DOI:
10.1039/D4RA04493B
(Paper)
RSC Adv., 2024,
14, 25918-25931
Mechanisms and selectivity of methanol oxidation on PtRuM3/C-MWCNT (M = Fe and Co) electrocatalysts†
Received
19th June 2024
, Accepted 3rd August 2024
First published on 19th August 2024
Abstract
Methanol oxidation efficiency and resistance to CO poisoning are the most challenging issues associated with direct methanol fuel cells. Much experimental effort has been undertaken, such as generating Pt-based binary and ternary nanoparticles, creating composite substrates, and fabricating nanoparticles with special shapes, to overcome these drawbacks. Our previous experiment showed that ternary PtRuM3/C-MWCNT (M = Fe and Co; C-MWCNT = carbon Vulcan-multiwalled carbon nanotube) electrocatalysts exhibited high methanol oxidation activity and tolerance to CO poisoning. However, reaction mechanisms on ternary PtRuM3/C-MWCNT (M = Fe and Co) electrocatalysts remain unknown. Therefore, this work is devoted to elucidating the problem using density functional theory calculations and thermodynamic models. Our present study showed that methanol oxidation proceeds via four possible reaction pathways on the surface of PtRuM3/C-MWCNTs, where the most favourable one follows a series of steps converting 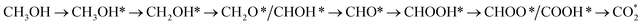 with a thermodynamic barrier of 0.513 eV for applied potentials of U = 0 V and 1.005 V on PtRuFe3/C-MWCNTs and 0.404 eV for U = 0 V and 0.167 eV for U = 1.005 V on PtRuCo3/C-MWCNTs. We also provide physical insights into the interaction between methanol oxidation intermediates and substrates' surface by analysing electronic properties. Our findings support the results of our previous experiment. The results of this study can be useful for rationally designing the anode for fuel cells.
with a thermodynamic barrier of 0.513 eV for applied potentials of U = 0 V and 1.005 V on PtRuFe3/C-MWCNTs and 0.404 eV for U = 0 V and 0.167 eV for U = 1.005 V on PtRuCo3/C-MWCNTs. We also provide physical insights into the interaction between methanol oxidation intermediates and substrates' surface by analysing electronic properties. Our findings support the results of our previous experiment. The results of this study can be useful for rationally designing the anode for fuel cells.
1 Introduction
Today, global energy shortages and environmental pollution problems have been increasing tremendously. Therefore, research on novel energy sources and technologies is an urgent requirement. Direct methanol fuel cells (DMFCs) are regarded as one of the most promising energy technologies owing to their high conversion efficiencies, low operating temperatures, and short recharge time.1,2 Platinum is hitherto the best electrocatalyst for methanol oxidation on the anode of DMFCs because of its outstanding catalytic efficiency, excellent electrical properties, and high corrosion resistance; however, CO poisoning is the major obstacle in increasing the performance of DMFCs to a practical level.1,2 Alloying is an effective way to enhance the activity of methanol oxidation and reduce the cost of DMFCs because of bifunctional mechanisms or electronic effects. Especially, the methanol oxidation ability of PtM (M = Fe, Co, Ni, Cu, Ru, etc.) binary alloys is higher than that of pure Pt,3–10 where PtRu exhibits the highest electrocatalytic performance due to its improved tolerances to CO poisoning. From the theoretical viewpoint, adding a third metal could change PtRu electronic properties and surface structure as well as improve catalytic performance.11,12 To date, many studies have shown that ternary Pt-based nanocrystals formed from various metals (Ru, Fe, Co, Ni, Pd, Rh, Cu, etc.) and support materials (carbon, carbon nanotubes, graphene, metal oxides, etc.) significantly enhance methanol oxidation activity and the durability of the anode of DMFCs.13–20 Among the inexpensive metals combined with PtRu alloy, earth-abundant transition metals with low cost and good catalytic performance, such as iron (Fe), cobalt (Co), and nickel (Ni), are superior candidates for developing the anode of DMFCs.21–30 Specifically, PtRuFe alloy displayed a higher onset potential and current intensity than PtRu and PtFe alloys.22–24 PtRuNi alloy on Vulcan carbon and carbon nanotubes can significantly improve the methanol oxidation performance and tolerance to CO poisoning compared to PtRu alloy.25–27 Meanwhile, PtRuCo alloy without supports also exhibited a superior ability to oxidize methanol and prevent CO poisoning compared to the PtRu alloy.28,29 Our research recently showed that the third metal component, M, enhanced the electrocatalytic activity of PtRuMx/C-MWCNTs (M = Fe, Co, Ni; x = 1, 3) compared to the binary PtRu/C-MWCNTs system. Moreover, PtRuCox/C-MWCNTs exhibited the best methanol oxidation activity, while PtRuFex/C-MWCNTs showed the highest tolerance to CO poisoning.31
Many theoretical studies on the interaction between methanol and catalysts have been carried out along with experiments. Using density functional theory (DFT) calculations, Zhao's group investigated the methanol oxidation on PtRu(111).32 They found that the most favourable reaction pathway of methanol oxidation on PtRu(111) is CH3OH → CH3O → CH2O → CHO → CHOOH → COOH → CO2. Moreover, on the PtRu(111) surface, most of the intermediates adsorbed around the Ru sites due to the lower electronegativity of Ru atoms. According to Orazi et al., the methanol molecule adsorbs most stably via its O atom on the top Co site of the PtCo(111) surface.33 The charge transfer from the C atom to the O atom of the methanol molecule and finally to the Co atoms changed the charge distribution around the adsorption site of methanol. Du et al. analysed the possible mechanisms of methanol decomposition on the Pt3Ni(111) surface by examining the initial scission of O–H, C–H, and C–O bonds.34 The DFT investigation of the reaction pathway for methanol on different Pt crystal planes and PtNi surface also suggested the Ni site as the favourable adsorption position for methanol oxidation.35 Furthermore, the density functional studies of the CO adsorption energy on Fe1−xPtRux nanocrystal surfaces showed that the Fe1−xPtRux alloy weakens the CO–Pt bond to enhance the anti-CO poisoning ability.36 Notably, the DFT calculations have focused only on studying the methanol oxidation on binary alloys, while there are only a few works for ternary alloy catalysts.
The experimental works confirmed that ternary PtRuM alloys without carbon supports exhibited higher methanol oxidation efficiency and CO poisoning resistance over binary Pt-based and pure metal catalysts.22–29,31 Remarkably, our previous experiment revealed an excellent catalytic activity of PtRuMx/C-MWCNTs (M = Fe, Ni, Co) and the atomic ratio of Pt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ru:M = 1:1:3 showed a better performance than that of 1:1:1, for the methanol oxidation.31 However, no studies are available to elucidate the methanol oxidation mechanisms of PtRuMx/C-MWCNTs, particularly for Pt:Ru:M = 1:1:3. Therefore, the present work is devoted to solving this issue by systematically searching for the possible intermediates and then clarifying the reaction pathways of methanol oxidation on PtRuM3/C-MWCNTs (M = Fe, Co) catalysts. Here, we selected two representatives of the electrocatalysts with M = Fe and Co because PtRuCo3/C-MWCNTs showed the best reaction activity, while PtRuFe3/C-MWCNTs exhibited the highest tolerance to CO poisoning among the considered third metal (Fe, Co, and Ni).31 The results of this work are completely new and will be valuable for optimally designing ternary Pt-based catalysts. To achieve the target, we used density functional theory calculations and the thermodynamic model.37,38
:Ru:M = 1:1:3 showed a better performance than that of 1:1:1, for the methanol oxidation.31 However, no studies are available to elucidate the methanol oxidation mechanisms of PtRuMx/C-MWCNTs, particularly for Pt:Ru:M = 1:1:3. Therefore, the present work is devoted to solving this issue by systematically searching for the possible intermediates and then clarifying the reaction pathways of methanol oxidation on PtRuM3/C-MWCNTs (M = Fe, Co) catalysts. Here, we selected two representatives of the electrocatalysts with M = Fe and Co because PtRuCo3/C-MWCNTs showed the best reaction activity, while PtRuFe3/C-MWCNTs exhibited the highest tolerance to CO poisoning among the considered third metal (Fe, Co, and Ni).31 The results of this work are completely new and will be valuable for optimally designing ternary Pt-based catalysts. To achieve the target, we used density functional theory calculations and the thermodynamic model.37,38
2 Catalyst model and computational details
PtRuFe3/C-MWCNTs and PtRuCo3/C-MWCNTs substrates were designed using the slab model with the (111) crystal surface framework and the (4 × 4) unit cell of five atomic layers. Furthermore, to maximize the role of precious metals (Pt and Ru), we designed the supercell with the layers of these metals close to the surface of the slab. As depicted in Fig. 1a and b (top and side views), the topmost and second layers consist of sixteen Pt and sixteen Ru atoms, respectively. The underneath three layers were composed of inexpensive Fe or Co metals. The above five atomic layers are grown on the support, which consists of the graphene nanosheet with a few extra C atoms adsorbed on the bottom side of the graphene to mimic the C-MWCNTs support of the PtRuM3/C-MWCNTs substrates (M = Fe and Co). The total number of atoms in each substrate is 116. We set up the substrates as described because the high-resolution transmission electron microscopy of our previous experiment exhibited that the scale of the MWCNTs is much broader than that of the nanoparticles dispersed on them. Therefore, the nanoparticle is regarded as located approximately on a nearly flat area on the surface of the MWCNT. Also, by ignoring the effects of the multiple walls, we can use a graphene sheet to mimic the MWCNT. The decorated C atoms on the bottom side of the graphene sheet mimic the Vulcan carbon in our previous experiment.31 We expect that our supercell model can capture the main physics of the methanol oxidation on the surface of the PtRuM3/C-MWCNTs catalysts. We have to emphasize that we do not intend to fully describe the MWCNTs, even though there were not many DFT calculations concerning the complexity of the model as in the present study. The ab initio studies have usually ignored carbon supports.32–34,39,40 The atomic positions and the unit cell size for the designed substrates were first optimized, and the structure of possible intermediates of methanol oxidation on the optimized substrates was then fully relaxed using the density functional theory calculations with the aid of the Vienna Ab initio Simulation Package (VASP).41 Our model does not include van der Waals interactions because the contribution toward chemical reactions on metal surfaces is usually ignorable.42,43 To optimize the unit cell size, we varied the lattice constant and calculated the total energy for each variation. The minimum point in the parabolic curve of the total energy versus the lattice constant determines the optimized size of the unit cell, as shown in Fig. S1 in ESI.† The optimized lattice constant of both substrates is 2.61 Å. Therefore, the dimensions of the unit cell in the slab model are 10.4 Å and 10.4 Å in the x and y axes and 23.4 Å in the surface normal direction with a vacuum space of about 12 Å. In all the optimization processes for the atomic positions, the atoms were allowed to move freely in all dimensions until the force acting on each atom reached 0.001 eV Å−1. We used a plane-wave basis set with a kinetic cut-off energy of 400 eV, the Perdew–Burke–Ernzerhof generalized gradient approximation for the exchange–correlation energy,44,45 and the projector augmented wave pseudopotentials for the ion-valence electron interaction.46 The k-point mesh sample of 5 × 5 × 1 with the special-point sampling technique of Monkhorst and Pack was used for the Brillouin zone integration.47 The cut-off energy and the k-point mesh sample have been tested for the convergence of the total energy. Spin polarization was appropriately constructed. To simulate periodic supercells, dipole corrections were set. The convergence speed of the calculations was supported by Methfessel–Paxton smearing48 of first order with the sigma value of 0.2 eV for geometry optimization and 0.05 eV for obtaining electronic properties.
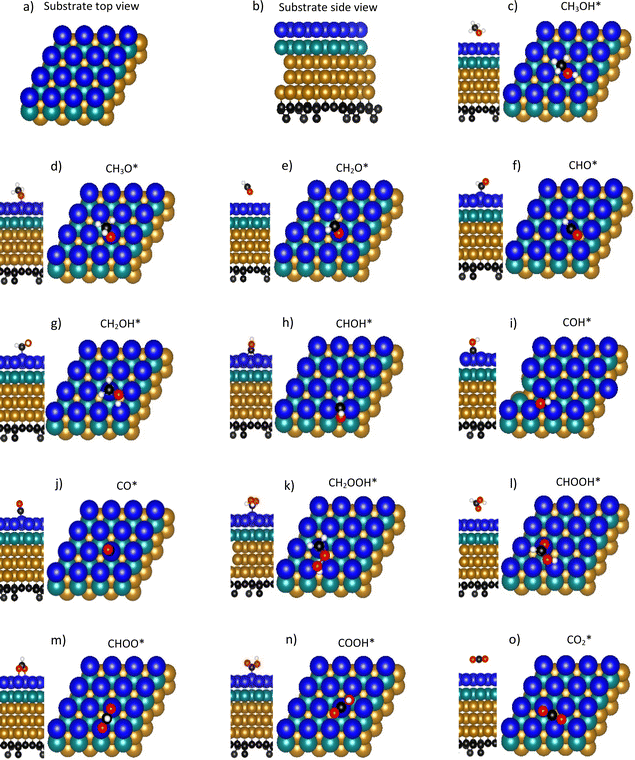 |
| | Fig. 1 The top and side views of the substrates (a and b) and the most stable configurations for the adsorption of methanol oxidation intermediates on the surface of PtRuM3/C-MWCNTs (M = Fe and Co) catalysts (c–o). Topmost-layer Pt (blue), Ru (green), Fe (golden), C (black), O (red), and H (white). | |
2.1 Adsorption energy
The binding strength of the various reaction intermediates on the possible adsorption sites of the substrates is expressed via the adsorption energy according to the following formula:| | |
Ea = E[sub+ads] − (Esub + Eads),
| (1) |
where E[sub+ads], Esub, and Eads are the total energy of the substrate–adsorbate system, the clean substrate, and the isolated adsorbate, respectively. This work ignored the influences of water medium on the adsorption of the methanol oxidation intermediates.
2.2 Charge density difference
The charge density difference (Δρ) is studied to elucidate the interaction between the adsorbent and adsorbate using the following equation:33,38| | |
Δρ = ρ[sub+ads] − ρsub − ρads,
| (2) |
where ρ[sub+ads], ρsub, and ρads are the charge density of the adsorbent/adsorbate system, substrate with the adsorbed state, and the adsorbed molecule, respectively.
2.3 Gibbs free energy
Free energy diagrams following the thermodynamic model37,49,50 were constructed to understand the thermodynamic stability of the reaction intermediates. The Gibbs free energy with electrode potential corrections was calculated as follows:Here, ΔG(0) = ΔE + ΔZPE − TΔS with ΔE and ΔZPE are reaction energy and the change of zero-point energies of intermediate reactions, estimated from the total energies and vibrational energies of our DFT calculations. The change in entropies (ΔS) was calculated from our DFT calculations using VASPKIT;51 n and U are the number of electrons in each reaction step and the electrode potential relative to the standard hydrogen electrode, respectively. According to our previous experimental results,31 the working potential of methanol oxidation was 0.8 V for the PtRuM3/C-MWCNTs (M = Fe, Co) electrocatalysts relative to an Ag/AgCl (3.5 M KCl) reference electrode. Therefore, the value of U for methanol oxidation in formula (3) is equivalent to 1.005 V, i.e., ERHE = EAg/AgCl + 0.205 V, relative to the standard hydrogen electrode in our thermodynamic model. For comparison, we also studied the reaction mechanisms with U = 0 V. The standard atmospheric pressure of 1 bar, room temperature of 300 K, and pH = 0 were used in this study. With the pH = 0 level, we can replace the proton and electron transfer by H+ + e− = ½H2, which is adequate with the hydrogen standard electrode model.
