13C is given in black). (b) Partial NICS1.7πZZ scans for α(OMe)R2′ with D–B–A path highlighted in orange. (Inset): Enhanced view of the central B ring. | ||
With the model α(OMe)R2′ series validated, we sought to probe how the R-groups impact the aromaticity of the α-NDT core. Nucleus independent chemical shift (NICS)72 calculations are a well-known computational means to assess the aromatic (or antiaromatic) character of heteroarene systems.73–75 Specifically, NICS-XY scans76,77 wherein the ghost atom is set to 1.7 Å above the planar α-NDT core were performed to ascertain if there are electronic effects in the core that were not apparent via traditional spectroscopic means. In Fig. 3b, we detail the results of the NICS-XY scans for the α(OMe)R2′ series referenced against a model compounds without R-groups, α(OMe)′. The α(OMe)R2′ cores are weakly paratropic no matter the R-group (NICS1.7πZZ values ≈4–6.5 ppm over the path of the probe); there are minor deviations in the ‘B’ ring that follow the Hammett parameter (σp specifically) of the R-group (i.e. there is an increase in aromatic character as the R-group becomes more electron deficient).
2.2 α(OHex)R2 series: photophysical properties
Our focus shifted to elucidating the photophysical properties of the α(OHex)R2 series upon completion of their structural authentication. UV-visible absorption spectra for the α(OHex)R2 series, shown in Fig. 4a, display defined (despite being broadened) vibronic structure in-line with known α-NDT containing materials.78 Table 1 highlights several key metrics of these data, where there is a slight bathochromic shift when progressing from α(OHex)Ph2 to stronger electron donating groups in α(OHex)Tol2 and α(OHex)(PhOMe)2 (Δλabs,max ≤5 nm). The lone A–D–A system, α(OHex)(PhCF3)2, is quite distinct with a 12 nm bathochromic shift relative to parent α(OHex)Ph2. The optical band gap (Eg,opt) was estimated from the tangent of the absorption onset, with α(OHex)(PhCF3)2 having the smallest gap (Eg,opt 2.89 eV). The emission spectra (Fig. 4b) have strong, detailed, vibronic structure.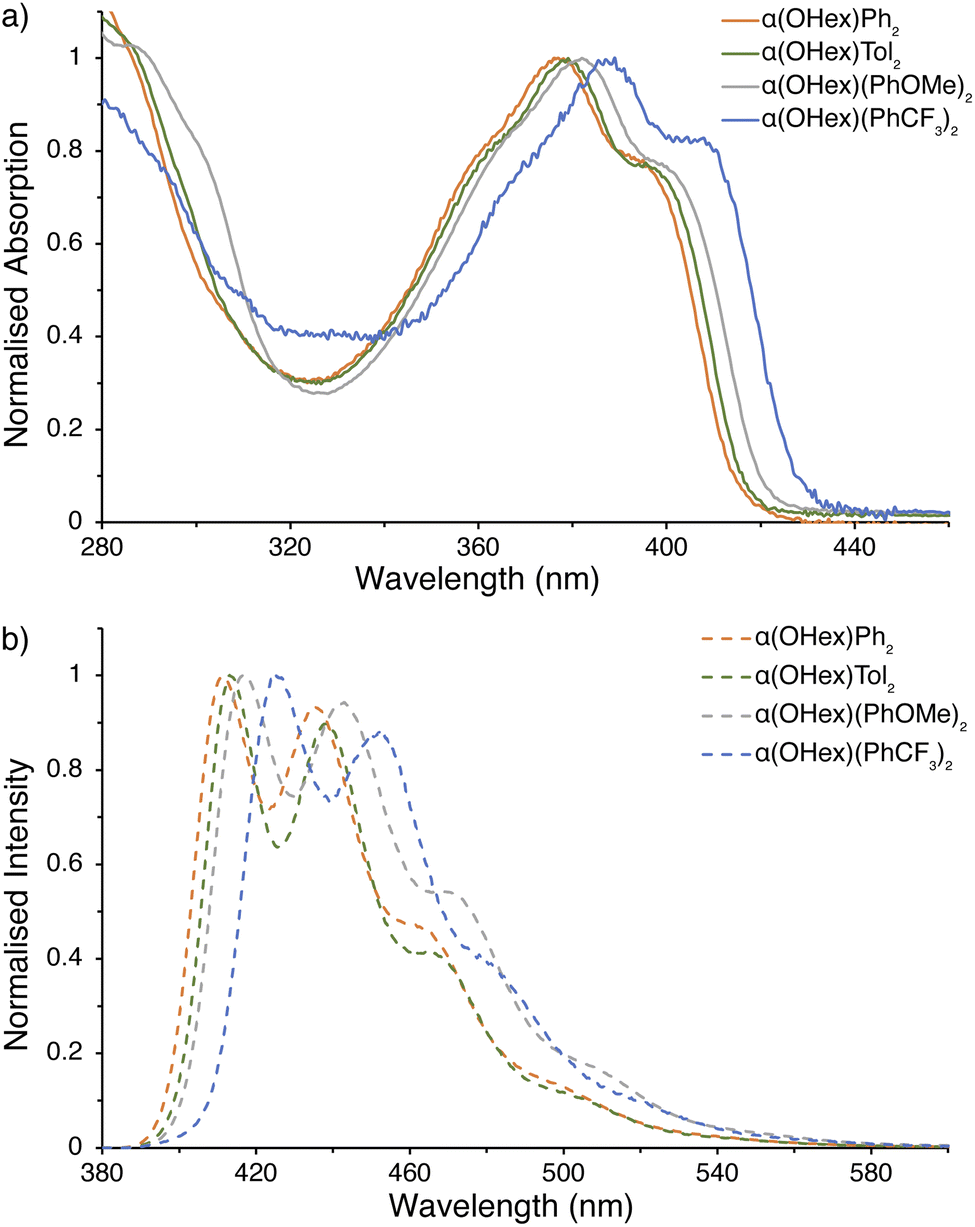 | ||
| Fig. 4 Photophysical measurements for α(OHex)R2 series. (a) UV-visible spectra; (b) emission spectra (λex = 365 nm). All samples were prepared under an inert atmosphere with degassed dry THF. | ||
| α(OR′)R2 | ε (M−1 cm−1) | λabs,max (nm) | Eg,optb (eV) | λemc (nm) | ΦPLc |
|---|---|---|---|---|---|
| a UV-visible and emission spectra were measured as solutions in dry, degassed THF under inert atmosphere.b Eg,opt estimated from the tangent of the absorption onset, λabs,onset.c λex = 365 nm. | |||||
| α(OHex)Ph2 | 3.78 × 104 | 377 | 2.99 | 412 | 0.25 |
| α(OHex)Tol2 | 3.67 × 104 | 379 | 2.97 | 416 | 0.25 |
| α(OHex)(PhOMe)2 | 3.40 × 104 | 382 | 2.94 | 419 | 0.40 |
| α(OHex)(PhCF3)2 | 1.18 × 104 | 389 | 2.89 | 427 | 0.24 |
The fine vibronic structure observed in the emission spectra are indicative of enhanced conjugation in the excited state through rigidification of the molecules (as compared to the relatively structure-less absorption spectra). For α-NDT containing molecules this implies stabilization (and an increase in) the quinoidal resonance contribution.79 Photoluminescence quantum yield (ΦPL) measurements were undertaken for each derivative under inert atmosphere in dry THF. α(OHex)(PhOMe)2 provided the highest quantum yield (ΦPL ≈ 0.40) in THF, while α(OHex)Ph2 (ΦPL ≈ 0.25), α(OHex)Tol2 (ΦPL ≈ 0.25), and α(OHex)(PhCF3)2 (ΦPL ≈ 0.24) are within the error of the technique (±10%). These results are in agreement with known phenylene end-capped α-NDTs α(PhBu)2 (ΦPL ≈ 0.28)20 and α(PhN(PhOMe)2)2 (ΦPL ≈ 0.55).16 Itami and co-workers posited that these photoactive molecules are suitable hole-transporting materials, which is an application we plan on pursuing in the future with solubilized α-NDTs. It is worth noting that the highly polarized all-donor systems containing anisole-based end-caps provided the highest quantum yields whether directly attached (α(OHex)(PhOMe)2) or connected via an amine linkage (α(PhN(PhOMe)2)2).
