
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and evaluation of sulfonamide derivatives of quinoxaline 1,4-dioxides as carbonic anhydrase inhibitors†
Galina I. Buravchenkoa,
Alexander M. Scherbakov b,
Stepan K. Krymova,
Diana I. Salnikovab,
George V. Zatonskya,
Dominique Scholsc,
Daniela Vullod,
Claudiu T. Supurand and
Andrey E. Shchekotikhin*a
b,
Stepan K. Krymova,
Diana I. Salnikovab,
George V. Zatonskya,
Dominique Scholsc,
Daniela Vullod,
Claudiu T. Supurand and
Andrey E. Shchekotikhin*a
aGause Institute of New Antibiotics, 11 B. Pirogovskaya Street, Moscow, 119021, Russia. E-mail: buravchenkogi@gmail.com; krymov.s.k@gmail.com; gzatonsk@gmail.com; shchekotikhin@mail.ru; shchekotikhin@gause-inst.ru
bDepartment of Experimental Tumor Biology, Institute of Carcinogenesis, Blokhin N.N. National Medical Research Center of Oncology, Kashirskoe sh. 24, 115522 Moscow, Russia. E-mail: dianasalnikova08@yandex.ru; alex.scherbakov@gmail.com
cRega Institute for Medical Research, KU Leuven, 3000 Leuven, Belgium. E-mail: dominique.schols@kuleuven.be
dDepartment of NEUROFARBA, Section of Pharmaceutical and Nutraceutical Sciences, University of Florence, Florence, Italy. E-mail: daniela.vullo@unifi.it; claudiu.supuran@unifi.it
First published on 23rd July 2024
Abstract
A series of sulfonamide-derived quinoxaline 1,4-dioxides were synthesized and evaluated as inhibitors of carbonic anhydrases (CA) with antiproliferative potency. Overall, the synthesized compounds demonstrated good inhibitory activity against four CA isoforms. Compound 7g exhibited favorable potency in inhibiting a CA IX isozyme with a Ki value of 42.2 nM compared to the reference AAZ (Ki = 25.7 nM). Nevertheless, most of the synthesized compounds have their highest activity against CA I and CA II isoforms over CA IX and CA XII. A molecular modeling study was used for an estimation of the binding mode of the selected ligand 7g in the active site of CA IX. The most active compounds (7b, 7f, 7h, and 18) exhibited significant antiproliferative activity against MCF-7, Capan-1, DND-41, HL60, and Z138 cell lines, with IC50 values in low micromolar concentrations. Moreover, derivatives 7a, 7e, and 8g showed similar hypoxic cytotoxic activity and selectivity compared to tirapazamine (TPZ) against adenocarcinoma cells MCF-7. The structure–activity relationships analysis revealed that the presence of a halogen atom or a sulfonamide group as substituents in the phenyl ring of quinoxaline-2-carbonitrile 1,4-dioxides was favorable for overall cytotoxicity against most of the tested cancer cell lines. Additionally, the presence of a carbonitrile fragment in position 2 of the heterocycle also had a positive effect on the antitumor properties of such derivatives against the majority of cell lines. The most potent derivative, 3-trifluoromethylquinoxaline 1,4-dioxide 7h, demonstrated higher or close antiproliferative activity compared to the reference agents, such as doxorubicin, and etoposide, with an IC50 range of 1.3–2.1 μM. Analysis of the obtained results revealed important patterns in the structure–activity relationship. Moreover, these findings highlight the potential of selected lead sulfonamides on the quinoxaline 1,4-dioxide scaffold for further in-depth evaluation and development of chemotherapeutic agents targeting carbonic anhydrases.
Introduction
Tumor progression and the development of resistance to chemotherapy and radiotherapy continue to pose significant threats to the lives of many cancer patients. Although many antitumor drugs demonstrate significant clinical efficacy, chemotherapy frequently leads to the emergence of resistance in tumor cells, either as a response to medication treatment or as a consequence of disease progression.1 Hypoxia is a distinctive feature of many types of malignant tumors, resulting from rapid cell division, reduced blood supply, and downgraded oxygen transport within the tumor node due to vascularization disorders and structural changes in tumor tissue.2 Hypoxia is considered one of the reasons for the resistance of solid tumors to chemotherapy and the development of an aggressive phenotype in subclones.3,4 In hypoxia conditions, adaptation to low oxygen levels of tumor cells occurs under the influence of hypoxia-induced factors (HIF).5 One of the outcomes of this adjustment is the overexpression of carbonic anhydrase IX (CA IX), which is observed in tumor cells of various histogenesis.6–10 Recent studies have shown that inhibiting CA IX helps overcome the resistance of tumor cells to apoptosis under hypoxic conditions.11,12Therefore, the design of new drugs capable of targeting cancer cells under hypoxic conditions is one of the priority directions in the development of advancing solid tumor chemotherapy. Carbonic anhydrase IX (CA IX) is selectively expressed in cancer cells and plays a crucial role in the formation of conditions that stimulate tumor growth and metastasis, including pH reduction, activation of survival mechanisms, reduction of adhesion, and stimulation of migration. Therefore, CA IX is considered a promising target for antitumor therapy.13–15
To date, several sulfonamides of carbo- and heterocyclic compounds capable of selectively inhibiting CA IX, which are prospective for the development of new antitumor agents, have been described (for example, derivatives 1–4, Fig. 1).16 One well-known example of a CA inhibitor is acetazolamide (AAZ, shown as 1 in Fig. 1), which has been used in clinical practice for over 40 years and can suppress tumor cell proliferation through CA inhibition.17,18 A promising antitumor CA IX inhibitor is the sulfonamide derivative 2 (SLC-0111), which has shown high efficacy in vivo in solid tumor models and low toxicity in Phase I clinical trials. Currently, it is undergoing Phase II clinical trials to further evaluate its efficacy and safety.19 Additionally, some sulfonamides exhibit potent antiproliferative activity (for example, compound 3, Fig. 1).20 Derivatives of this class inhibit CA IX at submicro- and nanomolar concentrations, confirming the significance of this enzyme in tumor progression.21
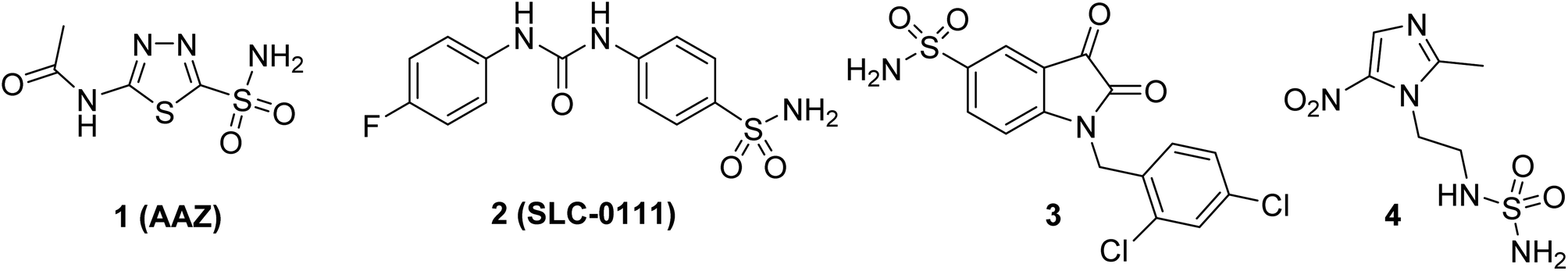 | ||
| Fig. 1 Perspective carbonic anhydrase inhibitors 1–4. | ||
Another promising class for the development of antitumor agents that selectively act on hypoxic tumors is quinoxaline 1,4-dioxide derivatives.22 It has been previously demonstrated that compounds from this series reduce the expression of HIF-1α in solid tumor cells under hypoxic conditions, effectively inhibiting their growth (for example, derivatives 5, 6, Fig. 2).23–26 The introduction of the sulfonamide moiety into the quinoxaline 1,4-dioxide scaffold can lead to derivatives with the ability to inhibit CA9 and could enhance their antitumor potential through multitargeted action on several pathways activated in tumor cells under hypoxic conditions.
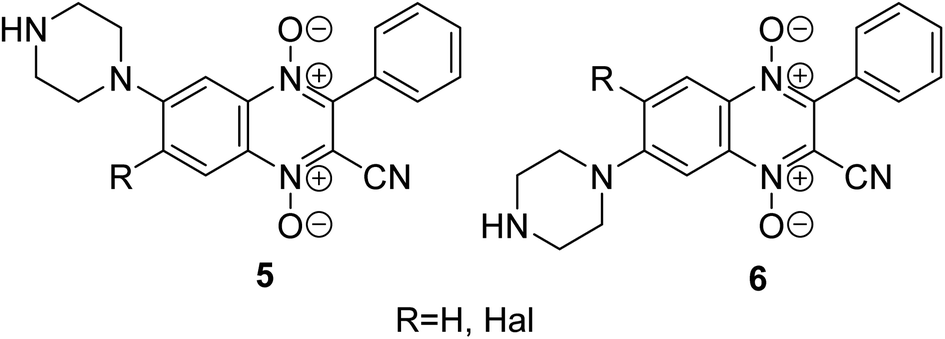 | ||
| Fig. 2 Hypoxia-selective derivatives 5, 6 based on quinoxaline 1,4-dioxide scaffold. | ||
Despite more than 50 years of active evaluation of quinoxaline 1,4-dioxides,22 the synthesis, and biological properties of their sulfonamide derivatives have not been described. Hence, the design, synthesis, and assessment of the anticancer activity of such sulfonamides based on quinoxaline 1,4-dioxide, including the study of their CA-activity profile, represents a promising direction in the development of novel chemotherapeutic agents.