3 Results and discussion
3.1 Adsorption configurations of intermediates
The total methanol oxidation reaction generates carbon dioxide as the following equation:| | |
CH3OH + H2O → CO2 + 3H2.
| (4) |
This equation can proceed in many possible pathways (see Fig. 2) with multi-intermediate steps from (1)–(18) and involve thirteen reaction intermediates. The intermediates of methanol oxidation are proposed by a continuous dehydrogenation process from the previously formed intermediates starting from CH3OH*, where the asterisk denotes the adsorbed state of the intermediates on the substrates' surface. The dehydrogenation can take place at the hydrogen atom near the O or C atoms of each intermediate. For example, in steps (1) and (2), CH3OH* can detach a hydrogen atom near the O atom to form CH3O* and near the C atom to generate CH2OH*, respectively. The other intermediates are also formed in the same manner. Noticeably, forming the double-oxygen intermediates such as CH2OOH*, CHOOH*, and COOH* in steps (6), (11), and (16) require the involvement of a water molecule, i.e., CH2O* + H2O → CH2OOH* + ½H2, CHO* + H2O → CHOOH* + ½H2, and CO* + H2O → COOH*, respectively. Here, CH2OOH*, CHOOH*, and COOH* were formed by combining CH2O*, CHO*, and CO* with the OH group, which stemmed from the water splitting reaction, H2O → OH + ½H2, respectively. To explore the possible intermediates of methanol oxidation reaction, we designed the structure of each intermediate on the topmost surface of the substrate with the initial position of the intermediate of about 3 Å with various directions above the surface. We then optimized all the atomic positions of the designed structure and obtained the adsorption energy. For each intermediate, we compared its adsorption energies at various adsorption sites and configurations to gauge the most favourable adsorption configuration with the most negative adsorption energy. Fig. 1c–o shows the most stable configuration for various adsorbed intermediates of the methanol oxidation reaction, i.e., methanol (CH3OH*), methoxy (CH3O*), hydroxymethyl (CH2OH*), formaldehyde (CH2O*), hydroxymethylene (CHOH*), hydroperoxymethyl (CH2OOH*), formyl (CHO*), isoformyl (COH*), formic acid (CHOOH*), carbon monoxide (CO*), formate (CHOO*), carboxyl (COOH*), and carbon dioxide 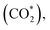 on the surface of the PtRuM3/C-MWCNTs (M = Fe, Co) substrates. Particularly, the adsorption site, the bond distance (dads-sur) from the nearest atom (carbon or oxygen atom, see Fig. 1c–o for each case) of the intermediates to the surface of the substrate, the adsorption energy (Eads), the zero-point energy (ZPE), and the total energy (Etotal, i.e., Eads plus ZPE) are also listed in Table 1. As shown in Fig. 1c–o, several intermediates such as CH3OH*, CH3O*, CH2O*, and CHOOH* adsorb most favourably with the end-on configuration on the top (T) site of the Pt atom, where the O atom is the nearest atom to the substrates' surface. The adsorption of the other intermediates, i.e., CH2OH*, CHOH*, CH2OOH*, CHO*, COH*, CO*, and COOH*, is also the most stable with the end-on configuration via their C atom; however, while CH2OH*, CH2OOH*, CHO*, COH*, CO*, and COOH* are on the top site, CHOH* is on the bridge (B) and COH* is on the hollow (H) with an underneath Ru atom. Exceptions are for CHOO* and
on the surface of the PtRuM3/C-MWCNTs (M = Fe, Co) substrates. Particularly, the adsorption site, the bond distance (dads-sur) from the nearest atom (carbon or oxygen atom, see Fig. 1c–o for each case) of the intermediates to the surface of the substrate, the adsorption energy (Eads), the zero-point energy (ZPE), and the total energy (Etotal, i.e., Eads plus ZPE) are also listed in Table 1. As shown in Fig. 1c–o, several intermediates such as CH3OH*, CH3O*, CH2O*, and CHOOH* adsorb most favourably with the end-on configuration on the top (T) site of the Pt atom, where the O atom is the nearest atom to the substrates' surface. The adsorption of the other intermediates, i.e., CH2OH*, CHOH*, CH2OOH*, CHO*, COH*, CO*, and COOH*, is also the most stable with the end-on configuration via their C atom; however, while CH2OH*, CH2OOH*, CHO*, COH*, CO*, and COOH* are on the top site, CHOH* is on the bridge (B) and COH* is on the hollow (H) with an underneath Ru atom. Exceptions are for CHOO* and 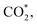 where they adsorb stably in the side-on configurations with the double top (T–T) site (each O atom on a T site) for CHOO* and the T–B site (one O atom on top and the other O atom on B) for
where they adsorb stably in the side-on configurations with the double top (T–T) site (each O atom on a T site) for CHOO* and the T–B site (one O atom on top and the other O atom on B) for 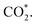 Especially, the
Especially, the 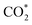 structure is in a straight configuration with an O–C–O angle of about 180°, parallel to the surface of the substrate. Table 1 also summarizes the most favourable adsorption site on the substrates' surface and the bond distance dads-sur (in columns 3 & 4). We found that the bond distance from the nearest atom of the intermediates to the surface is in the following order: hollow < bridge < top or top–top < top-bridge. The
structure is in a straight configuration with an O–C–O angle of about 180°, parallel to the surface of the substrate. Table 1 also summarizes the most favourable adsorption site on the substrates' surface and the bond distance dads-sur (in columns 3 & 4). We found that the bond distance from the nearest atom of the intermediates to the surface is in the following order: hollow < bridge < top or top–top < top-bridge. The 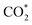 intermediate is located at the largest distance of about 3.4 Å from the surface of the PtRuM3/C-MWCNTs (M = Fe, Co) substrates. The adsorption energy of CH3OH*, CH2O*, CHOOH*, and
intermediate is located at the largest distance of about 3.4 Å from the surface of the PtRuM3/C-MWCNTs (M = Fe, Co) substrates. The adsorption energy of CH3OH*, CH2O*, CHOOH*, and 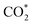 is positive (in column 5), which implies that these intermediates unfavourably adsorb and the other intermediates favourably adsorb with their negative adsorption energy on the surface of the PtRuFe3/C-MWCNTs. Particularly, the adsorption energy of all the intermediates (column 6) is negative on the PtRuCo3/C-MWCNTs catalyst. For each intermediate, it is more stable on PtRuCo3/C-MWCNTs than on PtRuFe3/C-MWCNTs because its adsorption energy is always more negative on M = Co than M = Fe in the range of about −0.135 eV (CHOH*) to −0.362 eV (CHOO*). The positive zero-point energy (in columns 7 & 8) competes with the negative adsorption energy and hence influences the stability of the intermediates adsorbed on the catalytic surface. We found the fluctuation (or zero-point energy) of each adsorption intermediate is rather large. Particularly, the fluctuation is stronger than the adsorption strength for CH3OH*, CH2O*, CHOOH*, and
is positive (in column 5), which implies that these intermediates unfavourably adsorb and the other intermediates favourably adsorb with their negative adsorption energy on the surface of the PtRuFe3/C-MWCNTs. Particularly, the adsorption energy of all the intermediates (column 6) is negative on the PtRuCo3/C-MWCNTs catalyst. For each intermediate, it is more stable on PtRuCo3/C-MWCNTs than on PtRuFe3/C-MWCNTs because its adsorption energy is always more negative on M = Co than M = Fe in the range of about −0.135 eV (CHOH*) to −0.362 eV (CHOO*). The positive zero-point energy (in columns 7 & 8) competes with the negative adsorption energy and hence influences the stability of the intermediates adsorbed on the catalytic surface. We found the fluctuation (or zero-point energy) of each adsorption intermediate is rather large. Particularly, the fluctuation is stronger than the adsorption strength for CH3OH*, CH2O*, CHOOH*, and 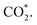 Therefore, the total energy Etotal of these intermediates becomes positive, which implies that CH3OH*, CH2O*, CHOOH*, and
Therefore, the total energy Etotal of these intermediates becomes positive, which implies that CH3OH*, CH2O*, CHOOH*, and 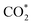 are unstable and subject to desorption from the substrates' surface. Emphasizing that the intermediates in the end-on via O configuration, such as CH3OH*, CH3O*, CH2O*, CHOOH*, and
are unstable and subject to desorption from the substrates' surface. Emphasizing that the intermediates in the end-on via O configuration, such as CH3OH*, CH3O*, CH2O*, CHOOH*, and 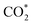 in the parallel structure have positive or least negative adsorption energy, indicating the adsorption via the O atom is not favourable, except for CHOO*. In contrast, the other intermediates remain in a stable state even after including the fluctuation on both substrates because of the negative Etotal. The isoformyl (COH*) is the most stable compared to all the other intermediates, with the most negative total energy on both substrates. We also found that, generally, the trend of the adsorption positions, bond distances, and adsorption energy of the intermediates are similar for both substrates, with the adsorption being more favourable on PtRuCo3/C-MWCNTs than on PtRuFe3/C-MWCNTs. Furthermore, the adsorption energy (Eads) of formaldehyde (CH2O*) on PtRuFe3/C-MWCNTs is insignificant, which is similar to the previous study for the adsorption of this intermediate on Pt(111).32,52 Also, the adsorption strength of carbon monoxide (CO*) is better on the PtRuCo3/C-MWCNTs surface with an adsorption energy of −1.335 eV compared to that of −1.097 eV on the PtRuFe3/C-MWCNTs surface. Interestingly, this result supports our previous experimental study, i.e., the jf/jr ratio of peak currents in forward and reverse directions demonstrated that the resistance to CO poisoning of PtRuFe3/C-MWCNTs is higher than that of PtRuCo3/C-MWCNTs, with jf/jr values of 6.9 and 4.3, respectively.31 The CO* adsorption via the C atom on the top of the Pt atom was also found in the experiment, which investigated the CO adsorption on the Pt(111) surface by the scanning tunnelling microscopy technique.53,54
in the parallel structure have positive or least negative adsorption energy, indicating the adsorption via the O atom is not favourable, except for CHOO*. In contrast, the other intermediates remain in a stable state even after including the fluctuation on both substrates because of the negative Etotal. The isoformyl (COH*) is the most stable compared to all the other intermediates, with the most negative total energy on both substrates. We also found that, generally, the trend of the adsorption positions, bond distances, and adsorption energy of the intermediates are similar for both substrates, with the adsorption being more favourable on PtRuCo3/C-MWCNTs than on PtRuFe3/C-MWCNTs. Furthermore, the adsorption energy (Eads) of formaldehyde (CH2O*) on PtRuFe3/C-MWCNTs is insignificant, which is similar to the previous study for the adsorption of this intermediate on Pt(111).32,52 Also, the adsorption strength of carbon monoxide (CO*) is better on the PtRuCo3/C-MWCNTs surface with an adsorption energy of −1.335 eV compared to that of −1.097 eV on the PtRuFe3/C-MWCNTs surface. Interestingly, this result supports our previous experimental study, i.e., the jf/jr ratio of peak currents in forward and reverse directions demonstrated that the resistance to CO poisoning of PtRuFe3/C-MWCNTs is higher than that of PtRuCo3/C-MWCNTs, with jf/jr values of 6.9 and 4.3, respectively.31 The CO* adsorption via the C atom on the top of the Pt atom was also found in the experiment, which investigated the CO adsorption on the Pt(111) surface by the scanning tunnelling microscopy technique.53,54
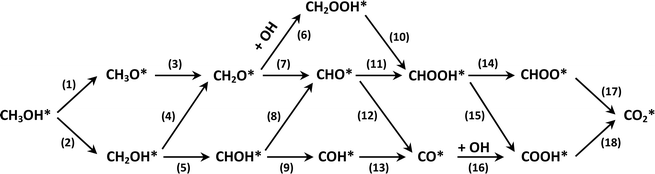 |
| | Fig. 2 Possible methanol decomposition pathways on catalyst surfaces. | |
Table 1 The bond distance (dads-sur) from the nearest atom (carbon or oxygen atom) of adsorbate molecules to the surface; adsorption energy (Eads), zero-point energy (ZPE), and total energy (Etotal, i.e., Eads plus ZPE) of the intermediates on the PtRuM3/C-MWCNT (M = Fe and Co) substrates. The top site of a Pt atom (T), the bridge of two Pt atoms (B), and the hollow with an underneath Ru atom (H)
| Molecule |
Chemical formula |
dads-sur (Å) |
Eads (eV) |
ZPE (eV) |
Etotal (eV) |
| M = Fe (site) |
M = Co (site) |
M = Fe |
M = Co |
M = Fe |
M = Co |
M = Fe |
M = Co |
| Methanol |
CH3OH* |
2.76 (T) |
2.66 (T) |
0.017 |
−0.305 |
1.375 |
1.379 |
1.392 |
1.074 |
| Methoxy |
CH3O* |
2.17 (T) |
2.15 (T) |
−1.194 |
−1.483 |
1.061 |
1.060 |
−0.133 |
−0.423 |
| Formaldehyde |
CH2O* |
2.97 (T) |
3.07 (T) |
0.072 |
−0.230 |
0.721 |
0.724 |
0.793 |
0.494 |
| Formyl |
CHO* |
2.36 (T) |
2.37 (T) |
−1.948 |
−2.129 |
0.470 |
0.471 |
−1.478 |
−1.658 |
| Carbon monoxide |
CO* |
2.17 (T) |
2.25 (T) |
−1.097 |
−1.335 |
0.203 |
0.205 |
−0.894 |
−1.130 |
| Hydroxymethyl |
CH2OH* |
2.45 (T) |
2.51 (T) |
−1.541 |
−1.793 |
1.109 |
1.106 |
−0.432 |
−0.687 |
| Hydroxymethylene |
CHOH* |
1.88 (B) |
1.94 (B) |
−3.650 |
−3.785 |
0.800 |
0.801 |
−2.850 |
−2.984 |
| Isoformyl |
COH* |
1.47 (H) |
1.47 (H) |
−3.516 |
−3.684 |
0.492 |
0.492 |
−3.024 |
−3.192 |
| Hydroperoxymethyl |
CH2OOH* |
2.52 (T) |
2.50 (T) |
−1.657 |
−1.835 |
1.162 |
1.171 |
−0.495 |
−0.664 |
| Formic acid |
CHOOH* |
2.93 (T) |
3.07 (T) |
0.038 |
−0.246 |
0.902 |
0.901 |
0.940 |
0.655 |
| Carboxyl |
COOH* |
2.35 (T) |
2.36 (T) |
−1.874 |
−2.139 |
0.616 |
0.616 |
−1.258 |
−1.523 |
| Formate |
CHOO* |
2.29 (T–T) |
2.32 (T–T) |
−1.947 |
−2.309 |
0.611 |
0.615 |
−1.336 |
−1.694 |
| Carbon dioxide |
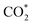 |
3.34 (T–B) |
3.45 (T–B) |
0.024 |
−0.206 |
0.316 |
0.316 |
0.340 |
0.110 |
Furthermore, we also explored the ORR intermediates on PtRuM3 substrates without C-MWCNTs support. The obtained results for the adsorption site and energy, Table S1 in ESI,† are similar to those of the PtRuM3/C-MWCNTs. The most noticeable finding for the role of the C-MWCNTs support is that it enhanced the adsorption energy of the less stable intermediates like CH3OH*, CH2O*, CHOOH*, and 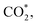 where the adsorption energy of these intermediates becomes more negative on PtRuM3/C-MWCNTs (Table 1) compared to the PtRuM3 substrates (Table S1†), respectively.
where the adsorption energy of these intermediates becomes more negative on PtRuM3/C-MWCNTs (Table 1) compared to the PtRuM3 substrates (Table S1†), respectively.
3.2 Electronic properties
One can reveal the nature of the interaction between the intermediates and the substrates via the electronic structure properties such as the Bader point charge, electronic density of states, and charge density difference. The point charge is determined by the Bader partition technique55,56 for the atoms of the substrate/intermediate systems; then, subtract the neutral charge of the atoms from the obtained point charge. Table 2 provides the Bader point charge of the C, O, and H atoms for each intermediate (columns 3–5), five atomic layers of metals (columns 6–10), and carbon layers of the substrate (column 11), where layer 1 is the topmost layer with the intermediates adsorbed on it. The total charge of each intermediate (column 12) is the charge summation of the C, H, and O atoms, while the total charge of the substrate (column 13) is calculated by the charge summation of the five atomic layers of metals and carbon layers of the substrate.
