DOI:
10.1039/D4RA03877K
(Paper)
RSC Adv., 2024,
14, 25908-25917
Molecular insight into binding affinities and blockade effects of selected flavonoid compounds on the PD-1/PD-L1 pathway†
Received
26th May 2024
, Accepted 5th August 2024
First published on 16th August 2024
Abstract
This study investigated the binding mechanisms of the flavonoids apigenin (Api), kaempferol (Kmp), and quercetin (Que) to the PD-L1 dimer using a combination of molecular modeling and experimental techniques. The binding free energy results demonstrated that the flavonoids could tightly bind to the PD-L1 dimer, with the binding abilities following the trend Que > Kmp > Api. Key residues Ile54, Tyr56, Met115, Ala121, and Tyr123 were identified as important for binding. The flavonoids primarily bind to the C-, F-, and G-sheet domains. The spontaneous formation of the complex systems was mainly driven by hydrophobic forces. Dynamic cross-correlation matrix and secondary structure analyses further indicated that the studied flavonoids could stably interact with the binding sites. ELISA results showed that the flavonoids could effectively block PD-1/PD-L1 interactions, although the inhibitory activity of Api was weaker. Therefore, flavonols might be more effective inhibitors compared to flavones. The findings of this study are expected to contribute to the development of novel flavonoids targeting the PD-1/PD-L1 pathway.
Introduction
Programmed cell death-1 (PD-1) and programmed cell death-ligand 1 (PD-L1) are pivotal negative immunoregulatory molecules in the tumor microenvironment.1,2 A series of cancer cells can exploit the PD-1/PD-L1 pathway to weaken the immune capacity of T cells and escape immune surveillance. Therefore, blocking this pathway can reverse immunosuppressive states and enhance the killing of cancer cells.3,4 Clinical success has been observed for several monoclonal antibodies targeting PD-1 (cemiplimab, nivolumab, and pembrolizumab) and PD-L1 (atezolizumab, durvalumab, and avelumab).5,6 However, antibodies are associated with several drawbacks, such as low oral bioavailability, limited tumor permeability, immune-related adverse reactions, and high cost.7,8 Therefore, it is urgent to explore other useful compounds for PD-1/PD-L1 blockade, including small molecules and natural products.9,10 Several synthetic small molecules from Bristol Myers Squibb (e.g., BMS1166 and BMS202) have been reported to exhibit promising inhibitory activities to interrupt PD-1/PD-L1 interactions. Co-crystallography studies have disclosed a mechanism of direct binding to PD-L1, which induces the dimerization of PD-L1.11,12 Since then, many highly efficient small molecule inhibitors that can induce the formation of the PD-L1 dimer have been successively designed.9,13
Recently, natural products have attracted great attention due to their lower toxicity and multiple pharmacological effects. Among them, polyphenols account for a relatively large proportion and can intervene in all stages of tumorigenesis and development by regulating multiple signaling pathways involved in different types of cancers.14,15 Interestingly, several polyphenols such as apigenin, curcumin, and resveratrol have been proven to be competitive inhibitors that block PD-1/PD-L1 interactions by binding to the PD-L1 dimer.16,17 However, there is a notable lack of comparative studies regarding the effect of polyphenol structures on their binding properties. Among polyphenols, flavonoids are the most prevalent, characterized by a C6–C3–C6 skeleton consisting of two phenyl rings (A and B) linked via an oxygen heterocycle (C).18–21 Structurally, flavonoids can be categorized into 3-hydroxy flavonoids and 3-desoxy flavonoids based on the presence or absence of a hydroxyl group at the 3-position on the C-ring.18–21 Flavones lack the 3-hydroxyl group, while flavonols can be considered the 3-hydroxy derivatives of flavones.18–21 To explore the impact of structures on binding characteristics, representative flavonoids apigenin (Api), kaempferol (Kmp), and quercetin (Que) were chosen.
In this study, molecular docking of the flavonoids with the PD-L1 dimer was carried out to build the complex systems (see Fig. 1 and 2), followed by various computational methods including molecular dynamics (MD) simulations and binding free energy calculations to determine the energy contributions of interface residues.22,23 Subsequently, the binding modes, contact numbers, principal component analysis (PCA), and secondary structure were investigated, thereby theoretically elucidating the binding mechanism of the flavonoids on the PD-L1 dimer.24,25 To further validate the inhibitory activities of the flavonoids on PD-1/PD-L1 interactions, ELISA (Enzyme-Linked Immunosorbent Assay) was performed. Generally, these findings may advance the development of more potent flavonoids and potentially facilitate the advancement of cancer immunotherapy.
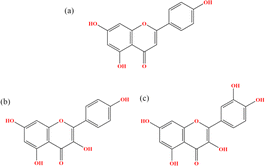 |
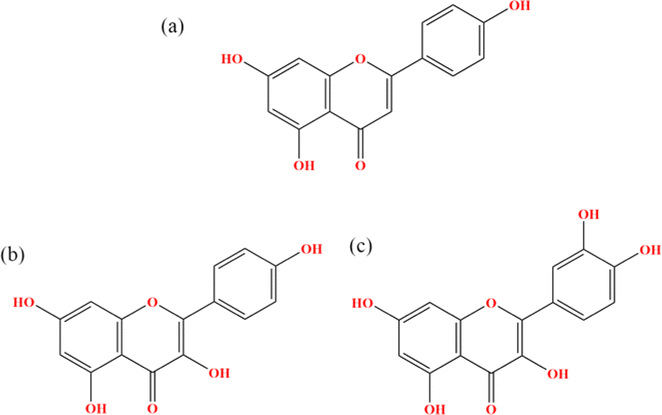
| | Fig. 1 Chemical structures of (a) Api, (b) Kmp and (c) Que. | |
 |
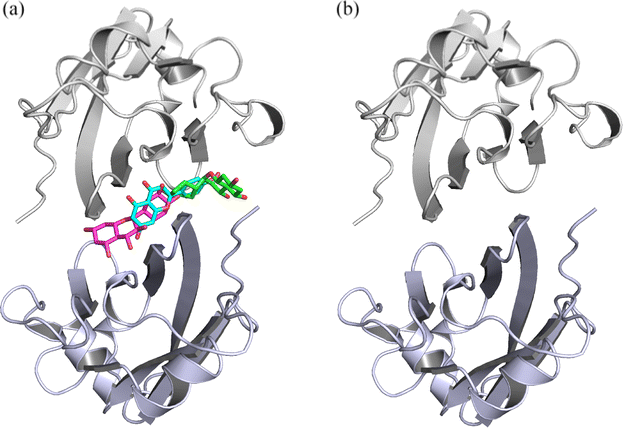
| | Fig. 2 The input structures for MD simulations of the (a) Api, Kmp, Que and (b) dimer systems. Api, Kmp and Que are shown in cyan, green and magenta sticks, respectively. | |
Experimental section
Materials and methods
Molecular docking. The molecular docking studies were carried out to obtain the complex systems of the PD-L1 dimer/flavonoids.26 The crystal structure of the PD-L1 dimer (PDB ID 5N2F) was downloaded from the PDB database. The 3D structures of the flavonoids were obtained from the PubChem database and optimized using the Gaussian 09 program27 with the B3LYP function and def-TZVP basis sets.28 Before docking, the PD-L1 dimer and flavonoids were converted to PDBQT format using AutoDockTools 1.5.6.29 The docking procedure was then performed using AutoDock Vina. The grid box size and grid spacing were set to (20 Å × 20 Å × 20 Å) and 1 Å, respectively. The center of the search space was determined based on the co-crystallized ligand. The docked poses with the strongest binding affinity were considered for subsequent MD simulations. The validation of the docking approach was performed by re-docking the original crystal ligand to the PD-L1 dimer. The RMSD between the docked complex and the solved structure was 0.77 Å.21,30 Moreover, the interaction profiles of the best-docked pose were similar to the original crystallized complex.21,30
Molecular dynamics simulation. MD simulations of the systems obtained from molecular docking were carried out for 150 ns using the GROMACS 2016.4 package with a time step of 2 fs.31 The AMBER ff99SB32 and general AMBER force field (GAFF)33 were employed to parameterize the PD-L1 dimer and flavonoids, respectively. The systems were centered in a cubic box with at least 10 Å from the edge and then dissolved in TIP3P water. Sodium and chloride ions were added to neutralize the system, followed by minimization using steepest descent and conjugate gradient methods to remove clashes. Subsequently, all systems were equilibrated at NVT and NPT ensembles for 1 ns to ensure converged systems for the further production run. The production run for the simulation was carried out at a constant temperature of 300 K and a pressure of 1 atm using the V-rescale and Parrinello–Rahman algorithms, respectively. The bond lengths involving hydrogen atoms were constrained using the LINCS algorithm. The Verlet scheme was used for the calculation of short-range non-bonded interactions with a cutoff of 1.0 nm. Particle Mesh Ewald (PME) was utilized to calculate the long-range electrostatic interactions. The trajectory and energy files were written every 10 ps for analysis.