Results and discussion
Design and synthesis of sulfonamidoquinoxaline 1,4-dioxides
For the preliminary assessment of sulfonamidoquinoxaline 1,4-dioxides ability to inhibit carbonic anhydrase IX (Protein Data Bank PDB 5SZ5), sulfonamide analogues of derivatives 5 and 6 (compounds 7a and 8a) were docked into the active site of the enzyme using the Molecular Operating Environment (MOE 2014) (Fig. 3). The results of molecular modeling suggest that the designed sulfonamides 7a and 8a may exhibit good affinity toward CA IX. Both isomeric ligands, 7a and 8a, fit well within the active site of the enzyme and form a coordination complex with the Zn2+ ion, a crucial cofactor for catalytic activity. Notably, based on the docking results, quinoxaline 1,4-dioxide 7a, with a sulfonamide group at position 7, is expected to bind to the target slightly more effectively than its counterpart 8a, which has a sulfonamide group at position 6 (Table 1 and Fig. 3). Consequently, the estimated binding energy (ΔGbind) values for isomer 7a with CA IX are approximately for 2.3 kcal mol−1 lower than those for 6-sulfonamide 8a (Table 1).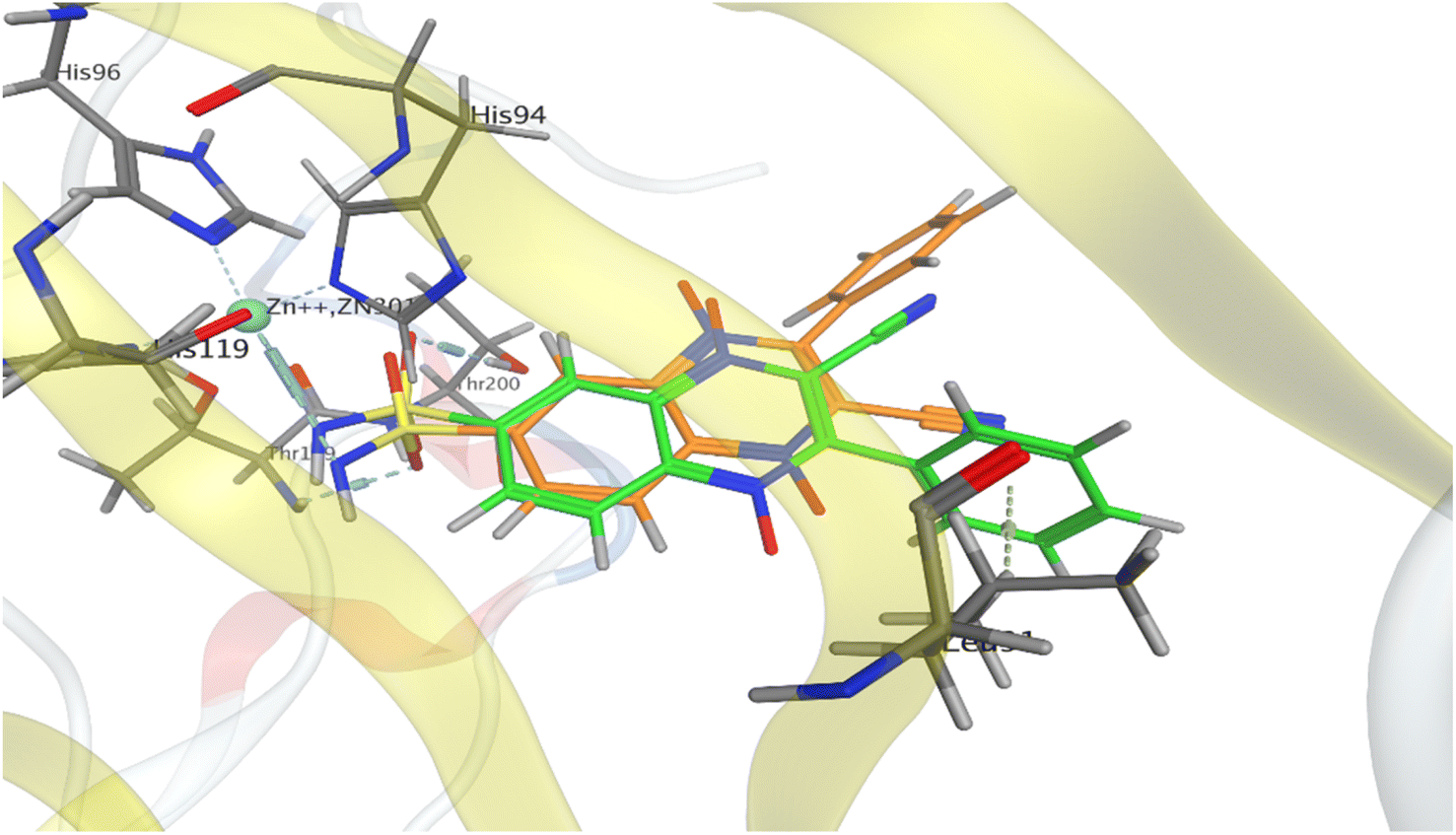 | ||
| Fig. 3 Models of binding of 6- and 7-sulfonamidequinoxaline 1,4-dioxide 7a (orange) and 8a (green) with the active site of carbonic anhydrase CA IX (PDB 5SZ5). | ||
| Compound | ΔGbind (kcal mol−1) | ΔGeq (kcal mol−1) | ΔGHbond (kcal mol−1) |
|---|---|---|---|
 |
−5.9 | −3.5 | −1.2 |
 |
−8.2 | −5.9 | −1.5 |
The binding modes of the isomeric quinoxaline 1,4-dioxides 7a and 8a in the active site of CA IX, as shown in Fig. 3, at first sight appear similar. In addition to forming bonds with Zn2+, the sulfonamide group of 7a and 8a also forms hydrogen bonds with Thr200 and Thr199 residues, respectively, maintaining the same orientation within the active site. However, despite these similarities, significant differences in the binding energies with the target were observed. The established coordination bond of ligand 8a, via the deprotonated sulfonamide group with the Zn2+ ion, has an energy value of −5.9 kcal mol−1, which is 2.4 kcal mol−1 more favourable than the analogous bond of ligand 7a (−3.5 kcal mol−1, Table 1). While the differences in interactions with the Thr199 and Thr200 residues were not as significant, ligand 8a still formed slightly more energetically favorable interactions with the target than ligand 7a (−1.5 and −1.2 kcal mol−1, respectively).
Furthermore, the distinct positioning of the nitrile and phenyl ring results in unequal interactions with the target. The 6-isomer, quinoxaline 1,4-dioxide 8a, engages in an additional hydrophobic interaction between the phenyl ring and Leu91, which is part of the hydrophobic region of the active site. In addition, despite the similarity of the simulated complexes, the binding energy for ligand 8a exceeded the ΔGbind for 7a, respectively (−8.2 versus −5.9 kcal mol−1).
Based on the results of docking studies, a series of 6(7)-sulfonamido-substituted quinoxaline 1,4-dioxides with varying substituents at the positions 2 and 3 of the heterocyclic nucleus was obtained by the Beirut reaction. The key 5-sulfonamidobenzofuroxan (12), required for the synthesis of the designed 6(7)-sulfonamidoquinoxaline 1,4-dioxides, had not been previously described. Mild oxidation of o-nitroanilines enables the synthesis of various functionalized benzofuroxans.27 Therefore, we adapted the previously described procedure for the preparation of the sulfamide analog 12 (Scheme 1). Initially, for their preparation, we developed a synthesis scheme starting from o-nitrochlorobenzene (9). Sulfochlorination of 9, followed by treatment with ammonia, yielded 4-chloro-3-nitrobenzenesulfonamide (10) in high yield (Scheme 1). The presence of two electron-withdrawing substituents in derivative 10 activated the chlorine atom for nucleophilic substitution. Therefore, heating compound 10 with ammonia in ethanol produced the key intermediate 4-amino-3-nitrosulfonamide (11) in high yield. Sulfonamido-substituted nitroaniline 11 is efficiently oxidized with sodium hypochlorite in the presence of KOH in DMF, yielding the desired benzofuroxan 12 in high yield, comparable to previously obtained benzofuroxans (Scheme 1).25,27
 | ||
| Scheme 1 (a) (1) ClSO3H, CHCl3, 0 °C, 30 min than 40 °C, 4 h; (2) NH4OH, THF, 0–5 °C (89%); (b) NH3, EtOH, 2–3 bar, 100 °C, 72 h (83%); (c) NaOCl, KOH(aq.), DMF, 0–5 °C, 30 min (74%). | ||
Despite the effectiveness and simplicity of the initially proven method (Scheme 1), an alternative scheme for synthesizing sulfonamidobenzofuroxan 12 was developed (Scheme 2) to overcome the challenges associated with the harsh conditions required for introducing the amino group in derivative 10. This method is based on the nitration of well-accessible sulfanilamide (13), taking into account the reactivity of the sulfonamide and aniline fragments. For nitration, it is necessary to protect the NH2 groups of sulfanilamide (13). Firstly, acetylation was used to protect the amino group of the aniline fragment in compound 13. It is worth noting that the interaction of sulfanilamide (13) with acetic anhydride proceeds extremely slowly and leads to the selective acylation of the amino group of the aniline. To enhance the acylation rate, we used the addition of catalytic amounts of DMAP. Consequently, N-acetylation rapidly proceeds when sulfonamide 13 is treated with acetic anhydride in refluxing acetic acid in the presence of DMAP, yielding derivative 14 in high yield (Scheme 2).28
 | ||
| Scheme 2 (a) Ac2O, AcOH, DMAP, 120 °C, 4 h (98%); (b) DMF–DMA, DMF, rt, 1 h (99%); (c) HNO3 (100%), H2SO4, 0–5 °C, 2 h (93%); (d) HCl (20%), 100 °C, 2 h (91%); (e) NaOCl, KOH(aq.), DMF, 0–5 °C, 30 min (90%). | ||
It is also known that the interaction of N-unsubstituted sulfonamide derivatives with nitric acid leads to N-nitration, resulting in the formation of unstable N-nitroamide as the major product of the reaction.29 This circumstance requires the protection of the NH2 group of the sulfonamide moiety for a nitration reaction. The amidine group was chosen for this purpose.30 Treatment of acetanilide 14 with N,N-dimethylformamide dimethyl acetal (DMF–DMA) in N,N-dimethylformamide (DMF) at room temperature yielded the key sulfamidine 15 in high yield. Due to the electron-withdrawing effect of the sulfonamide group, the reactivity of the aromatic ring in sulfanilamide derivative 15 in electrophilic substitution reactions is significantly reduced. It seemed appropriate to use a mixture of concentrated nitric and sulfuric acids for the preparation of the target nitro derivative 11.31 Treating acetamide 15 with concentrated HNO3 in H2SO4 at 0–5 °C leads to the nitro derivative 16 in good yield. Deprotection of compound 16 in reflux hydrochloric acid gives 4-amino-3-nitrobenzenesulfonamide (11, Scheme 2). The oxidative cyclization of nitroaniline 11, under previously optimized conditions, by treatment with NaOCl in the presence of KOH in DMF, proved suitable for scaling up the synthesis and provided the target sulfamidobenzofuroxan 12 (Scheme 2).
Furthermore, we examined the possibilities of synthesizing the target sulfamidoquinoxaline 1,4-dioxides through the cyclization of the corresponding benzofuroxan with 1,3-dicarbonyl compounds. However, it was found that the heterocyclization of 5-sulfonamidobenzofuroxan (12) with 1,3-dicarbonyl compounds26,32–38 did not proceed under previously described procedures for the Beirut reaction. In experiments for optimization of reaction conditions we tested several bases (K2CO3, Cs2CO3, pyridine, triethylamine, N,N-diisopropylethylamine, morpholine) in different solvents (CHCl3, MeCN, EtOH, MeOH, THF). It was found that the condensation of benzofuroxan 12 with 1,3-dicarbonyl compounds proceeds quite efficiently in THF in the presence of triethylamine at 50 °C. Nevertheless, the yields of the target quinoxalines 7–8a–h in the Beirut reaction proved to be considerably lower than for previously described monosubstituted quinoxaline 1,4-dioxides.23,25,38 This is attributed to the formation of deoxygenation by-products, as well as difficulties encountered during the purification process of the final compounds. However, despite these challenges, a series of targeted sulfamoylamidoquinoxaline 1,4-dioxides 7–8a–h, with varying substituents in positions 2 and 3 of the heterocycle, were obtained in sufficient quantities to study their properties (Scheme 3 and Table 2). It should be noted that the interaction of sulfonamide-substituted benzofuroxan 12 with 1,3-dicarbonyl compounds leads to a mixture of isomers with different position of the sulfonamide group (derivatives 7a–h and 8a–h).25,27
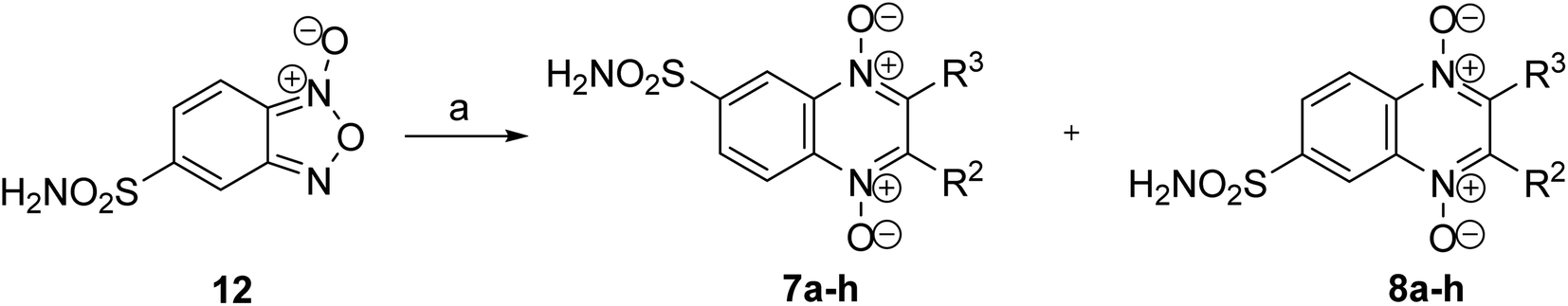 | ||
| Scheme 3 (a) R1COCH2R2, TEA, THF, 50 °C, 5–8 h. | ||
| R2 | R3 | Products | Yields of products, % | |
|---|---|---|---|---|
| 6-Isomers 7a–h | 7-Isomers 8a–h | |||
| CN | Ph | 7a, 8a | 37 | 28 |
| CN | 4-ClC6H4 | 7b, 8b | 42 | 34 |
| CN | 2-Furanyl | 7c, 8c | 11 | 4 |
| CN | 2-Thiophenyl | 7d, 8d | 9 | — |
| CO2Et | Ph | 7e, 8e | 20 | — |
| CO2Et | Me | 7f, 8f | 7 | — |
| COMe | Me | 7g, 8g | 13 | 8 |
| COPh | CF3 | 7h, 8h | 12 | — |
It has been previously demonstrated that the Beirut reaction between monosubstituted benzofurans with electron-withdrawing groups and benzoylacetonitrile gives a mixture of regioisomers, with a predominance of 6-isomers.27 It was revealed that quinoxaline 1,4-dioxides with a sulfamide group in position 6 also predominate in the condensation between sulfamidobenzofuroxan 12 and 1,3-dicarbonyl compounds (Scheme 3). The obtained regioisomers 7a–h and 8a–h demonstrated almost identical spectral characteristics (except for 13C NMR spectra). However, it should be noted that the chromatographic mobility of the components of the mixture of regioisomers significantly depends on the substituents in position 2 of quinoxaline. For example, the isomeric products exhibit close Rf values on TLC and cannot be separated using chromatographic methods in the case of derivatives 7d–f and 7h respectively. Nevertheless, in some examples, namely for derivatives 7a–c, 7g and 8a–c, 8g isomeric mixtures were separated by column chromatography on silica gel and subsequent crystallization. In other examples, biological assessments and physicochemical characterizations were performed for the main 6-isomers of corresponding compounds 7d–f and 7h.