2.3 β(Oi-Pent)R2 series: synthesis and structural analysis
The poor solubility of the α(OHex)R2 series led us to reconfigure the solubilizing groups for the β-NDT series to a short, branched alkoxy group that promotes solubility but still allows for crystallization. Alkylation of 4,5-dibromocatechol with iso-pentyl bromide efficiently affords 1(Oi-Pent) as a colorless oil that is converted to 3(Oi-Pent) via Suzuki cross-coupling with 2-thiophene boronic acid in modest yield on the gram scale (Scheme 2). This is quite astonishing considering this boronic acid coupling partner is known to undergo moderate protodeborylation under traditional cross-coupling conditions.80 Blocking of the unfunctionalized α-positions of the thienyl groups is required prior to planarization to mitigate polymerization (or rearrangement).78,81 With this in mind, bromination82 of the residual α-positions of 3(Oi-Pent) affords 4(Oi-Pent) in modest yield. In our hands traditional Scholl oxidation conditions were inefficient (yields < 10%) to planarize the brominated species, but organic oxidant and acidic conditions (DDQ, methanesulfonic acid, DCM)82,83 proved to be far superior in accessing β(Oi-Pent)Br2 in modest yield (>40% isolated yields on 500 mg scale).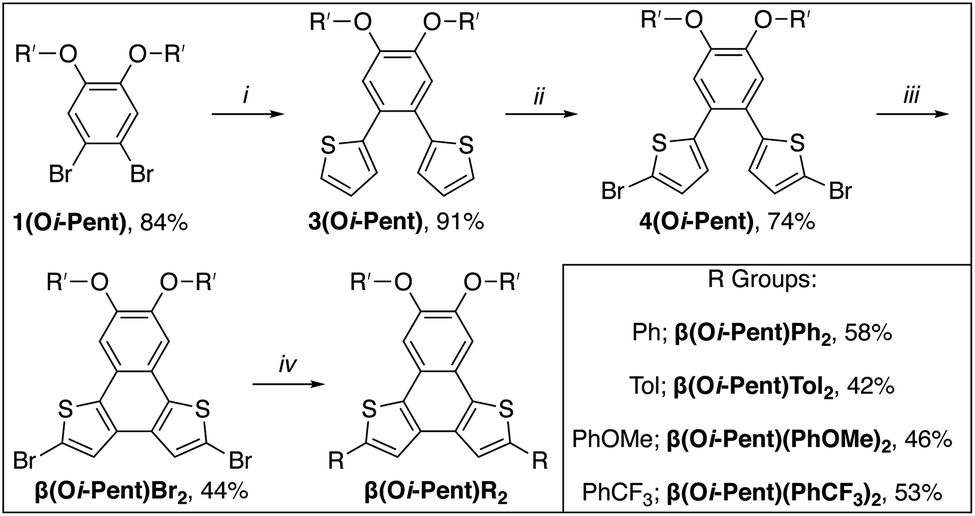 | ||
| Scheme 2 Synthesis of β(Oi-Pent)R2 series. (i) 2-Thiophene boronic acid, Pd(PPh3)2Cl2, 2 M K2CO3, THF, 80 °C, overnight; (ii) NBS, CHCl3, RT, 2 d; (iii) DDQ, MeSO3H, DCM, 0 °C to RT, 1 h; (iv) R boronic acid, Pd(PPh3)2Cl2, 2 M K2CO3, THF, 80 °C, overnight. Inset: isolated yields for β(Oi-Pent)R2 series. | ||
End-capping of β(Oi-Pent)Br2 with the aryl boronic acid derivatives yields the β(Oi-Pent)R2 series as off-white to pale yellow solids. Exchanging the solubilizing groups for iso-pentyloxy resulted in an appreciable improvement in solubility, with all β(Oi-Pent)R2 being soluble in common organic solvents (e.g. 50 mM in CDCl3). The central goal of using the iso-pentyloxy groups is to thread the needle between solubility and the ability to grow single crystals for analysis. After several attempts with all derivatives, single crystals suitable for X-ray diffraction were grown via vapor diffusion of hexanes into a saturated solution of β(Oi-Pent)Tol2 in THF. The crystal obtained for β(Oi-Pent)Tol2 is dimeric, wherein the two units are aligned in the same direction with the β-NDT cores offset (i.e. a slip-stacked arene–arene stacking arrangement) with a distance between the dimers of ≈ 3.6 Å (see ESI†). The inter-ring C–C bond length for β(Oi-Pent)Tol2 is 1.46 Å, nearly identical to the same metrics obtained in the α(OMe)R2 series. However the crystallograhic packing of β(Oi-Pent)Tol2 dimers into columnar stacks is distinct in comparison to the α(OMe)R2 series. This difference in crystallographic packing can brought back to the branch alkoxy solubilizing group of β(Oi-Pent)Tol2, which upon crystallization intercalate with adjacent stacks, thus allowing for columnar stacks of the dimers to form.