Analysis details. The trajectories generated by MD simulations were used to analyze the root mean square deviation (RMSD), root mean square fluctuation (RMSF), contact numbers, and secondary structure of the systems using standard GROMACS tools. The interactions between the target residues and the flavonoids were calculated using the Protein–Ligand Interaction Profiler (PLIP) tool. Hydrogen bond occupancy was analyzed using Visual Molecular Dynamics (VMD) 1.9.3 software.34 Principal component analysis (PCA) was carried out via the gmx covar tool to gain insights into the collective motions of the PD-L1 dimer.35 The dynamic cross-correlation matrix (DCCM) provided information on the relevant movements of each residue on a simulation scale.
Binding free energy calculation. The binding free energy (ΔGbind) between the PD-L1 dimer and flavonoids was assessed using the molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) method with the g_mmpbsa tool.36 For each system, a total of 300 snapshots were collected at intervals of 100 ps from the last equilibrated 30 ns of the MD trajectory for the calculation.| | |
ΔG = Gcomplex − (Gprotein + Gligand)
| (1) |
| | |
ΔGbind = ΔEMM + ΔGsol
| (2) |
The Gcomplex, Gprotein and Gligand are denoted as the free energies of the complex, PD-L1 dimer, and flavonoids, respectively (eqn (1)). ΔGbind is obtained by computing the molecular mechanics energy (ΔEMM) and solvation free energy (ΔGsol) (eqn (2)). ΔEMM is the sum of electrostatic energy (ΔEele) and van der Waals energy (ΔEvdW). ΔGsol consists of polar solvation energy (ΔGPB) and nonpolar solvation energy (ΔGSA) (eqn (3)). Among them, ΔGPB and ΔGSA were calculated based on the Poisson–Boltzmann (PB) equation and solvent-accessible surface area (SASA), respectively (eqn (4)). The contribution of each residue was also obtained by decomposing the binding free energy using the MM-PBSA approach.
ELISA. The PD-1/PD-L1 blockade effect of the flavonoids was determined using a PD-1 ELISA assay kit.37 Human PD-L1 was coated onto a 96-well plate and incubated overnight at 4 °C. The plate was then washed with PBS buffer, blocked for 30 min at 37 °C, and washed again. Subsequently, the flavonoids were added and incubated for 30 min before adding human PD-1. BMS-202 was used as a positive control compound. After five washes, streptavidin–horseradish peroxidase was added, and the plate was incubated for 30 min. Then, the substrate was added and incubated for 10 min at 37 °C. After incubation, the stop solution was added, followed by measuring the absorbance optical density values at a wavelength of 450 nm.
Results and discussion
RMSD
To evaluate the structural stability of each system during the 150 ns simulation period, RMSD was calculated initially.38 As shown in Fig. 3, the complex systems reached equilibrium after 5 ns, exhibiting relatively small fluctuations. In contrast, larger fluctuations were observed in the simulation of the PD-L1 dimer system, especially during the first 60 ns, which aligns with our previous studies.21,30 Subsequently, these curves stabilized, indicating minimal conformational changes. The average RMSD values for the PD-L1 dimer, apigenin (Api), kaempferol (Kmp), and quercetin (Que) systems were 3.11 Å, 2.70 Å, 2.38 Å, and 2.31 Å, respectively. The higher stability observed in the complex systems may be attributed to the binding of the flavonoids. Additionally, the Api system showed larger RMSD values compared to the other two complex systems. These results illustrate that the structures of these systems maintained similarity to their initial frames throughout the simulation time, confirming stable trajectories for subsequent analysis.
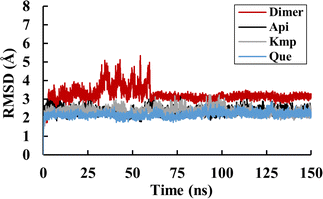 |
| | Fig. 3 RMSD values of the dimer, Api, Kmp and Que systems. | |
RMSF
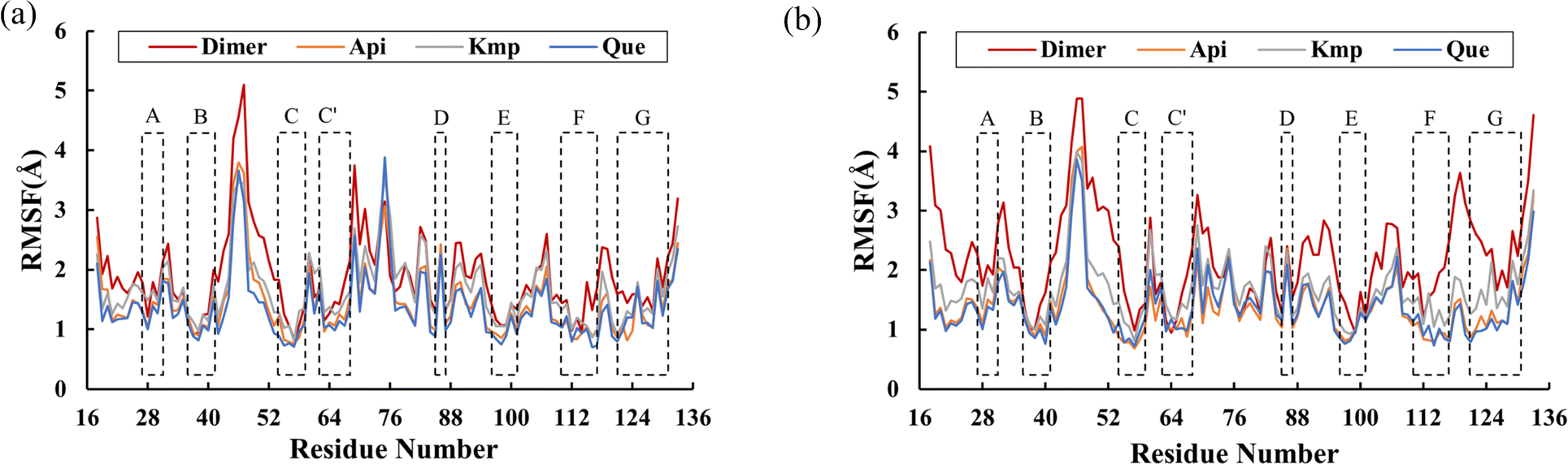
RMSF was further examined to reveal the local fluctuations of residues in the PD-L1 dimer. As depicted in Fig. 4, all systems exhibited a similar trend in RMSF with minor variations. The N-terminal, C-terminal, and loop areas showed larger RMSF in both the PD-L1 dimer and complex systems, consistent with previous MD simulation findings.9 Specifically, the BC loop of the PD-L1 dimer system exhibited an RMSF value of 5.1 Å. In contrast, residues in the beta sheets (A–G sheets) connected by loop domains showed comparatively less fluctuation in both unbound and bound states. Notably, the flexibility of residues in the C, F, and G sheets was notably reduced in the presence of the flavonoids. Moreover, the RMSF values of the beta sheets did not significantly differ among the complex systems. These RMSF observations are consistent with the RMSD results.
