
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotal synthesis of naturally occurring abietane diterpenoids via a late-stage Fe(III)-bTAML catalysed Csp3–H functionalization†
Mintu Munda a,
Debasmita Chatterjeeb,
Moumita Majhib,
Souvik Biswasb,
Debopam Palb and
Alakesh Bisai*ab
a,
Debasmita Chatterjeeb,
Moumita Majhib,
Souvik Biswasb,
Debopam Palb and
Alakesh Bisai*ab
aDepartment of Chemistry, Indian Institute of Science Education and Research Bhopal, Bhauri, Bhopal-462 066, Madhya Pradesh, India
bDepartment of Chemical Sciences, Indian Institute of Science Education and Research Kolkata, Mohanpur, Nadia-741 246, West Bengal, India. E-mail: alakesh@iiserkol.ac.in
First published on 26th June 2024
Abstract
The synthesis of diverse trans-fused decalins, including the abietane diterpenoids scaffold, using an efficient selective oxidation strategy is described. The abietane core was demonstrated to be a versatile scaffold that can be site-selectively functionalized. The utility of this novel oxidation strategy was showcased in a concise total synthesis of six abietane congeners.
Introduction
The use of iron catalyzed reactions to construct complex structures remains a powerful strategy in chemical synthesis.1 In nature, both heme and nonheme metalloenzymes mediated oxidation of alkanes occurs in excellent selectivity and operate under mild reaction conditions.2 Inspired by the high catalytic efficiency of the enzymatic model systems, chemists have developed numerous synthetic iron-based complexes for use in C–H bond oxygenation reactions.3 Despite significant progress, achieving site-selective oxidation of unactivated C(sp3)–H bonds, which constitute the most prevalent structural motifs in complex natural products, remains a significant challenge. Recent efforts have demonstrated C(sp3)–H functionalization,4 including iron-5,6 and manganese-7 and copper catalyzed8 and photochemical,9 methods that are compatible with diverse C–H substrates as the limiting reagent. Although these transformations can be performed in a stoichiometric fashion by organic peracids,10 dioxiranes,11 and oxaziridines,12 but a catalytic method involving H2O2 or O2 as cheap stoichiometric oxidants is highly desirable.In this context, abietane diterpenoids are structurally versatile, exhibits tricyclic ring system (Fig. 1) with several stereocenters and a wide grade of oxygenation pattern on the skeleton invites synthetic chemists to be creative and efficient. Majusanic acid D (1) and majusanin B (2), belongs to a novel class of abietane-type diterpenoids (3–6, Fig. 1), that have been isolated in recent years from the roots of Illicium majus in 2013.13 Structurally, they share densely functionalized trans-decalin framework. Moreover, the decalin core contains three contiguous stereocenters, two of which are quaternary centers.
 | ||
| Fig. 1 Representative abietane-diterpenoids. | ||
Biologically, angustanoic acid E (6) demonstrates significant cytotoxicities against five human tumor cells (HCT-8, Bel-7402, BGC-823, A549, and A2780), with IC50 value 2.47 ± 0.43 μM.14 Stimulated by their impressive structures and bioactivities, we initiated a synthesis study of majusanic acid D (1) and its derivatives (2–6, Fig. 1).