The partial aromatic region shown in Fig. 5 reveals the interior 2b proton resonance (as well as the 13C resonance) is responsive to remote functionalization. These perturbations of the 2b proton and carbon resonances are quite similar to those observed in the α(OHex)R2 series, with the β(Oi-Pent)R2 series having Δδ ≈ 0.2 ppm for 1H and Δδ ≈ 2 ppm for 13C. Fitting these chemical shifts to both σp and σm Hammett parameters reveals a quality correlation to σp, albeit not as pure of a fit as with the α(OHex)R2 series. Although the β(Oi-Pent)R2 derivates are quite soluble, one concern for the poor fit (r2 0.7) for the 1H spectra is aggregation. The orientation of the thiophene rings in the β-NDT isomer give the molecules a pronounced bent shape (the approximate bite angle of the terminal substituents is 110°) in comparison to the α series (bite angle ≈ 125°), which may impact their propensity for aggregation. Solutions of β(Oi-Pent)Tol2 and β(Oi-Pent)(PhOMe)2 in CDCl3 were prepared and analyzed across a range of concentrations (50–1 mM, see ESI†) to probe this concern. Only chemical shifts in which Δδ ≥ 0.05 ppm were included in the fitting analysis,33,34 revealing Kd values of 1.1 M and 1.5 M for β(Oi-Pent)Tol2 and β(Oi-Pent)(PhOMe)2, respectively. These values are in-line with the α(OHex)R2 series, highlighting that the minor geometric differences in the bent NDTs do not impart vastly different aggregation potential.
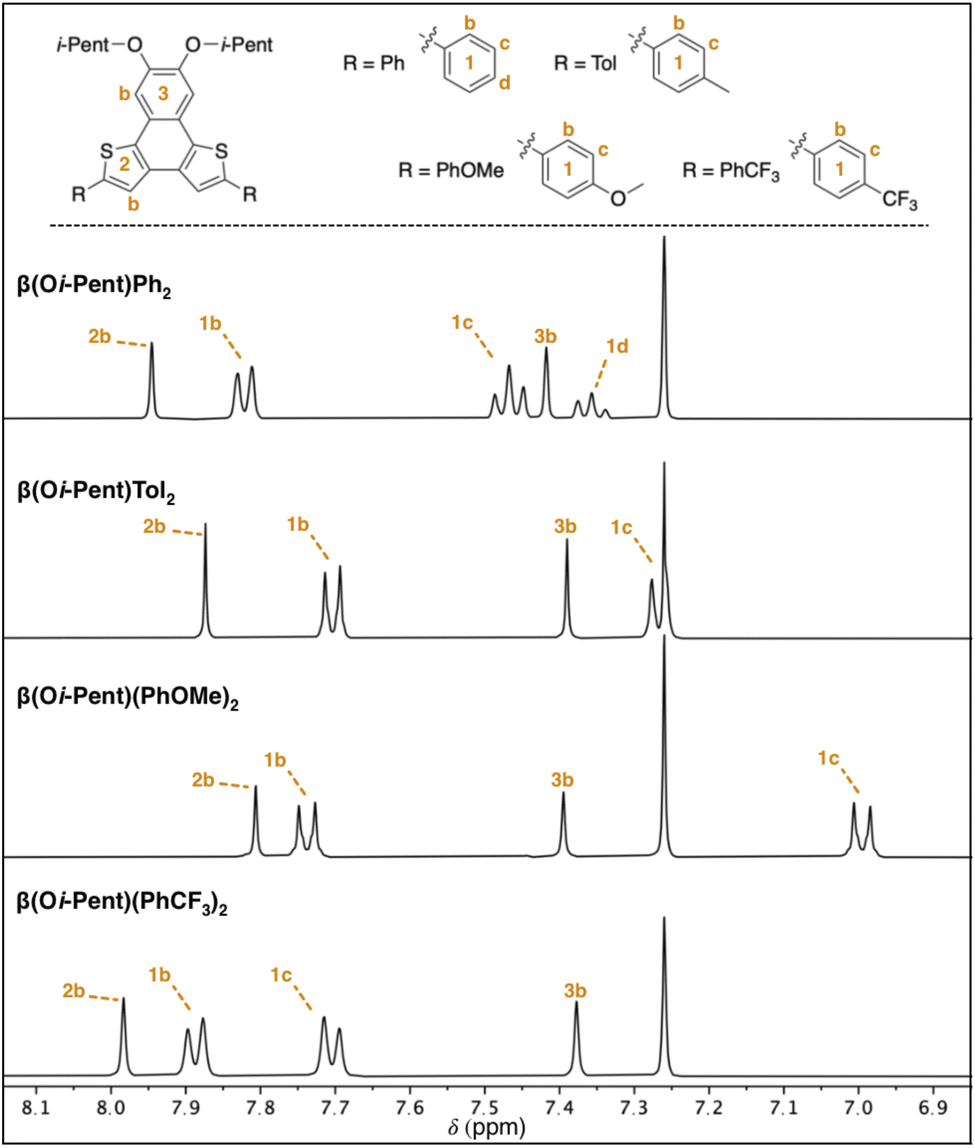 | ||
| Fig. 5 Partial 1H NMR spectra for the β(Oi-Pent)R2 series with assignment of aromatic resonances (400 MHz, CDCl3). See ESI† for full spectra and complete chemical shift assignments. | ||
Following the same computational methodology as the α-NDT series, model compounds bearing methoxy groups were used to calculate both the NMR spectra and perform NICS-XY scans. The model β-NDT series, β(OMe)R2′, reveals that the experimental 1H and 13C resonances obtained from the 1H–13C HSQC spectra for each derivative yield quality fits (r2 > 0.9) with the calculated isotropic shielding values (see ESI†). NICS-XY scans across the β-NDT core yields a similiar pattern to the α(OMe)R2′ series, with the central differences being the NICS1.7πZZ values of the ‘A’ ring β(OMe)R2′ are ≈0.4 ppm lower than α(OMe)R2′). The ‘B’ ring follows the same trend with σp, although the differences between each derivative are minute (ΔNICS1.7πZZ values ≈0.10 ppm, see ESI†).