Table 2 The Bader charge transfer (e−) of the substrate/intermediate systems. Positive and negative values indicate the charge gain and loss, respectively. The charge summation over all the atoms of each species in the intermediate is denoted by Σ. The error of the charge calculation is 0.005e−
| Radical |
PtRuM3/C-MWCNTs |
C |
ΣO |
ΣH |
Layer 1 (Pt) |
Layer 2 (Ru) |
Layer 3 (Fe or Co) |
Layer 4 (Fe or Co) |
Layer 5 (Fe or Co) |
Carbon layers |
Intermediate |
Substrate |
| CH3OH* |
M = Fe |
−0.572 |
1.647 |
−1.134 |
2.939 |
−0.519 |
−2.222 |
−0.381 |
−3.769 |
4.011 |
−0.059 |
0.059 |
| M = Co |
−0.643 |
1.669 |
−1.066 |
2.929 |
−1.930 |
−0.898 |
−0.345 |
−2.619 |
2.903 |
−0.040 |
0.040 |
| CH3O* |
M = Fe |
−0.692 |
1.159 |
−0.113 |
2.512 |
−0.493 |
−2.201 |
−0.373 |
−3.769 |
3.970 |
0.354 |
−0.354 |
| M = Co |
−0.705 |
1.160 |
−0.079 |
2.517 |
−1.887 |
−0.906 |
−0.349 |
−2.622 |
2.871 |
0.376 |
−0.376 |
| CH2O* |
M = Fe |
−1.580 |
1.734 |
−0.155 |
2.909 |
−0.545 |
−2.217 |
−0.384 |
−3.768 |
4.006 |
−0.001 |
0.001 |
| M = Co |
−1.632 |
1.756 |
−0.121 |
2.883 |
−1.929 |
−0.895 |
−0.344 |
−2.620 |
2.902 |
0.003 |
−0.003 |
| CHO* |
M = Fe |
−1.671 |
1.793 |
−0.112 |
2.793 |
−0.387 |
−2.217 |
−0.396 |
−3.767 |
3.964 |
0.010 |
−0.010 |
| M = Co |
−1.583 |
1.799 |
−0.138 |
2.731 |
−1.790 |
−0.907 |
−0.349 |
−2.621 |
2.858 |
0.078 |
−0.078 |
| CO* |
M = Fe |
−1.760 |
1.880 |
— |
2.665 |
−0.394 |
−2.216 |
−0.368 |
−3.768 |
3.961 |
0.120 |
−0.120 |
| M = Co |
−1.736 |
1.870 |
— |
2.688 |
−1.825 |
−0.906 |
−0.334 |
−2.621 |
2.864 |
0.134 |
−0.134 |
| CH2OH* |
M = Fe |
−0.773 |
1.810 |
−1.156 |
2.910 |
−0.403 |
−2.218 |
−0.391 |
−3.765 |
3.986 |
−0.119 |
0.119 |
| M = Co |
−0.729 |
1.811 |
−1.127 |
2.839 |
−1.804 |
−0.902 |
−0.347 |
−2.620 |
2.879 |
−0.045 |
0.045 |
| CHOH* |
M = Fe |
−0.862 |
1.915 |
−1.112 |
2.776 |
−0.308 |
−2.225 |
−0.391 |
−3.766 |
3.973 |
−0.059 |
0.059 |
| M = Co |
−0.846 |
1.937 |
−1.125 |
2.780 |
−1.733 |
−0.917 |
−0.345 |
−2.621 |
2.870 |
−0.034 |
0.034 |
| COH* |
M = Fe |
−0.921 |
2.042 |
−1.000 |
2.529 |
−0.250 |
−2.234 |
−0.395 |
−3.768 |
3.997 |
0.121 |
−0.121 |
| M = Co |
−0.865 |
2.045 |
−1.000 |
2.512 |
−1.685 |
−0.937 |
−0.337 |
−2.623 |
2.890 |
0.180 |
−0.180 |
| CH2OOH* |
M = Fe |
−0.741 |
1.850 |
−1.183 |
2.876 |
−0.392 |
−2.217 |
−0.386 |
−3.766 |
3.959 |
−0.074 |
0.074 |
| M = Co |
−0.625 |
1.840 |
−1.234 |
2.833 |
−1.794 |
−0.909 |
−0.346 |
−2.620 |
2.855 |
−0.019 |
0.019 |
| CHOOH* |
M = Fe |
−2.723 |
3.802 |
−1.084 |
2.924 |
−0.547 |
−2.219 |
−0.380 |
−3.767 |
3.994 |
−0.005 |
0.005 |
| M = Co |
−2.675 |
3.768 |
−1.089 |
2.898 |
−1.939 |
−0.891 |
−0.341 |
−2.621 |
2.890 |
0.004 |
−0.004 |
| COOH* |
M = Fe |
−2.691 |
3.870 |
−1.000 |
2.619 |
−0.407 |
−2.222 |
−0.377 |
−3.767 |
3.975 |
0.179 |
−0.179 |
| M = Co |
−2.643 |
3.860 |
−1.000 |
2.599 |
−1.821 |
−0.902 |
−0.345 |
−2.620 |
2.872 |
0.217 |
−0.217 |
| CHOO* |
M = Fe |
−2.783 |
3.366 |
−0.093 |
2.412 |
−0.542 |
−2.201 |
−0.362 |
−3.768 |
3.971 |
0.490 |
−0.490 |
| M = Co |
−2.742 |
3.369 |
−0.128 |
2.401 |
−1.906 |
−0.909 |
−0.337 |
−2.620 |
2.872 |
0.499 |
−0.499 |
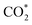 |
M = Fe |
−4.000 |
4.024 |
— |
2.922 |
−0.543 |
−2.220 |
−0.379 |
−3.767 |
3.963 |
0.024 |
−0.024 |
| M = Co |
−4.000 |
4.030 |
— |
2.891 |
−1.925 |
−0.896 |
−0.342 |
−2.622 |
2.864 |
0.030 |
−0.030 |
As seen in Table 2, for all the intermediates on both substrates, the C and H atoms always donate, and the O atoms always accumulate the negative charge (in the e− unit). Also, the layer-resolved charge shows that, under the adsorption of the intermediates, layer 1 of the Pt atoms and carbon layers of both substrates always gain, and the other layers of Ru, Fe, and Co atoms always lose the charge. Although the charge accumulation and donation have such a simple rule, the total charge of the intermediate and substrate shows that the gain and loss vary for different systems. The charge exchange of CH2O* and CHOOH* with the substrates is ignorable because of the same magnitude, with a calculation error of 0.005e−. The CH3O*, CO*, COH*, COOH*, and CHOO* intermediates on both substrates and CH2OH* on the PtRuFe3/C-MWCNTs surface exhibit large charge exchanges with the substrates (more than 10% of e− like CO* and COH*, to 36% of e− as CH3O*, and up to 50% of e− such as CHOO*), in the magnitude order: CHOO* > CH3O* > COOH* > COH* > CO* > CH2OH* for both substrates. Also, the small charge exchange (more than 0.005e− and less than 10% of e−) is found for the remaining molecules, i.e., CH3OH*, CHO*, CHOH*, CH2OOH*, and 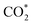 on both substrates and CH2OH* on the PtRuCo3/C-MWCNTs. Particularly, while CH3OH*, CH2OH*, CHOH*, and CH2OOH* donate, the other intermediates gain the charge from the substrates. Notably, the weak charge exchange intermediates, i.e., CH3OH*, CH2O*, CHOOH*, and
on both substrates and CH2OH* on the PtRuCo3/C-MWCNTs. Particularly, while CH3OH*, CH2OH*, CHOH*, and CH2OOH* donate, the other intermediates gain the charge from the substrates. Notably, the weak charge exchange intermediates, i.e., CH3OH*, CH2O*, CHOOH*, and 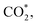 have small adsorption energies, as shown in Table 1.
have small adsorption energies, as shown in Table 1.
From the above analysis, we can state that the interaction between the methanol oxidation reaction intermediates and the substrates is the charge exchange; except for CH2O* and CHOOH*, the interaction should be due to the charge attraction (no charge exchange).
The subtle physical insights into the interaction of the intermediates and the substrates are also revealed via analysing the electronic density of states (DOS). We first present the orbital-projected DOS of the isolated intermediates to identify the location of peaks (Fig. 3) before considering their interaction with the substrates. We then show the orbital-projected DOS of the adsorbed intermediates to determine at which peaks the modification occurs the most to understand the changes in the DOS due to the substrate–adsorbate interaction (Fig. 4).
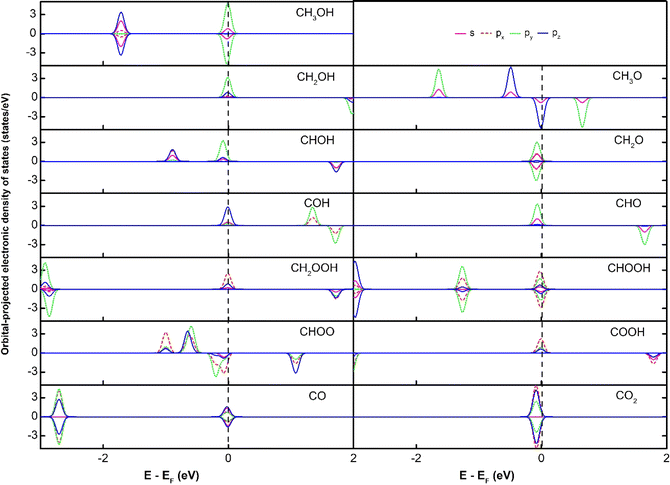 |
| | Fig. 3 The orbital-projected electronic density of states of the isolated adsorbates. | |
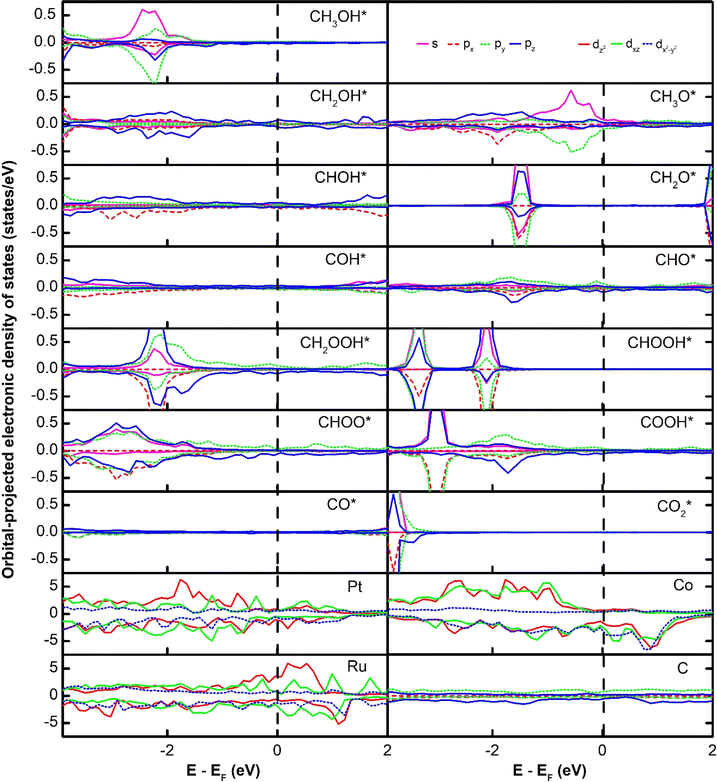 |
| | Fig. 4 The orbital-projected electronic density of states for the systems of the PtRuCo3/C-MWCNT substrate and the adsorbed intermediate: dxz is identical to dyz, and dxy is identical to dx2−y2. For the PtRuFe3/C-MWCNT substrate, see Fig. S2 in ESI.† | |
Fig. 3 shows that all the isolated intermediates have a peak near or at the Fermi level, which can be the spin-up or spin-down components or both. The px orbital dominates this peak for the double O intermediates such as CH2OOH, CHOOH, CHOO, COOH, and CO2. For the other intermediates, the dominated orbital can be the py orbital for CH3OH, CH2OH, CHOH, CH2O, and CHO, and the pz orbital for CH3O, COH, and CO. The s orbital contributes to the peak at the Fermi level for most of the intermediates, except for CO and CO2. However, this contribution is much smaller than the p orbitals.
The orbital-projected electronic density of states of the substrate/intermediate systems in Fig. 4 and S2, ESI,† will assist in substantially understanding the nature of the interaction between the intermediates and the substrates. In Fig. 4 and S2,† we plotted only the most significant orbitals near the Fermi level, in the energy range from −4.0 to 2.0 eV. We presented the identical orbitals with the same colour, i.e., dxz and dyz (green) and dxy and dx2−y2 (black). We found that the spin-up dxz, dyz, and dz2 orbitals of the Pt, Ru, Co, and Fe metal atoms and all the spin-down d orbitals (i.e., dxy, dx2−y2, dxz, dyz, dz2) of the metal atoms dominate the DOS of the PtRuCo3/C-MWCNTs and PtRuFe3/C-MWCNTs substrates around the Fermi level. Notably, the orbitals of the carbon support are insignificant. The interaction between an adsorbate and an adsorbent is established by the attraction between the occupied states of the adsorbate and the unoccupied states of the adsorbent and vice versa. Comparing the orbital-projected DOS of the adsorbed intermediates in Fig. 4 and S2† to that of the isolated intermediates in Fig. 3, we found that the peak at the Fermi level of the isolated intermediates disappears in the adsorbed intermediates (Fig. 4) and rearranges clearly inside the valence and conduction bands. Tracking the DOS of the isolated intermediates at the Fermi level, we can conclude that the interaction between the p orbitals, i.e., (px, py, pz) of all the intermediates with the z-axis projected spin-up d orbitals and all the spin-down d orbitals of the Pt, Ru, Co, and Fe atoms is the physical reason for the adsorption of the intermediates on the PtRuM3/C-MWCNTs substrates.
Fig. 5 depicts the charge density difference of the substrate/intermediate systems, where the cyan and yellow colours present the charge depletion and accumulation, respectively. We found that the topmost Pt layer (layer 1 in Table 2) and the O atom of the intermediates always gain charge. Therefore, if the CH3OH*, CH3O*, CH2O*, CHOOH*, 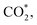 and CHOO* intermediates approach the substrate surface, their O atom will compete with the Pt layer to gain the charge. Therefore, the interface region between the O atom and the surface will form the depletion cloud (cyan colour). This cyan cloud couples with the accumulation clouds to form charge dipoles while it simultaneously couples with the charge gain cloud of the O atom of the intermediates to form another charge dipole. Therefore, the intermediates and the substrates can establish the interaction via these dipoles. If the depletion cloud in the interface region is broad, the intermediates such as CH3OH*, CH3O*, CH2O*, CHOOH*, and
and CHOO* intermediates approach the substrate surface, their O atom will compete with the Pt layer to gain the charge. Therefore, the interface region between the O atom and the surface will form the depletion cloud (cyan colour). This cyan cloud couples with the accumulation clouds to form charge dipoles while it simultaneously couples with the charge gain cloud of the O atom of the intermediates to form another charge dipole. Therefore, the intermediates and the substrates can establish the interaction via these dipoles. If the depletion cloud in the interface region is broad, the intermediates such as CH3OH*, CH3O*, CH2O*, CHOOH*, and 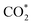 compete strongly with the Pt layer. Hence, their interaction with the substrate surface becomes weak, resulting in unstable adsorption, as analysed in Subsection 3.1. Although CHOO* also adsorbs via the O atoms, its charge dipoles are arranged more harmonically and equitably. The other strongly adsorbed intermediates also have characteristics similar to those of the charge arrangement of CHOO*.
compete strongly with the Pt layer. Hence, their interaction with the substrate surface becomes weak, resulting in unstable adsorption, as analysed in Subsection 3.1. Although CHOO* also adsorbs via the O atoms, its charge dipoles are arranged more harmonically and equitably. The other strongly adsorbed intermediates also have characteristics similar to those of the charge arrangement of CHOO*.