The position of the substituent in compound 7a was confirmed by 2D NMR spectroscopy using HSQC, HMBC, and selective NOESY experiments. The presence of a key four-bond correlation in the HMBC spectra between the H-8 and C-2 signals confirms the structure of derivative 7a (ESI, Fig. S69 and S70†). The structure of compound 7a was confirmed based on a selective NOE experiment, in which the ortho-protons of the phenyl group were selectively inverted. As a result, the signal at 8.87 ppm (H-5) was the only one that increased upon the Overhauser effect, revealing the proximity of the CH group at position 5 of 7a to the phenyl residue at position 3 of the heterocyclic scaffold (ESI, Fig. S70†).
An analogue with a phenylsulfonamide group in position 3 of the heterocycle was synthesized to analyze the role of the location of the sulfonamide group in the quinoxaline 1,4-dioxide core. The previously described hypoxia-selective derivative, 3-phenylquinoxaline-2-carbonitrile 1,4-dioxide 17,23,24 used as the starting compound for this modification. We tested the possibilities of directly introducing the sulfamide group by sulfochlorination of the phenyl ring of derivative 17 and subsequent amidation to obtain the key derivative 18. It was found that the reaction of quinoxaline-2-carbonitrile 1,4-dioxide 17 with chlorosulfonic acid followed by treatment with ammonia led to derivative 18 with a sulfamide group in position 3 of the phenyl moiety (Scheme 4).
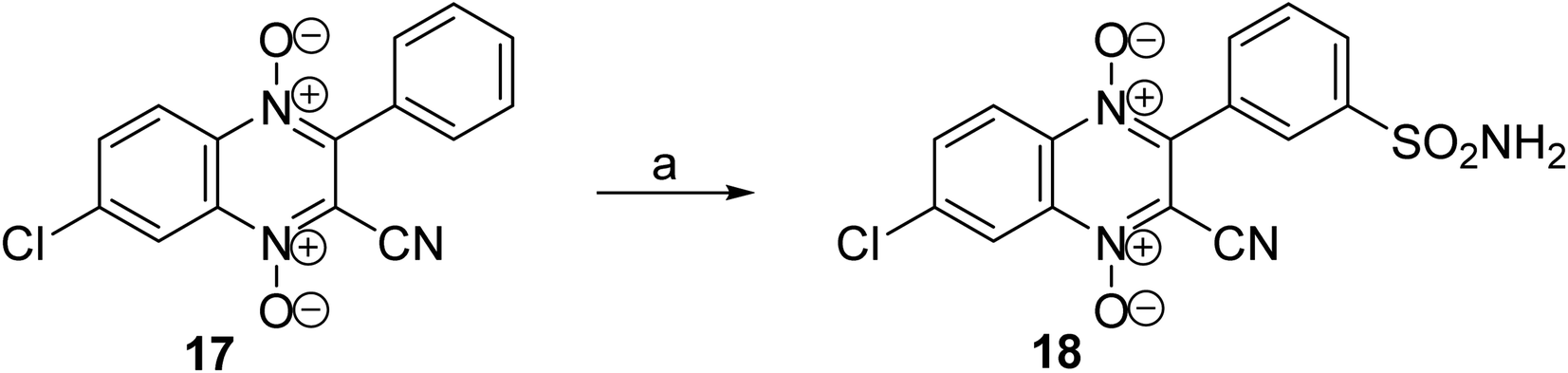 | ||
| Scheme 4 (a) (1) ClSO3H, CHCl3, 60 °C, 4 h; (2) NH4OH, THF, 5–10 °C (34%). | ||
The revealed regioselectivity, as well as the relatively harsh conditions of the sulfochlorination reaction, can be explained by the strong electron-withdrawing character of the quinoxaline 1,4-dioxide nucleus, which has a deactivating meta-orienting effect on the conjugated phenyl in the electrophilic substitution reaction. Also noteworthy is the acceptable stability of the labile quinoxaline 1,4-dioxide ring to treatment with chlorosulfonic acid and ammonia, which allows the obtainment of product 18 in a satisfactory yield.
Biology
All sulfonamide derivatives of quinoxaline 1,4-dioxide 7a–h, 8a, and 18, with varying substituents in positions 2 and 3, were tested for cytotoxicity in vitro against nine human cancer cell lines, including breast adenocarcinoma (MCF-7), pancreatic adenocarcinoma (Capan-1), colorectal carcinoma (HCT116), glioblastoma (LN229), lung carcinoma (NCI-H1975), acute lymphoblastic leukemia (DND-41), acute myeloid leukemia (HL-60), chronic myeloid leukemia (K562), and non-Hodgkin's lymphoma (Z138). Tirapazamine (TPZ) was used as a reference agent for testing hypoxic cytotoxicity (Table 3). Positive controls for the screening of antiproliferative activity included doxorubicin (DOX), and etoposide (Table 4). Acetazolamide (AAZ) was used as a reference agent for assessing inhibitory activity against various carbonic anhydrase isoforms (Table 5).39 The results of the evaluation of hypoxic cytotoxicity, spectrum of antiproliferative activity properties of the new series of quinoxaline 1,4-dioxide derivatives, as well as their ability to inhibit the enzymatic activity of CA I, CA II, CA IX, and CA XII, are presented in Tables 3–5, respectively.| Compound | IC50 (μM) | HCRd | |
|---|---|---|---|
| Nb | Hc | ||
| a IC50, μM (mean ± S.D. of 3 experiments).b N = normoxia: 21% of oxygen.c H = hypoxia: 1% of oxygen.d HCR, hypoxic cytotoxicity ratio: IC50(N)/IC50(H). | |||
| 7a | 3.5 ± 0.2 | 0.9 ± 0.3 | 3.9 |
| 7b | 1.8 ± 0.2 | 0.8 ± 0.2 | 2.2 |
| 7c | >25 | >25 | — |
| 7d | >25 | >25 | — |
| 7e | 4.0 ± 0.6 | 0.9 ± 0.06 | 4.7 |
| 7f | 10.5 ± 1.2 | 4.3 ± 0.3 | 2.4 |
| 7g | 7.1 ± 0.2 | 6.9 ± 0.3 | 1.1 |
| 7h | 1.1 ± 0.1 | 0.8 ± 0.05 | 1.4 |
| 8a | 4.8 ± 0.2 | 1.3 ± 0.3 | 3.7 |
| 8g | 11.7 ± 1.2 | 2.8 ± 0.4 | 4.2 |
| 18 | 8.9 ± 0.9 | 2.8 ± 0.3 | 3.1 |
 |
24.2 ± 3.3 | 4.5 ± 1.1 | 5.4 |
| DOXO | 0.3 ± 0.03 | 0.4 ± 0.03 | 0.7 |
| Cmpnd | IC50 (μM) | |||||||
|---|---|---|---|---|---|---|---|---|
| Capan-1 | HCT-116 | LN229 | NCI-H1975 | DND-41 | HL-60 | K562 | Z138 | |
| a IC50, μM (mean ± S.D. of 3 experiments). | ||||||||
| 7a | 7.5 ± 0.4 | 47.9 ± 2.3 | 38.8 ± 1.9 | 19.9 ± 0.9 | 0.8 ± 0.05 | 2.3 ± 0.1 | 10.8 ± 0.5 | 8.7 ± 0.4 |
| 7b | 1.8 ± 0.1 | 34.3 ± 1.7 | 24.0 ± 1.2 | 13.9 ± 0.7 | 5.4 ± 0.3 | 2.0 ± 0.1 | 29.7 ± 1.5 | 2.5 ± 0.2 |
| 7c | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 7d | >100 | >100 | 98.8 ± 4.9 | >100 | 44.8 ± 2.2 | 72.4 ± 3.6 | >100 | >100 |
| 7e | 6.0 ± 0.3 | >100 | 87.0 ± 4.3 | 47.4 ± 2.3 | 28.2 ± 1.4 | 52.7 ± 2.6 | 52.6 ± 2.1 | 15.3 ± 0.8 |
| 7f | 1.7 ± 0.1 | 2.7 ± 0.1 | 35.9 ± 1.8 | 33.3 ± 1.7 | 10.3 ± 0.5 | 32.6 ± 1.6 | 26.0 ± 1.3 | 3.3 ± 1.7 |
| 7g | 38.3 ± 1.9 | 69.6 ± 3.5 | >100 | 66.1 ± 3.3 | 66.9 ± 3.3 | 85.9 ± 4.3 | 94.8 ± 4.7 | 44.3 ± 2.2 |
| 7h | 1.4 ± 0.07 | 1.3 ± 0.08 | 1.4 ± 0.03 | 1.9 ± 0.1 | 2.1 ± 0.2 | 1.8 ± 0.1 | 1.9 ± 0.2 | 1.9 ± 0.1 |
| 18 | 1.5 ± 0.01 | 15.1 ± 0.8 | 7.5 ± 0.4 | 2.2 ± 0.1 | 2.4 ± 0.1 | 2.5 ± 0.1 | 8.1 ± 0.4 | 3.4 ± 0.2 |
| Etoposide | 0.13 ± 0.01 | 2.3 ± 0.1 | 1.7 ± 0.1 | 1.1 ± 0.1 | 0.29 ± 0.01 | 0.7 ± 0.04 | 1.2 ± 0.06 | 0.3 ± 0.02 |
| Cmpnd | Kia (nM) | |||
|---|---|---|---|---|
| hCA I | hCA II | hCA IX | hCA XII | |
| a Mean from three experiment using a stopped flow CO2 hydrase assay. | ||||
| 7a | 49.1 ± 3 | 2.7 ± 0.2 | 2396 ± 96 | 89.0 ± 7 |
| 7b | 53.7 ± 1.5 | 5.1 ± 0.3 | 429 ± 21 | 178 ± 9 |
| 7c | 38.0 ± 1.1 | 4.4 ± 0.2 | >10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
>10000 |
| 7d | 41.7 ± 2 | 4.2 ± 0.1 | >10000 |
178 ± 13 |
| 7e | 42.4 ± 1.9 | 5.4 ± 0.3 | 257 ± 14 | 252 ± 14 |
| 7f | 63.0 ± 3.1 | 8.0 ± 0.3 | >10000 |
56.4 ± 4 |
| 7g | 65.7 ± 2.4 | 7.4 ± 0.2 | 42.2 ± 4 | 240 ± 13 |
| 7h | 51.5 ± 3.2 | 4.8 ± 0.3 | >10000 |
143 ± 11 |
| 8a | 60.7 ± 2.6 | 4.4 ± 0.1 | >10000 |
111 ± 9.7 |
| 8b | 40.3 ± 3.1 | 5.0 ± 0.4 | >10000 |
133 ± 12 |
| 18 | 90 ± 4.8 | 41.5 ± 2 | >10000 |
127 ± 6.8 |
| AAZ | 250 ± 13 | 12.1 ± 0.2 | 25.7 ± 1.8 | 5.7 ± 0.3 |
Antiproliferative activity
Evaluation of the antiproliferative activity against breast cancer cells (MCF-7) of quinoxaline 1,4-dioxides 7a–h, 8a, g, and 18 under hypoxic and normoxic conditions showed that most of the obtained compounds inhibit the growth of tumor cells at micromolar concentrations (Table 3). Furthermore, the cytotoxicity of most synthesized derivatives increased by 1.5–4.7 times under hypoxic conditions. Among all the series of sulfonamido derivatives, 2-carboethoxy-6-sulfonamido-3-phenylquinoxaline 1,4-dioxide (7e) was the most active and hypoxia-selective with IC50 values of 4.0 and 0.9 μM under normoxia and hypoxia, respectively.Analysis of the obtained data revealed the significant role of the nitrile function at position 2 of the heterocyclic ring of quinoxaline 1,4-dioxide in the cytotoxic properties of these compounds. When the cyano group (derivatives 7a–d) was replaced with acyl or ethoxycarbonyl groups, which have similar electronic influences, it led to a decrease in the activity of these compounds (derivatives 7e, 7f, respectively). The trifluoromethyl group at position 3 of quinoxaline also significantly potentiated the antiproliferative properties. For instance, the trifluoromethyl derivative 7h exhibited significantly higher activity against most tumor cells (1.5–20 times) compared to other analogs. However, the introduction of a trifluoromethyl group reduced the hypoxic selectivity index. It was observed that introducing an aromatic fragment at position 3 of quinoxaline generally had a positive effect on both the antitumor properties of these derivatives (compounds 7a–b, 7e; Table 3) and their selectivity under hypoxic conditions. In contrast, replacing the phenyl with its bioisosteric analogs, such as furyl and thienyl (compounds 7c and 7d, respectively), led to a complete loss of activity. Another critical factor affecting the ability of these compounds to inhibit tumor cell growth is the position of the key sulfonamide group on the benzene ring of the heterocycle. Shifting the sulfonamide group from position 6 to 7 of quinoxaline (derivatives 7a and 8a, 7g and 8g) reduced the antiproliferative activity of these compounds by approximately 1.5 times under normoxic conditions and by 1.4–5.6 times under hypoxia (Table 3). It is interesting to note that the introduction of a sulfonamide group into the phenyl ring at position 3 of quinoxaline had a negative impact on the cytotoxicity of compound 18 under both normoxic and hypoxic conditions (IC50 = 8.9 and 2.8 μM, respectively). Surprisingly, this modification did not affect the value of the hypoxic selectivity index (HCR = 3.1, Table 3). Therefore, the position of the sulfonamide group in the benzene ring of quinoxaline 1,4-dioxide also significantly influences the antitumor properties of these derivatives.