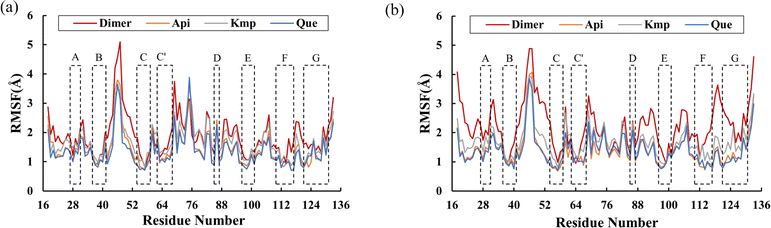 |
| | Fig. 4 RMSF values of residues on (a) APD-L1 and (b) BPD-L1 of the dimer, Api, Kmp and Que systems. | |
Binding free energy
To analyze the energy differences of the PD-L1 dimer upon binding to flavonoids, the MM-PBSA method was employed to calculate the binding free energy and contributions of energy components (see Table 1). As shown, the average binding free energies between the PD-L1 dimer and apigenin (Api), kaempferol (Kmp), and quercetin (Que) are −21.49 ± 0.09, −32.66 ± 0.12, and −35.39 ± 0.34 kcal mol−1, respectively. This indicates that the binding abilities of Kmp and Que to the PD-L1 dimer are similar to that of curcumin but stronger than resveratrol and epigallocatechin gallate.21 Structurally, the binding free energies between flavonols and the PD-L1 dimer appear stronger compared to flavones, possibly due to the 3-hydroxyl group on the C ring. Previous reports have suggested that the activities of flavonoids largely depend on the positions of hydroxyl groups relative to the total number of hydroxyl groups.16 Furthermore, the binding free energy for the PD-L1 dimer system alone is 36.11 ± 0.89 kcal mol−1, indicating that flavonoids play a crucial role in the formation of the PD-L1 dimer. Upon analyzing different components of the complex systems, it is evident that the binding of flavonoids to the PD-L1 dimer is predominantly mediated by van der Waals energy. The van der Waals interaction energy for the Que system is the strongest at −51.30 ± 0.31 kcal mol−1, while for Api and Kmp systems, it is −40.63 ± 0.60 and −47.55 ± 0.38 kcal mol−1, respectively. Electrostatic interaction energy also contributes favorably, with Que exhibiting a stronger electrostatic interaction energy (−6.44 ± 1.47 kcal mol−1) compared to Api (−1.14 ± 1.39 kcal mol−1) and Kmp (−4.98 ± 1.64 kcal mol−1) when binding to the PD-L1 dimer, possibly due to the 3′-hydroxyl group on the B ring. In summary, polar solvation interactions can impair binding, whereas nonpolar solvation interactions are favorable for the binding process.
Table 1 Binding free energies of the dimer, Api, Kmp and Que systems (kcal mol−1)
| Contribution |
Api |
Kmp |
Que |
Dimer |
| van der Waals interaction energy. Electrostatic energy. Polar solvent effect energy. Nonpolar solvent effect energy. Polar binding free energy. Nonpolar binding free energy. Binding free energy. |
| ΔEvdwa |
−40.63 ± 0.60 |
−47.55 ± 0.38 |
−51.30 ± 0.31 |
−44.59 ± 9.85 |
| ΔEeleb |
−1.14 ± 1.39 |
−4.98 ± 1.64 |
−6.44 ± 1.47 |
−124.35 ± 23.36 |
| ΔEPBc |
23.51 ± 2.05 |
23.54 ± 1.33 |
25.99 ± 1.58 |
211.28 ± 17.07 |
| ΔESAd |
−3.24 ± 0.08 |
−3.66 ± 0.07 |
−3.64 ± 0.06 |
−6.23 ± 0.37 |
| ΔEpolar,totale |
22.37 ± 0.75 |
18.56 ± 0.32 |
19.55 ± 0.52 |
86.94 ± 10.00 |
| ΔEnonpolar,totalf |
−43.86 ± 0.68 |
−51.21 ± 0.44 |
−54.94 ± 0.36 |
−50.82 ± 10.06 |
| ΔGg |
−21.49 ± 0.09 |
−32.66 ± 0.12 |
−35.39 ± 0.34 |
36.11 ± 0.89 |
Per-residue energy decomposition
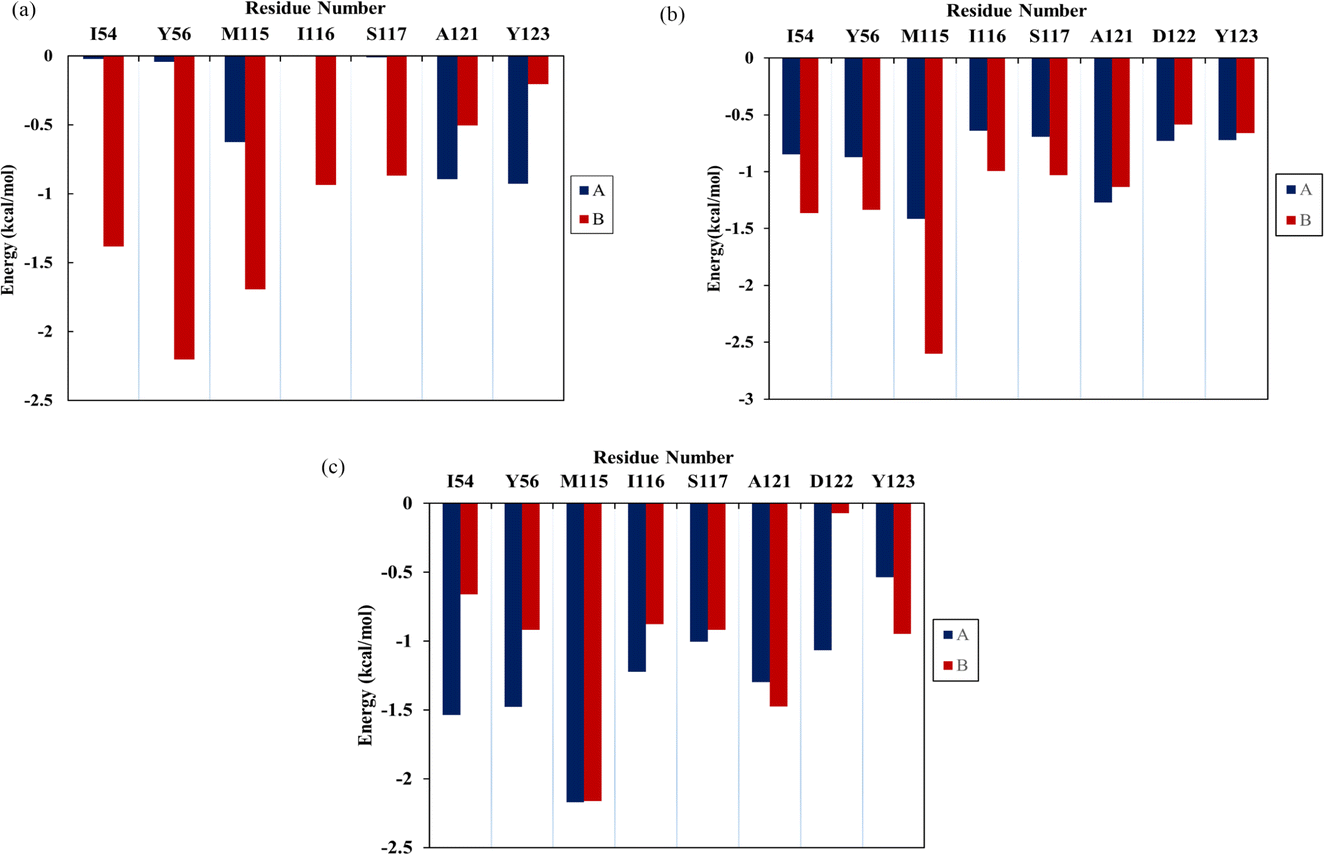
For a comprehensive analysis of key residues involved in the binding of flavonoids to the PD-L1 dimer, per-residue energy decomposition was performed. As depicted in Fig. 5, In the Api system (Fig. 5a), nine residues showed binding free energy contributions exceeding −0.5 kcal mol−1. Among these, BIle54, BTyr56, and BMet115 were major contributors, with BTyr56 exerting the most significant negative impact at −2.20 kcal mol−1. In the Kmp system (Fig. 5b), 16 residues interacted strongly with the PD-L1 dimer, each contributing more than −0.5 kcal mol−1. Notably, AMet115, AAla121, BIle54, BTyr56, BMet115, BIle116, BSer117, and BAla121 contributed over −1 kcal mol−1. The total interaction energies of these residues with Kmp ranged from −1.00 to −2.60 kcal mol−1. Compared to the Api system, ATyr123, BIle54, and BTyr56 in the Kmp system showed slightly attenuated contributions (by 0.21 to 0.86 kcal mol−1), while contributions of other residues, particularly AIle54 and ATyr56, were enhanced. In the Que system (Fig. 5c), 15 residues had binding free energy contributions exceeding −0.5 kcal mol−1. Key residues like AIle54, ATyr56, AMet115, AIle116, ASer117, AAla121, AAsp122, BMet115, BAla121, and BY56 contributed over −1 kcal mol−1. Notably, AMet115 showed an enhanced interaction with Que by 0.76 kcal mol−1 compared to Kmp. The role of Met115 in small-molecule binding has been underscored in previous studies.9,39,40 Comparative analysis indicated that residues Ile54, Tyr56, Met115, Ala121, and Tyr123 play crucial roles in binding to flavonoids, with varying contributions across different systems. These residues are known to be involved in constructing active binding sites between PD-1 and PD-L1, as reported in experimental and computational studies.9,39,40 Additionally, compared to the Que system, the Api and Kmp systems showed a more concentrated distribution of key residues in the binding domain of BPD-L1. In summary, these results suggest that flavonoids effectively bind to the PD-L1 dimer, thereby potentially inhibiting PD-1/PD-L1 interactions.