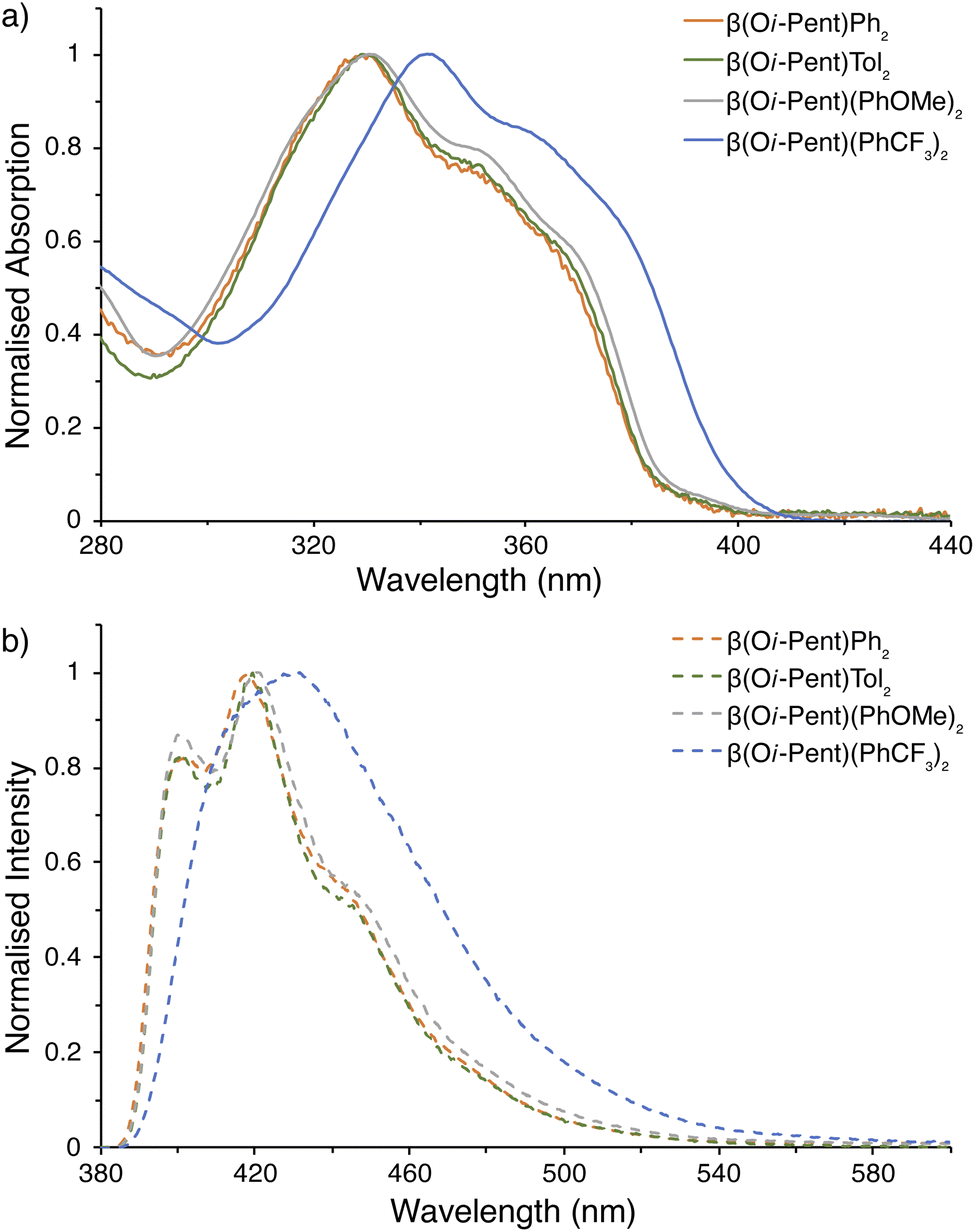 | ||
| Fig. 6 Photophysical measurements for β(Oi-Pent)R2 series. (a) UV-visible spectra; (b) emission spectra (λex = 365 nm). All samples were prepared under inert atmosphere with degassed dry THF. | ||
Critically, there are no discernible differences in the photophysical properties between the β(Oi-Pent)Ph2, β(Oi-Pent)Tol2 and β(Oi-Pent)(PhOMe)2 derivatives (Table 2). These derivatives have broadened vibronic structure in both the emission and excitation spectra. In contrast to their α-NDT series counterparts the β(Oi-Pent)Ph2, β(Oi-Pent)Tol2 and β(Oi-Pent)(PhOMe)2 derivatives have larger band gaps (Eg,opt > 3.10 eV) and decreased ΦPL, with β(Oi-Pent)(PhOMe)2 having the largest difference between isomers at 20% below α(OHex)(PhOMe)2. While there are, to the best of our knowledge, no directly comparable quantum yields in the literature for β-NDTs, this drop in quantum yield has been observed in scrambled bent NDTs (compound 2l ΦPL = 0.03)20 and with Miyazaki and co-workers 2,3-naphthalimide-fused benzodithiophenes (ΦPL ≈ 0.03–0.09).22 However, β(Oi-Pent)(PhCF3)2 is distinct in this analysis. The UV-visible spectrum is bathochromically shifted (λabs,max = 11 nm) in comparison to the rest of the β(Oi-Pent)R2 series with similar vibronic structure, but the emission spectrum is broadened and ill-defined (Fig. 6b).
| β(Oi-Pent)R2 | ε (M−1 cm−1) | λabs,max (nm) | Eg,optb (eV) | λemc (nm) | ΦPLc |
|---|---|---|---|---|---|
| a UV-visible and emission spectra were measured as solutions in dry, degassed THF under inert atmosphere.b Eg,opt estimated from the tangent of the absorption onset, λabs,onset.c λex = 365 nm. | |||||
| β(Oi-Pent)Ph2 | 4.98 × 104 | 331 | 3.22 | 418 | 0.16 |
| β(Oi-Pent)Tol2 | 4.31 × 104 | 330 | 3.22 | 420 | 0.16 |
| β(Oi-Pent)(PhOMe)2 | 6.25 × 104 | 331 | 3.21 | 421 | 0.20 |
| β(Oi-Pent)(PhCF3)2 | 4.40 × 104 | 342 | 3.10 | 432 | 0.23 |
This is in stark contrast to the rest of the β(Oi-Pent)R2 series (and the α(OHex)R2 series), where the emission spectra have detailed structure (in THF). A series of variable polarity solvents was used to elucidate the extent, and type, of solvatochromism. The absorption spectra of β(Oi-Pent)(PhCF3)2 display a weak hypsochromic shift (Δλabs = 4 nm), with a larger (albeit still weak) bathochromic shift (Δλem = 9 nm) observed in the emission spectra (Fig. 7). This solvatochromism is well-correlated with the solvent polarity parameter, ET(30),86 with r2 values of 0.98 and 0.87 for the absorption and emission spectra respectively. It should be noted that ET(30) values for binary solvent mixtures such as THF·MeCN, used here as the maximum polarity solvent, are quite complex87 and the value used here (based on mole fractions) is an approximation. However, it is apparent that this solvatochromism trend is due to the stabilization of the excited state in polar solvent, which in turn yields a larger Stokes shift as solvent polarity increases (Table 3). The A–D–A alignment of β(Oi-Pent)(PhCF3)2 in tandem with the modest Stokes shift (≈10–20 nm) observed imply that the excited state geometry is quite rigid due to the push–pull of the intramolecular charge transfer.88 Photoluminescence quantum yields for each solvent are given in Table 3 for β(Oi-Pent)(PhCF3)2, which are in-line with the positive fluorosolvatochromism observed in Miyazaki and co-workers systems.22
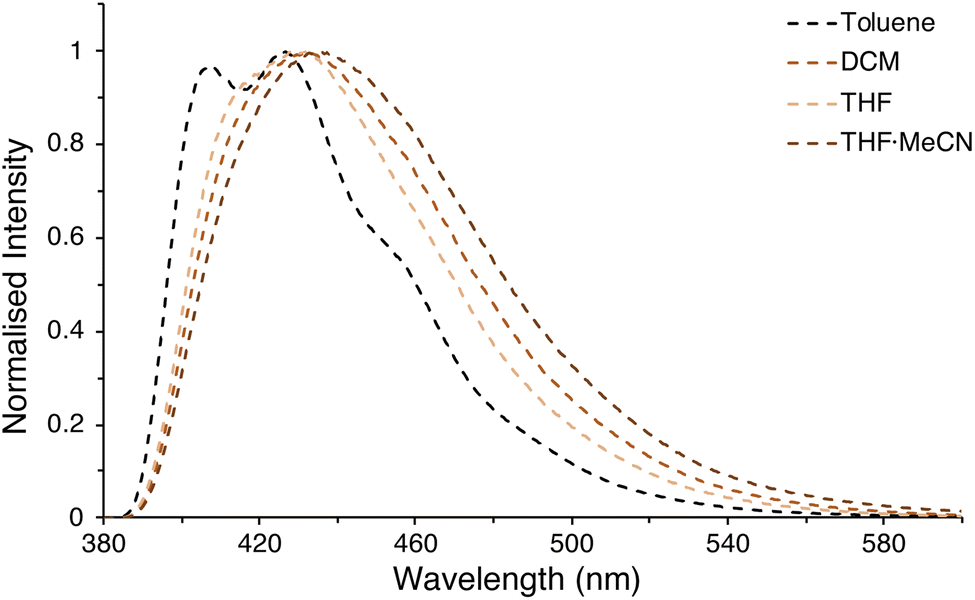 | ||
| Fig. 7 Fluorosolvatochromism observed for β(Oi-Pent)(PhCF3)2. | ||
| Solvent | ET(30) (kcal mol−1) | ε (M−1 cm−1) | λabs,max (nm) | λemb (nm) | ΦPLb |
|---|---|---|---|---|---|
a UV-visible and emission spectra were measured as solutions in dry, degassed solvents under inert atmosphere.b λex = 365 nm.c Weighed by mole fraction of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 solvent mixture. :1 solvent mixture. |
|||||
| Tol | 33.9 | 4.37 × 104 | 343 | 426 | 0.18 |
| DCM | 40.7 | 4.68 × 104 | 340 | 432 | 0.17 |
| THF | 37.4 | 4.35 × 104 | 341 | 432 | 0.23 |
| THF·MeCN | 42.4c | 4.93 × 104 | 339 | 435 | 0.15 |
3 Conclusions