The spectrum of antiproliferative properties of new quinoxaline 1,4-dioxide derivatives 7a–h and 18 was studied, comparing them to etoposide, against an expanded panel of tumor cells, including eight lines of different histogenesis under normoxic conditions (Table 4). The screening results revealed that pancreatic adenocarcinoma cells Capan-1 were the most sensitive to sulfonamide-substituted quinoxaline 1,4-dioxides, while glioblastoma cells LN229 and colon adenocarcinoma cells HCT116 were relatively sustainable to the obtained derivatives. Thus, for compounds 7a–h and 18, the IC50 value for these cell lines differed in 10–20 times (Table 4).
It was observed that the presence of a halogen atom in the phenyl ring at position 3 of quinoxaline 1,4-dioxide 7b generally enhances the activity of this derivative (Table 4). Additionally, the halogen atom at this position significantly contributes to the activity of the synthesized derivatives. For instance, the introduction of a chlorine atom enhances the cytotoxicity of derivative 7b against all tested cell lines except DND-41 by 1.2 to 6.8 times compared to its unsubstituted analogue 7a. Replacement of the phenyl group at position 3 of quinoxaline (compound 7a) with its bioisosteric heteroaromatic analogues, such as furyl and thienyl (compounds 7c, 7d), leads to a complete or partial loss of activity for all tested cell lines. This observation aligns with the results obtained for breast cancer cells (MCF-7) (Table 4). Interestingly, the introduction of a sulfonamide group into the phenyl ring at the C3 carbon atom of the heterocycle (derivative 18) generally enhances the ability to inhibit the growth of tumor cells with various histogenesis. Compound 18 inhibits the growth of all tested tumor cells within the micromolar to low micromolar concentration range (IC50 = 1.5–15.1 μM), which is comparable to the activity of the reference drug etoposide (IC50 = 0.13–2.3 μM).
It is worth noting the important role of the cyano group at position 2 of quinoxaline (in compounds 7a–d, 18) in the cytotoxic properties of these derivatives. Replacing it with an ethoxycarbonyl group with similar electronic effects (compounds 7e, 7f) leads to a noticeable increase in the IC50 value (2–35 times) against all cell lines, except for pancreatic cancer cells (Capan-1). Equally critical is the modification of the substituent at position 3 of quinoxaline-1,4-dioxide. For instance, replacing the phenyl group in compound 7e with a methyl group (derivative 7f) significantly enhances (1.4–40 times) the ability of compound 7f to inhibit the growth of tumor cells. Furthermore, a comparison between the activity of the 2-acetyl derivative 7g and its 2-ethoxycarbonyl analogue 7f reveals that the presence of an acetyl residue at position 2 of the heterocycle results in a 2–25-fold decrease in the activity of compound 7g. In contrast, the introduction of a trifluoromethyl group at position 2 of quinoxaline 1,4-dioxide increased the cytotoxic properties of derivatives of this series. So, compound 7h, which effectively suppressed the growth of all tumor cells at low micromolar concentrations (IC50 = 1.3–2.1 μM), emerged as the most active in the series of quinoxaline 1,4-dioxides. It exhibits a similar activity to the topoisomerase II inhibitor etoposide (IC50 = 0.13–2.3 μM). Thus, the results of the antiproliferative activity evaluation on a broad panel of tumor cell lines show that substituents at positions 2 and 3 significantly affect the cytotoxicity of sulfonamide derivatives of quinoxaline 1,4-dioxide.
Simultaneously, the introduction of methyl, trifluoromethyl, fluorophenyl, or nitrile group leads to a significant increase in activity against certain tumor cell lines, primarily Capan-1 pancreatic adenocarcinoma, as well as LN229, DND-41, HL-60, and K562 cells (Table 4). The data obtained from SAR analysis may potentially promote the effective modulation of the spectrum of antitumor properties in future quinaxoline-1,4-dioxide derivatives.
Carbonic anhydrase inhibition assay
Targeting CA IX represents a promising oncological approach, with the goal of overcoming the progression of the most aggressive tumors, including those characterized by extensive hypoxic regions. The inhibition of carbonic anhydrases CAIX and CAXII, which are expressed in response to hypoxia and activated by the transcription factor HIF-1α, is one of sulfonamides.40 Therefore, we assessed the action of sulfonamidoquinoxaline 1,4-dioxides 7a–h, 8a, b and 18 in comparison to acetazolamide (AAZ) on the catalytic activity of pharmacologically relevant human CA (hCA) isoforms, including cytosolic hCA I and hCA II, as well as membrane-bound tumor-associated isoforms hCA IX and hCA XII (Table 5).The inhibition constant values (Ki) demonstrated that quinoxaline 1,4-dioxides 7a–h, 8a, b and 18 can inhibit CA isoforms in range of the low micromolar to nanomolar concentrations (Table 5). However, the majority of derivatives exhibited higher activity against hCA I and hCA II isoforms than against hCA XII and hCA IX. Thus, the cytosolic isoforms were inhibited with Ki values in the range of 33.6–65.7 nM (hCA I) and 2.7–8.0 nM (hCA II), respectively. For the membrane-bound isoforms hCA IX and hCA XII, the Ki values ranged from 42 nM to >10 μM. As a result, the sulfonamides 7a–h and 8a, b are 2–8 times more potent against CA I and CA II than the reference drug AAZ. Additionally, the CA XII isoform has higher susceptibility to quinoxaline 1,4-dioxides compared to CA IX. Notably, the introduction of a halogen atom at the para-position of the benzene ring in position 3 of quinoxaline-2-carbonitrile 1,4-dioxides results in a slight decrease in inhibitory activity for derivative 7b against the tested carbonic anhydrase isoforms when compared to their unsubstituted analog 7a.
When comparing the inhibitory potential of the obtained sulfonamide derivatives of quinoxaline 1,4-dioxide, it is evident that the presence of a phenyl ring in position 3 of the heterocycle enhances the ability of this chemotype to inhibit CAIX activity. This enhancement can be attributed to additional hydrophobic interactions in the active site of the enzyme. It was also discovered that replacing of the phenyl group in position 3 of quinoxaline with furyl- and thienyl group led to a complete loss of activity for compounds 7c and 7d against CA IX (Table 5). However, these substitutions retain high activity against CA I and CA II isoforms. Additionally, 6-sulfonamido-3-furylquinoxaline-2-carbonitrile 1,4-dioxide (7c) was also found to be inactive against the CA XII isoform. Interestingly, when a sulfonamide group was introduced into the phenyl ring at the C3 carbon atom of quinoxaline, it had a negative effect on the inhibitory ability of derivative 18 compared to a series of 6(7)-sulfonamide derivatives of quinoxaline 1,4-dioxide toward all CA isoforms.
Among the tested series of quinoxaline 1,4-dioxides, the most active inhibitor of CA IX was 2-acetyl-3-methyl-6-sulfonamidoquinoxaline 1,4-dioxide (7g). This compound demonstrated activity comparable to the reference drug acetazolamide (AAZ) against CA IX, with Ki values of 42.2 and 12.1 nM, respectively. Notably, 6-sulfonamido-2-carboethoxy-3-methylquinoxaline 1,4-dioxide (7f) emerged as the most active inhibitor of CA XII among the series of obtained derivatives, but it did not exhibit any inhibitory activity against the CA IX isoform. In summary, it is worth noting that some compounds not only exhibit cytotoxic activity but also have the ability to inhibit CA activity.
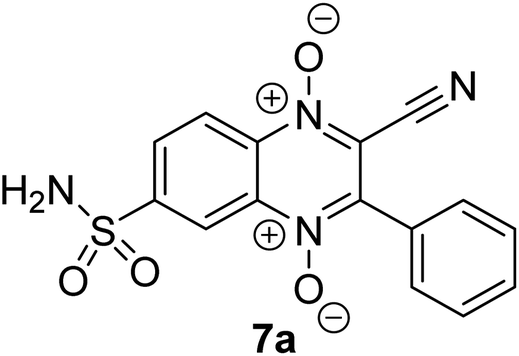
Molecular docking studies
Based on the results of screening of CA inhibition activity of sulfonamidoquinoxaline 1,4-dioxides, we docked the most active inhibitor, CA IX (compound 7g), into the active site (Fig. 4). According to the docking simulations, quinoxaline 1,4-dioxide 7g forms a coordination bond with the Zn2+ ion and a hydrogen bond with the Thr200 residue. | ||
| Fig. 4 Binding mode of compound 7g in the hCA IX active site. | ||
In comparison with nitrile 7a (Fig. 3), the acetyl derivative 7g lacks π–H interaction with the hydrophobic residue Leu91. Instead, derivative 7g establishes an additional hydrogen bond with the side carboxamide residue of Gln67 and N-oxide group of the ligand. The obtained model indicates the presence of a strong coordination bond between 7g and Zn2+ ion with a value of −6.2 kcal mol−1. The establishment of a new hydrogen bond contributed to the affinity of the ligand to the active center of CA IX and to the final value of ΔGbind = −8.6 kcal mol−1. The results of in vitro screening and docking models suggest that not only the structure and nature of the substituents but also their positioning within the quinoxaline core may be determining factors in the inhibition of CA IX. This facilitates better binding of the sulfonamide group to the Zn2+ ion and other key amino acid residues in the active site of CA IX.
These observations are consistent with previously published data, where the most active CA IX inhibitor also formed a hydrogen bond with the Gln67 residue.41 Thus, the affinity of 6- and 7-sulfonamides of quinoxaline 1,4-dioxide highly depends on substituents at positions 2 and 3, forming interactions with amino acid residues of CA IX.