To this end, research in our group along with that in other synthetic groups (e.g., Corey,15 Alvarez-Manzaneda,16 Matsumoto,17 Tada,18 Burnell,19 Gonzalez,20) focused on the development of new synthetic strategies to access abietane-type diterpenoids. Synthetic challenges include stereoselective installation of three adjacent carbon stereocenters and formation of the polyoxygenated moiety. Biosynthetically, the abietane scaffold can be constructed from farnesyl diphosphate (FPP) via a cascade polyene cyclization.21 We envisioned a strategy to advance the abietane scaffold by synthesizing callitrisic acid (7) from abietic acid (14) using a modified literature procedure.22 Then we focused our attention to perform selective oxidation of activated 3 °C–H bonds and activated 2 °C–H bonds via NO2[Fe-b-TAML]5 complex (12) in the presence of the oxidant m-CPBA (Fig. 2c). It is worth mentioning that Matsushita et al. has developed Co-catalyzed aerobic benzylic oxidation of 8,11,13-abietatrienes in the presence of N-hydroxyphthalimide combined with AIBN derivative and its application in total synthesis of diterpenoids.23
 | ||
| Fig. 2 Site-selective functionalization strategies enabled by versatile reactivity of abietane derivatives. | ||
Initially, we wanted to synthesize the core structure of abietane diterpenoids, such as callitrisic acid22 (7). On the basis of previous reports by two independent groups, Pelletier et al. and Alvarez-Manzaneda et al.,24 we have started our synthesis from commercially available (+)-abietic acid (14). By subjecting abietic acid to a temperature of 200 °C for a period of 4 hours resulting in the formation of dehydroabietic acid (14a) (see ESI†). Subsequently, the resultant product underwent methylation utilizing dimethyl sulfate [(MeO)2SO2], resulting in the formation of (15). The subsequent compound (15) was subjected to reduction using LiAlH4, leading to the formation of the primary alcohol (16) with a yield of 96%. Treatment of (16) with PCC afforded the aldehyde (17) which, when treated with m-chloroperbenzoic acid, gave the formate (see ESI† for details), which after saponification with refluxing 2,6-lutidine yielded (18), whose physical and spectroscopic properties were identical to those reported in the literature.20b The C-4 hydroxyl group in (19) was installed by treatment of (18) with BH3·SMe2 and oxidation of the resultant alkyl borane with H2O2 to generate (19) regio- and diastereoselectively in 72% yield and its primary alcohol was converted to the corresponding aldehyde (20) by PCC oxidation in 82% yield. The C-4 quaternary chiral center was installed by treating aldehyde (20) with tBuOK in THF, and the resultant enolate was reacted with MeI followed by a Pinnick oxidation introduced a carboxylic acid group in callitrisic acid (7). Subsequently, callitrisic acid (7) was methylated using dimethyl sulfate [(MeO)2SO2], resulting in the formation of methyl callitrisate (8) in 98% yield (Scheme 1).
 | ||
| Scheme 1 Synthesis of callitrisic acid (7). | ||
We then began to evaluate the proposed iron complex catalyzed selective C–H bond oxidation from callitrisic acid methyl ester (8). Finally, substrate bearing activated methine and benzylic C–H bonds were explored. We then performed catalytic reaction by introducing m-CPBA (5 equiv) via a syringe pump at a rate of 100–200 μL h−1 into a solution containing (12) (1 mol%) and substrate (8) (0.2 mmol scale) in an 80% CH3CN- 20% K2HPO4 (aq) solvent system (Table 1).5 Following this protocol for 15 minutes yielded the benzylic ketone intermediate (22) with a 70% yield (entry 1, Table 1). Extending the reaction time to 1 hour with ketone (22) produced the over-oxidized product (13) in an 85% yield (see ESI† for details). Similarly, callitrisic acid methyl ester (8) was converted directly to the over-oxidized product (13) in 82% yield under the same conditions for 1.5 h (entry 5, Table 1). Notably, the 2° benzylic C–H bonds were observed to be preferentially oxidized over the statistically more significant 3° benzylic C–H bonds.
|
|||
|---|---|---|---|
| Entry | Solvent | Time (min) | Yielda,b,c (%) 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :22:SM :22:SM |
| a All reactions were conducted on a 0.2 mmol scale under 1 mol% catalyst.b Isolated yield reported after column chromatography.c 5 equivalents of mCPBA was used.d The reaction can be performed conveniently in 100 mg scale. | |||
| 1 | CH3CN/H2O (4:1) |
15 | 0:70:30 |
| 2 | CH3CN/H2O (4:1) |
30 | 20:57:23 |
| 3 | THF/H2O (4:1) |
30 | 46:25:29 |
| 4 | DMF/H2O (4:1) |
30 | 32:20:48 |
| 5d | CH3CN/H2O | 90 | 82![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :0:0 :0:0 |
| 6 | CH3CN/H2O (2:1) |
90 | 75:15:0 |
| 7 | DMF/H2O (2:1) |
90 | 61:0:0 |
| 8 | H2O | 90 | 0:23:77 |
| 9 | CH3CN | 90 | 0:31:69 |
 |
|||

The ketone formation can be rationalized through a two-step oxidation process of the C–H bond, involving a two-electron transfer mechanism. The second step, specifically the conversion of alcohol to ketone, occurs approximately 100 times faster than the oxidation of alcohol originating from C–H bonds. We posit that the generation of the ketone in the benzylic position of (22) can be explained by this mechanism. Taken together, the synthetic utility and versatility of our methodology was successfully showcased, resulting in first asymmetric total synthesis of majusanic acid D (1) in significant yield (Scheme 4).
Our next synthetic target was majusanin B (2) and its derivatives (3–6, Fig. 1). With (13) in hand, we next explored Wolff–Kishner reduction25 with hydrazine hydrate (N2H4·H2O) provided the tertiary alcohol in (23) in 77% yield, which underwent elimination by reaction with MsCl in the presence of Et3N in CH2Cl2 to give (24) in 88% yield.