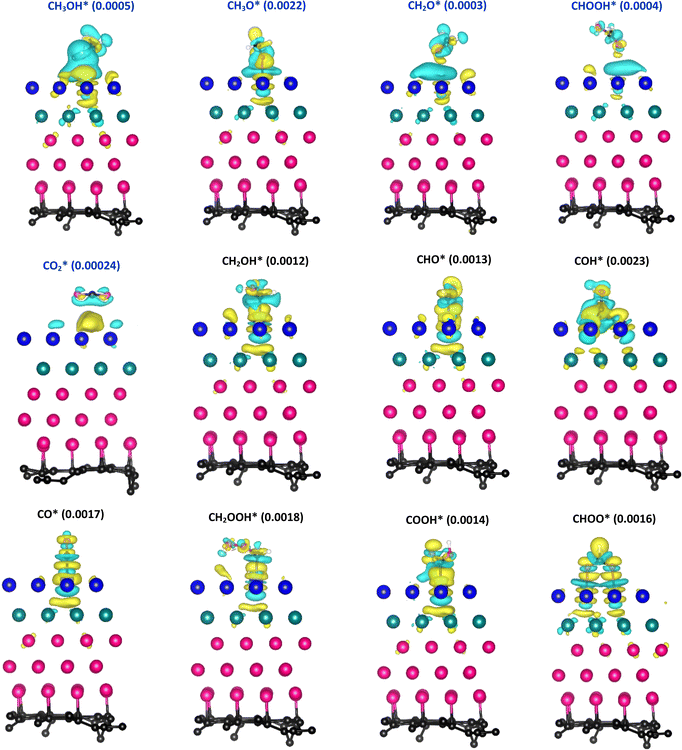 |
| | Fig. 5 The charge density difference of the systems of the adsorbed intermediate on the PtRuCo3/C-MWCNT substrate. The isosurface values are inside parentheses, with the unit being e− Å−3. | |
Crystal orbital Hamilton population (COHP) can also provide a better understanding of the chemical bonding strength of the intermediates to the substrate surface.57–59 Fig. 6 shows the COHP, the O–Pt bond, between the oxygen atoms of the representative intermediates (CH3OH*, CH3O*, and CH2O*) and the PtRuCo3/C-MWCNTs substrate. The positive and negative values of the COHP describe the binding and antibonding energy regions, respectively. The results implied that the O–Pt interaction is the strongest for CH3O* and weakest for CH3OH* and CH2O*, which agrees well with the trend of their adsorption energies and the Bader charge transfers.
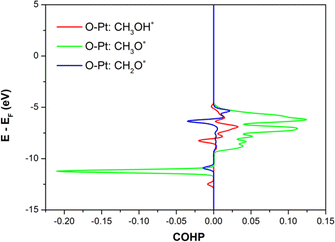 |
| | Fig. 6 The crystal orbital Hamilton population of the O–Pt bonding interaction between the intermediates (CH3OH*, CH3O*, and CH2O*) and the PtRuCo3/C-MWCNT substrate. | |
3.3 Methanol oxidation reaction mechanisms
Fig. 2 presents the possible reaction pathways on the surface of the PtRuM3/C-MWCNTs substrates, which are expressed in detail through the following intermediate reactions:| | |
CH3OH* + H2O ⇄ CH3O* + ½H2 + H2O,
| (R1) |
| | |
CH3OH* + H2O ⇄ CH2OH* + ½H2 + H2O,
| (R2) |
| | |
CH3O* + ½H2 + H2O ⇄ CH2O* + H2 + H2O,
| (R3) |
| | |
CH2OH* + ½H2 + H2O ⇄ CH2O* + H2 + H2O,
| (R4) |
| | |
CH2OH* + ½H2 + H2O ⇄ CHOH* + H2 + H2O,
| (R5) |
| | |
CH2O* + H2 + H2O ⇄ CH2OOH* + 3/2H2,
| (R6) |
| | |
CH2O* + H2 + H2O ⇄ CHO* + 3/2H2 + H2O,
| (R7) |
| | |
CHOH* + H2 + H2O ⇄ CHO* + 3/2H2 + H2O,
| (R8) |
| | |
CHOH* + H2 + H2O ⇄ COH* + 3/2H2 + H2O,
| (R9) |
| | |
CH2OOH* + 3/2H2 ⇄ CHOOH* + 2H2,
| (R10) |
| | |
CHO* + 3/2H2 + H2O ⇄ CHOOH* + 2H2,
| (R11) |
| | |
CHO* + 3/2H2 + H2O ⇄ CO* + 2H2 + H2O,
| (R12) |
| | |
COH* + 3/2H2 + H2O ⇄ CO* + 2H2 + H2O,
| (R13) |
| | |
CHOOH* + 2H2 ⇄ CHOO* + 5/2H2,
| (R14) |
| | |
CHOOH* + 2H2 ⇄ COOH* + 5/2H2,
| (R15) |
| | |
CO* + 2H2 + H2O ⇄ COOH* + 5/2H2,
| (R16) |
| |
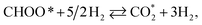 | (R17) |
| |
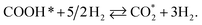 | (R18) |
The formula to calculate the Gibbs free energy for each intermediate step from (R1)–(R18) is presented as follows60
| | |
ΔGi(U) = GiR(U) − GiL(U) = ΔGi(0) + eU,
| (5) |
| | |
ΔGi(0) = GiR(0) − GiL(0).
| (6) |
where i runs from
(R1)–(R18). L and R denote the left and right sides of each intermediate step, from
(R1)–(R18).
Gi and Δ
Gi indicate the Gibbs free energy for one side of the reaction and the whole intermediate step, respectively.
To identify the favourable reaction pathways, we calculated the Gibbs free energy (ΔG)61 for all intermediate steps from (R1)–(R18) with zero-point energies provided in Table 1. To do that, we first have to attain the values of the Gibbs free energy GiL and GiR as listed in Table 3 for the applied potential U = 0 V. We then calculate ΔGi(0), following eqn (6), for the applied potential U = 0 V, and determine ΔGi(U), following eqn (5), for U = 1.005 V. Fig. 7 and 8 depict the Gibbs free energy diagrams for methanol oxidation reaction on the PtRuFe3/C-MWCNTs and PtRuCo3/C-MWCNTs substrates at the applied potentials U = 0 and 1.005 V, respectively.
Table 3 The Gibbs free energy GiL and GiR (eV) of the intermediate steps on the PtRuM3/C-MWCNTs (M = Fe and Co) substrates at an applied potential (U) of 0 V, pressure of 1 bar, and temperature of 300 K
| Intermediate step |
PtRuFe3/C-MWCNTs |
PtRuCo3/C-MWCNTs |
| CH3OH* + H2O |
0.513 |
0.167 |
| CH3O* + 1/2H2 + H2O |
1.357 |
1.037 |
| CH2OH* + 1/2H2 + H2O |
0.653 |
0.388 |
| CH2O* + H2 + H2O |
1.084 |
0.792 |
| CHOH* + H2 + H2O |
1.127 |
0.641 |
| CHO* + 3/2H2 + H2O |
0.508 |
0.398 |
| COH* + 3/2H2 + H2O |
0.879 |
0.716 |
| CH2OOH* + 3/2H2 |
3.696 |
3.503 |
| CO* + 2H2 + H2O |
0.003 |
−0.260 |
| CHOOH* + 2H2 |
0.835 |
0.548 |
| CHOO* + 5/2H2 |
0.997 |
0.634 |
| COOH* + 5/2H2 |
0.789 |
0.453 |
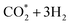 |
0.329 |
0.033 |
 |
| | Fig. 7 Gibbs free energy diagram for the methanol oxidation reaction on (a) PtRuFe3/C-MWCNT and (b) PtRuCo3/C-MWCNT substrates at an applied potential (U) of 0 V, a pressure of 1 bar, and a temperature of 300 K. For simplicity, the hydrogen and water molecules in each step have been omitted in the diagrams. Relative Gibbs free energy (eV) is in parenthesis. | |
 |
| | Fig. 8 Gibbs free energy diagram for the methanol oxidation reaction on (a) PtRuFe3/C-MWCNT and (b) PtRuCo3/C-MWCNT substrates at an applied potential (U) of 1.005 V, pressure of 1 bar, and temperature of 300 K. For simplicity, the hydrogen and water molecules in the reaction equations have been omitted in the diagrams. Relative Gibbs free energy (eV) is in parenthesis. | |
We first analyse the reaction mechanisms of methanol oxidation on the PtRuFe3/C-MWCNTs substrate at the applied potential U = 0 V, see Fig. 7a and Table 4. Note that we did not study transition states for the intermediate steps in the present work. In the forward direction, Fig. 7 shows the possible pathways: path 1 converts 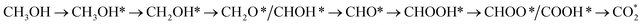 going uphill from CH3OH → CH3OH* → CH2OH* → CH2O* and CHO* → CHOOH* → CHOO*, where the rate-limiting step occurs at the methanol adsorption step (CH3OH → CH3OH*) with the thermodynamic barrier of 0.513 eV (see Table 4). Path 2 transforms
going uphill from CH3OH → CH3OH* → CH2OH* → CH2O* and CHO* → CHOOH* → CHOO*, where the rate-limiting step occurs at the methanol adsorption step (CH3OH → CH3OH*) with the thermodynamic barrier of 0.513 eV (see Table 4). Path 2 transforms 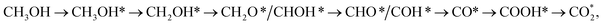 which goes uphill from CH3OH → CH3OH* → CH2OH* → CH2O*/CHOH* and CO* → COOH*, where the rate-limiting step happens at the carbon monoxide to carboxyl converting step (CO* → COOH*) with the thermodynamic barrier of 0.786 eV on PtRuFe3/C-MWCNTs (see Table 4). Path 3:
which goes uphill from CH3OH → CH3OH* → CH2OH* → CH2O*/CHOH* and CO* → COOH*, where the rate-limiting step happens at the carbon monoxide to carboxyl converting step (CO* → COOH*) with the thermodynamic barrier of 0.786 eV on PtRuFe3/C-MWCNTs (see Table 4). Path 3: 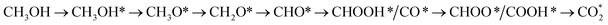 with the rate-limiting step taking place at the methanol to methoxy transforming step (CH3OH* → CH3O*) with a thermodynamic barrier of 0.844 eV (Table 4). Path 4:
with the rate-limiting step taking place at the methanol to methoxy transforming step (CH3OH* → CH3O*) with a thermodynamic barrier of 0.844 eV (Table 4). Path 4: 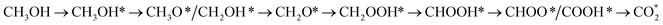 with the rate-limiting step occurs at the formaldehyde to hydroperoxymethyl converting step (CH2O* → CH2OOH*) with a thermodynamic barrier of 2.612 eV. We can see that the four reaction pathways are found not only on the PtRuFe3/C-MWCNTs but also on the PtRuCo3/C-MWCNTs substrate. However, the rate-limiting step of path 1 occurs at the hydroxymethyl to formaldehyde transforming step (CH2OH* → CH2O*) on the PtRuCo3/C-MWCNTs substrate, which is different from that on the PtRuFe3/C-MWCNTs surface. The highest thermodynamic barrier (Eb) for path 1, path 2, path 3, and path 4 on the PtRuCo3/C-MWCNTs substrate is 0.404, 0.713, 0.870, and 2.711 eV, respectively. According to the Arrhenius equation for calculating the reaction-rate constant (R), R ∼ exp(−Eb/RT), the lower the barrier, the higher the favourable order of the reaction pathway should be. We found that the favourable order of reaction pathways is path 1 > path 2 > path 3 > path 4 for both substrates. By comparing the energy barrier for each path, we found that path 1 and path 2 of the methanol oxidation reaction are more favourable on PtRuCo3/C-MWCNTs than on PtRuFe3/C-MWCNTs, while path 3 and path 4 are slightly less favourable on PtRuCo3/C-MWCNTs. Notably, path 4 has a very high thermodynamic barrier, which perhaps prohibits the methanol oxidation reaction from proceeding on both substrates.
with the rate-limiting step occurs at the formaldehyde to hydroperoxymethyl converting step (CH2O* → CH2OOH*) with a thermodynamic barrier of 2.612 eV. We can see that the four reaction pathways are found not only on the PtRuFe3/C-MWCNTs but also on the PtRuCo3/C-MWCNTs substrate. However, the rate-limiting step of path 1 occurs at the hydroxymethyl to formaldehyde transforming step (CH2OH* → CH2O*) on the PtRuCo3/C-MWCNTs substrate, which is different from that on the PtRuFe3/C-MWCNTs surface. The highest thermodynamic barrier (Eb) for path 1, path 2, path 3, and path 4 on the PtRuCo3/C-MWCNTs substrate is 0.404, 0.713, 0.870, and 2.711 eV, respectively. According to the Arrhenius equation for calculating the reaction-rate constant (R), R ∼ exp(−Eb/RT), the lower the barrier, the higher the favourable order of the reaction pathway should be. We found that the favourable order of reaction pathways is path 1 > path 2 > path 3 > path 4 for both substrates. By comparing the energy barrier for each path, we found that path 1 and path 2 of the methanol oxidation reaction are more favourable on PtRuCo3/C-MWCNTs than on PtRuFe3/C-MWCNTs, while path 3 and path 4 are slightly less favourable on PtRuCo3/C-MWCNTs. Notably, path 4 has a very high thermodynamic barrier, which perhaps prohibits the methanol oxidation reaction from proceeding on both substrates.
Table 4 The thermodynamic barrier (eV) of the intermediate steps on the PtRuM3/C-MWCNTs (M = Fe, Co) substrates at the applied potential U = 0 V, pressure of 1 bar, and temperature of 300 K. The rate-limiting step is marked by bold font. The highest thermodynamic barrier for each pathway is presented in parentheses
| Intermediate step |
PtRuFe3/C-MWCNTs |
PtRuCo3/C-MWCNTs |
| CH3OH + H2O → CH3OH* + H2O |
0.513 (path 1) |
0.167 |
| CH3OH* + H2O → CH3O* + 1/2H2 + H2O |
0.844 (path 3) |
0.870 (path 3) |
| CH3OH* + H2O → CH2OH* + 1/2H2 + H2O |
0.140 |
0.221 |
| CH3O* + 1/2H2 + H2O → CH2O* + H2 + H2O |
−0.273 |
−0.245 |
| CH2OH* + 1/2H2 + H2O → CH2O* + H2 + H2O |
0.431 |
0.404 (path 1) |
| CH2OH* + 1/2H2 + H2O → CHOH* + H2 + H2O |
0.474 |
0.253 |
| CH2O* + H2 + H2O → CH2OOH* + 3/2H2 |
2.612 (path 4) |
2.711 (path 4) |
| CH2O* + H2 + H2O → CHO* + 3/2H2 + H2O |
−0.576 |
−0.394 |
| CHOH* + H2 + H2O → CHO* + 3/2H2 + H2O |
−0.619 |
−0.243 |
| CHOH* + H2 + H2O → COH* + 3/2H2 + H2O |
−0.248 |
0.075 |
| CH2OOH* + 3/2H2 → CHOOH* + 2H2 |
−2.861 |
−2.955 |
| CHO* + 3/2H2 + H2O → CHOOH* + 2H2 |
0.327 |
0.150 |
| CHO* + 3/2H2 + H2O → CO* + 2H2 + H2O |
−0.505 |
−0.658 |
| COH* + 3/2H2 + H2O → CO* + 2H2 + H2O |
−0.876 |
−0.976 |
| CHOOH* + 2H2 → CHOO* + 5/2H2 |
0.162 |
0.086 |
| CHOOH* + 2H2 → COOH* + 5/2H2 |
−0.046 |
−0.095 |
| CO* + 2H2 + H2O → COOH* + 5/2H2 |
0.786 (path 2) |
0.713 (path 2) |
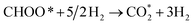 |
−0.668 |
−0.601 |
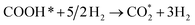 |
−0.460 |
−0.420 |
From the above analysis, our results exhibited that, for U = 0 V, path 1 is the most favourable mechanism: CH3OH → CH3OH* → CH2OH* → CH2O*/CHOH* → CHO* → CHOOH* → CHOO*/COOH* → CO2 on both substrates. Compared to the literature, our result is different from the work of Lv et al.,35 where they found that the most favourable mechanism for the methanol oxidation on Pt and PtNi catalysts should proceed via the CO* formation, i.e., 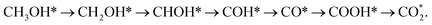 Besides, the most favourable pathway on PtRu/Pt(111) in the research of Ding et al.,42 is
Besides, the most favourable pathway on PtRu/Pt(111) in the research of Ding et al.,42 is 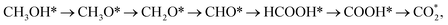 where forming CH3O* does not result in the most favourable mechanism for the PtRuCo3/C-MWCNTs and PtRuFe3/C-MWCNTs substrates in our present work. The most favourable reaction pathway (path 1) in this study supports the previous experiments that PtRuCo3/C-MWCNTs and PtRuFe3/C-MWCNTs catalysts have higher tolerances to CO poisoning compared to the Pt-based bimetallic alloys.31,62
where forming CH3O* does not result in the most favourable mechanism for the PtRuCo3/C-MWCNTs and PtRuFe3/C-MWCNTs substrates in our present work. The most favourable reaction pathway (path 1) in this study supports the previous experiments that PtRuCo3/C-MWCNTs and PtRuFe3/C-MWCNTs catalysts have higher tolerances to CO poisoning compared to the Pt-based bimetallic alloys.31,62
At the experimental condition, U = 1.005 V relative to the hydrogen standard electrode, Fig. 8 shows that all of path 1, path 2, and path 3 are going downhill after the uphill step at CH3OH → CH3OH*. Therefore, all three pathways have the same rate-limiting step at CH3OH → CH3OH* and the same thermodynamic barrier of 0.513 eV and 0.167 eV on both PtRuCo3/C-MWCNTs and PtRuFe3/C-MWCNTs substrates, respectively. The barrier on PtRuCo3/C-MWCNTs is three times lower than that on PtRuFe3/C-MWCNTs, implying that the third metal component Co offers a significantly better performance over Fe. Interestingly, this result is consistent with our experiment,31 where PtRuCo3/C-MWCNTs showed a considerably higher methanol oxidation peak and significantly lower electron transfer resistance compared to PtRuFe3/C-MWCNTs. Besides, path 4 has the rate-limiting step occurring at the converting step from formaldehyde to hydroperoxymethyl (CH2O* → CH2OOH*), which is the same as that at the applied potential U = 0 V; however, the activation barrier of 1.607 and 1.705 eV on the PtRuCo3/C-MWCNTs and PtRuFe3/C-MWCNTs surfaces, respectively. Also, the activation barrier for path 4 at U = 1.005 V is significantly lower than that at U = 0 V.