Mechanism of tumor cell death
The growth of tumor cells under hypoxic conditions is accompanied by significant metabolic changes.42 Hypoxia impacts the activity of crucial enzymes that play a role in maintaining cell survival.43 These alterations lead to the development of a resistant phenotype and a decrease in the efficacy of chemotherapy.44 Consequently, the identification of compounds exhibiting selectivity for tumor cells in hypoxic conditions stands as a top priority in cancer pharmacology.45,46 For an in-depth investigation under reduced oxygen conditions (1% O2), the most promising inhibitor of CA IX was chosen (compound 7g). Subsequent experiments focused on aggressive epidermoid cancer A431 cells, known for developing a resistant phenotype under hypoxia.47–49 So, A431 cells were treated with compound 7g, and their survival was analyzed after 72 h using the MTT assay. As shown in Fig. 5, the antiproliferative potency of compound 7g was limited in normoxia. At low concentrations, the compound 7g slightly inhibited the growth of A431 cells. However, increasing its concentration to 50 μM resulted in an approximately 40% decrease in cell survival, with an IC50 value higher than 50 μM in normoxia. Interestingly, transferring A431 cells treated with compound 7g to hypoxic conditions significantly impacted their survival rate. In hypoxia, the compound 7g exhibited considerable antiproliferative effects at concentrations higher than 10 μM, with an IC50 value of approximately 11 μM. This suggests that compound 7g demonstrates high activity under hypoxic conditions.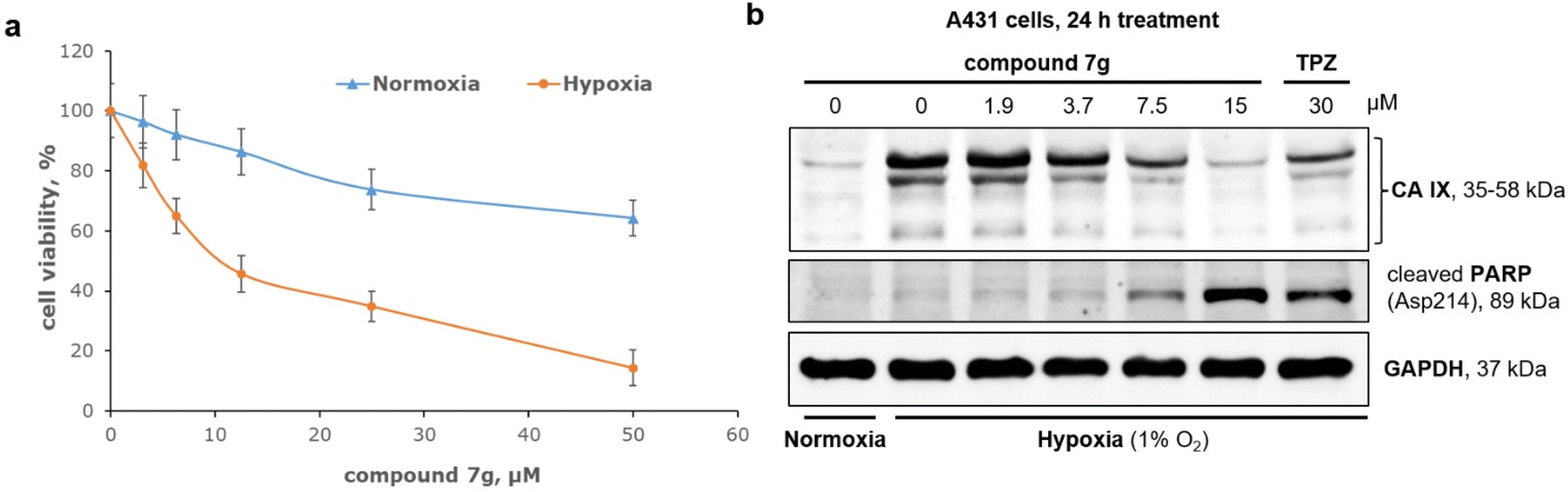 | ||
| Fig. 5 Activity of compound 7g against skin cancer cells A431 in normoxia and hypoxia. (a) Antiproliferative effects of compound 7g on A431 skin cancer cells; A431 cells were treated with compound 7g for 72 h and then cell viability was assessed by the MTT assay. (b) Effect of compound 7g on CA IX and cleaved PARP expression; TPZ – tirapazamine (a reference drug), GAPDH – a loading control. | ||
Hypoxia induces the expression of various hypoxic factors crucial for cellular adaptation to stress.50 Among these factors, CA IX plays a key role by facilitating a gradual acidification of the extracellular environment.51 As illustrated in Fig. 5b, hypoxia led to a notable increase in CA IX expression in A431 cells. The compound 7g resulted in a dose-dependent reduction in CA IX expression under hypoxic conditions. Furthermore, treatment with compound 7g led to the accumulation of cleaved PARP, a well-established marker of apoptosis. For comparison, the TPZ,52 a well-known hypoxic cytotoxin, was used at a concentration of 30 μM as a reference drug. Interestingly, the effects of compound 7g on both CA IX expression and the apoptosis marker PARP were more pronounced compared to TPZ.
In conclusion, the comprehensive data from molecular modeling, screening, and immunoblotting strongly suggest that compound 7g not only inhibits CA IX but also significantly suppresses its expression in tumor cells under hypoxia, possibly similar to other derivatives of quinoxaline 1,4-dioxide by blocking HIF-1α.23–25,27 Inhibition of CA IX in A431 cells is associated with PARP cleavage, indicating the induction of apoptosis.
Experimental section
Chemistry
All solutions were evaporated at a reduced pressure on a Buchi-R200 rotary evaporator at temperature below 50 °C. All products were dried under vacuum at room temperature. All solvents, chemicals, and reagents were obtained from Sigma-Aldrich (unless specified otherwise) and used without purification. The purity of all synthesized compounds was >95% as determined by HPLC analysis.
:1). The crude mixture of isomers was separated by column chromatography on silica gel: for derivatives 7–8a–d, the eluting solvent toluene–diethyl ether mixture (5:1) was used, while for derivatives 7–8e–h, the eluting solvent chloroform–acetone (6:1) was used. Crystallization of the obtained products from a toluene-ethylacetate mixture yielded the pure 6-isomer and 7-isomer.
3-Phenyl-6-sulfamoylquinoxaline-2-carbonitrile 1,4-dioxide (7a) and 2-phenyl-6-sulfamoylquinoxaline-2-carbonitrile 1,4-dioxide (8a). This compound was prepared from benzofuroxan 12 and benzoylacetonitrile according to the general procedure.
3-Phenyl-6-sulfamoylquinoxaline-2-carbonitrile 1,4-dioxide (7a). The yield of 7a was 0.28 g (37%) as deep yellow powder, mp 209–210 °C. Rf = 0.5 (CHCl3–EtOAc, 1
:2). HPLC (LW = 300 nm, gradient B 20/80% (45 min)) tR = 16.94 min, purity 96.6%. λmax, EtOH: 243, 276, 295, 375, 433 nm. 1H NMR (400 MHz, DMSO-d6) δ 8.87 (1H, s, H-5); 8.73 (1H, d, J = 8.6, H-8); 8.41 (1H, d, J = 8.6, H-7); 8.01 (2H, br. s, SO2NH2); 7.77–7.74 (2H, m, HAr); 7.66–7.63 (3H, m, HAr). 13C NMR (100 MHz, DMSO-d6) δ 147.4 (6-C); 144.1 (3-C); 140.2 (10-C); 136.8 (9-C); 131.4 (4′-CH); 130.4 (7-CH); 130.1 (2 × 2′-CH); 128.7 (2 × 3′-CH); 127.4 (1′-C); 122.6 (8-CH); 121.4 (2-C); 117.8 (5-CH); 110.9 (CN). HRMS (ESI) calculated for C15H11N4O4S+ [M + H]+ 343.0496, found 343.0586.
3-Phenyl-7-sulfamoylquinoxaline-2-carbonitrile 1,4-dioxide (8a). The yield of 8a was 0.09 g (28%) as yellow powder, mp 234–235 °C. Rf = 0.3 (CHCl3–EtOAc, 1
:2). HPLC (LW = 300 nm, gradient B 20/80% (45 min)) tR = 16.82 min, purity 95.1%. λmax, EtOH: 246, 277, 299, 394 nm. 1H NMR (400 MHz, DMSO-d6) δ 8.91 (1H, s, H-8); 8.72 (1H, d, J = 8.6, H-5); 8.37 (1H, dd, J3 = 8.6, J4 = 1.6, H-6); 7.98 (2H, br. s, SO2NH2); 7.76–7.73 (2H, m, HAr); 7.66–7.64 (3H, m, HAr). 13C NMR (100 MHz, DMSO-d6) δ 148.6 (7-C); 143.7 (3-C); 138.9 (9-C); 138.1 (10-C); 131.3 (4′-CH); 130.0 (2 × 2′-CH); 128.8 (6-CH); 128.7 (2 × 3′-CH); 127.4 (1′-C); 122.1 (5-CH); 121.6 (2-C); 118.3 (8-CH); 110.9 (CN). HRMS (ESI) calculated for C15H11N4O4S+ [M + H]+ 343.0496, found 343.0508.
3-(4-Chlorophenyl)-6-sulfamoylquinoxaline-2-carbonitrile 1,4-dioxide (7b) and 3-(4-chlorophenyl)-6-sulfamoylquinoxaline-2-carbonitrile 1,4-dioxide (8b). This compound was prepared from benzofuroxan 12 and 3-(4-chlorophenyl)-3-oxopropanenitrile according to the general procedure.
3-(4-Chlorophenyl)-6-sulfamoylquinoxaline-2-carbonitrile 1,4-dioxide (7b). The yield of 7b was 157 mg (42%) as yellow powder, mp 160–162 °C. Rf = 0.6 (CHCl3–EtOAc, 1
:3). HPLC (LW = 300 nm, gradient B 30/80% (45 min)) tR = 16.9 min, purity 99.0%. λmax, EtOH: 226, 244, 278, 301, 377 nm. 1H NMR (400 MHz, DMSO-d6) δ 8.89 (1H, s, H-5); 8.72 (1H, d, J = 8.9, H-8); 8.37 (1H, dd, J3 = 8.9, J4 = 1.8, H-7); 7.99 (2H, br. s, SO2NH2); 7.78–7.74 (4H, m, HAr). 13C NMR (100 MHz, DMSO-d6) δ 148.7 (6-C); 142.7 (3-C); 138.8 (9-C); 138.0 (10-C); 136.2 (4′-CCl); 131.9 (2 × 2′-CH); 128.9 (7-CH); 128.8 (2 × 3′-CH); 126.1 (1′-C); 122.1 (8-CH); 121.4 (2-C); 118.1 (5-CH); 110.7 (CN). HRMS (ESI) calculated for C15H8ClN4O4S− [M − H]− 374.9960, found 374.9823.
3-(4-Chlorophenyl)-7-sulfamoylquinoxaline-2-carbonitrile 1,4-dioxide (8b). The yield of 8b was 127 mg (34%) as yellow powder, mp 237–238 °C. Rf = 0.5 (CHCl3–EtOAc, 1
:3). HPLC (LW = 300 nm, gradient B 30/80% (45 min)) tR = 16.8 min, purity 100.0%. λmax, EtOH: 227, 245, 278, 303, 382 nm. 1H NMR (400 MHz, DMSO-d6) δ 8.86 (1H, d, J = 1.8, H-8); 8.73 (1H, d, J = 8.9, H-5); 8.42 (1H, dd, J3 = 8.9, J4 = 1.8, H-6); 8.00 (2H, br. s, SO2NH2); 7.78–7.74 (4H, m, HAr). 13C NMR (100 MHz, DMSO-d6) δ 147.5 (7-C); 143.1 (3-C); 140.0 (10-C); 136.7 (9-C); 136.2 (4′-CCl); 131.9 (2 × 2′-CH); 130.4 (6-CH); 128.9 (2 × 3′-CH); 126.2 (1′-C); 122.5 (5-CH); 121.2 (2-C); 117.7 (8-CH); 110.7 (CN). HRMS (ESI) calculated for C15H10ClN4O4S+ [M + H]+ 377.0106, found 377.0159.
3-(Furan-2-yl)-6-sulfamoylquinoxaline-2-carbonitrile 1,4-dioxide (7c) and 2-(furan-2-yl)-6-sulfamoylquinoxaline-2-carbonitrile 1,4-dioxide (8c). This compound was prepared from benzofuroxan 12 and 3-(furan-2-yl)-3-oxopropanenitrile according to the general procedure.
3-(Furan-2-yl)-6-sulfamoylquinoxaline-2-carbonitrile 1,4-dioxide (7c). The yield of 7c was 34 mg (11%) as yellow powder, mp 174–176 °C. Rf = 0.6 (CHCl3–Me2CO, 1
:3). HPLC (LW = 300 nm, gradient B 40/80% (45 min)) tR = 9.86 min, purity 95.7%. λmax, EtOH: 229, 255, 307, 394 nm. 1H NMR (400 MHz, DMSO-d6) δ 8.46 (1H, s, H-5); 8.37 (1H, d, J = 8.9, H-8); 8.23 (1H, dd, J3 = 8.9, J4 = 1.5, H-7); 8.21–8.19 (1H, m, HAr); 7.84 (2H, br. s, SO2NH2); 7.71 (1H, d, J = 3.9, HAr); 6.91–6.89 (1H, m, HAr). 13C NMR (100 MHz, DMSO-d6) δ 148.3 (1′-C); 148.1 (6-C); 147.4 (4′-CH); 144.3 (3-C); 140.7 (10-C); 140.1 (9-C); 130.7 (5-CH); 127.4 (2-C); 127.1 (7-CH); 125.9 (8-CH); 116.3 (CN); 115.9 (2′-CH); 113.3 (3′-CH). HRMS (ESI) calculated for C13H9N4O5S+ [M + H]+ 333.0288, found 333.0393.
3-(Furan-2-yl)-7-sulfamoylquinoxaline-2-carbonitrile 1,4-dioxide (8c). The yield of 8c was 12 mg (4%) as yellow powder, mp 210–212 °C. Rf = 0.5 (CHCl3–Me2CO, 1
:3). HPLC (LW = 300 nm, gradient B 40/80% (45 min)) tR = 9.58 min, purity 99.4%. λmax, EtOH: 229, 254, 302, 391 nm. 1H NMR (400 MHz, DMSO-d6) δ 8.48 (1H, s, H-8); 8.33–8.31 (2H, br. m, H-5, H-6); 8.21–8.20 (1H, br. m, HAr); 7.80 (2H, br. s, SO2NH2); 7.72 (1H, d, J = 3.7, HAr); 6.92–6.90 (1H, br. m, HAr). 13C NMR (100 MHz, DMSO-d6) δ 148.4 (1′-C); 147.5 (4′-CH); 145.7 (7-C); 144.6 (3-C); 142.4 (10-C); 138.3 (9-C); 130.2 (8-CH); 129.8 (5-CH); 127.1 (2-C); 126.4 (6-CH); 116.3 (CN); 116.2 (2′-CH); 113.3 (3′-CH). HRMS (ESI) calculated for C13H9N4O5S+ [M + H]+ 333.0288, found 333.0300.