Subsequent oxidative cleavage of (24) with ozonolysis to furnish aromatic acetophenone (25) in 76% moderate yield. Dihydroxylation of the olefin group of the styrene derivative in (24) with OsO4/NMO26 gave diol (26) in 92% yield (Scheme 2).
 | ||
| Scheme 2 Synthesis of sesquiterpenoids scaffold. | ||
Later, we moved ahead for the collective total synthesis of abietane diterpenoids 1–6 shown in Fig. 1. Reduction of (23) and (26) via LiAlH4-reduction ensures the total synthesis of angustanol (5) and majusanin B (2) (Scheme 3). Lastly, hydrolysis of methyl ester of (13), (23), (24), and (25) using LiOH and KOH in hot methanol27 completed the total synthesis of majusanic acid D (1), angustanoic acid F (4), angustanoic acid E (6) and angustanoic acid G (3) respectively (Scheme 4).
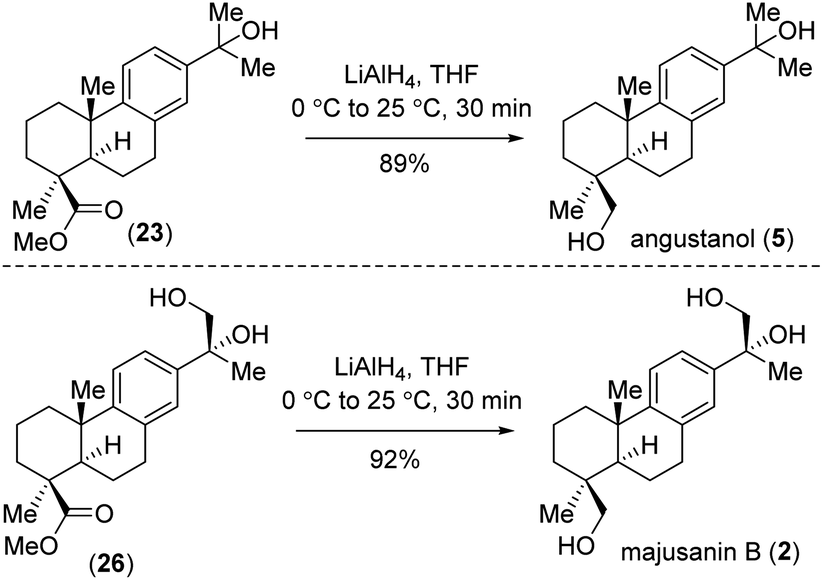 | ||
| Scheme 3 Synthesis of angustanol (5) and majusanin B (2). | ||
 | ||
| Scheme 4 Majusanic acid D (1), angustanoic acid F (4), angustanoic acid E (6) and angustanoic acid G (3). | ||
Conclusions
We have completed the total syntheses of all known abietane diterpenoids (1–6) through a unified strategy inspired by our hypothesis for their biogenesis. Key features of our syntheses include: (1) (+)-callitrisic acid (7) synthesis accomplished in 10 steps from abietic acid (14) with 32% overall yield, (2) we demonstrate synthetic utility and versatility of Fe-bTAML complex catalyzed biomimetic C–H bonds oxidation strategy in the total synthesis of complex natural products.Data availability
Experimental details and spectral analysis are available free of charge from the ESI† available with this article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the SERB [CRG/2023/000782], [SCP/2022/000486] and CSIR [02(403)/21/EMR-II], is gratefully acknowledged. MM thanks the CSIR for a research fellowship (SRF). SB, and MM, thank the CSIR for research fellowships (JRFs). DP thanks the INSPIRE fellowship. AB is a SERB-STAR Fellow and sincerely acknowledges the SERB [STR/2020/000061] for generous support. Our sincere thanks to Prof. Sayam Sen Gupta for scientific discussions particularly for Fe(III)-catalyzed C–H functionalization process.Notes and references
- (a) A. Furstner, ACS Cent. Sci., 2016, 2, 778–789 CrossRef PubMed; (b) I. Bauer and H. J. Knolker, Chem. Rev., 2015, 115, 3170–3387 CrossRef CAS PubMed.
- (a) M. Costas, M. P. Mehn, M. P. Jensen and L. Q. Jr, Chem. Rev., 2004, 104, 939–986 CrossRef CAS PubMed; (b) P. R. O. D. Montellano, Chem. Rev., 2010, 110, 932–948 CrossRef PubMed; (c) L. Que Jr and W. B. Tolman, Nature, 2008, 455, 333–340 CrossRef PubMed; (d) G. B. Shulpin, Org. Biomol. Chem., 2010, 8, 4217–4228 RSC.