In summary, two isomeric sets of bent naphthodithiophenes have been synthesized and characterized as photoactive small molecules. In both the α(OR′)R2 and β(Oi-Pent)R2 series structural authentication in the solid-state show that the end-caps do not impact inter-ring bond lengths, yet solution-state measurements made via NMR spectroscopy show minor perturbations of interior resonances far removed from the R-groups. In the α(OHex)R2 series spectrophotometry measurements detail fine vibronic structure across all derivatives with modest photoluminescence quantum yields (ΦPL ≈ 0.24–0.40). Alternatively, the β(Oi-Pent)R2 series are far less luminescent due to the limited conjugation path brought on by the orientation of the thiophene moieties. Attachment of electron donating groups has negligible impact on the properties of the fluorophores, but electron deficient groups have a distinct response to their A–D–A motif. β(Oi-Pent)(PhCF3)2 displays positive fluorosolvatochromism and becomes broadened as solvent polarity increases, indicative of increased intramolecular charge transfer character. The photophysical characterization of these isomeric bent NDTs highlights how these simple fluorophores have unique applications as photoactive small molecules. Efforts to maximize the effective conjugation path of these isomeric bent NDTs in small molecule systems and determine their applications in organic electronics are underway.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Z. J. K. conceptualized the project and acquired funding to carry out the experimental portion of the research. E. B. A. A, C. D. G., M. C., D. A., J. P., and Z. J. K. carried out the synthesis, characterization, and spectral analysis of compounds. V. C. and A. R. performed the computations to support the experimental findings and elucidate other parameters of interest. M. Z. carried out the crystallographic analysis. E. B. A. A. and Z. J. K wrote the original draft, with all authors participating in the review and editing process. All authors have given approval to the final version of the manuscript, their respective contributions, and have agreed to publish this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Z. J. K., E. B. A. A, C. D. G., M. C., D. A., and J. P. acknowledge support by the National Science Foundation (CHE-2316854) and Oakland University. A. A. R. thanks the Biomedical Research Center at Oakland University for support through the Research Excellence Program. High-performance computing facilities at Oakland University were provided via collaboration between the Oakland University Research Office and University Technology Services. M. Z. acknowledges the National Science Foundation Major Research Instrumentation grant (CHE-1625543) for funds used to purchase the single crystal X-ray diffractometer.Notes and references
- J. E. Anthony, Functionalized Acenes and Heteroacenes for Organic Electronics, Chem. Rev., 2006, 106, 5028–5048 CrossRef CAS PubMed.
- W. Jiang, Y. Li and Z. Wang, Heteroarenes as high performance organic semiconductors, Chem. Soc. Rev., 2013, 42, 6113–6127 RSC.
- X. Guo, M. Baumgarten and K. Müllen, Designing π-conjugated polymers for organic electronics, Prog. Polym. Sci., 2013, 38, 1832–1908 CrossRef CAS.
- Main Group Strategies towards Functional Hybrid Materials, ed. T. Baumgartner and F. Jäkle, John Wiley & Sons, Inc., 1st edn, 2018 Search PubMed.
- K. Takimiya, I. Osaka and M. Nakano, π-building blocks for organic electronics: Revaluation of “inductive” and “resonance” Effects of π-electron deficient units, Chem. Mater., 2014, 26, 587–593 CrossRef CAS.
- S. C. Rasmussen, S. J. Evenson and C. B. McCausland, Fluorescent thiophene-based materials and their outlook for emissive applications, Chem. Commun., 2015, 51, 4528–4543 RSC.
- S. Shinamura, I. Osaka, E. Miyazaki, A. Nakao, M. Yamagishi, J. Takeya and K. Takimiya, Linear- and angular-shaped naphthodithiophenes: Selective synthesis, properties, and application to organic field-effect transistors, J. Am. Chem. Soc., 2011, 133, 5024–5035 CrossRef CAS.
- K. Takimiya and I. Osaka, Naphthodithiophenes: Emerging building blocks for organic electronics, Chem. Rec., 2015, 15, 175–188 CrossRef CAS.
- I. Osaka, S. Shinamura, T. Abe and K. Takimiya, Naphthodithiophenes as building units for small molecules to polymers; A case study for in-depth understanding of structure–property relationships in organic semiconductors, J. Mater. Chem. C, 2013, 1, 1297–1304 RSC.