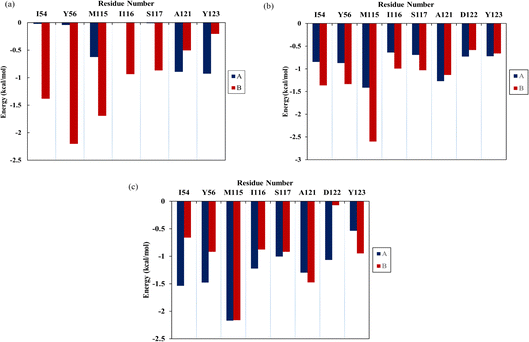 |
| | Fig. 5 The contribution of residues of the PD-L1 dimer in complex with three flavonoids, (a) Api, (b) Kmp and (c) Que. | |
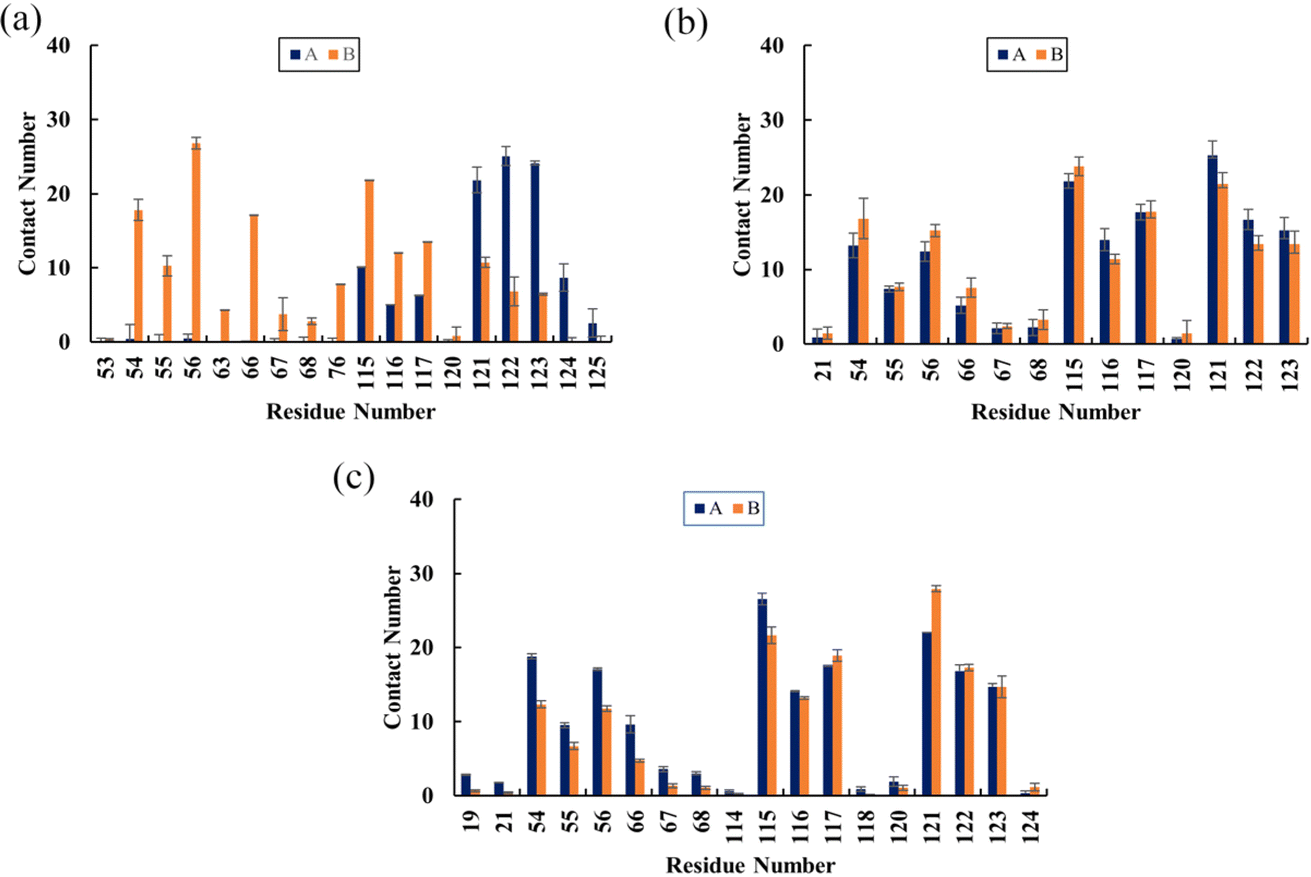
Contact numbers
To determine the binding sites of flavonoids on the PD-L1 dimer, the average number of contacts between each residue and flavonoids was calculated.41 As depicted in Fig. 6, Api (Fig. 6a) predominantly binds to the residues AMet115, AAla121, AAsp122, ATyr123, BIle54, BVal55, BTyr56, BGln66, BIle116, BSer117, and BAla121, each with an average contact number greater than 10, though the specific values vary slightly. Notably, BTyr56 shows the highest contact number (27 contacts) among all residues, particularly in the C sheet, with Api. Kmp (Fig. 6b) and Que (Fig. 6c) exhibit relatively high contact numbers with the residues including Ile54, Tyr56, Met115, Ile116, Ser117, Ala121, Asp122, and Tyr123 in both APD-L1 and BPD-L1. AAla121 and BAla121 have the highest contact numbers with Kmp and Que, respectively. Additionally, Que preferentially contacts AGln66 and AVal55. Kmp and Que also show significant interactions with AIle54, ATyr56, BAla121, BAsp122, and BTyr123, especially AIle54 and ATyr56, compared to Api. Overall, Kmp and Que exhibit notably higher contact numbers (ranging from 310 to 329) with the PD-L1 dimer compared to Api, primarily due to interactions in the β-sheet regions. These findings are consistent with the calculated binding free energies presented in Table 1 and Fig. 5. Interestingly, residues with high contact numbers are located in the C sheet (residues 54–56), C′ sheet (residues 66–68), F sheet (residues 115–117), and G sheet (residues 121–124), which are crucial for forming and stabilizing the PD-1/PD-L1 binding interface. This suggests that flavonoids can bind closely to the PD-L1 dimer, thereby potentially blocking PD-1/PD-L1 interactions.