- (a) W. Nam, Acc. Chem. Res., 2007, 40, 522–531 CrossRef CAS PubMed; (b) M. Puri and L. Que Jr, Acc. Chem. Res., 2015, 48, 2443–2452 CrossRef CAS PubMed; (c) W. N. Oloo and L. Que Jr, Acc. Chem. Res., 2015, 48, 2612–2621 CrossRef CAS PubMed; (d) M. Christmann, Angew. Chem., Int. Ed., 2008, 47, 2740–2742 CrossRef CAS PubMed; (e) A. C. Lindhorst, S. Haslinger and F. E. Kuhn, Chem. Commun., 2015, 51, 17193–17212 RSC; (f) B. Meunier, Chem. Rev., 1992, 92, 1411–1456 CrossRef CAS; (g) M. Costas, M. P. Mehn, M. P. Jensen and L. Que Jr, Chem. Rev., 2004, 104, 939–986 CrossRef CAS PubMed; (h) J. T. Groves, R. C. Haushalter, M. Nakamura, T. E. Nemo and B. J. Evans, J. Am. Chem. Soc., 1981, 103, 2884–2886 CrossRef CAS.
- L. Tanwar, J. Borgel and T. Ritter, J. Am. Chem. Soc., 2019, 141, 17983–17988 CrossRef CAS PubMed.
- S. Jana, M. Ghosh, M. Ambule and S. S. Gupta, Org. Lett., 2017, 19, 746–749 CrossRef CAS PubMed.
- (a) A. Sharma and J. F. Hartwig, Nature, 2015, 517, 600–604 CrossRef CAS PubMed; (b) R. R. Karimov, A. Sharma and J. F. Hartwig, ACS Cent. Sci., 2016, 2, 715–724 CrossRef CAS PubMed.
- (a) J. R. Clark, K. Feng, A. Sookezian and M. C. White, Nat. Chem., 2018, 10, 583–591 CrossRef CAS PubMed; (b) X. Huang, T. M. Bergsten and J. T. Groves, J. Am. Chem. Soc., 2015, 137, 5300–5303 CrossRef CAS PubMed.
- (a) S. E. Suh, S. J. Chen, M. Mandal, I. A. Guzei, C. J. Cramer and S. S. Stahl, J. Am. Chem. Soc., 2020, 142, 11388–11393 CrossRef CAS PubMed; (b) P. Li, Y. Wang, X. Wang, Y. Wang, Y. Liu, K. Huang, J. Hu, L. Duan, C. Hu and J. Liu, J. Org. Chem., 2020, 85, 3101–3109 CrossRef CAS PubMed.
- (a) K. A. Margrey, W. L. Czaplyski, D. A. Nicewicz and E. J. Alexanian, J. Am. Chem. Soc., 2018, 140, 4213–4217 CrossRef CAS PubMed; (b) Y. Wang, G. X. Li, G. Yang, G. He and G. Chen, Chem. Sci., 2016, 7, 2679–2683 RSC; (c) Y. Wang, N. Wang, J. Zhao, M. Sun, H. You, F. Fang and Z. Q. Liu, ACS Catal., 2020, 10, 6603–6612 CrossRef CAS.
- H. J. Schneider and W. Muller, J. Org. Chem., 1985, 50, 4609–4615 CrossRef CAS.
- R. Mello, M. Fiorentino, C. Fusco and R. Curci, J. Am. Chem. Soc., 1989, 111, 6749–6757 CrossRef CAS.
- D. D. DesMarteau, A. Donadelli, V. Montanari, V. Montanari, V. A. Petrov and G. Resnati, J. Am. Chem. Soc., 1993, 115, 4897–4898 CrossRef CAS.
- (a) L. K. Sy and G. D. Brown, J. Nat. Prod., 1998, 61, 907–912 CrossRef CAS PubMed; (b) Y. Li, G. T. T. Wang, J. Zhao, X. Shi, S. C. Hu and K. Gao, Food Chem., 2012, 131, 972–976 CrossRef CAS; (c) Y. D. Wang, G. J. Zhang, J. Qu, Y. H. Li, J. D. Jiang, Y. B. Liu, S. G. Ma, Y. Li, H. N. Lv and S. S. Yu, J. Nat. Prod., 2013, 76, 1976–1983 CrossRef CAS PubMed; (d) L. P. Hua, Y. Q. Zhang, M. Ye, W. Xu, X. Y. Wang, Y. H. Fu and W. Xu, Nat. Prod. Res., 2018, 32, 2577–2582 CrossRef CAS PubMed.