- K. Zhang, J. Zhang, X. Zhang, G. Yu and M. S. Wong, Synthesis and characterization of novel push-pull oligomer based on naphthodithiophene-benzothiodiazole for OFETs application, Tetrahedron Lett., 2018, 59, 641–644 CrossRef CAS.
- L. Zhao, Y. Cao, H. Qin, X. He, Z. Zhao, Y. Guo and H. Chen, Synthesis and charge-transport properties of novel π-conjugated polymers incorporating core-extended naphtho[2,1-b:3,4-b′] dithiophene diimides, Polym. Chem., 2024, 15, 59–70 RSC.
- H. D. Pham, H. Hu, F. L. Wong, C. S. Lee, W. C. Chen, K. Feron, S. Manzhos, H. Wang, N. Motta, Y. M. Lam and P. Sonar, Acene-based organic semiconductors for organic light-emitting diodes and perovskite solar cells, J. Mater. Chem. C, 2018, 6, 9017–9029 RSC.
- M. Löbert, A. Mishra, C. Uhrich, M. Pfeiffer and P. Bäuerle, Synthesis and characterization of benzo- and naphtho[2,1-b:3,4-b′] dithiophene-containing oligomers for photovoltaic applications, J. Mater. Chem. C, 2014, 2, 4879–4892 RSC.
- J. Zhu, Z. Ke, Q. Zhang, J. Wang, S. Dai, Y. Wu, Y. Xu, Y. Lin, W. Ma, W. You and X. Zhan, Naphthodithiophene-Based Nonfullerene Acceptor for High-Performance Organic Photovoltaics: Effect of Extended Conjugation, Adv. Mater., 2018, 30, 1–7 Search PubMed.
- X. Wang, L. Guo, P. F. Xia, F. Zheng, M. S. Wong and Z. Zhu, Dye-sensitized solar cells based on organic dyes with naphtho[2,1-b:3,4-b′] dithiophene as the conjugated linker, J. Mater. Chem. A, 2013, 1, 13328–13336 RSC.
- H. A. Lin, N. Mitoma, L. Meng, Y. Segawa, A. Wakamiya and K. Itami, Hole-transporting materials based on thiophene-fused arenes from sulfur-mediated thienannulations, Mater. Chem. Front., 2018, 2, 275–280 RSC.
- C. C. Hsu, K. M. Lee, X. W. Wu, L. Lin, W. L. Yu and C. Y. Liu, Hole-Transporting Materials based on Oligo(hetero)aryls with a Naphthodithiophene Core – Succinct Synthesis by Twofold Direct CH Olefination, Chem.–Eur. J., 2024, 30, e202302552 CrossRef CAS.
- X. Shen, X. Lai, H. Lai, T. Zhao, Y. Zhu, M. Pu, H. Wang, P. Tan and F. He, Isomerism Strategy to Optimize Aggregation and Morphology for Superior Polymer Solar Cells, Macromolecules, 2022, 55, 6384–6393 CrossRef CAS.
- M. Su, M. Lin, S. Mo, J. Chen, X. Shen, Y. Xiao, M. Wang, J. Gao, L. Dang, X.-c. Huang, F. He and Q. Wu, Manipulating the Alkyl Chains of Naphthodithiophene Imide-Based Polymers to Concurrently Boost the Efficiency and Stability of Organic Solar Cells, ACS Appl. Mater. Interfaces, 2023, 15, 37371–37380 CrossRef CAS PubMed.
- L. Meng, T. Fujikawa, M. Kuwayama, Y. Segawa and K. Itami, Thiophene-Fused -Systems from Diarylacetylenes and Elemental Sulfur, J. Am. Chem. Soc., 2016, 138, 10351–10355 CrossRef CAS PubMed.
- A. Meyer, E. Sigmund, F. Luppertz, G. Schnakenburg, I. Gadaczek, T. Bredow, S. S. Jester and S. Höger, Syntheses and properties of thienyl-substituted dithienophenazines, Beilstein J. Org. Chem., 2010, 6, 1180–1187 CrossRef CAS PubMed.
- T. Miyazaki, T. Tsutsumi and O. Hayashida, Fluorosolvatochromic Behavior of 2,3-Naphthalimides Expanded by Double Fusion with Benzothiophene and Benzofuran Units, ChemistrySelect, 2023, 8, 6–11 CrossRef.
- M. Klikar, P. Solanke, J. Tydlitát and F. Bureš, Alphabet-Inspired Design of (Hetero)Aromatic Push–Pull Chromophores, Chem. Rec., 2016, 1886–1905 CrossRef CAS PubMed.
- F. Bureš, Fundamental aspects of property tuning in push–pull molecules, RSC Adv., 2014, 4, 58826–58851 RSC.
- D. Chaudhuri, E. Sigmund, A. Meyer, L. Röck, P. Klemm, S. Lautenschlager, A. Schmid, S. R. Yost, T. Vanvoorhis, S. Bange, S. Höger and J. M. Lupton, Metal-free OLED triplet emitters by side-stepping Kasha's rule, Angew. Chem., Int. Ed., 2013, 52, 13449–13452 CrossRef CAS PubMed.
- W. Ratzke, L. Schmitt, H. Matsuoka, C. Bannwarth, M. Retegan, S. Bange, P. Klemm, F. Neese, S. Grimme, O. Schiemann, J. M. Lupton and S. Höger, Effect of Conjugation Pathway in Metal-Free Room-Temperature Dual Singlet-Triplet Emitters for Organic Light-Emitting Diodes, J. Phys. Chem. Lett., 2016, 7, 4802–4808 CrossRef CAS PubMed.
- J. P. Gallivan and G. B. Schuster, Aggregates of Hexakis(n-hexyloxy)triphenylene Self-Assemble in Dodecane Solution: Intercalation of (-)-Menthol 3,5-Dinitrobenzoate Induces Formation of Helical Structures, J. Org. Chem., 1995, 60, 2423–2429 CrossRef CAS.
- M. Kohn, Bromination of Catechol, J. Am. Chem. Soc., 1951, 73, 480 CrossRef CAS.
- A. A. Sarhan and C. Bolm, Iron(iii) chloride in oxidative C–C coupling reactions, Chem. Soc. Rev., 2009, 38, 2730–2744 RSC.
- C. Hansch, A. Leo and R. W. Taft, A Survey of Hammett Substituent Constants and Resonance and Field Parameters, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- B. Wang, J. Zhang, H. L. Tam, B. Wu, W. Zhang, M. S. Chan, F. Pan, G. Yu, F. Zhu and M. S. Wong, Impact of alkyl side chains on the photovoltaic and charge mobility properties of naphthodithiophene-benzothiadiazole copolymers, Polym. Chem., 2014, 5, 836–843 RSC.
- G. Barbarella, M. Zambianchi, A. Bongini and L. Antolini, The Deformability of the Thiophene Ring: A Key to the Understanding of the Conformational Properties of Oligo- and Polythiophenes, Adv. Mater., 1993, 5, 834–838 CrossRef CAS.