6-Sulfamoyl-3-(thiophen-2-yl)quinoxaline-2-carbonitrile 1,4-dioxide (7d). This compound was prepared from benzofuroxan 12 and 3-oxo-3-(thiophen-2-yl)propanenitrile according to the general procedure. The yield of 7d was 31 mg (9%) as yellow powder, mp 203–204 °C. Rf = 0.55 (CHCl3–Me2CO, 3
:1). HPLC (LW = 300 nm, gradient B 40/80% (45 min)) tR = 13.8 min, purity 98.9%. λmax, EtOH: 228, 253, 296, 389 nm. 1H NMR (400 MHz, DMSO-d6) δ 8.45 (1H, s, H-5); 8.38–8.31 (2H, m, HAr); 8.22 (1H, d, J = 8.6, H-7); 8.05 (1H, d, J = 8.6, H-8); 7.84 (2H, br. s, SO2NH2); 7.38 (1H, d, J = 4.1, HAr). 13C NMR (100 MHz, DMSO-d6) δ 148.1 (6-C); 140.7 (10-C); 140.1 (9-C); 133.7 (3-C); 133.5 (4′-CH); 130.7 (2′-CH); 130.5 (5-CH); 130.2 (1′-C); 129.9 (2-C); 129.4 (7-CH); 127.1 (3′-CH); 125.8 (8-CH); 116.9 (CN). HRMS (ESI) calculated for C13H9F3N4O4S2+ [M + H]+ 349.0060, found 349.0091.
2-(Ethoxycarbonyl)-3-phenyl-6-sulfamoylquinoxaline 1,4-dioxide (7e). This compound was prepared from benzofuroxan 12 and ethyl 3-oxo-3-phenylpropanoate according to the general procedure. The yield of 7e was 40 mg (20%) as an orange powder, mp 210–211 °C. Rf = 0.5 (CHCl3–Me2CO, 1
:3). HPLC (LW = 270 nm, gradient B 30/80% (45 min)) tR = 12.61 min, purity 96.7%. λmax, EtOH: 238, 271, 289, 393 nm. 1H NMR (400 MHz, DMSO-d6) δ 8.85 (1H, d, J = 2.0, H-5); 8.68 (1H, d, J = 8.8, H-8); 8.32 (1H, dd, J3 = 8.8, J4 = 2.0, H-7); 7.93 (2H, br. s, SO2NH2); 7.58–7.54 (5H, br. m, C6H5); 4.16 (2H, q, J = 7.3, OCH2CH3); 0.95 (3H, t, J = 7.3, OCH2CH3). 13C NMR (100 MHz, DMSO-d6) δ 159.3 (CO); 147.4 (6-C); 140.0 (3-C); 138.7 (10-C); 137.3 (9-C); 136.7 (2-C); 131.0 (4′-CH); 130.3 (2 × 2′-CH); 129.3 (7-CH); 128.9 (2 × 3′-CH); 128.2 (1′-C); 122.8 (8-CH); 118.2 (5-CH); 63.2 (CH3CH2O); 13.8 (CH3CH2O). HRMS (ESI) calculated for C17H16N3O6S+ [M + H]+ 390.0754, found 390.0650.
2-(Ethoxycarbonyl)-3-methyl-6-sulfamoylquinoxaline 1,4-dioxide (7f). This compound was prepared from benzofuroxan 12 and acetoacetic ester according to the general procedure. The yield of 7f was 10 mg (3%) as light yellow powder, mp 108–110 °C. Rf = 0.4 (CHCl3–EtOAc, 1
:3). HPLC (LW = 254 nm, gradient B 20/80% (45 min)) tR = 10.5 min, purity 94.1%. λmax, EtOH: 238, 270, 383 nm. 1H NMR (400 MHz, DMSO-d6) δ 8.64 (1H, d, J = 8.9, H-5); 8.28 (1H, dd, J3 = 8.9, J4 = 1.5, H-6); 7.88 (2H, br. s, SO2NH2); 7.79 (1H, d, J = 1.5, H-8); 4.52 (2H, q, J = 7.3, OCH2CH3); 2.45 (3H, s, CH3); 1.36 (3H, t, J = 7.3, OCH2CH3). 13C NMR (100 MHz, DMSO-d6) δ 159.4 (CO); 146.3 (6-C); 140.0 (3-C); 138.8 (10-C); 136.2 (9-C); 135.7 (2-C); 128.7 (7-CH); 121.6 (5-CH); 117.6 (8-CH); 63.3 (CH3CH2O); 14.2 (CH3); 13.7 (CH3CH2O). HRMS (ESI) calculated for C12H14N3O6S+ [M + H]+ 328.0598, found 328.0593.
2-Acetyl-3-methyl-6-sulfamoylquinoxaline 1,4-dioxide (7g) and 2-acetyl-3-methyl-7-sulfamoylquinoxaline 1,4-dioxide (8g). This compound was prepared from benzofuroxan 12 and acetylacetone according to the general procedure.
2-Acetyl-3-methyl-6-sulfamoylquinoxaline 1,4-dioxide (7g). The yield of 7g was 40 mg (13%) as light yellow powder, mp 170–172 °C. Rf = 0.6 (CHCl3–Me2CO, 1
:3). HPLC (LW = 385 nm, gradient B 10/50% (45 min)) tR = 14.18 min, purity 95.6%. λmax, EtOH: 238, 269, 386 nm. 1H NMR (400 MHz, DMSO-d6) δ 8.87 (1H, s, H-5); 8.62 (1H, d, J = 9.1, H-8); 8.26 (1H, d, J = 9.1, H-7); 7.90 (2H, br. s, SO2NH2); 2.65 (3H, s, CH3); 2.39 (3H, s, CH3). 13C NMR (100 MHz, DMSO-d6) δ 195.2 (CO); 147.2 (6-C); 140.6 (2-C); 139.6 (3-C); 137.6 (9-C); 137.2 (10-C); 127.6 (7-CH); 121.5 (8-CH); 117.6 (5-CH); 29.5 (COCH3); 13.6 (CH3). HRMS (ESI) calculated for C11H12N3O5S+ [M + H]+ 298.0492, found 298.0527.
2-Acetyl-3-methyl-7-sulfamoylquinoxaline 1,4-dioxide (8g). The yield of 8g was 22 mg (8%) as light yellow powder, mp 195–196 °C. Rf = 0.5 (CHCl3–Me2CO, 1
:3). HPLC (LW = 385 nm, gradient B 10/50% (45 min)) tR = 14.13 min, purity 95.0%. λmax, EtOH: 239, 269, 385 nm. 1H NMR (400 MHz, DMSO-d6) δ 8.81 (1H, d, J = 1.5, H-8); 8.66 (1H, d, J = 9.5, H-5); 8.29 (1H, dd, J3 = 9.5, J4 = 1.5, H-6); 7.88 (2H, br. s, SO2NH2); 2.66 (3H, s, CH3); 2.39 (3H, s, CH3). 13C NMR (100 MHz, DMSO-d6) δ 195.1 (CO); 146.3 (6-C); 140.3 (2-C); 140.0 (3-C); 138.5 (10-C); 136.1 (9-C); 128.6 (6-CH); 121.6 (5-CH); 117.5 (8-CH); 29.5 (COCH3); 13.7 (CH3). HRMS (ESI) calculated for C11H12N3O5S+ [M + H]+ 298.0492, found 298.0494.
2-Benzoyl-6-sulfamoyl-3-(trifluoromethyl)quinoxaline 1,4-dioxide (7h). This compound was prepared from benzofuroxan 12 and 4,4,4-trifluoro-1-phenylbutane-1,3-dione according to the general procedure. The yield of 7h was 48 mg (12%) as an orange powder, mp 236–237 °C. Rf = 0.5 (CHCl3–Me2CO, 1
:3). HPLC (LW = 275 nm, gradient B 40/80% (45 min)) tR = 14.41 min, purity 97.9%. λmax, EtOH: 241, 275, 401 nm. 1H NMR (400 MHz, DMSO-d6) δ 8.89 (1H d, J = 1.5, H-8); 8.59 (1H, d, J = 8.8, H-5); 8.39 (1H, dd, J3 = 8.8, J4 = 1.5, H-6); 8.15 (2H, d, J = 7.3, C6H5); 8.02 (2H, br. s, SO2NH2); 7.79 (1H, t, J = 7.3, C6H5); 7.61 (2H, d, J = 7.3, C6H5). 13C NMR (100 MHz, DMSO-d6) δ 183.9 (CO); 147.8 (7-C); 139.9 (10-C); 139.1 (9-C); 138.3 (3-C); 137.9 (2-C); 135.4 (4′-CH); 133.9 (1′-C); 129.9 (6-CH); 129.4 (2 × 2′-CH); 129.2 (2 × 3′-CH); 121.7 (5-CH); 118.9 (CF3, J = 276); 117.8 (8-CH). HRMS (ESI) calculated for C16H11F3N3O5S+ [M + H]+ 414.0366, found 414.0224.
4-Chloro-3-nitrobenzenesulfonamide (10). A solution of 1-chloro-2-nitrobenzene (9, 15.8 g, 0.1 mol) in CHCl3 (70 mL) was cooled to −5 °C. Once this temperature was reached, chlorosulfonic acid (0.8 mol, 50 mL) was added to the solution through a dropping funnel with vigorous stirring. The temperature of the reaction mixture was carefully maintained below 0 °C throughout. The mixture was stirred at this temperature for 30 min, then heated to 40 °C for 4 h. The flask was cooled in an ice bath, and the reaction mixture was poured into ice-water mixture (300 g). The resulting precipitate was filtered, washed with water (3 × 50 mL), and then air-dried. Next, obtained crude 4-chloro-3-nitrobenzenesulfonyl chloride (25.0 g, 0.1 mol) was dissolved in THF (50 mL), and aqueous NH4OH (100 mL, 25% w/v) was added dropwise to the stirred solution at 0 °C. The reaction was monitored by TLC, and when it was completed, the reaction mixture was poured into ice (100 g). The formed precipitate was filtered and washed with water (3 × 50 mL). The resulting product was air-dried and used for further transformations without additional purification. The yield of derivative 8 was 21.1 g (89%), yellow powder. mp 176–178 °C.
4-Amino-3-nitrobenzenesulfonamide (11). In a sealed flask, a mixture of 2-chloronitrobenzene 10 (10.0 g, 0.04 mol) and a solution of anhydrous ammonia in ethanol (50 mL, 15% w/v) was stirred for 72 h at 100 °C. After the reaction was completed (confirmed by TLC), the mixture was cooled, and water (50 mL) was added. It was then heated until the precipitate was completely dissolved and subsequently cooled. The resulting crystalline product was filtered and air-dried. If necessary, the product can be recrystallized from a mixture of ethanol and water (3
:1). The yield of nitroaniline 11 was 8.1 g (91%), light yellow crystals, mp 198–200 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.40 (1H, s, HAr); 7.94 (2H, s, NH2); 7.72 (1H, d, J = 8.7, HAr); 7.30 (2H, s, SO2NH2); 7.12 (1H, d, J = 8.7, HAr). 13C NMR (100 MHz, DMSO-d6) δ 147.9 (CSO2NH2); 132.0 (CH); 130.4 (CNO2); 128.6 (CNH2); 124.2 (CH); 119.9 (CH).
4-Amino-3-nitrobenzenesulfonamide (11, Scheme 2). In an aqueous solution (20%) of hydrochloric acid (32 mL), acetanilide 16 (5.0 g, 0.016 mol) was added, and mixture refluxed with stirring for 1 h. The reaction mixture was poured into water (100 mc). The resulting yellow precipitate, formed upon cooling, was filtered, crystallized from aqueous ethanol (1
:1), and air-dried. The yield of benzenesulfonamide 11 was 3.1 g (91%), yellow crystals, mp 199–201 °C. HRMS (ESI) calculated for C6H6N3O4S− [M − H]− 216.0079, found 216.0066.