- Z. F. Fang, G. J. Zhang, H. Chen, J. Bai, S. S. Yu, Y. Liu, W. J. Wang, S. G. Ma, J. Qu and S. Xu, Planta Med., 2013, 79, 142–149 CAS.
- K. Surendra, G. Rajendar and E. J. Corey, J. Am. Chem. Soc., 2014, 136, 642–645 CrossRef CAS PubMed.
- (a) E. J. Alvarez-Manzaneda, R. Chahboun, J. J. Guardia, M. Lachkar, A. Dahdouh, A. Lara and I. Messouri, Tetrahedron Lett., 2006, 47, 2577–2580 CrossRef CAS; (b) E. Alvarez-Manzaneda, R. Chahboun, E. Cabrera, E. Alvarez, R. Alvarez-Manzaneda, M. Lachkar and I. Messouri, Synlett, 2007, 15, 2425–2429 CrossRef; (c) E. Alvarez-Manzaneda, R. Chahboun, E. Alvarez, J. M. Ramos, J. J. Guardia, I. Messouri, I. Chayboun, A. I. Mansour and A. Dahdouh, Synthesis, 2010, 20, 3493–3503 CrossRef.
- (a) T. Matsumoto and S. Usui, Bull. Chem. Soc. Jpn., 1979, 52, 212–215 CrossRef CAS; (b) T. Matsumoto, S. Imai, S. Takeda and M. Mitsuki, Bull. Chem. Soc. Jpn., 1983, 56, 2013–2017 CrossRef CAS; (c) T. Matsumoto, S. Imai, S. Miuchi and H. Sugibayashi, Bull. Chem. Soc. Jpn., 1985, 58, 340–345 CrossRef CAS; (d) T. Matsumoto, H. Terao and S. Imai, Bull. Chem. Soc. Jpn., 1991, 64, 2855–2856 CrossRef CAS; (e) T. Matsumoto, Y. Tanaka, H. Terao, Y. Takeda and M. Wada, Bull. Chem. Soc. Jpn., 1993, 66, 3053–3057 CrossRef CAS.
- M. Tada and K. Ishimaru, Chem. Pharm. Bull., 2006, 54, 1412–1417 CrossRef CAS PubMed.
- R. H. Burnell, C. Cote and M. Girard, J. Nat. Prod., 1993, 56, 461–472 CrossRef CAS.
- (a) M. A. Gonzalez and R. J. Zaragoza, J. Nat. Prod., 2014, 77, 2114–2117 CrossRef CAS PubMed; (b) R. J. Zaragoza and M. A. Gonzalez, RSC Adv., 2020, 10, 15015–15022 RSC.
- J. W. Yang and Y. Orihara, Tetrahedron, 2002, 58, 1265–1270 CrossRef CAS.
- (a) K. Mori and M. Matsui, Tetrahedron, 1968, 24, 6573–6575 CrossRef CAS; (b) S. C. Welch and J. H. Kim, Synth. Commun., 1976, 6, 27–31 CrossRef CAS; (c) X. Wu, M. S. Ejneby, N. E. Ottosson, F. Elinder and P. Konradsson, Eur. J. Org Chem., 2018, 1730–1734 CrossRef CAS.
- Y. Matsushita, K. Sugamoto, Y. Iwakiri, S. Yoshida, T. Chaen and T. Matsui, Tetrahedron Lett., 2010, 51, 3931–3934 CrossRef CAS.
- For two different approaches for the C4-stereogenic centre inversion of dehydroabietic acid, see; (a) S. W. Pelletier and D. L. Herald, Chem. Commun., 1971, 10–11 RSC; (b) E. Alvarez-Manzaneda, R. Chahboun, E. Alvarez, J. M. Ramos, J. J. Guardia, I. Messouri, I. Chayboun, A. I. Mansour and A. Dahdouh, Synthesis, 2010, 3493–3503 CrossRef CAS.
- D. Todd, Org. React., 1948, 4, 378–423 CAS.
- S. De, M. K. Das, A. Roy and A. Bisai, J. Org. Chem., 2016, 81, 12258–12274 CrossRef CAS PubMed.
- Y. J. Zhao and T. P. Loh, Org. Lett., 2008, 10, 2143–2145 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra03791j |
| This journal is © The Royal Society of Chemistry 2024 |