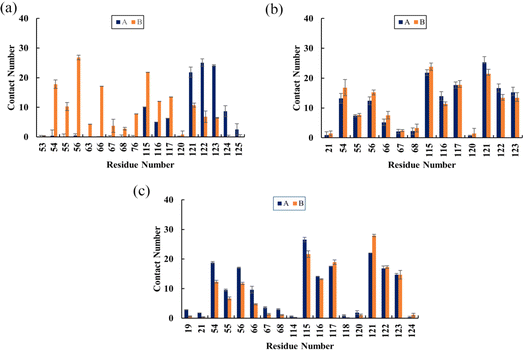 |
| | Fig. 6 The average contact numbers between the PD-L1 dimer and (a) Api, (b) Kmp and (c) Que. The error bars represent the standard of the mean. | |
Non-bonded interactions
In the protein–ligand systems, intermolecular hydrogen bonds contribute significantly to stabilizing ligands within binding pockets.26 The hydrogen bond occupancies between the PD-L1 dimer and flavonoids throughout the equilibration period were calculated and summarized in Table 2. For Api, the NZ atom of ALys124 forms one hydrogen bond with an occupancy of 16.28%. This interaction is crucial as ALys124 acts as a hydrogen donor. In the Kmp system, the O atom of the negatively charged AAsp122 forms two hydrogen bonds, one with the O27 atom (51.16% occupancy) and another with the O29 atom (50.16% occupancy) of Kmp, serving as hydrogen acceptors. These bonds are pivotal for maintaining the binding stability of this system due to their occupancies exceeding 50%.42 In the Que system, the highest hydrogen bond occupancy (85.05%) occurs between the OE1 atom of AGln66 and the O29 atom of Que. Additionally, the O atom of AIle116 forms a stable hydrogen bond with the O27 atom of Que with an occupancy of 59.14%. This strong interaction may be attributed to more hydroxyl groups in the B ring of Que compared to Api and Kmp. These results align with the weaker electrostatic contribution to the binding energy observed in the Api system (ΔEele in Table 1) compared to the other complex systems. Importantly, these residues have been identified in previous studies as critical for binding to the PD-L1 dimer.9,39,40 Therefore, these stable hydrogen bonds likely contribute significantly to the inhibitory activity of flavonoids against the PD-1/PD-L1 pathway.
Table 2 Hydrogen bond occupancies of the Api, Kmp and Que systems
| Donor |
Donor H |
Acceptor |
Occupancy (%) |
| ALys124@NZ |
HZ1 |
Api@OAB |
16.28 |
| Kmp@O27 |
HO27 |
AAsp122@O |
51.16 |
| Kmp@O29 |
HO29 |
AAsp122@O |
50.17 |
| Que@O29 |
HO29 |
AGln66@OE1 |
85.05 |
| Que@O27 |
HO27 |
AIle116@O |
59.14 |
The binding modes of the representative complex structures were analyzed, as depicted in Fig. 7, revealing predominantly hydrophobic interactions. Here are the specific interactions observed, residues ATyr123, BTyr56, and BMet115 are involved in binding with Api. Additionally, a hydrogen bond is formed between Api and ALys124. Residues AIle54, AMet115, AAla121, BIle54, BTyr56, BMet115, BAla121, and BTyr123 are directly involved in binding with Kmp. Six hydrogen bonds are observed between Kmp and AIle116, AAla121, AAsp122, BIle54, and BMet115. Residues ATyr56, AMet115, AAla121, ATyr123, and BAla121 are directly involved in binding with Que. Six hydrogen bonds are formed between Que and AIle54, AGln66, AIle116, AAsp122, and BTyr123. In summary, while all studied flavonoids bind to the PD-L1 dimer, the specific residues involved in these interactions differ. Notably, these interactions predominantly involve small polar interactions and relatively large hydrophobic interactions. Moreover, the Kmp and Que systems exhibit stronger hydrogen bond interactions compared to the Api system, which can be attributed to their higher number of hydroxyl groups. These findings underscore the diverse binding mechanisms and the potential differences in binding affinity among flavonoids, highlighting their distinct modes of interaction with the PD-L1 dimer.
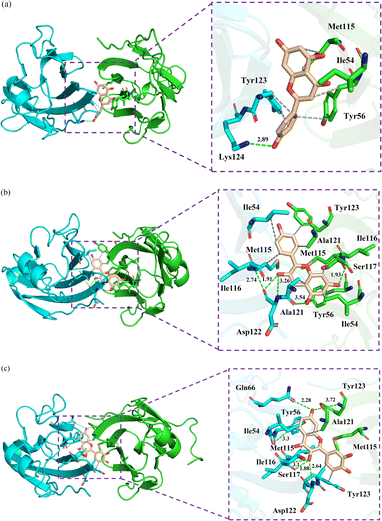 |
| | Fig. 7 The binding modes of the PD-L1 dimer in complex with (a) Api, (b) Kmp and (c) Que. The residues are shown as sticks with APD-L1 and BPD-L1 coloured cyan and green, respectively. The hydrophobic interactions and hydrogen bonds are shown as grey and green dashes, respectively. | |
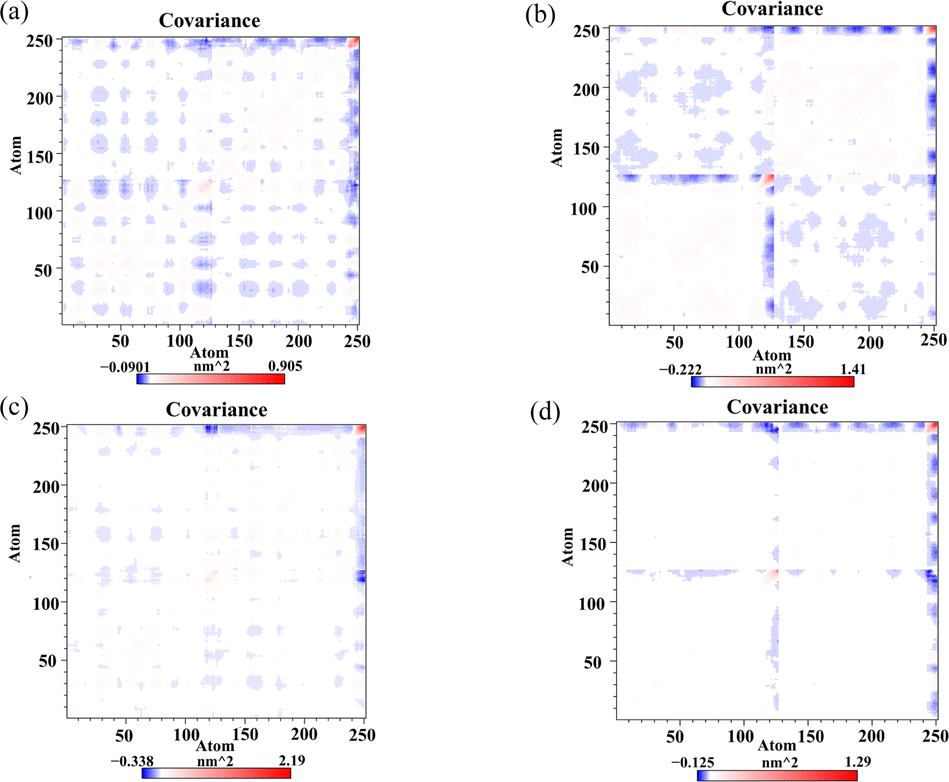
Dynamic cross-correlation matrix analysis
Dynamic cross-correlation matrix (DCCM) analysis was conducted to examine the dynamical motions among Cα atoms of the PD-L1 dimer.43 In the DCCM, positive areas (red) indicate correlated motions among residues, while negative areas (blue) indicate anti-correlated movements. The intensity of colors reflects the degree of correlation. As illustrated in Fig. 8, the N-terminal and C-terminal regions, such as residues AAla0 to AAsp8 (corresponding to residues AAla18 to AAsp26), show a red block within each system, suggesting correlated motions. These areas are typically distant from the binding sites and may exhibit similar dynamics across different systems. The unbound PD-L1 dimer shows the highest anti-correlated movements, depicted by extensive blue regions. This indicates diverse and independent motions of residues in the absence of ligand. The Api-bound PD-L1 dimer displays higher correlated motions compared to the unbound dimer, particularly evident in most areas of the protein. This suggests that Api binding induces more coordinated motions among residues, possibly due to specific interactions stabilizing the complex. Both Kmp and Que bound to the PD-L1 dimer exhibit maximum correlated motions compared to Api. This enhanced correlation can be attributed to their tighter interactions with the PD-L1 dimer, resulting in more concerted movements among residues. In summary, the DCCM analysis provides insights into how flavonoid binding affects the dynamical motions of the PD-L1 dimer. It underscores the differential impact of Api, Kmp, and Que on the correlated motions within the PD-L1 dimer, reflecting their varying degrees of binding affinity and stabilization mechanisms.