5-Sulfamoylbenzofuroxan (12). The benzenesulfonamide 11 (5.0 g, 0.02 mol) was dissolved in DMF (30 mL), and aqueous solution (50%) of KOH (0.05 mL, 1.3 mmol) was added. The mixture was cooled to 0–5 °C, and a solution (13%) of sodium hypochlorite (25 mL, 0.4 mol) was added dropwise with vigorous stirring. After the addition was complete, the reaction mixture was stirred for 10 min. The resulting solution was poured into cold water (100 mL), acidified with dilute (5%) hydrochloric acid until a neutral reaction was achieved (pH = 7.0). The product was then extracted with ethyl acetate (3 × 50 mL). The combined extract was washed with water, a solvent was evaporated under vacuum, and the product was precipitated from hexane–dichloromethane mixture (5
:1), yielded benzofuroxan 10 (3.7 g, 94%) as a yellow powder. mp 140–141 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.03 (1H, s, H-4); 7.89 (1H, d, J = 9.5, H-6); 7.71 (1H, d, J = 9.5, H-7); 7.66 (2H, br. s, SO2NH2). 13C NMR (100 MHz, DMSO-d6) δ 146.8 (br. s, CSO2NH2); 128.3 (br. s, 4-CH); 118.1 (br. s, 6-CH); 114.2 (br. s, 7-CH). HRMS (ESI) calculated for C6H6N3O4S+ [M – H]+ 216.0074, found 216.0085.
N-(4-sulfamoylphenyl)acetamide (14). 4-Aminobenzenesulfonamide (13, 10 g, 0.06 mol) was dissolved in acetic acid (50 mL), acetic anhydride (10 mL, 0.1 mol) and DMAP (0.35 g, 2.9 mmol) were added dropwise to the solution, and the mixture was refluxed for 4 h. After the reaction was complete, the reaction mixture was poured onto ice (100 g), and the precipitate was filtered and dried. The yield of acetanilide 14 was 12.2 g (98%), white crystals, mp 213–215 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.27 (1H, s, NHCOMe); 7.77–7.72 (4H, m, HAr); 7.24 (2H, s, SO2NH2); 2.08 (3H, s, CH3). 13C NMR (100 MHz, DMSO-d6) δ 169.2 (CO); 142.5 (CSO2NH2); 138.3 (C-NH); 126.9 (2×CH); 118.8 (2×CH); 24.4 (CH3). HRMS (ESI) calculated for C8H11N2O3S+ [M + H]+ 215.0485, found 215.0470.
(E)-N-(4-(N-((dimethylamino)methylene)sulfamoyl)phenyl)acetamide (15). To a solution of N-(4-sulfamoylphenyl)acetamide 14 (5 g, 23.4 mmol) in DMF (25 mL) DMF–DMA (4.1 mL, 30.6 mmol) was added dropwise at room temperature, and the reaction mixture was stirred for 1 h. After the reaction was complete, the mixture was poured into water (100 mL). The resulting precipitate was filtered, washed with cold water (50 mL), and then dried under vacuum. The yield of acetamide 15 was 6.2 g (99%), white crystals, mp 185–187 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.25 (NHCO); 8.18 (1H, s, CHN(CH3)2); 7.73–7.68 (4H, m, HAr); 3.12 (3H, s, N(CH3)2); 2.89 (3H, s, N(CH3)2); 2.07 (3H, s, CH3CO). 13C NMR (100 MHz, DMSO-d6) δ 169.1 (CONH); 159.8 (CHN(CH3)2); 142.4 (CSO2-); 137.1 (CNHCOMe); 127.2 (2×CH); 118.8 (2×CH); 41.1 (N(CH3)2); 35.2 (N(CH3)2); 24.3 (COCH3). HRMS (ESI) calculated for C11H15N3O3S+ [M + H]+ 270.0907, found 270.0862.
(E)-N-(4-(N-((dimethylamino)methylene)sulfamoyl)-2-nitrophenyl)acetamide (16). The mixture of concentrated HNO3 (7.0 mL, 0.17 mol) and concentrated H2SO4 (10.0 mL, 0.18 mol) was cooled to 0 °C. Subsequently, (E)-N-(4-(N-((dimethylamino)methylene)sulfamoyl)phenyl)acetamide 15 (5.0 g, 0.02 mol) was added in small portions with stirring, maintaining the temperature 0–5 °C. The reaction mixture was stirred at 5–7 °C for 2 h, and then poured onto ice (100 g). The resulting precipitate was filtered, washed with water (100 mL), and air-dried. The yield of acetanilide derivative 16 was 5.8 g (93%), light yellow crystals, mp 176–177 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.56 (NHCO); 8.25 (1H, s, CHN(CH3)2); 8.22 (1H, s, HAr); 8.06 (1H, dd, J3 = 8.6, J4 = 2.1, HAr); 7.81 (1H, d, J = 8.6, HAr); 3.15 (3H, s, N(CH3)2); 2.92 (3H, s, N(CH3)2); 2.11 (3H, s, CH3CO). 13C NMR (100 MHz, DMSO-d6) δ 168.2 (CONH); 160.2 (CHN(CH3)2); 141.2 (CSO2–); 138.9 (CNHCOMe); 134.0 (CNO2); 131.2 (CH); 125.5 (CH); 122.8 (CH); 41.1 (N(CH3)2); 35.2 (N(CH3)2); 23.6 (COCH3). HRMS (ESI) calculated for C11H14N4O5S+ [M + H]+ 315.0758, found 315.0741.
6-Chloro-2-(3-sulfamoylphenyl)quinoxaline-2-carbonitrile 1,4-dioxide (18). Chlorosulfonic acid (0.5 mL, 0.78 g, 6.7 mmol) was slowly added to a solution of 7-chloroquinoxaline-2-carbonitrile 1,4-dioxide (17, 0.5 g, 1.7 mmol)53 in CHCl3 (10 mL), while maintaining the temperature of the reaction mixture at about 60 °C. The mixture was stirred for 4 h at 60 °C. Subsequently, the solvent was evaporated under vacuum, and the resulting reaction mixture was poured onto ice (50 g). The formed precipitate of sulfonyl chloride was filtered and dried in air. The resulting product was dissolved in THF (5 mL) and added dropwise to a stirred solution of an aqueous NH4OH solution (1 mL, 25% w/v) at 5–10 °C. The insoluble precipitate was filtered, washed with water (3 × 10 mL), and dried in air. The residue was further purified using column chromatography (toluene–diethyl ether mixture, 5
:2) and then precipitated from a hexane–dichloromethane mixture (4:1) to yield product 18. The yield of 18 was 34%, yellow-orange powder. mp 184–185 °C. HPLC (LW = 300 nm, gradient B 30/80% (45 min)) tR = 13.61 min, purity 97.2%. λmax, EtOH: 221, 241, 291, 366 nm. 1H NMR (400 MHz, DMSO-d6) δ 8.57 (1H, s, H-8); 8.55 (1H, d, J = 9.5, H-5); 8.22 (1H, s, HAr); 8.17 (1H, d, J = 9.5, H-6); 8.09 (1H, d, J = 7.4, HAr); 7.93 (1H, d, J = 7.4, HAr); 7.87 (1H, t, J = 7.4, HAr); 7.64 (2H, br. s, SO2NH2). 13C NMR (100 MHz, DMSO-d6) δ 144.6 (3-C); 141.9 (3′-CSO2NH2); 138.2 (10-C); 137.9 (9-C); 137.6 (7-CCl); 134.8 (4′-CH); 133.4 (6-CH); 129.8 (2′-CH); 128.4 (6′-CH); 128.1 (1′-C); 127.4 (5′-CH); 122.8 (5-CH); 121.1 (2-C); 119.2 (8-CH); 110.8 (CN). HRMS (ESI) calculated for C15H8ClN4O4S [M − H]− 374.9955, found 375.0132.
Biology
000 (A431) or 40000 (MCF-7) cells per well. After 24 h, the compounds were added to the wells; an appropriate solvent volume was added to the control cells. The hypoxia (1% O2) conditions were simulated in Binder multigas incubator (Binder GmbH, Tuttlingen, Germany), as described.20 The IC50 values of the compounds obtained were calculated using GraphPad Prism 8.0 (GraphPad Software, Boston, MA, USA).Cell lysates were separated in 10% SDS-PAGE under reducing conditions, transferred to a nitrocellulose membrane (GE HealthCare, Chicago, IL, USA), and processed according to a standard protocol. To prevent nonspecific absorption, the membranes were treated with 5% nonfat milk solution in a TBS buffer (20 mM Tris, 500 mM NaCl, and pH 7.5) with 0.1% Tween-20 and then incubated with primary antibodies overnight at 4 °C.
CA IX and cleaved PARP antibodies were obtained from Cell Signaling Technology (Beverly, MA, USA); the antibodies against GAPDH (Cell Signaling Technology, Beverly, MA, USA) were added to standardize loading. Goat antirabbit IgGs (Jackson ImmunoResearch, West Grove, PA, USA) conjugated to horseradish peroxidase were used as secondary antibodies. Signals were detected using the ECL reagent as described in Mruk and Cheng's protocol57 and an ImageQuant LAS4000 system (GE HealthCare, Chicago, IL, USA).
Conclusions
An original approach to synthesizing previously unknown sulfoamido-substituted quinoxaline 1,4-dioxides was developed, giving insights into their chemical and biological properties. The designed compounds were evaluated for their inhibitory activity against cytosolic isoforms (hCA I, hCA II) and membrane-bound CA isozymes (hCA IX, hCA XII). Most of the synthesized derivatives exhibited more than a twofold higher potency than the reference compound AAZ against CA I and CA II isoforms, with Ki values of 5.65 and 12 nM, respectively. Nevertheless, one derivative 7g demonstrated potent inhibition of the hCA IX isozyme, with a Ki of 42.2 nM, comparable to AAZ (Ki = 25.7 nM). Screening of the anticancer potency of the sulfonamides of quinoxaline 1,4-dioxides against cancer cell lines of various histogenesis revealed that most of the synthesized compounds were active in low micromolar concentrations (Tables 3 and 4). Among the tested derivatives, the most active was 3-trifluoromethylquinoxaline 1,4-dioxide 7h, inhibiting all cell lines with IC50 values in the range from 1.1 to 2.1 μM and having a similar or higher activity profile than the reference drugs etoposide, and doxorubicin. Regarding hypoxia selectivity, it was observed that compounds 7a, 7e and 8g exhibited both comparable selectivity and activity to the reference drug TPZ against the adenocarcinoma cell line MCF-7 under hypoxic conditions.A comprehensive analysis of structure–activity relationships revealed that, generally, compounds containing 4-halogeno- and 3-sulfonamidosubstituted phenyl groups at position 3 of the quinoxaline ring, along with a 2-carbonitrile moiety (derivatives 7–8a–b, 18), displayed the highest potency against the majority of tested tumor cell lines. Consequently, both the structure and the positioning of substituents in the heterocyclic ring have a beneficial influence on the biological properties of quinoxaline 1,4-dioxides, enabling the modulation of their activity. The obtained data regarding the antiproliferative and CA-inhibiting activities of new sulfamido derivatives of quinoxaline 1,4-dioxide allowed the identification of the key role played by not only the structure of individual functional groups but also their position within the heterocyclic ring in the ability of this chemotype of compounds to inhibit tumor cell growth. Molecular docking simulations showed that the lead compound 7g accepted favorable binding patterns in the hCA IX isoform, involving the fitting of the sulfonamide moiety into the base of the CA active site through the chelation with the Zn2+ ion and hydrogen bond interactions with the key amino acids Thr200 and Gln67. Furthermore, the mechanism study revealed that derivative 7g induced apoptosis in A431 cells and exhibited significant potency as a CA IX blocker. So, this research has identified sulfoamido-substituted quinoxaline 1,4-dioxides as promising scaffold for further development of anticancer hypoxic cytotoxins with CA inhibition potencies.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization and methodology, A. E. S., G. I. B., A. M. S. and C. T. S.; chemical synthesis, G. I. B.; NMR investigation, G. V. Z.; in vitro investigation, D. I. S., A. M. S. D. S., D. V.; molecular docking studies S. K. K., data curation, A. M. S., A. E. S. and C. T. S.; writing original draft preparation, review, editing, and visualization, A. E. S. and G. I. B.; resources, A. E. S.; supervision, A. E. S., A. M. S. and C. T. S.; funding acquisition, A. E. S. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been partially financially supported by the Russian Science Foundation (grant 20-13-00402, https://rscf.ru/project/23-13-45035/). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We are grateful to Fedor B. Bogdanov for assistance in antiproliferative assays and Danila V. Sorokin for assistance in immunoblotting (Blokhin N.N. National Medical Research Center of Oncology). We thank Alvina Khamidullina and the Center for Precision Genome Editing and Genetic Technologies for Biomedicine, Institute of Gene Biology, Russian Academy of Sciences, for providing the equipment and research facilities (microplate spectrophotometer).References
- K. D. Miller, R. L. Siegel, C. C. Lin, A. B. Mariotto, J. L. Kramer, J. H. Rowland, K. D. Stein, R. Alteri and A. Jemal, Ca-Cancer J. Clin., 2016, 66, 271–289, DOI:10.3322/caac.21349.
- L. Schito and G. L. Semenza, Trends Cancer, 2016, 2, 758–770, DOI:10.1016/j.trecan.2016.10.016.
- J. M. Brown and W. R. Wilson, Nat. Rev. Cancer, 2004, 4, 437–447, DOI:10.1038/nrc1367.