We also calculated the Gibbs free energy and studied Gibbs free energy diagrams at the applied potentials of U = 0 V and 1.005 V for the methanol oxidation reaction on PtRuM3 catalysts without the C-MWCNTs support. The Gibbs free energy diagrams are also similar to those on the PtRuM3/C-MWCNTs; however, the activation barrier differs. Comparing Tables 4 and S2† shows that, in the presence of the C-MWCNTs support, this value at U = 0 V increases for path 1, path 2 and decreases for path 3, path 4 on PtRuM3/C-MWCNTs (M = Fe and Co). At U = 1.005 V, the energy barrier for the three most favourable pathways (path 1, path 2, and path 3; three paths have the same energy barrier for each metal M = Fe and Co) on PtRuM3 substrates is 0.417 eV (Fe) and 0.376 eV (Co), and that for the most unfavourable one (via CH2OOH*) is 1.732 eV (Fe) and 1.953 eV (Co). We found that the C-MWCNTs support increases the activation barrier for the three most favourable paths on PtRuFe3/C-MWCNTs, while it decreases for all the other cases. Therefore, the C-MWCNTs support plays a better role for the PtRuCo3/C-MWCNTs.
4 Conclusions
Using density functional theory calculations, we searched for the possible intermediates of methanol oxidation reaction on the surface of PtRuM3/C-MWCNTs (M = Fe and Co) substrates. Most intermediates stably adsorb except for methanol, formaldehyde, formic acid, and carbon dioxide. The interaction between the intermediates and the substrates is due to the charge exchange for all intermediates, excluding the electrostatic attraction with CH2O and CHOOH, which mainly stems from the p orbitals of the intermediates interacting with the z-axis projected spin-up components of and all the spin-down component of the d orbitals of the Pt, Ru, Co, and Fe atoms. The Gibbs free energy diagram showed four possible reaction pathways for the methanol oxidation on the PtRuM3/C-MWCNTs (M = Fe and Co) substrates. At the applied potential of 0 V, three most favourable mechanisms with different activation barriers on each substrate are (path 1) 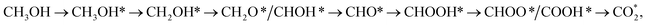 (path 2)
(path 2) 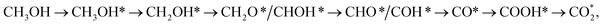 and (path 3)
and (path 3) 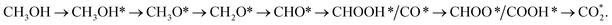 However, at the working potential of 1.005 V, these pathways have the same downhill behaviour, with the highest thermodynamic barrier occurring at the methanol adsorption step, CH3OH → CH3OH*. Three pathways have the same energy barrier of 0.513 and 0.167 eV for the PtRuFe3/C-MWCNTs and PtRuCo3/C-MWCNTs substrates, respectively. This result implied that the performance of PtRuCo3/C-MWCNTs is better than that of PtRuFe3/C-MWCNTs. This study only considered the low coverage of reaction intermediates. However, further investigation of the effects of coverages is necessary for understanding a full physical picture of the interaction between the intermediates and the substrate surface, which will be a research topic for future studies.
However, at the working potential of 1.005 V, these pathways have the same downhill behaviour, with the highest thermodynamic barrier occurring at the methanol adsorption step, CH3OH → CH3OH*. Three pathways have the same energy barrier of 0.513 and 0.167 eV for the PtRuFe3/C-MWCNTs and PtRuCo3/C-MWCNTs substrates, respectively. This result implied that the performance of PtRuCo3/C-MWCNTs is better than that of PtRuFe3/C-MWCNTs. This study only considered the low coverage of reaction intermediates. However, further investigation of the effects of coverages is necessary for understanding a full physical picture of the interaction between the intermediates and the substrate surface, which will be a research topic for future studies.
Data availability
The data supporting this article have been included as part of the ESI.†
Author contributions
Conceptualization (DNS), formal analysis (DNS, DLQ), investigation (DLQ, DNS), resources (DNS, VC), supervision (DNS, VC), validation (DNS, DLQ, VC), visualization (DLQ), writing manuscript (DNS, DLQ), reviewing and editing (DNS, VC).
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This research is funded by Vietnam National University Ho Chi Minh City (VNU-HCM) under grant number VL2022-20-01.
References
- A. Yuda, A. Ashok and A. Kumar, Catal. Rev., 2022, 64, 126–228 CrossRef CAS.
- Q. Lu, X. Zhao, R. Luque and K. Eid, Coord. Chem. Rev., 2023, 493, 215280 CrossRef CAS.
- J. Xu, K. Hua, G. Sun, C. Wang, X. Lv and Y. Wang, Electrochem. Commun., 2006, 8, 982–986 CrossRef CAS.
- R. Baronia, J. Goel, S. Tiwari, P. Singh, D. Singh, S. P. Singh and S. K. Singhal, Int. J. Hydrogen Energy, 2017, 42, 10238–10247 CrossRef CAS.
- A. B. A. A. Nassr, I. Sinev, W. Grünert and M. Bron, Appl. Catal., B, 2013, 142, 849–860 CrossRef.
- J. Sun, Y. Hou, X. Wang, T. Kou, N. Liu, R. Zhang and Z. Zhang, RSC Adv., 2021, 11, 14970–14979 RSC.
- J. Maya-Cornejo, A. Garcia-Bernabé and V. Compañ, Int. J. Hydrogen Energy, 2018, 43, 872–884 CrossRef CAS.
- S. Das and P. P. Kundu, RSC Adv., 2015, 5, 93539–93546 RSC.
- Q. Lu, J. Huang, C. Han, L. Sun and X. Yang, Electrochim. Acta, 2018, 266, 305–311 CrossRef CAS.
- H. Zhao, W. Qi, X. Zhou, H. Wu and Y. Li, Chin. J. Catal., 2018, 39, 342–349 CrossRef CAS.
- V. Stamenkovic, B. S. Mun, K. J. J. Mayrhofer, P. N. Ross, N. M. Markovic, J. Rossmeisl, J. Greeley and J. K. Nørskov, Angew. Chem., 2006, 45, 2897–2901 CrossRef CAS PubMed.
- P. Strasser, Q. Fan, M. Devenney, W. H. Weinberg, P. Liu and J. K. Nørskov, J. Phys. Chem. B, 2003, 107, 11013–11021 CrossRef CAS.
- M. E. Scofield, C. Koenigsmann, L. Wang, H. Liu and S. S. Wong, Energy Environ. Sci., 2015, 8, 350–363 RSC.
- Q. Zhao, G. Zhang, G. Xu, Y. Li, B. Liu, X. Gong, D. Zheng, J. Zhang and Q. Wang, Appl. Surf. Sci., 2016, 389, 181–189 CrossRef CAS.
- Y. Yang, L. M. Luo, R. H. Zhang, J. J. Du, P. C. Shen, Z. X. Dai, C. Sun and X. W. Zhou, Electrochim. Acta, 2016, 222, 1094–1102 CrossRef CAS.
- C. Shang, Y. Guo and E. Wang, Nano Res., 2018, 11, 4348–4355 CrossRef CAS.
- K. Deng, Y. Xu, C. Li, Z. Wang, H. Xue, X. Li, L. Wang and H. Wang, Langmuir, 2018, 35, 413–419 CrossRef PubMed.
- H. Wang, Y. Wu, X. Luo, L. Jiao, X. Wei, W. Gu, D. Du, Y. Lin and C. Zhu, Nanoscale, 2019, 11, 10575–10580 RSC.
- Y. Pan, H. Li, Z. Wang, Y. Han, Z. Wu, X. Zhang, J. Lai, L. Wang and S. Feng, Chem. Commun., 2020, 56, 9028–9031 RSC.
- W. Zhao, F. Xie, M. Gan, L. Ma, Y. Zhang, X. Li, L. Wang and X. Hua, Mater. Today Chem., 2022, 24, 100788 CrossRef CAS.
- X. Ren, Q. Lv, L. Liu, B. Liu, Y. Wang, A. Liu and G. Wu, Sustainable Energy Fuels, 2020, 4, 15–30 RSC.
- M. K. Jeon, J. Y. Won, K. R. Lee and S. I. Woo, Electrochem. Commun., 2007, 9, 2163–2166 CrossRef CAS.
- M. K. Jeon, K. R. Lee, H. Daimon, A. Nakahara and S. I. Woo, Catal. Today, 2008, 132, 123–126 CrossRef CAS.
- Z. Cai, Y. Kuang, X. Qi, P. Wang, Y. Zhang, Z. Zhang and X. Sun, J. Mater. Chem. A, 2015, 3, 1182–1187 RSC.
- Z. B. Wang, G. P. Yin, Y. Y. Shao, B. Q. Yang, P. F. Shi and P. X. Feng, J. Power Sources, 2007, 165, 9–15 CrossRef CAS.
- Y. Zhao, L. Fan, J. Ren and B. Hong, Int. J. Hydrogen Energy, 2014, 39, 4544–4557 CrossRef CAS.
- H. Lee, S. Park and H. Kim, Chem. Eng. J., 2021, 414, 128792 CrossRef CAS.
- T. Huang, J. Liu, R. Li, W. Cai and A. Yu, Electrochem. Commun., 2009, 11, 643–646 CrossRef CAS.
- T. Huang, X. Wang, J. Zhuang, W. B. Cai and A. Yu, Electrochem. Solid-State Lett., 2009, 12, B112–B115 CrossRef CAS.
- M. Semasko, L. Tamasauskaite-Tamasiunaite, V. Kepeniene, A. Balciunaite, J. Vaiciuniene, A. Drabavicius and E. Norkus, ECS Trans., 2015, 68, 55–62 CrossRef CAS.
- D. L. Quan, P. T. Hai, P. H. Le and D. N. Son, Res. Chem. Intermed., 2023, 49, 3987–4007 CrossRef CAS.
- L. Zhao, S. Wang, Q. Ding, W. Xu, P. Sang, Y. Chi, X. Lu and W. Guo, J. Phys. Chem. C, 2015, 119, 20389–20400 CrossRef CAS.
- V. Orazi, P. Bechthold, P. V. Jasen, R. Faccio, M. E. Pronsato and E. A. Gonzalez, Appl. Surf. Sci., 2017, 420, 383–389 CrossRef CAS.
- P. Du, P. Wu and C. Cai, J. Phys. Chem. C, 2017, 121, 9348–9360 CrossRef CAS.
- Q. Lv, X. Ren, L. Liu, W. Guan and A. Liu, Ionics, 2020, 26, 1325–1336 CrossRef CAS.
- D. Y. Wang, H. L. Chou, Y. C. Lin, F. J. Lai, C. H. Chen, J. F. Lee, B. J. Hwang and C. C. Chen, J. Am. Chem. Soc., 2012, 134, 10011–10020 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- H. T. Thanh, O. K. Le, V. Chihaia and D. N. Son, Phys. Chem. Chem. Phys., 2024, 26, 3963–3973 RSC.
- D. N. Son, H. Nakanishi, M. Y. David and H. Kasai, J. Phys. Soc. Jpn., 2009, 78, 114601 CrossRef.
- D. N. Son, P. N. Thanh, N. D. Quang, K. Takahashi and M. P. Pham-Ho, J. Appl. Electrochem., 2017, 47, 747–754 CrossRef CAS.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- Q. Ding, W. Xu, P. Sang, J. Xu, L. Zhao, X. He and W. Guo, Appl. Surf. Sci., 2016, 369, 257–266 CrossRef CAS.
- F. Chiter, V. B. Nguyen, N. Tarrat, M. Benoit, H. Tang and C. Lacaze-Dufaure, Mater. Res. Express, 2016, 3, 046501 CrossRef.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671–6687 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- M. Methfessel and A. T. Paxton, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 3616–3621 CrossRef CAS PubMed.
- D. N. Son and K. Takahashi, J. Phys. Chem. C, 2012, 116, 6200–6207 CrossRef CAS.
- D. N. Son, O. K. Le, V. Chihaia and K. Takahashi, J. Phys. Chem. C, 2015, 119, 24364–24372 CrossRef CAS.
- V. Wang, N. Xu, J. C. Liu, G. Tang and W. T. Geng, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS.
- J. Greeley and M. Mavrikakis, J. Am. Chem. Soc., 2004, 126, 3910–3919 CrossRef CAS PubMed.
- J. A. Stroscio and D. M. Eigler, Science, 1991, 254, 1319–1326 CrossRef CAS PubMed.
- M. Ø. Pedersen, M. L. Bocquet, P. Sautet, E. Lægsgaard, I. Stensgaard and F. Besenbacher, Chem. Phys. Lett., 1999, 299, 403–409 CrossRef CAS.
- G. Henkelman, A. Arnaldsson and H. Jonsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- D. N. Son, T. T. T. Huong and V. Chihaia, RSC Adv., 2018, 8, 38648–38655 RSC.
- S. Xia and S. Bobev, J. Alloys Compd., 2007, 427, 67–72 CrossRef CAS.
- X. Li and F. Peng, Phys. Chem. Chem. Phys., 2019, 21, 15609–15614 RSC.
- A. V. Kovalenko, A. V. Bandura, D. D. Kuruch, S. I. Lukyanov and R. A. Evarestov, Phys. Status Solidi B, 2021, 258, 2100072 CrossRef CAS.
- O. M. Na, N. T. X. Huynh, P. T. Thi, V. Chihaia and D. N. Son, RSC Adv., 2020, 10, 8460–8469 RSC.
- T. P. Dung, V. Chihaia and D. N. Son, RSC Adv., 2023, 13, 8523–8534 RSC.