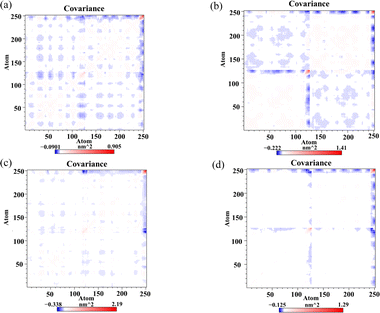 |
| | Fig. 8 Dynamic cross-correlation matrix of the (a) dimer, (b) Api, (c) Kmp and (d) Que systems. | |
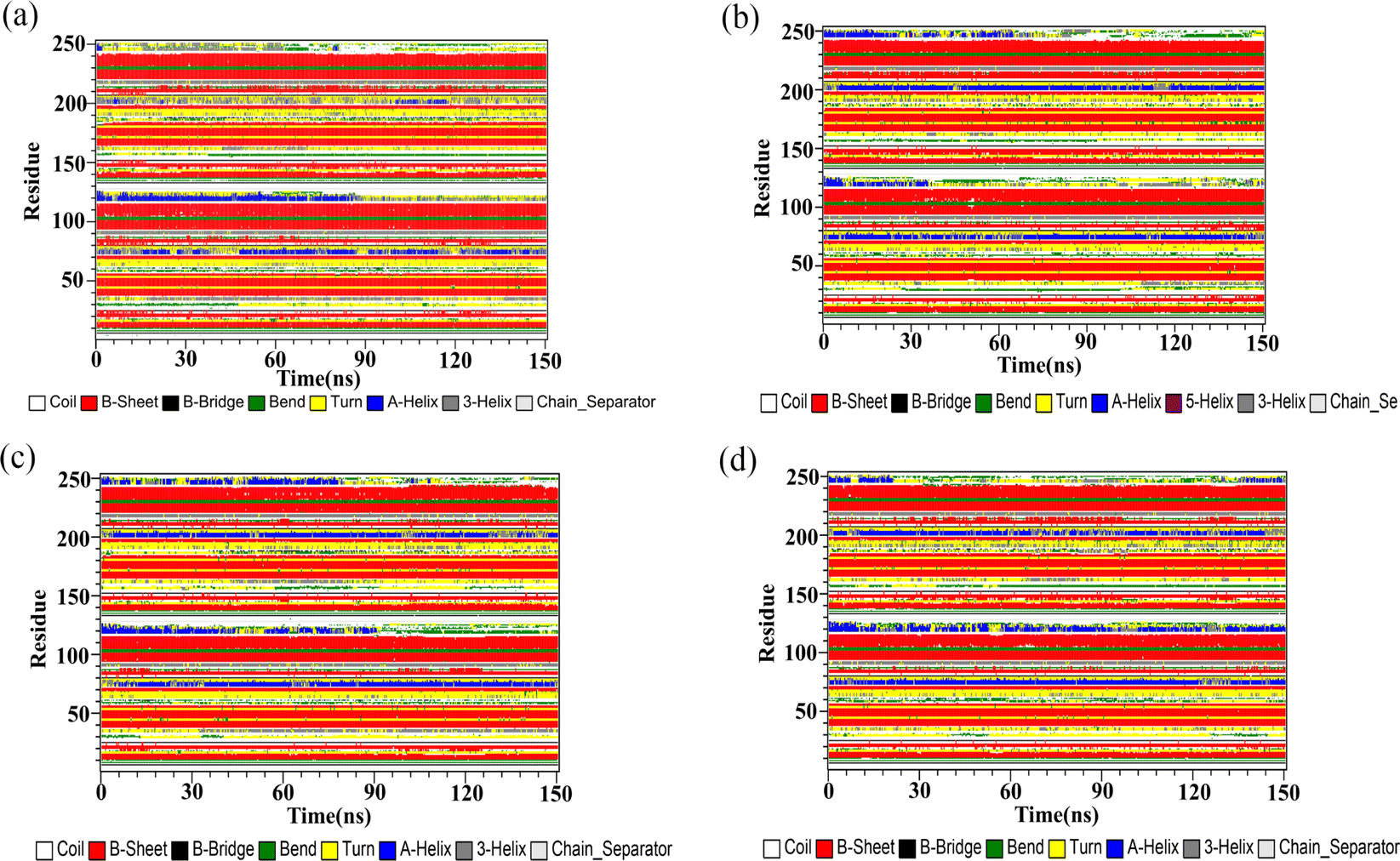
Secondary structure
The DSSP program was used to gain insight into the effect of the flavonoids on the secondary structures of the PD-L1 dimer. According to Fig. 9, no apparent changes were observed in the secondary structure of the PD-L1 dimer when binding with the flavonoids compared to the dimer system. The beta-sheet conformation of the PD-L1 dimer remained almost entirely stable throughout the 150 ns MD simulations. Importantly, the C, F, and G sheet domains are crucial for the recognition of the flavonoids, as mentioned earlier. Additionally, as the simulation progressed, the secondary structure of other domains mainly transformed between coil, α-helix, bend, and turn. In general, secondary structure analysis clearly indicated the formation of stable and compact complexes.
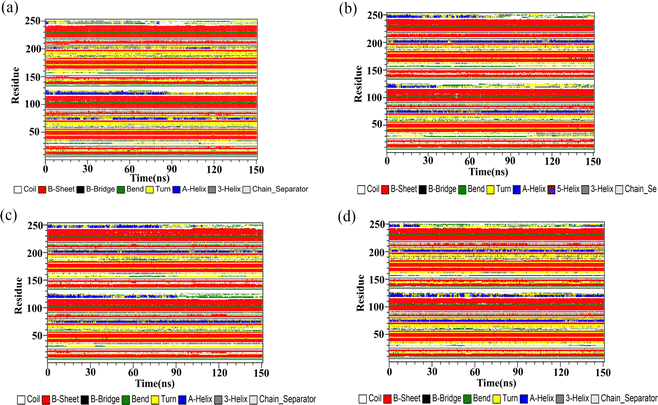 |
| | Fig. 9 The secondary structures of the PD-L1 dimer in the (a) dimer, (b) Api, (c) Kmp and (d) Que systems. | |
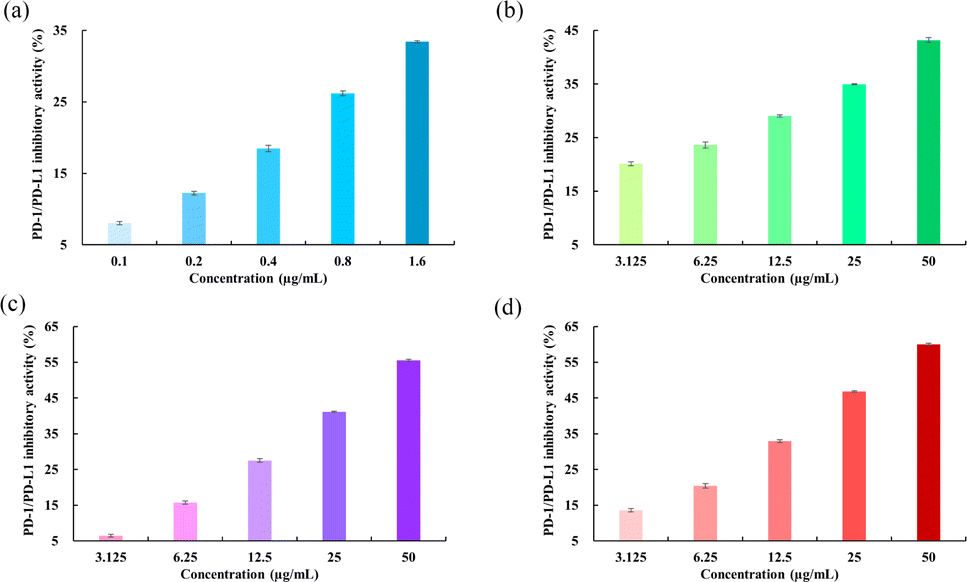
PD-1/PDL-1 inhibitory activity
The blockade effects of the flavonoids on PD-1/PD-L1 interactions were tested using an ELISA assay. The PD-1/PD-L1 small-molecule inhibitor BMS-202, which recognizes the extracellular domain of PD-L1 and blocks PD-1/PD-L1 interactions, was used as a positive control.44 The results showed that BMS-202 dose-dependently inhibited the interaction between PD-1 and PD-L1 with an IC50 value of 9.94 μg mL−1. To determine whether the flavonoids possess inhibitory effects on PD-1/PD-L1 interactions, a comparative study was performed. As illustrated in Fig. 10, Api showed a smaller blockade effect with an IC50 of 134.85 μg mL−1. Moreover, Kmp and Que also revealed inhibitory impacts on PD-1/PD-L1 binding, with activities of 39.72 and 29.93 μg mL−1, respectively, following a similar trend to the binding free energy of the complex systems as mentioned above. These findings indicated that Api, Kmp, and Que are effective PD-1/PD-L1 blockaders, and further studies can focus on flavonoids with similar structural skeletons.