- BindFit v0.5, http://app.supramolecular.org/bindfit/.
- D. Brynn Hibbert and P. Thordarson, The death of the Job plot, transparency, open science and online tools, uncertainty estimation methods and other developments in supramolecular chemistry data analysis, Chem. Commun., 2016, 52, 12792–12805 RSC.
- K. Wolinski, J. F. Hinton and P. Pulay, Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations, J. Am. Chem. Soc., 1990, 112, 8251–8260 CrossRef CAS.
- J. R. Cheeseman, G. W. Trucks, T. A. Keith and M. J. Frisch, A comparison of models for calculating nuclear magnetic resonance shielding tensors, J. Chem. Phys., 1996, 104, 5497–5509 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision C.01, Gaussian Inc. Wallingford CT, 2016 Search PubMed.
- J. Vícha, S. Komorovsky, M. Repisky, R. Marek and M. Straka, Relativistic Spin–Orbit Heavy Atom on the Light Atom NMR Chemical Shifts: General Trends Across the Periodic Table Explained, J. Chem. Theory Comput., 2018, 14, 3025–3039 CrossRef.
- J. Vıcha, J. Novotný, S. Komorovsky, M. Straka, M. Kaupp and R. Marek, Relativistic Heavy-Neighbor-Atom Effects on NMR Shifts: Concepts and Trends Across the Periodic Table, Chem. Rev., 2020, 120, 7065–7103 CrossRef PubMed.
- I. G. Shenderovich, The Scope of the Applicability of Non-relativistic DFT Calculations of NMR Chemical Shifts in Pyridine-Metal Complexes for Applied Applications, ChemPhysChem, 2024, 25, e202300986 CrossRef CAS PubMed.
- I. L. Rusakova and Y. Y. Rusakov, Relativistic Effects from Heavy Main Group p-Elements on the NMR Chemical Shifts of Light Atoms: From Pioneering Studies to Recent Advances, Magnetochemistry, 2023, 9, 24 CrossRef CAS.
- P. Pyykkö, A. Görling and N. Rösch, A transparent interpretation of the relativistic contribution to the N.M.R. ‘heavy atom chemical shift’, Mol. Phys., 1987, 61, 195–205 CrossRef.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- Z. S. Safi and N. Wazzan, DFT calculations of 1H- and 13C-NMR chemical shifts of 3-methyl-1-phenyl-4-(phenyldiazenyl)-1H-pyrazol-5-amine in solution, Sci. Rep., 2022, 12, 17798 CrossRef CAS PubMed.
- J. Li, J.-K. Liu and W.-X. Wang, GIAO 13C NMR Calculation with Sorted Training Sets Improves Accuracy and Reliability for Structural Assignation, J. Org. Chem., 2020, 85, 11350–11358 CrossRef CAS.
- P. Gao, J. Zhang, Q. Peng, J. Zhang and V.-A. Glezakou, General Protocol for the Accurate Prediction of Molecular 13C/1H NMR Chemical Shifts via Machine Learning Augmented DFT, J. Chem. Inf. Model., 2020, 60, 3746–3754 CrossRef CAS PubMed.
- D. Xin, C. A. Sader, O. Chaudhary, P.-J. Jones, K. Wagner, C. S. Tautermann, Z. Yang, C. A. Busacca, R. A. Saraceno, K. R. Fandrick, N. C. Gonnella, K. Horspool, G. Hansen and C. H. Senanayake, Development of a 13C NMR Chemical Shift Prediction Procedure Using B3LYP/cc-pVDZ and Empirically Derived Systematic Error Correction Terms: A Computational Small Molecule Structure Elucidation Method, J. Org. Chem., 2017, 82, 5135–5145 CrossRef CAS PubMed.
- A. Bose, G. A. Valdivia-Berroeta and N. C. Gonnella, Predicting Autoxidation of Sulfides in Drug-like Molecules Using Quantum Mechanical/Density Functional Theory Methods, J. Chem. Inf. Model., 2024, 64, 128–137 CrossRef CAS.
- D. Xin, C. A. Sader, U. Fischer, K. Wagner, P.-J. Jones, M. Xing, K. R. Fandrick and N. C. Gonnella, Systematic investigation of DFT-GIAO 15N NMR chemical shift prediction using B3LYP/cc-pVDZ: application to studies of regioisomers, tautomers, protonation states and N-oxides, Org. Biomol. Chem., 2017, 15, 928–936 RSC.
- J. Li, J. Liang, Z. Wang, A. L. Ptaszek, X. Liu, B. Ganoe, M. Head-Gordon and T. Head-Gordon, Highly Accurate Prediction of NMR Chemical Shifts from Low-Level Quantum Mechanics Calculations Using Machine Learning, J. Chem. Theory Comput., 2024, 20, 2152–2166 CrossRef.
- M. A. Iron, Evaluation of the Factors Impacting the Accuracy of 13C NMR Chemical Shift Predictions using Density Functional Theory–The Advantage of Long-Range Corrected Functionals, J. Chem. Theory Comput., 2017, 13, 5798–5819 CrossRef CAS.
- G. L. Stoychev, A. A. Auer and F. Neese, Efficient and Accurate Prediction of Nuclear Magnetic Resonance Shielding Tensors with Double-Hybrid Density Functional Theory, J. Chem. Theory Comput., 2018, 14, 4756–4771 CrossRef CAS.
- S. Li, W. Zhou, H. Gao and Z. Zhou, Density functional theory study of 13C NMR chemical shift of chlorinated compounds, Magn. Reson. Chem., 2012, 50, 106–113 CrossRef CAS.
- R. Jain, T. Bally and P. R. Rablen, Calculating Accurate Proton Chemical Shifts of Organic Molecules with Density Functional Methods and Modest Basis Sets, J. Org. Chem., 2009, 74, 4017–4023 CrossRef CAS.
- T. Bally and P. R. Rablen, Quantum-chemical simulation of 1H NMR spectra. 2. Comparison of DFT-based procedures for computing proton-proton coupling constants in organic molecules, J. Org. Chem., 2011, 76, 4818–4830 CrossRef CAS PubMed.
- A. Bagno, F. Rastrelli and G. Saielli, Predicting 13C NMR Spectra by DFT Calculations, J. Phys. Chem. A, 2003, 107, 9964–9973 CrossRef CAS.
- D. Flaig, M. Maurer, M. Hanni, K. Braunger, L. Kick, M. Thubauville and C. Ochsenfeld, Benchmarking Hydrogen and Carbon NMR Chemical Shifts at HF, DFT, and MP2 Levels, J. Chem. Theory Comput., 2014, 10, 572–578 CrossRef CAS.
- A. M. Teale, O. B. Lutnæs, T. Helgaker, D. J. Tozer and J. Gauss, Benchmarking density-functional theory calculations of NMR shielding constants and spin–rotation constants using accurate coupled-cluster calculations, J. Chem. Phys., 2013, 138, 024111 CrossRef.