- P. Wardman, Br. J. Radiol., 2009, 82, 89–104, DOI:10.1259/bjr/60186130.
- G. L. Semenza, Nat. Rev. Cancer, 2003, 3, 721–732, DOI:10.1038/nrc1187.
- N. Robertson, C. Potter and A. L. Harris, Cancer Res., 2004, 64, 6160–6165, DOI:10.1158/0008-5472.can-03-2224.
- C. T. Supuran, Nat. Rev. Drug Discovery, 2008, 7, 168–181, DOI:10.1038/nrd2467.
- C. T. Supuran, Expert Opin. Invest. Drugs, 2018, 27, 963–970, DOI:10.1080/13543784.2018.1548608.
- G. De Simone and C. T. Supuran, Expert Opin. Ther. Pat., 2024, 15, 1–3, DOI:10.1080/13543776.2024.2353625.
- R. Ronca and C. T. Supuran, Biochim. Biophys. Acta, Rev. Cancer, 2024, 1879, 189120, DOI:10.1016/j.bbcan.2024.189120.
- Z. Li, L. Jiang, S. H. Chew, T. Hirayama, Y. Sekido and S. Toyokuni, Redox Biol., 2019, 26, 101297, DOI:10.1016/j.redox.2019.101297.
- E. O. Pettersen, P. Ebbesen, R. G. Gieling, K. J. Williams, L. Dubois, P. Lambin, C. Ward, J. Meehan, I. H. Kunkler, S. P. Langdon, A. H. Ree, K. Flatmark, H. Lyng, M. J. Calzada, L. D. Peso, M. O. Landazuri, A. Görlach, H. Flamm, J. Kieninger, G. Urban, A. Weltin, D. C. Singleton, S. Haider, F. M. Buffa, A. L. Harris, A. Scozzafava, C. T. Supuran, I. Moser, G. Jobst, M. Busk, K. Toustrup, J. Overgaard, J. Alsner, J. Pouyssegur, J. Chiche, N. Mazure, I. Marchiq, S. Parks, A. Ahmed, M. Ashcroft, S. Pastorekova, Y. Cao, K. M. Rouschop, B. G. Wouters, M. Koritzinsky, H. Mujcic and D. Cojocari, J. Enzyme Inhib. Med. Chem., 2015, 30, 689–721, DOI:10.3109/14756366.2014.966704.
- P. C. McDonald, J. Y. Winum, C. T. Supuran and S. Dedhar, Oncotarget, 2012, 3, 84–97, DOI:10.18632/oncotarget.422.
- C. T. Supuran, Expert Opin. Invest. Drugs, 2017, 12, 61–88, DOI:10.1080/17460441.2017.1253677.
- B. Z. Kurt, F. Sonmez, D. Ozturk, A. Akdemir, A. Angeli and C. T. Supuran, Eur. J. Med. Chem., 2019, 183, 111702, DOI:10.1016/j.ejmech.2019.111702.
- S. G. Nerella, P. S. Thacker, M. Arifuddin and C. T. Supuran, Eur. J. Med. Chem., 2024, 10, 100131, DOI:10.1016/j.ejmcr.2024.100131.
- D. Tsikas, J. Enzyme Inhib. Med. Chem., 2024, 39, 2291336, DOI:10.1080/14756366.2023.2291336.
- R. Mohammadpour, S. Safarian, F. Ejeian, Z. Sheikholya-Lavasani, M. H. Abdolmohammadi and N. Sheinabi, Cell Biol. Int., 2014, 38, 228–238, DOI:10.1002/cbin.10197.
- P. C. McDonald, S. Chia, P. L. Bedard, Q. Chu, M. Lyle, L. Tang, M. Singh, Z. Zhang, C. T. Supuran, D. J. Renouf and S. Dedhar, Am. J. Clin. Oncol., 2020, 43, 484–490, DOI:10.1097/COC.0000000000000691.
- S. K. Krymov, A. M. Scherbakov, D. I. Salnikova, D. V. Sorokin, L. G. Dezhenkova, I. V. Ivanov, D. Vullo, V. De Luca, C. Capasso, C. T. Supuran and A. E. Shchekotikhin, Eur. J. Med. Chem., 2022, 228, 113997, DOI:10.1016/j.ejmech.2021.113997.
- C. T. Supuran and J. Y. Winum, Future Med. Chem., 2015, 7, 1407–1414, DOI:10.4155/fmc.15.71.
- G. I. Buravchenko and A. E. Shchekotikhin, Pharmaceuticals, 2023, 16, 1174, DOI:10.3390/ph16081174.
- A. M. Scherbakov, A. M. Borunov, G. I. Buravchenko, O. E. Andreeva, I. A. Kudryavtsev, L. G. Dezhenkova and A. E. Shchekotikhin, Cancer Invest., 2018, 36, 199–209, DOI:10.1080/07357907.2018.1453072.
- G. I. Buravchenko, A. M. Scherbakov, L. G. Dezhenkova, E. E. Bykov, S. E. Solovieva, A. A. Korlukov, D. N. Sorokin, L. M. Fidalgo and A. E. Shchekotikhin, Bioorg. Chem., 2020, 104, 104324, DOI:10.1016/j.bioorg.2020.104324.
- G. I. Buravchenko, A. M. Scherbakov, L. G. Dezhenkova, L. Monzote and A. E. Shchekotikhin, RSC Adv., 2021, 11, 38782–38795, 10.1039/D1RA07978F.
- G. I. Buravchenko, D. A. Maslov, M. S. Alam, N. E. Grammatikova, S. G. Frolova, A. A. Vatlin, X. Tian, I. V. Ivanov, O. B. Bekker, M. A. Kryakvin, O. A. Dontsova, V. N. Danilenko, T. Zhang and A. E. Shchekotikhin, Pharmaceuticals, 2022, 15, 155, DOI:10.3390/ph15020155.
- G. I. Buravchenko, A. M. Scherbakov, A. D. Korlukov, P. V. Dorovatovskii and A. E. Shchekotikhin, Curr. Org. Chem., 2020, 17, 29–39, DOI:10.2174/1570179416666191210100754.
- U. Ragnarssona and L. Grehn, Acc. Chem. Res., 1998, 31, 494–501, DOI:10.1021/ar980001k.
- W. D. Emmons and J. P. Freeman, J. Am. Chem. Soc., 1955, 77, 6061–6062, DOI:10.1021/ja01627a083.
- A. Nocentini, E. Trallori, S. Singh, C. L. Lomelino, G. Bartolucci, L. D. C. Mannelli, C. Ghelardini, R. McKenna, P. Gratteri and C. T. Supuran, J. Med. Chem., 2018, 61, 10860–10874 CrossRef CAS PubMed.
- P. M. Heertjes, Recl. Trav. Chim. Pays-Bas, 1958, 77, 693–713, DOI:10.1002/recl.19580770802.
- Y. Hu, Q. Xia, S. Shangguan, X. Liu, Y. Hu and R. Sheng, Molecules, 2012, 17, 9683–9696, DOI:10.3390/molecules17089683.
- B. Zarranz, A. Jaso, I. Aldana, A. Monge, S. Maurel, E. Deharo, V. Jullian and M. Sauvain, Arzneim.-Forsch./Drug Res., 2004, 55, 754–761, DOI:10.1055/s-0031-1296926.
- S.-K. Lin and Q.-Z. Cong, J. Mol. Struct., 1987, 159, 279–286, DOI:10.1016/0022-2860(87)80046-7.
- E. Vicente, S. Pérez-Silanes, L. M. Lima, S. Ancizu, A. Burguete, B. Solano, R. Villar, I. Aldana and A. Monge, Bioorg. Med. Chem., 2009, 17, 385–389, DOI:10.1016/j.bmc.2008.10.086.
- E. Vicente, R. Villar, A. Burguete, B. Solano, S. Perez-Silanes, I. Aldana, J. A. Maddry, A. J. Lenaerts, S. G. Franzblau, S.-h. Cho, A. Monge and R. C. Goldman, Antimicrob. Agents Chemother., 2008, 52, 3321–3326, DOI:10.1128/aac.00379-08.
- A. Jaso, B. Zarranz, I. Aldana and A. Monge, J. Med. Chem., 2005, 48, 2019–2025, DOI:10.1021/jm049952w.
- S. G. Frolova, A. A. Vatlin, D. A. Maslov, B. Yusuf, G. I. Buravchenko, O. B. Bekker, K. M. Klimina, S. V. Smirnova, L. M. Shnakhova, I. K. Malyants, A. I. Lashkin, X. Tian, Md S. Alam, G. V. Zatonsky, T. Zhang, A. E. Shchekotikhin and V. N. Danilenko, Pharmaceuticals, 2023, 16, 1565, DOI:10.3390/ph16111565.
- L. Dubois, S. Peeters, N. G. Lieuwes, N. Geusens, A. Thiry, S. Wigfield, F. Carta, A. McIntyre, A. Scozzafava, J. M. Dogné, C. T. Supuran, A. L. Harris, B. Masereel and P. Lambin, Radiother. Oncol., 2011, 99, 424–431, DOI:10.1016/j.radonc.2011.05.045.
- C. T. Supuran, Bioorg. Med. Chem. Lett., 2023, 93, 129411, DOI:10.1016/j.bmcl.2023.129411.
- S. K. Krymov, A. M. Scherbakov, L. G. Dezhenkova, D. I. Salnikova, S. E. Solov’eva, D. V. Sorokin, D. Vullo, V. De Luca, C. Capasso, C. T. Supuran and A. E. Shchekotikhin, Pharmaceuticals, 2022, 15, 1453, DOI:10.3390/ph15121453.
- B. Muz, P. de la Puente, F. Azab and A. K. Azab, Hypoxia, 2015, 11(3), 83–92, DOI:10.2147/HP.S93413.
- D. V. Sorokin, A. M. Scherbakov, I. A. Yakushina, S. E. Semina, M. V. Gudkova and M. A. Krasil′nikov, Bull. Exp. Biol. Med., 2016, 160, 555–559, DOI:10.1007/s10517-016-3217-5.
- B. A. Teicher, Cancer Metastasis Rev., 1994, 13, 139–168, DOI:10.1007/BF00689633.
- Y. Li, L. Zhao and X.-F. Li, Front. Oncol., 2021, 11, 700407, DOI:10.3389/fonc.2021.700407.
- S. Mathur, S. Chen and K. A. Rejniak, npj Syst. Biol. Appl., 2024, 10, 1, DOI:10.1038/s41540-023-00327-z.
- T. T. Kwok and R. M. Sutherland, Radiat. Res., 1989, 119, 261–267, DOI:10.2307/3577618.
- A. Misra, C. Pandey, S. K. Sze and T. Thanabalu, PLoS One, 2012, 7, e49766, DOI:10.1371/journal.pone.0049766.
- Y. Ren, P. Hao, B. Dutta, E. S. Cheow, K. H. Sim, C. S. Gan, S. K. Lim and S. K. Sze, Mol. Cell. Proteomics, 2013, 12, 485–498, DOI:10.1074/mcp.M112.018325.
- V. L. Dengler, M. Galbraith and J. M. Espinosa, Crit. Rev. Biochem. Mol. Biol., 2014, 49, 1–15, DOI:10.3109/10409238.2013.838205.
- S. Pastorekova and R. J. Gillies, Cancer Metastasis Rev., 2019, 38, 65–77, DOI:10.1007/s10555-019-09799-0.
- S. B. Reddy and S. K. Williamson, Expert Opin. Invest. Drugs, 2009, 18, 77–87, DOI:10.1517/13543780802567250.
- S. Alavi, M. H. Mosslemin, R. Mohebat and A. R. Massah, Res. Chem. Intermed., 2017, 43, 4549–4559, DOI:10.1007/s11164-017-2895-6.
- A. M. Scherbakov, R. Yu. Balakhonov, D. I. Salnikova, D. V. Sorokin, A. V. Yadykov, A. I. Markosyan and V. Z. Shirinian, Org. Biomol. Chem., 2021, 19, 7670–7677, 10.1039/D1OB01362A.
- M. V. Zapevalova, E. S. Shchegravina, I. P. Fonareva, D. I. Salnikova, D. V. Sorokin, A. M. Scherbakov, A. A. Maleev, S. K. Ignatov, I. D. Grishin, A. N. Kuimov, M. V. Konovalova, E. V. Svirshchevskaya and A. Yu. Fedorov, Int. J. Mol. Sci., 2022, 23, 10854, DOI:10.3390/ijms231810854.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254, DOI:10.1006/abio.1976.9999.
- D. D. Mruk and C. Y. Cheng, Spermatogenesis, 2011, 1, 121–122, DOI:10.4161/spmg.1.2.16606.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04548c |
| This journal is © The Royal Society of Chemistry 2024 |