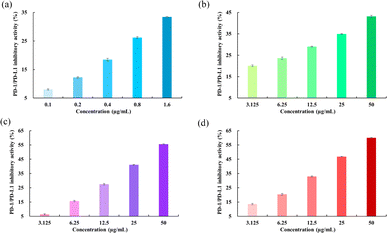 |
| | Fig. 10 Inhibitory activities of (a) BMS-202, (b) Api, (c) Kmp and (d) Que on PD-1/PD-L1 interactions. | |
Conclusions
In this paper, molecular modeling and experimental approaches were employed to explore the interaction mechanism between the PD-L1 dimer and the flavonoids Api, Kmp, and Que. The binding free energy calculations revealed that the binding of flavonoids to the PD-L1 dimer was spontaneous, with Kmp and Que showing stronger binding abilities than Api. Notably, the residues Ile54, Tyr56, Met115, Ala121, and Tyr123 contributed significantly to the binding. Furthermore, the structural basis, specifically the C-, F-, and G-sheet regions, for the binding of the flavonoids with the PD-L1 dimer was elucidated via contact number analysis. The interaction analysis showed that the active cavity is mainly driven by hydrophobic interactions. The analysis of DCCM and secondary structure further indicated that the flavonoids could stably interact with the binding regions. Consistent with the dynamics data, the ELISA results demonstrated that the flavonoids inhibited PD-1/PD-L1 interactions in a concentration-dependent manner, with Kmp and Que showing stronger activities than Api. In summary, this study offers useful information for the further development of flavonoids targeting the PD-1/PD-L1 pathway.
Data availability
Autodock Vina and MGLTools were used for molecular docking, which can be obtained free of charge: https://vina.scripps.edu/downloads/ and https://ccsb.scripps.edu/mgltools/downloads/. GROMACS 2016.4 was used for MD simulations at 300 K, which can be obtained free of charge: https://manual.gromacs.org/2016.4/download.html. AmberTools23 was used to parameterize the force field of the flavonoids, which can be obtained from https://ambermd.org/GetAmber.php. VMD 1.9.3 was used to compute the occupancies of intermolecular H bonds, which can be obtained free of charge: https://www.ks.uiuc.edu/Research/vmd/vmd-1.9.3/. PyMOL 2.5 was used for visualization, which can be obtained free of charge: https://pymol.org/2/. The WHAT IF online server was used to complete the PD-L1 dimer, which can be obtained free of charge: https://swift.cmbi.umcn.nl/servers/html/index.html. PLIP was used to compute non-bonded interactions, which can be obtained free of charge: https://plip-tool.biotec.tu-dresden.de/plip-web/plip/index.
Author contributions
Yan Guo: writing – original draft, methodology, investigation, formal analysis, and conceptualization. Jinchang Tong: investigation, and investigation. Kaixin Shi, Xinyue Song, and Zichao Guo: investigation, and date analysis. Jianhuai Liang: conceptualization, formal analysis, and writing – review and editing. Boping Liu and Jianguo Xu: conceptualization, and writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was funded by the Natural Science Foundation (202203021212392) of Shanxi Province and Technological Innovation Program in Higher Education Institutions (No. 2022L262) of Shanxi Province.
Notes and references
- J. J. Park, E. P. Thi and V. H. Carpio, et al., Checkpoint inhibition through small molecule-induced internalization of programmed death-ligand 1, Nat. Commun., 2021, 12, 1222 CrossRef CAS PubMed.
- V. Kumar, A. Bahuguna and M. Kim, Molecular insights into binding of bioactive compounds from essential oil of Trachyspermum ammi with human programmed cell death protein 1, J. Biomol. Struct. Dyn., 2023, 1–11 Search PubMed.
- Q. Wu, L. Jiang and S. Li, et al., Small molecule inhibitors targeting the PD-1/PD-L1 signaling pathway, Acta Pharmacol. Sin., 2021, 42, 1–9 CrossRef CAS.
- G. R. Sartori, A. O. Albuquerque and A. H. Santos-Costa, et al., In silico mapping of the dynamic interactions and structure-activity relationship of flavonoid compounds against the immune checkpoint programmed-cell death 1 pathway, Front. Drug Discov., 2022, 2, 1032587 CrossRef.
- C. Liu, N. P. Seeram and H. Ma, Small molecule inhibitors against PD-1/PD-L1 immune checkpoints and current methodologies for their development: a review, Cancer Cell Int., 2021, 21, 239 CrossRef CAS.
- H. Li, N. P. Seeram and C. Liu, et al., Further investigation of blockade effects and binding affinities of selected natural compounds to immune checkpoint PD-1/PD-L1, Front. Oncol., 2022, 12, 995461 CrossRef CAS PubMed.
- E. Domingo-Contreras, J. R. Tormo and V. Gonzalez-Menendez, et al., Discovery of bioactive natural products of microbial origin as inhibitors of the PD-1/PD-L1 protein-protein interaction, Int. J. Biol. Macromol., 2024, 264, 130458 CrossRef CAS.
- H. Zheng, G. Wang and M. Liu, et al., Traditional Chinese medicine inhibits PD-1/PD-L1 axis to sensitize cancer immunotherapy: a literature review, Front. Oncol., 2023, 13, 1168226 CrossRef CAS.
- X. Xu, S. Luo and X. Zhao, et al., Computational analysis of PD-L1 dimerization mechanism induced by small molecules and potential dynamical properties, Int. J. Biol. Macromol., 2024, 265, 130921 CrossRef CAS PubMed.
- N. S. Alharthi, M. S. Alwethaynani and A. Y. Alhazmi, et al., In silico assessment of a natural small molecule as an inhibitor of programmed death ligand 1 for cancer immunotherapy: a computational approach, J. Biomol. Struct. Dyn., 2024, 1–21 CrossRef.
- K. M. Zak, P. Grudnik and K. Guzik, et al., Structural basis for small molecule targeting of the programmed death ligand 1 (PD-L1), Oncotarget, 2016, 7, 30323 CrossRef PubMed.
- L. Skalniak, K. M. Zak and K. Guzik, et al., Small-molecule inhibitors of PD-1/PD-L1 immune checkpoint alleviate the PD-L1-induced exhaustion of T-cells, Oncotarget, 2017, 8, 72167 CrossRef.
- Y. OuYang, J. Gao and L. Zhao, et al., Design, synthesis, and evaluation of o-(biphenyl-3-ylmethoxy) nitrophenyl derivatives as PD-1/PD-L1 inhibitors with potent anticancer efficacy in vivo, J. Med. Chem., 2021, 64, 7646–7666 CrossRef CAS.