- H. Li, Y. Pan, D. Zhang, Y. Han, Z. Wang, Y. Qin, S. Lin, X. Wu, H. Zhao, J. Lai, B. Huang and L. Wang, J. Mater. Chem. A, 2020, 8, 2323–2330 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Mechanisms and selectivity of methanol oxidation reaction on PtRuM3/C-MWCNTs (M = Fe, Co) electrocatalysts. See DOI: https://doi.org/10.1039/d4ra04493b |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence d and
Do Ngoc Son
d and
Do Ngoc Son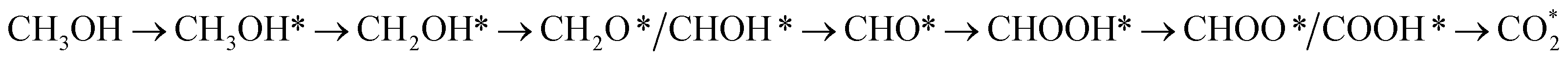 with a thermodynamic barrier of 0.513 eV for applied potentials of U = 0 V and 1.005 V on PtRuFe3/C-MWCNTs and 0.404 eV for U = 0 V and 0.167 eV for U = 1.005 V on PtRuCo3/C-MWCNTs. We also provide physical insights into the interaction between methanol oxidation intermediates and substrates' surface by analysing electronic properties. Our findings support the results of our previous experiment. The results of this study can be useful for rationally designing the anode for fuel cells.
with a thermodynamic barrier of 0.513 eV for applied potentials of U = 0 V and 1.005 V on PtRuFe3/C-MWCNTs and 0.404 eV for U = 0 V and 0.167 eV for U = 1.005 V on PtRuCo3/C-MWCNTs. We also provide physical insights into the interaction between methanol oxidation intermediates and substrates' surface by analysing electronic properties. Our findings support the results of our previous experiment. The results of this study can be useful for rationally designing the anode for fuel cells.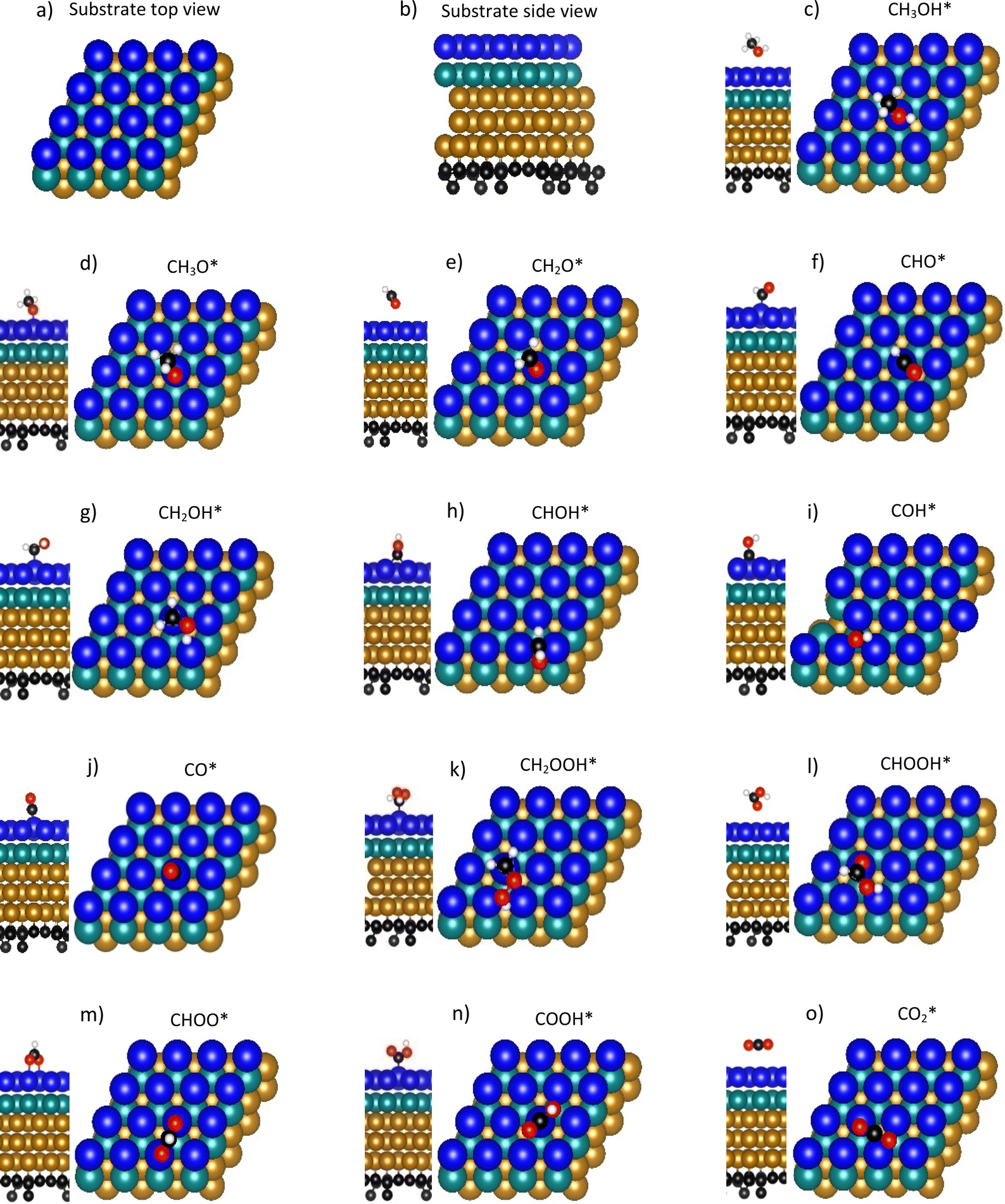
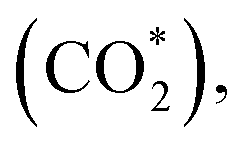 on the surface of the PtRuM3/C-MWCNTs (M = Fe, Co) substrates. Particularly, the adsorption site, the bond distance (dads-sur) from the nearest atom (carbon or oxygen atom, see Fig. 1c–o for each case) of the intermediates to the surface of the substrate, the adsorption energy (Eads), the zero-point energy (ZPE), and the total energy (Etotal, i.e., Eads plus ZPE) are also listed in Table 1. As shown in Fig. 1c–o, several intermediates such as CH3OH*, CH3O*, CH2O*, and CHOOH* adsorb most favourably with the end-on configuration on the top (T) site of the Pt atom, where the O atom is the nearest atom to the substrates' surface. The adsorption of the other intermediates, i.e., CH2OH*, CHOH*, CH2OOH*, CHO*, COH*, CO*, and COOH*, is also the most stable with the end-on configuration via their C atom; however, while CH2OH*, CH2OOH*, CHO*, COH*, CO*, and COOH* are on the top site, CHOH* is on the bridge (B) and COH* is on the hollow (H) with an underneath Ru atom. Exceptions are for CHOO* and
on the surface of the PtRuM3/C-MWCNTs (M = Fe, Co) substrates. Particularly, the adsorption site, the bond distance (dads-sur) from the nearest atom (carbon or oxygen atom, see Fig. 1c–o for each case) of the intermediates to the surface of the substrate, the adsorption energy (Eads), the zero-point energy (ZPE), and the total energy (Etotal, i.e., Eads plus ZPE) are also listed in Table 1. As shown in Fig. 1c–o, several intermediates such as CH3OH*, CH3O*, CH2O*, and CHOOH* adsorb most favourably with the end-on configuration on the top (T) site of the Pt atom, where the O atom is the nearest atom to the substrates' surface. The adsorption of the other intermediates, i.e., CH2OH*, CHOH*, CH2OOH*, CHO*, COH*, CO*, and COOH*, is also the most stable with the end-on configuration via their C atom; however, while CH2OH*, CH2OOH*, CHO*, COH*, CO*, and COOH* are on the top site, CHOH* is on the bridge (B) and COH* is on the hollow (H) with an underneath Ru atom. Exceptions are for CHOO* and 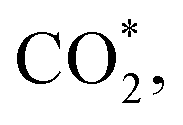 where they adsorb stably in the side-on configurations with the double top (T–T) site (each O atom on a T site) for CHOO* and the T–B site (one O atom on top and the other O atom on B) for
where they adsorb stably in the side-on configurations with the double top (T–T) site (each O atom on a T site) for CHOO* and the T–B site (one O atom on top and the other O atom on B) for 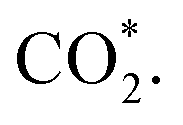 Especially, the
Especially, the 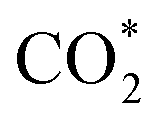 structure is in a straight configuration with an O–C–O angle of about 180°, parallel to the surface of the substrate. Table 1 also summarizes the most favourable adsorption site on the substrates' surface and the bond distance dads-sur (in columns 3 & 4). We found that the bond distance from the nearest atom of the intermediates to the surface is in the following order: hollow < bridge < top or top–top < top-bridge. The
structure is in a straight configuration with an O–C–O angle of about 180°, parallel to the surface of the substrate. Table 1 also summarizes the most favourable adsorption site on the substrates' surface and the bond distance dads-sur (in columns 3 & 4). We found that the bond distance from the nearest atom of the intermediates to the surface is in the following order: hollow < bridge < top or top–top < top-bridge. The 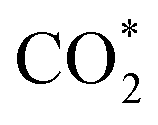 intermediate is located at the largest distance of about 3.4 Å from the surface of the PtRuM3/C-MWCNTs (M = Fe, Co) substrates. The adsorption energy of CH3OH*, CH2O*, CHOOH*, and
intermediate is located at the largest distance of about 3.4 Å from the surface of the PtRuM3/C-MWCNTs (M = Fe, Co) substrates. The adsorption energy of CH3OH*, CH2O*, CHOOH*, and 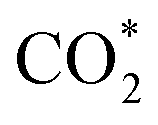 is positive (in column 5), which implies that these intermediates unfavourably adsorb and the other intermediates favourably adsorb with their negative adsorption energy on the surface of the PtRuFe3/C-MWCNTs. Particularly, the adsorption energy of all the intermediates (column 6) is negative on the PtRuCo3/C-MWCNTs catalyst. For each intermediate, it is more stable on PtRuCo3/C-MWCNTs than on PtRuFe3/C-MWCNTs because its adsorption energy is always more negative on M = Co than M = Fe in the range of about −0.135 eV (CHOH*) to −0.362 eV (CHOO*). The positive zero-point energy (in columns 7 & 8) competes with the negative adsorption energy and hence influences the stability of the intermediates adsorbed on the catalytic surface. We found the fluctuation (or zero-point energy) of each adsorption intermediate is rather large. Particularly, the fluctuation is stronger than the adsorption strength for CH3OH*, CH2O*, CHOOH*, and
is positive (in column 5), which implies that these intermediates unfavourably adsorb and the other intermediates favourably adsorb with their negative adsorption energy on the surface of the PtRuFe3/C-MWCNTs. Particularly, the adsorption energy of all the intermediates (column 6) is negative on the PtRuCo3/C-MWCNTs catalyst. For each intermediate, it is more stable on PtRuCo3/C-MWCNTs than on PtRuFe3/C-MWCNTs because its adsorption energy is always more negative on M = Co than M = Fe in the range of about −0.135 eV (CHOH*) to −0.362 eV (CHOO*). The positive zero-point energy (in columns 7 & 8) competes with the negative adsorption energy and hence influences the stability of the intermediates adsorbed on the catalytic surface. We found the fluctuation (or zero-point energy) of each adsorption intermediate is rather large. Particularly, the fluctuation is stronger than the adsorption strength for CH3OH*, CH2O*, CHOOH*, and 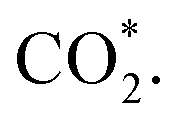 Therefore, the total energy Etotal of these intermediates becomes positive, which implies that CH3OH*, CH2O*, CHOOH*, and
Therefore, the total energy Etotal of these intermediates becomes positive, which implies that CH3OH*, CH2O*, CHOOH*, and 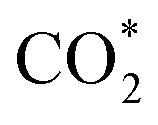 are unstable and subject to desorption from the substrates' surface. Emphasizing that the intermediates in the end-on via O configuration, such as CH3OH*, CH3O*, CH2O*, CHOOH*, and
are unstable and subject to desorption from the substrates' surface. Emphasizing that the intermediates in the end-on via O configuration, such as CH3OH*, CH3O*, CH2O*, CHOOH*, and 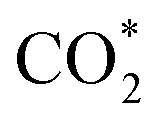 in the parallel structure have positive or least negative adsorption energy, indicating the adsorption via the O atom is not favourable, except for CHOO*. In contrast, the other intermediates remain in a stable state even after including the fluctuation on both substrates because of the negative Etotal. The isoformyl (COH*) is the most stable compared to all the other intermediates, with the most negative total energy on both substrates. We also found that, generally, the trend of the adsorption positions, bond distances, and adsorption energy of the intermediates are similar for both substrates, with the adsorption being more favourable on PtRuCo3/C-MWCNTs than on PtRuFe3/C-MWCNTs. Furthermore, the adsorption energy (Eads) of formaldehyde (CH2O*) on PtRuFe3/C-MWCNTs is insignificant, which is similar to the previous study for the adsorption of this intermediate on Pt(111).32,52 Also, the adsorption strength of carbon monoxide (CO*) is better on the PtRuCo3/C-MWCNTs surface with an adsorption energy of −1.335 eV compared to that of −1.097 eV on the PtRuFe3/C-MWCNTs surface. Interestingly, this result supports our previous experimental study, i.e., the jf/jr ratio of peak currents in forward and reverse directions demonstrated that the resistance to CO poisoning of PtRuFe3/C-MWCNTs is higher than that of PtRuCo3/C-MWCNTs, with jf/jr values of 6.9 and 4.3, respectively.31 The CO* adsorption via the C atom on the top of the Pt atom was also found in the experiment, which investigated the CO adsorption on the Pt(111) surface by the scanning tunnelling microscopy technique.53,54
in the parallel structure have positive or least negative adsorption energy, indicating the adsorption via the O atom is not favourable, except for CHOO*. In contrast, the other intermediates remain in a stable state even after including the fluctuation on both substrates because of the negative Etotal. The isoformyl (COH*) is the most stable compared to all the other intermediates, with the most negative total energy on both substrates. We also found that, generally, the trend of the adsorption positions, bond distances, and adsorption energy of the intermediates are similar for both substrates, with the adsorption being more favourable on PtRuCo3/C-MWCNTs than on PtRuFe3/C-MWCNTs. Furthermore, the adsorption energy (Eads) of formaldehyde (CH2O*) on PtRuFe3/C-MWCNTs is insignificant, which is similar to the previous study for the adsorption of this intermediate on Pt(111).32,52 Also, the adsorption strength of carbon monoxide (CO*) is better on the PtRuCo3/C-MWCNTs surface with an adsorption energy of −1.335 eV compared to that of −1.097 eV on the PtRuFe3/C-MWCNTs surface. Interestingly, this result supports our previous experimental study, i.e., the jf/jr ratio of peak currents in forward and reverse directions demonstrated that the resistance to CO poisoning of PtRuFe3/C-MWCNTs is higher than that of PtRuCo3/C-MWCNTs, with jf/jr values of 6.9 and 4.3, respectively.31 The CO* adsorption via the C atom on the top of the Pt atom was also found in the experiment, which investigated the CO adsorption on the Pt(111) surface by the scanning tunnelling microscopy technique.53,54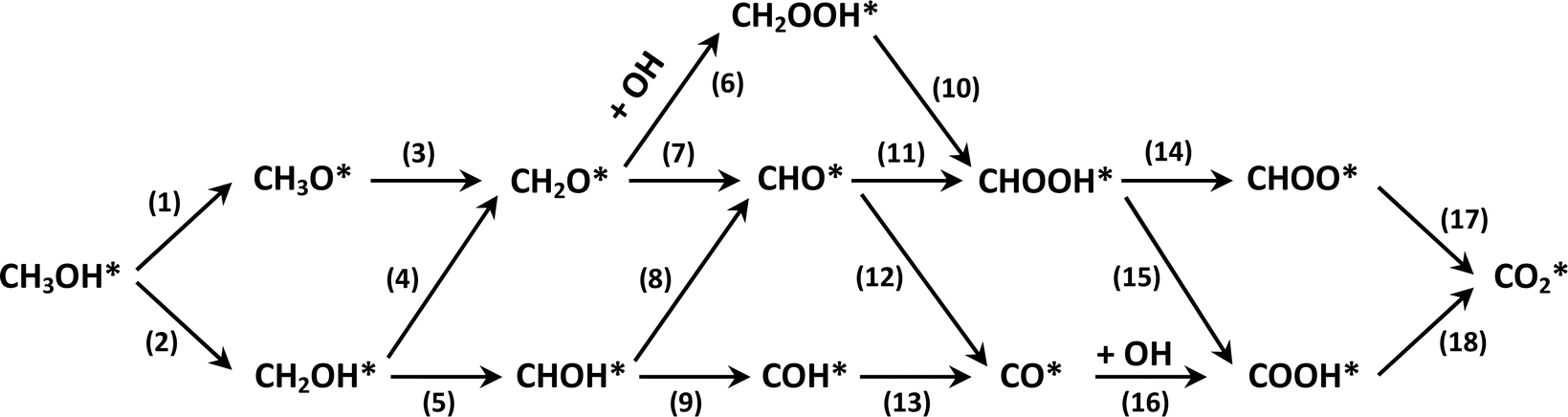
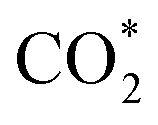
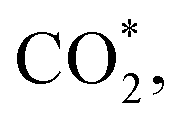 where the adsorption energy of these intermediates becomes more negative on PtRuM3/C-MWCNTs (Table 1) compared to the PtRuM3 substrates (Table S1†), respectively.
where the adsorption energy of these intermediates becomes more negative on PtRuM3/C-MWCNTs (Table 1) compared to the PtRuM3 substrates (Table S1†), respectively.