- R. J. Iuliucci, J. D. Hartman and G. J. O. Beran, Do Models beyond Hybrid Density Functionals Increase the Agreement with Experiment for Predicted NMR Chemical Shifts or Electric Field Gradient Tensors in Organic Solids?, J. Phys. Chem. A, 2023, 127, 2846–2858 CrossRef CAS.
- W.-J. Ai, J. Li, D. Cao, S. Liu, Y.-Y. Yuan, Y. Li, G.-S. Tan, K.-P. Xu, X. Yu, F. Kang, Z.-X. Zou and W.-X. Wang, A Very Deep Graph Convolutional Network for 13C NMR Chemical Shift Calculations with Density Functional Theory Level Performance for Structure Assignment, J. Nat. Prod., 2024, 87, 743–752 CrossRef CAS.
- I. M. Novitskiy and A. G. Kutateladze, DU8ML: Machine Learning-Augmented Density Functional Theory Nuclear Magnetic Resonance Computations for High-Throughput In Silico Solution Structure Validation and Revision of Complex Alkaloids, J. Org. Chem., 2022, 87, 4818–4828 CrossRef CAS PubMed.
- W. Yan and X. Xu, Accurate Prediction of Nuclear Magnetic Resonance Parameters via the XYG3 Type of Doubly Hybrid Density Functionals, J. Chem. Theory Comput., 2022, 18, 2931–2946 CrossRef CAS.
- J. B. K. Büning and S. Grimme, Computation of CCSD(T)-Quality NMR Chemical Shifts via Δ-Machine Learning from DFT, J. Chem. Theory Comput., 2023, 19, 3601–3615 CrossRef PubMed.
- F. Jensen, How Large is the Elephant in the Density Functional Theory Room?, J. Phys. Chem. A, 2017, 121, 6104–6107 CrossRef CAS.
- B. Nagy and F. Jensen, Reviews in Computational Chemistry, Reviews in Computational Chemistry, 2018, pp. 93–149 Search PubMed.
- F. Jensen, Computational Chemistry: The Exciting Opportunities and the Boring Details, Isr. J. Chem., 2022, 62, year CrossRef.
- F. Jensen, Unifying General and Segmented Contracted Basis Sets. Segmented Polarization Consistent Basis Sets, J. Chem. Theory Comput., 2014, 10, 1074–1085 CrossRef CAS PubMed.
- F. Jensen, Segmented Contracted Basis Sets Optimized for Nuclear Magnetic Shielding, J. Chem. Theory Comput., 2015, 11, 132–138 CrossRef CAS.
- A. E. A. Fouda and N. A. Besley, Assessment of basis sets for density functional theory-based calculations of core-electron spectroscopies, Theor. Chem. Acc., 2017, 137, 6 Search PubMed.
- R. T. Ireland and L. K. McKemmish, On the specialization of Gaussian basis sets for core-dependent properties, J. Chem. Phys., 2023, 159, 064102 CrossRef CAS PubMed.
- G. I. Warren, J. E. Barker, L. N. Zakharov and M. M. Haley, Enhancing the Antiaromaticity of s-Indacene through Naphthothiophene Fusion, Org. Lett., 2021, 23, 5012–5017 CrossRef CAS PubMed.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta, V. Ragué and P. Schleyer, Nucleus-independent chemical shifts (NICS) as an aromaticity criterion, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS.
- R. Gershoni-Poranne and A. Stanger, Magnetic criteria of aromaticity, Chem. Soc. Rev., 2015, 44, 6597–6615 RSC.
- A. Stanger, NICS – Past and Present, Eur. J. Org Chem., 2020, 2020, 3120–3127 CrossRef CAS.
- A. Rahalkar and A. Stanger, AROMA Package, 2014, http://chemistry.technion.ac.il/members/amnon-stanger/ Search PubMed.
- R. Gershoni-Poranne and A. Stanger, The NICS-XY-scan: Identification of local and global ring currents in multi-ring systems, Chem.–Eur. J., 2014, 20, 5673–5688 CrossRef CAS PubMed.
- J. D. Tovar and T. M. Swager, Poly(naphthodithiophene)s: Robust, Conductive Electrochromics via Tandem Cyclization–Polymerizations, Adv. Mater., 2001, 13, 1775–1780 CrossRef CAS.
- D. Xia, A. Keerthi, C. An and M. Baumgarten, Synthesis of a quinoidal dithieno[2,3-d;2,3-d benzo[2,1-b;3,4-b′]-dithiophene] based open-shell singlet biradicaloid, Org. Chem. Front., 2017, 4, 18–21 RSC.
- X. A. Cook, A. de Gombert, J. McKnight, L. R. Pantaine and M. C. Willis, The 2-Pyridyl Problem: Challenging Nucleophiles in Cross-Coupling Arylations, Angew. Chem., Int. Ed., 2021, 60, 11068–11091 CrossRef CAS PubMed.
- J. D. Tovar, A. Rose and T. M. Swager, Functionalizable polycyclic aromatics through oxidative cyclization of pendant thiophenes, J. Am. Chem. Soc., 2002, 124, 7762–7769 CrossRef CAS.
- D. Waghray, C. De Vet, K. Karypidou and W. Dehaen, Oxidative transformation to naphthodithiophene and thia[7] helicenes by intramolecular scholl reaction of substituted 1,2-bis(2-thienyl)benzene precursors, J. Org. Chem., 2013, 78, 11147–11154 CrossRef CAS PubMed.
- W. Wang, X. Li, Z. Qi, B. Ji, Z. Wang, S. Wang and J. Xiao, Synthesis, Crystal Analysis, and Physical Properties of Double [6] helicene-Containing Heteroarenes with Circularly Polarized Luminescence, Org. Lett., 2023, 25, 1343–1347 CrossRef CAS PubMed.
- E. Clar, The Aromatic Sextet, J. Wiley, 1972 Search PubMed.
- M. Solà, Forty years of Clar's aromatic π-sextet rule, Front. Chem., 2013, 1, 4–11 Search PubMed.
- C. Reichardt, Solvatochromic dyes as solvent polarity indicators, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS.
- S. Nigam and S. Rutan, Principles and Applications of Solvatochromism, Appl. Spectrosc., 2001, 55, 362A–370A CrossRef CAS.
- P. Vázquez-Domínguez, J. F. Rizo, J. F. Arteaga, D. Jacquemin, L. Favereau, A. Ros and U. Pischel, Azaborahelicene fluorophores derived from four-coordinate N,C-boron chelates: synthesis, photophysical and chiroptical properties, Organic, Chem. Front., 2023, 11, 843–853 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR and photophysical spectra, supplemental figures and analysis referred to in the text, computational data, and crystallographic results. CCDC 2361476–2361480. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra04850d |
| This journal is © The Royal Society of Chemistry 2024 |