- D. Tewari, P. Patni and A. Bishayee, et al., Natural products targeting the PI3K-Akt-mTOR signaling pathway in cancer: a novel therapeutic strategy, Semin. Cancer Biol., 2022, 80, 1–17 CrossRef PubMed.
- M. Moloudizargari, M. H. Asghari and S. F. Nabavi, et al., Targeting Hippo signaling pathway by phytochemicals in cancer therapy, Semin. Cancer Biol., 2022, 80, 183–194 CrossRef CAS PubMed.
- J. G. Choi, Y. S. Kim and J. H. Kim, et al., Anticancer effect of Salvia plebeia and its active compound by improving T-cell activity via blockade of PD-1/PD-L1 interaction in humanized PD-1 mouse model, Front. Immunol., 2020, 11, 598556 CrossRef CAS PubMed.
- Y. Guo, J. Liang and B. Liu, et al., Molecular mechanism of food-derived polyphenols on PD-L1 dimerization: a molecular dynamics simulation study, Int. J. Mol. Sci., 2021, 22, 10924 CrossRef CAS.
- C. Papuc, G. V. Goran and C. N. Predescu, et al., Plant polyphenols as antioxidant and antibacterial agents for shelf-life extension of meat and meat products: classification, structures, sources, and action mechanisms, Compr. Rev. Food Sci. Food Saf., 2017, 16, 1243–1268 CrossRef CAS PubMed.
- A. Silva, V. Silva and G. Igrejas, et al., Phenolic compounds classification and their distribution in winemaking by-products, Eur. Food Res. Technol., 2023, 249, 207–239 CrossRef CAS.
- L. Li, X. Yan and F. Chen, et al., A comprehensive review of the metabolism of citrus flavonoids and their
binding to bitter taste receptors, Compr. Rev. Food Sci. Food Saf., 2023, 22, 1763–1793 CrossRef.
- D. Shah, M. Gandhi and A. Kumar, et al., Current insights into epigenetics, noncoding RNA interactome and clinical pharmacokinetics of dietary polyphenols in cancer chemoprevention, Crit. Rev. Food Sci., 2023, 63, 1755–1791 CrossRef CAS PubMed.
- A. Belhassan, S. Chtita and H. Zaki, et al., In silico detection of potential inhibitors from vitamins and their derivatives compounds against SARS-CoV-2 main protease by using molecular docking, molecular dynamic simulation and ADMET profiling, J. Mol. Struct., 2022, 1258, 132652 CrossRef CAS.
- Y. Liao, Y. Ding and L. Yu, et al., Exploring the mechanism of Alisma orientale for the treatment of pregnancy induced hypertension and potential hepato-nephrotoxicity by using network pharmacology, network toxicology, molecular docking and molecular dynamics simulation, Front. Pharmacol, 2022, 13, 1027112 CrossRef CAS.
- Z. Yang, X. Fu and Y. Zhao, et al., Molecular insights into the inhibition mechanism of harringtonine against essential proteins associated with SARS-CoV-2 entry, Int. J. Biol. Macromol., 2023, 240, 124352 CrossRef CAS PubMed.
- Z. A. Bhat, M. M. Khan and A. Rehman, et al., MD simulations indicate Omicron P132H of SARS-CoV-2 Mpro is a potential allosteric mutant involved in modulating the dynamics of catalytic site entry loop, Int. J. Biol. Macromol., 2024, 130077 CrossRef CAS.
- P. Dashtestani and L. Karami, The molecular mechanism of the effects of the anti-neuropathic ligands on the modulation of the sigma-2 receptor: an in-silico study, Int. J. Biol. Macromol., 2024, 254, 127925 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, et al., Gaussian 09, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- F. Liang, K. Meng and X. Pu, et al., Deciphering the binding behavior and interaction mechanism of apigenin and α-glucosidase based on multi-spectroscopic and molecular simulation studies, Int. J. Biol. Macromol., 2024, 130535 CrossRef CAS.
- Y. Guo, Y. Jin and B. Wang, et al., Molecular mechanism of small-molecule inhibitors in blocking the PD-1/PD-L1 pathway through PD-L1 dimerization, Int. J. Mol. Sci., 2021, 22, 4766 CrossRef CAS PubMed.
- S. L. Jiang, Z. Y. Hu and W. J. Wang, et al., Investigation on the binding behavior of human α1-acid glycoprotein with Janus kinase inhibitor baricitinib: multi-spectroscopic and molecular simulation methodologies, Int. J. Biol. Macromol., 2023, 125096 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola and R. Schulz, et al., GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers, SoftwareX, 2015, 1, 19–25 CrossRef.
- J. Wang, R. M. Wolf and J. W. Caldwell, et al., Development and testing of a general amber force field, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- J. Liu, B. Li and Z. Li, et al., Deciphering the alkaline stable mechanism of bacterial laccase from Bacillus pumilus by molecular dynamics simulation can improve the decolorization of textile dyes, J. Hazard. Mater., 2023, 443, 130370 CrossRef CAS PubMed.
- Y. Pan, C. Zhao and W. Fu, et al., Comparative analysis of structural dynamics and allosteric mechanisms of RecA/Rad51 family proteins: integrated atomistic MD simulation and network-based analysis, Int. J. Biol. Macromol., 2024, 261, 129843 CrossRef CAS PubMed.
- B. Pandey and A. Grover, Mechanistic and structural insight into R2R3-MYB transcription factor in plants: molecular dynamics based binding free energy analysis, J. Biomol. Struct. Dyn., 2024, 42, 2632–2642 CrossRef CAS PubMed.
- H. F. Li, N. P. Seeram and C. Liu, et al., Further investigation of blockade effects and binding affinities of selected natural compounds to immune checkpoint PD-1/PD-L1, Front. Oncol., 2022, 12, 995461 CrossRef CAS PubMed.
- J. F. Cao, X. Yang and L. Xiong, et al., Exploring the mechanism of action of dapansutrile in the treatment of gouty arthritis based on molecular docking and molecular dynamics, Front. Physiol., 2022, 13, 990469 CrossRef.
- C. Sun, Y. Cheng and X. Liu, et al., Novel phthalimides regulating PD-1/PD-L1 interaction as potential immunotherapy agents, Acta Pharm. Sin. B, 2022, 12, 4446–4457 CrossRef CAS PubMed.
- R. Butera, M. Ważyńska and K. Magiera-Mularz, et al., Design, synthesis, and biological evaluation of imidazopyridines as PD-1/PD-L1 antagonists, ACS Med. Chem. Lett., 2021, 12, 768–773 CrossRef CAS PubMed.
- X. Li, Y. Zhang and Z. Yang, et al., The Inhibition Effect of Epigallocatechin-3-Gallate on the Co-Aggregation of Amyloid-β and Human Islet Amyloid Polypeptide Revealed by Replica Exchange Molecular Dynamics Simulations, Int. J. Mol. Sci., 2024, 25, 1636 CrossRef CAS PubMed.
- S. H. Mirmajidi, C. Irajie and A. Savardashtaki, et al., Identification of potential RapJ hits as sporulation pathway inducer candidates in Bacillus coagulans via structure-based virtual screening and molecular dynamics simulation studies, J. Mol. Model., 2023, 29, 256 CrossRef PubMed.
- D. Zhan, S. Guan and H. Jin, et al., Stereoselectivity of phosphotriesterase with paraoxon derivatives: a computational study, J. Biomol. Struct. Dyn., 2016, 34, 600–611 CrossRef CAS PubMed.
- K. Guzik, K. M. Zak and P. Grudnik, et al., Small-Molecule inhibitors of the programmed cell death-1/programmed death-ligand 1 (PD-1/PD-L1) interaction via transiently induced protein states and dimerization of PD-L1, J. Med. Chem., 2017, 60, 5857–5867 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Jinchang Tonga,
Jianhuai Liangb,
Kaixin Shia,
Xinyue Songa,
Zichao Guoa,
Boping Liu*b and
Jianguo Xu
a,
Jinchang Tonga,
Jianhuai Liangb,
Kaixin Shia,
Xinyue Songa,
Zichao Guoa,
Boping Liu*b and
Jianguo Xu