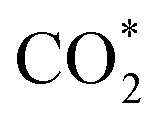 on both substrates and CH2OH* on the PtRuCo3/C-MWCNTs. Particularly, while CH3OH*, CH2OH*, CHOH*, and CH2OOH* donate, the other intermediates gain the charge from the substrates. Notably, the weak charge exchange intermediates, i.e., CH3OH*, CH2O*, CHOOH*, and
on both substrates and CH2OH* on the PtRuCo3/C-MWCNTs. Particularly, while CH3OH*, CH2OH*, CHOH*, and CH2OOH* donate, the other intermediates gain the charge from the substrates. Notably, the weak charge exchange intermediates, i.e., CH3OH*, CH2O*, CHOOH*, and 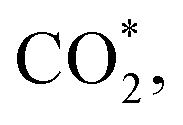 have small adsorption energies, as shown in Table 1.
have small adsorption energies, as shown in Table 1.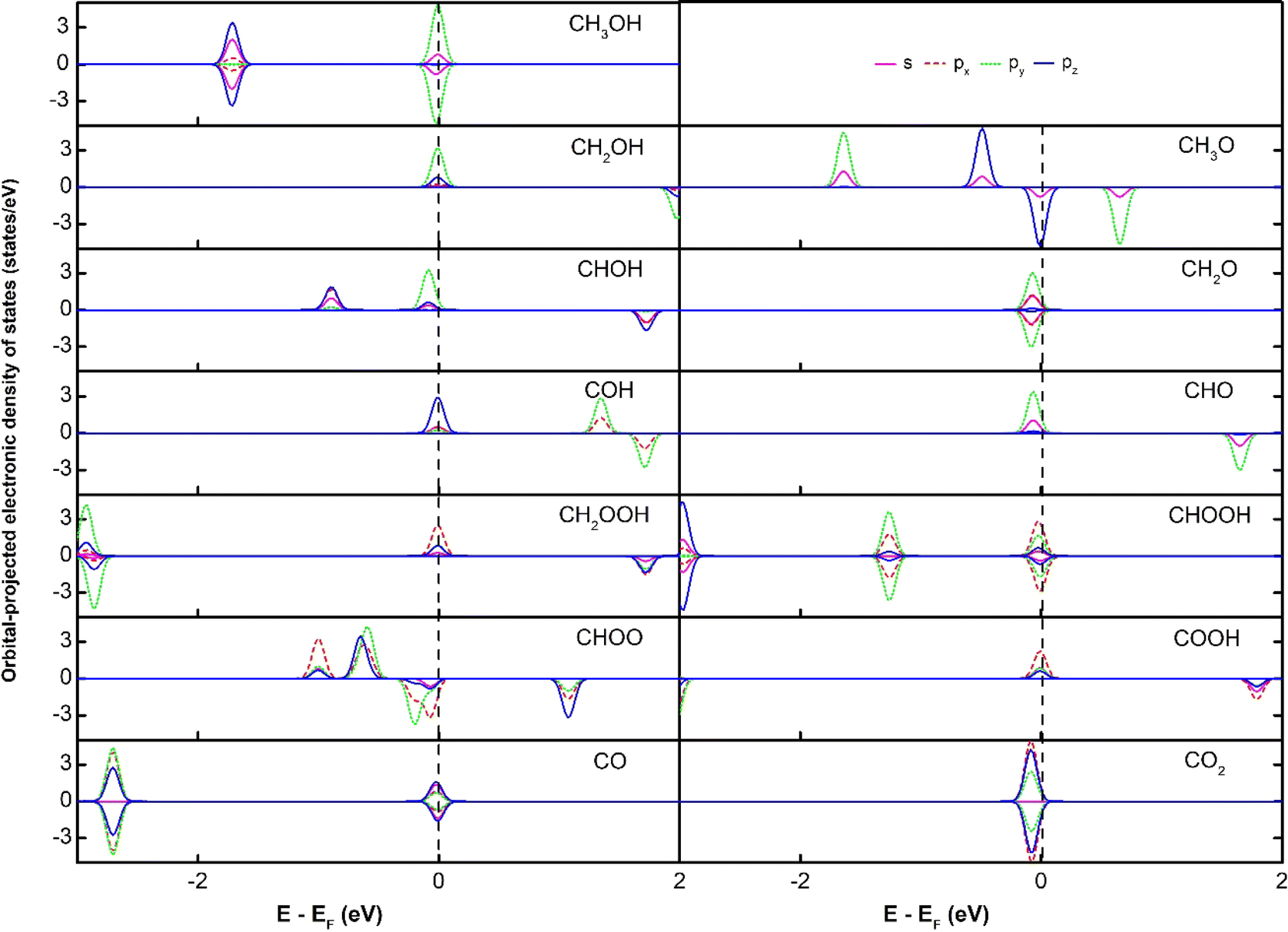
 and CHOO* intermediates approach the substrate surface, their O atom will compete with the Pt layer to gain the charge. Therefore, the interface region between the O atom and the surface will form the depletion cloud (cyan colour). This cyan cloud couples with the accumulation clouds to form charge dipoles while it simultaneously couples with the charge gain cloud of the O atom of the intermediates to form another charge dipole. Therefore, the intermediates and the substrates can establish the interaction via these dipoles. If the depletion cloud in the interface region is broad, the intermediates such as CH3OH*, CH3O*, CH2O*, CHOOH*, and
and CHOO* intermediates approach the substrate surface, their O atom will compete with the Pt layer to gain the charge. Therefore, the interface region between the O atom and the surface will form the depletion cloud (cyan colour). This cyan cloud couples with the accumulation clouds to form charge dipoles while it simultaneously couples with the charge gain cloud of the O atom of the intermediates to form another charge dipole. Therefore, the intermediates and the substrates can establish the interaction via these dipoles. If the depletion cloud in the interface region is broad, the intermediates such as CH3OH*, CH3O*, CH2O*, CHOOH*, and 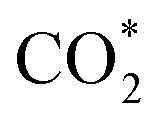 compete strongly with the Pt layer. Hence, their interaction with the substrate surface becomes weak, resulting in unstable adsorption, as analysed in Subsection 3.1. Although CHOO* also adsorbs via the O atoms, its charge dipoles are arranged more harmonically and equitably. The other strongly adsorbed intermediates also have characteristics similar to those of the charge arrangement of CHOO*.
compete strongly with the Pt layer. Hence, their interaction with the substrate surface becomes weak, resulting in unstable adsorption, as analysed in Subsection 3.1. Although CHOO* also adsorbs via the O atoms, its charge dipoles are arranged more harmonically and equitably. The other strongly adsorbed intermediates also have characteristics similar to those of the charge arrangement of CHOO*.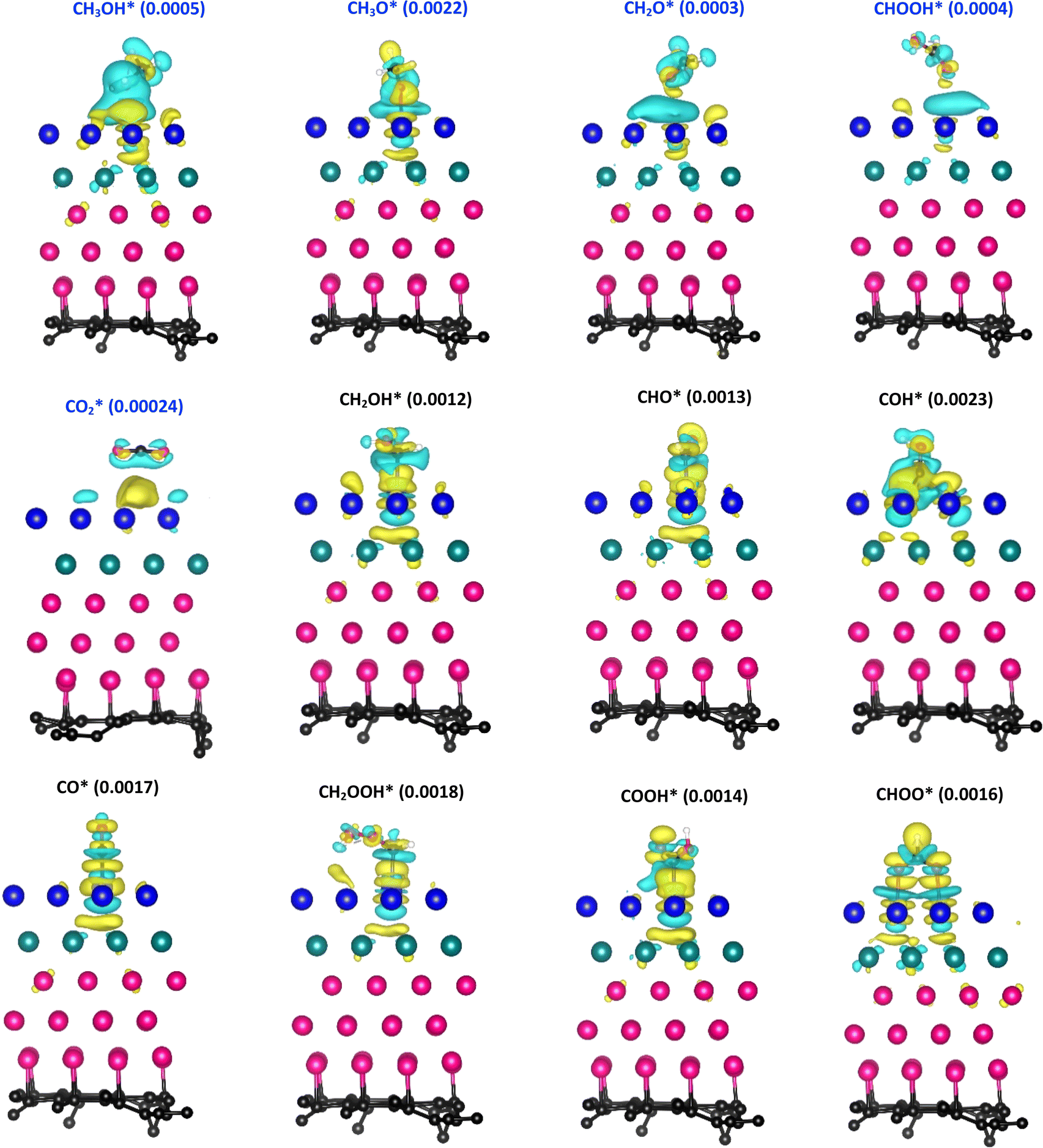
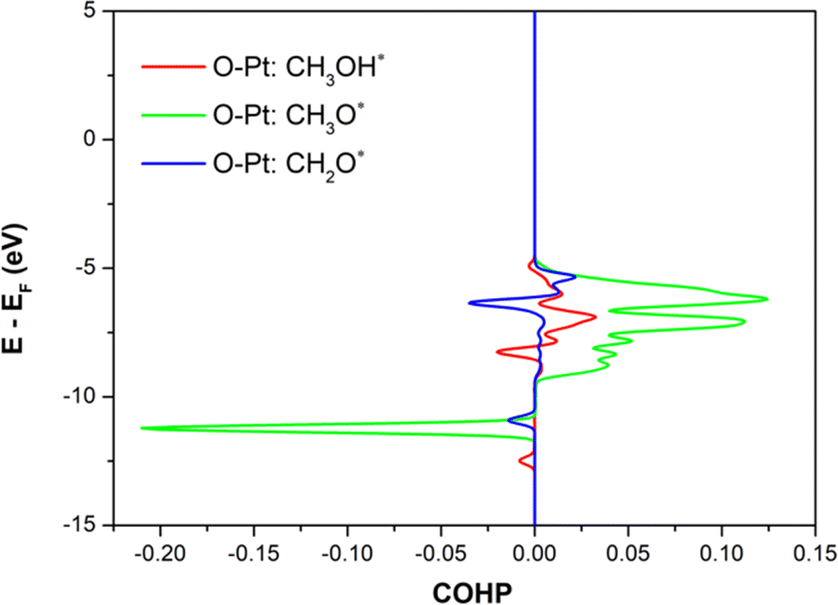
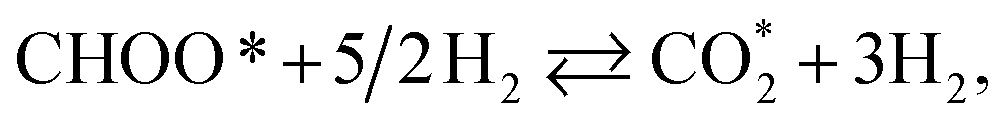
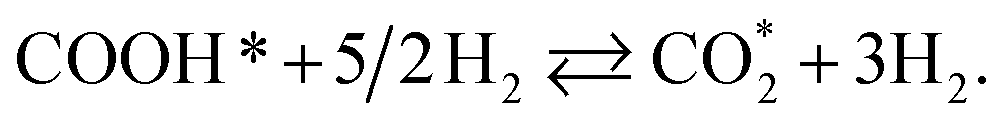
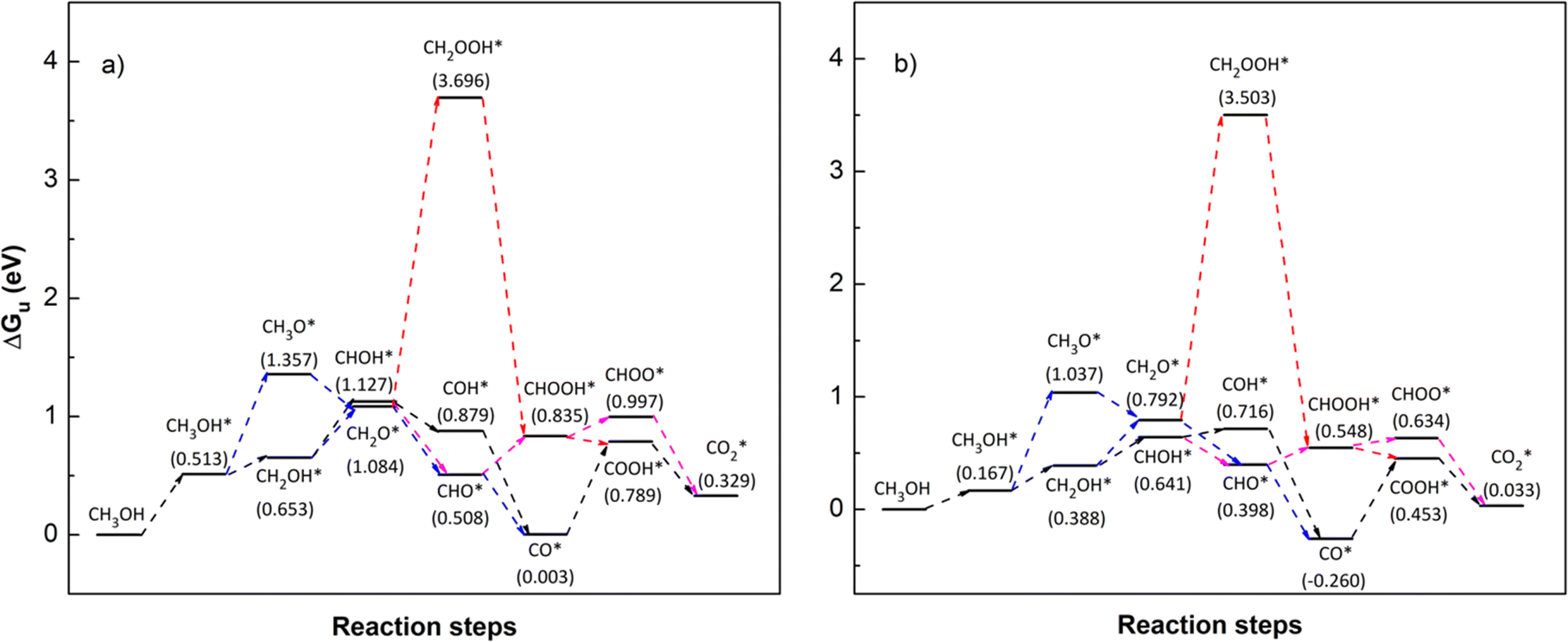
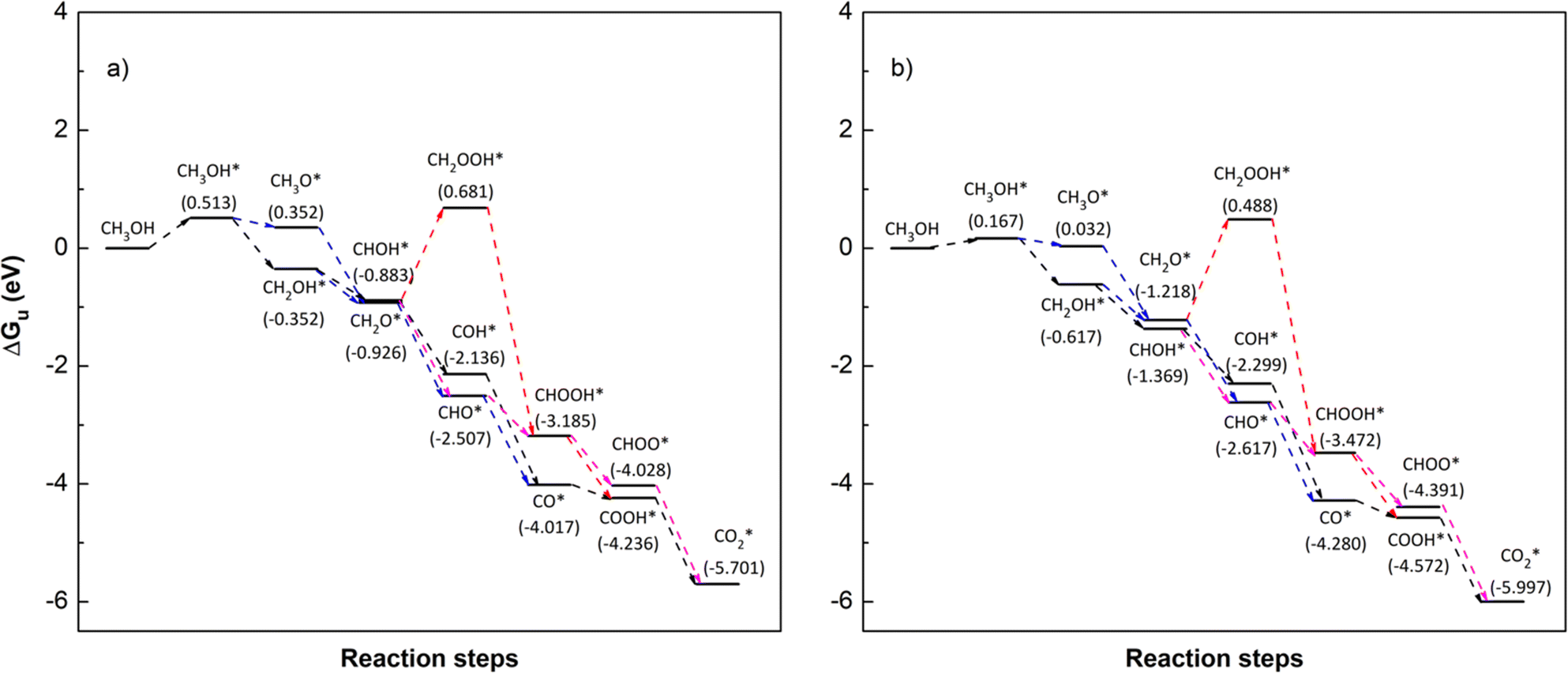
 going uphill from CH3OH → CH3OH* → CH2OH* → CH2O* and CHO* → CHOOH* → CHOO*, where the rate-limiting step occurs at the methanol adsorption step (CH3OH → CH3OH*) with the thermodynamic barrier of 0.513 eV (see Table 4). Path 2 transforms
going uphill from CH3OH → CH3OH* → CH2OH* → CH2O* and CHO* → CHOOH* → CHOO*, where the rate-limiting step occurs at the methanol adsorption step (CH3OH → CH3OH*) with the thermodynamic barrier of 0.513 eV (see Table 4). Path 2 transforms  which goes uphill from CH3OH → CH3OH* → CH2OH* → CH2O*/CHOH* and CO* → COOH*, where the rate-limiting step happens at the carbon monoxide to carboxyl converting step (CO* → COOH*) with the thermodynamic barrier of 0.786 eV on PtRuFe3/C-MWCNTs (see Table 4). Path 3:
which goes uphill from CH3OH → CH3OH* → CH2OH* → CH2O*/CHOH* and CO* → COOH*, where the rate-limiting step happens at the carbon monoxide to carboxyl converting step (CO* → COOH*) with the thermodynamic barrier of 0.786 eV on PtRuFe3/C-MWCNTs (see Table 4). Path 3:  with the rate-limiting step taking place at the methanol to methoxy transforming step (CH3OH* → CH3O*) with a thermodynamic barrier of 0.844 eV (Table 4). Path 4:
with the rate-limiting step taking place at the methanol to methoxy transforming step (CH3OH* → CH3O*) with a thermodynamic barrier of 0.844 eV (Table 4). Path 4: 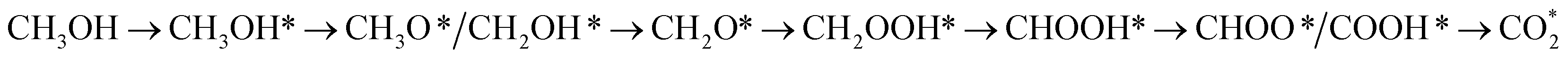 with the rate-limiting step occurs at the formaldehyde to hydroperoxymethyl converting step (CH2O* → CH2OOH*) with a thermodynamic barrier of 2.612 eV. We can see that the four reaction pathways are found not only on the PtRuFe3/C-MWCNTs but also on the PtRuCo3/C-MWCNTs substrate. However, the rate-limiting step of path 1 occurs at the hydroxymethyl to formaldehyde transforming step (CH2OH* → CH2O*) on the PtRuCo3/C-MWCNTs substrate, which is different from that on the PtRuFe3/C-MWCNTs surface. The highest thermodynamic barrier (Eb) for path 1, path 2, path 3, and path 4 on the PtRuCo3/C-MWCNTs substrate is 0.404, 0.713, 0.870, and 2.711 eV, respectively. According to the Arrhenius equation for calculating the reaction-rate constant (R), R ∼ exp(−Eb/RT), the lower the barrier, the higher the favourable order of the reaction pathway should be. We found that the favourable order of reaction pathways is path 1 > path 2 > path 3 > path 4 for both substrates. By comparing the energy barrier for each path, we found that path 1 and path 2 of the methanol oxidation reaction are more favourable on PtRuCo3/C-MWCNTs than on PtRuFe3/C-MWCNTs, while path 3 and path 4 are slightly less favourable on PtRuCo3/C-MWCNTs. Notably, path 4 has a very high thermodynamic barrier, which perhaps prohibits the methanol oxidation reaction from proceeding on both substrates.
with the rate-limiting step occurs at the formaldehyde to hydroperoxymethyl converting step (CH2O* → CH2OOH*) with a thermodynamic barrier of 2.612 eV. We can see that the four reaction pathways are found not only on the PtRuFe3/C-MWCNTs but also on the PtRuCo3/C-MWCNTs substrate. However, the rate-limiting step of path 1 occurs at the hydroxymethyl to formaldehyde transforming step (CH2OH* → CH2O*) on the PtRuCo3/C-MWCNTs substrate, which is different from that on the PtRuFe3/C-MWCNTs surface. The highest thermodynamic barrier (Eb) for path 1, path 2, path 3, and path 4 on the PtRuCo3/C-MWCNTs substrate is 0.404, 0.713, 0.870, and 2.711 eV, respectively. According to the Arrhenius equation for calculating the reaction-rate constant (R), R ∼ exp(−Eb/RT), the lower the barrier, the higher the favourable order of the reaction pathway should be. We found that the favourable order of reaction pathways is path 1 > path 2 > path 3 > path 4 for both substrates. By comparing the energy barrier for each path, we found that path 1 and path 2 of the methanol oxidation reaction are more favourable on PtRuCo3/C-MWCNTs than on PtRuFe3/C-MWCNTs, while path 3 and path 4 are slightly less favourable on PtRuCo3/C-MWCNTs. Notably, path 4 has a very high thermodynamic barrier, which perhaps prohibits the methanol oxidation reaction from proceeding on both substrates.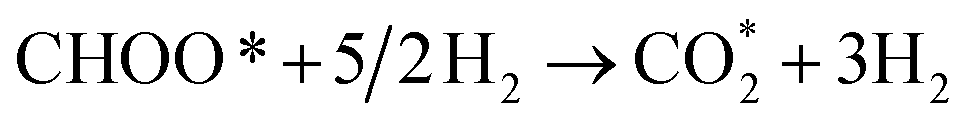
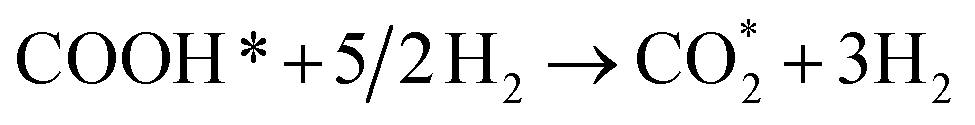
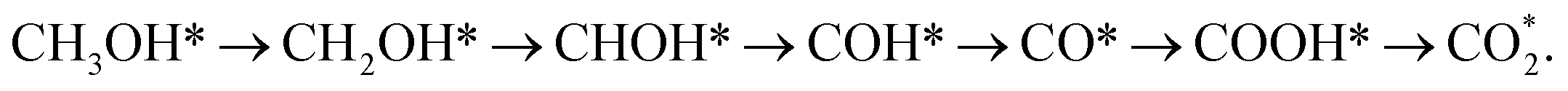 Besides, the most favourable pathway on PtRu/Pt(111) in the research of Ding et al.,42 is
Besides, the most favourable pathway on PtRu/Pt(111) in the research of Ding et al.,42 is 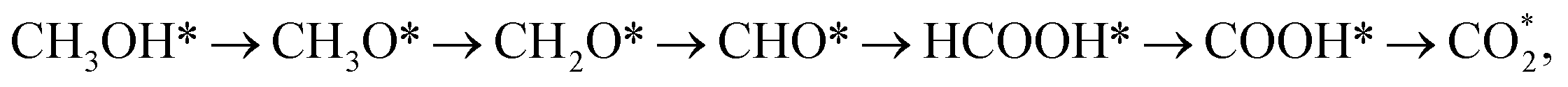 where forming CH3O* does not result in the most favourable mechanism for the PtRuCo3/C-MWCNTs and PtRuFe3/C-MWCNTs substrates in our present work. The most favourable reaction pathway (path 1) in this study supports the previous experiments that PtRuCo3/C-MWCNTs and PtRuFe3/C-MWCNTs catalysts have higher tolerances to CO poisoning compared to the Pt-based bimetallic alloys.31,62
where forming CH3O* does not result in the most favourable mechanism for the PtRuCo3/C-MWCNTs and PtRuFe3/C-MWCNTs substrates in our present work. The most favourable reaction pathway (path 1) in this study supports the previous experiments that PtRuCo3/C-MWCNTs and PtRuFe3/C-MWCNTs catalysts have higher tolerances to CO poisoning compared to the Pt-based bimetallic alloys.31,62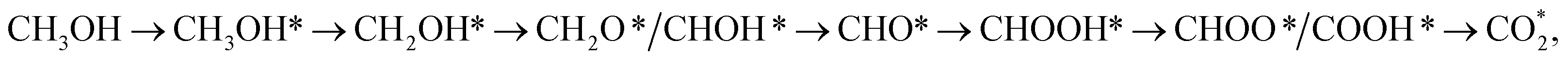 (path 2)
(path 2) 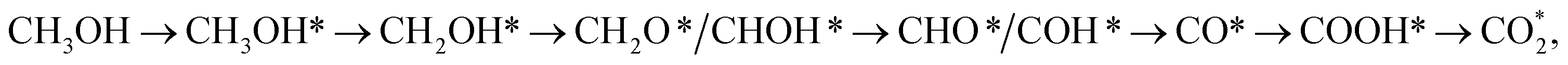 and (path 3)
and (path 3) 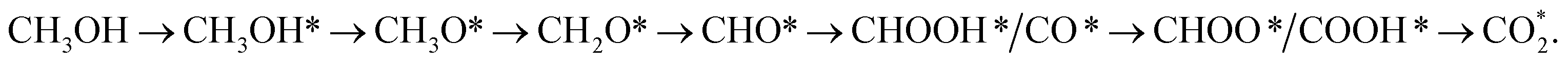 However, at the working potential of 1.005 V, these pathways have the same downhill behaviour, with the highest thermodynamic barrier occurring at the methanol adsorption step, CH3OH → CH3OH*. Three pathways have the same energy barrier of 0.513 and 0.167 eV for the PtRuFe3/C-MWCNTs and PtRuCo3/C-MWCNTs substrates, respectively. This result implied that the performance of PtRuCo3/C-MWCNTs is better than that of PtRuFe3/C-MWCNTs. This study only considered the low coverage of reaction intermediates. However, further investigation of the effects of coverages is necessary for understanding a full physical picture of the interaction between the intermediates and the substrate surface, which will be a research topic for future studies.
However, at the working potential of 1.005 V, these pathways have the same downhill behaviour, with the highest thermodynamic barrier occurring at the methanol adsorption step, CH3OH → CH3OH*. Three pathways have the same energy barrier of 0.513 and 0.167 eV for the PtRuFe3/C-MWCNTs and PtRuCo3/C-MWCNTs substrates, respectively. This result implied that the performance of PtRuCo3/C-MWCNTs is better than that of PtRuFe3/C-MWCNTs. This study only considered the low coverage of reaction intermediates. However, further investigation of the effects of coverages is necessary for understanding a full physical picture of the interaction between the intermediates and the substrate surface, which will be a research topic for future studies.