
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImpact of POM's coordination mode and Mo-hybrid constituents on the binding, stability, and catalytic properties of hybrid (pre)catalysts†
Jana Pisk a,
Višnja Vrdoljak*a,
Mirna Mandarića,
Tomica Hrenara,
Dominique Agustinbc and
Mirta Rubčića
a,
Višnja Vrdoljak*a,
Mirna Mandarića,
Tomica Hrenara,
Dominique Agustinbc and
Mirta Rubčića
aUniversity of Zagreb, Faculty of Science, Department of Chemistry, Horvatovac 102a, 10000 Zagreb, Croatia. E-mail: visnja.vrdoljak@chem.pmf.hr
bLCC-CNRS (Laboratoire de Chimie de Coordination), 205 Route de Narbonne, BP44099, CEDEX 4, 31077 Toulouse, France
cDepartment of Chemistry, IUT Paul Sabatier, Université Paul Sabatier, University of Toulouse, Av. G. Pompidou, CS20258, 81104 Castres, France
First published on 13th June 2024
Abstract
The assembly of MoVIO22+ and methoxy-substituted salicylaldehyde nicotinoyl hydrazone ligands afforded two classes of hybrid polyoxometalates (POMs). In the Class I architectures, [MoO2(HL1−3)(D)]2[Mo6O19]·xCH3COCH3 (D = CH3COCH3 or H2O, x = 0 or 2, and L1–3 = ligands bearing the OMe group at position 3, 4 and 5, respectively), the main driving force for self-assembly is the electrostatic interaction between the components. Class II architectures are composed of a POM anion covalently linked to two Mo-complex units through the terminal Ot or bridging μ2-OPOM oxygen atoms, as found in Lindqvist-based hybrids [{MoO2(HL1–3)}2Mo6O19]·xCH3CN (x = 0 or 2) and the asymmetrical β-octamolybdate-based hybrid [{Mo2O4(HL2)(H2L)}{MoO2(HL2)}2Mo8O26]·CH3CN·H2O. Quantum chemical calculations were applied to evaluate the impact of the POM hybrid constituents on the hybrid-type stability, showing that it strongly depends on the ligand substituent position and ancillary ligand nature. Hybrids were tested as catalysts for cyclooctene epoxidation using tert-butyl hydroperoxide (TBHP in water or decane) and with or without the addition of acetonitrile (CH3CN) as an organic solvent. The catalytic results provided by the use of TBHP in decane are the best ones and classify all the prepared catalysts as very active, with the conversion of cyclooctene >90%, and high selectivity towards epoxide, >80%. We also examined the influence of the ligand structure, POM's hybrid type, and coordination mode on the Mo-hybrid activity and selectivity.
Introduction
Polyoxometalates (POMs) have attracted attention in many fields of science due to their structural diversity, compositional adjustability, and numerous chemical features.1–5 They have particularly received attention in catalysis,6,7 contributing to the atomically efficient and energetically sustainable objectives. Namely, POMs are highly effective catalysts in various relevant processes, such as in oxidation reactions of olefins,8–10 alcohols,11,12 aldehydes,13 or amines.14 The development of new POM functional materials with unique properties is therefore essential for the continuous progress in that research field. In this context, their derivatization by organic components15–19 or transition metal complexes20–25 has proven to be a convenient method.Among various catalysts, MoVI complexes have been particularly useful for the epoxidation of cyclic olefins.26–30 The metallosupramolecular species with long Mo–Nligand bonds were particularly effective.31–34 It was, therefore, reasonable to combine MoVI coordination compounds with POMs into organic–inorganic hybrid architectures. In principle, POMs should be a suitable substitute for any ligand, but due to steric hindrance effects, one might expect their coordination to Mo with a long Mo–O bond. However, their controlled assembly in organic media is less straightforward. To date, only two examples of crystalline species [{MoO2(HL)}2{Mo6O19}] have been structurally characterized and in both of them, POM links two Mo-complex fragments through POM terminal oxygen atoms.35,36
The coordination of Lindqvist anion to Mo-complex through the bridging μ2-OPOM is not yet known, although the bridging oxygen atoms are more nucleophilic and basic than the Ot terminal sites.37 This drawback may be due to steric hindrance imposed by POM and organic ligands. Only one example of the Lindqvist μ2-OPOM coordination to Cu-complex has been reported so far.38 Additionally, a few examples of such α-Keggin-type hybrid structures with transition metal complex moieties are known.39–42 Furthermore, according to the Cambridge Structural Database,43 the formation of asymmetrically anchored Mo hybrid systems has never been reported.
So far, more results have been achieved in the synthesis of various MoVI POM hybrid salts. POM anions tend to associate with cationic Mo-complexes via multiple non-covalent interactions and give ionic hybrids (Class I).44–46 However, investigation of their potential was limited by their low stability. Coordination of POM to metal centres offers a way to overcome this issue and allows the discovery of new POM coordination modes.
Herein, we report the successful self-assembly of individual building units into ionic hybrids [MoO2(HL1–3)(D)]2[Mo6O19]·xsolv (Class I) consisting of coordination cations and POM anions, and their conversion into the corresponding covalently linked architectures [{MoO2(HL1–3)}2Mo6O19]·xsolv (Class II). As organic ligands, we selected methoxy-substituted salicylaldehyde nicotinoyl hydrazones H2L1–3, (Scheme 1), considering the feasibilities for the fine-tuning of structural features and their tendency towards reversible protonation and deprotonation.
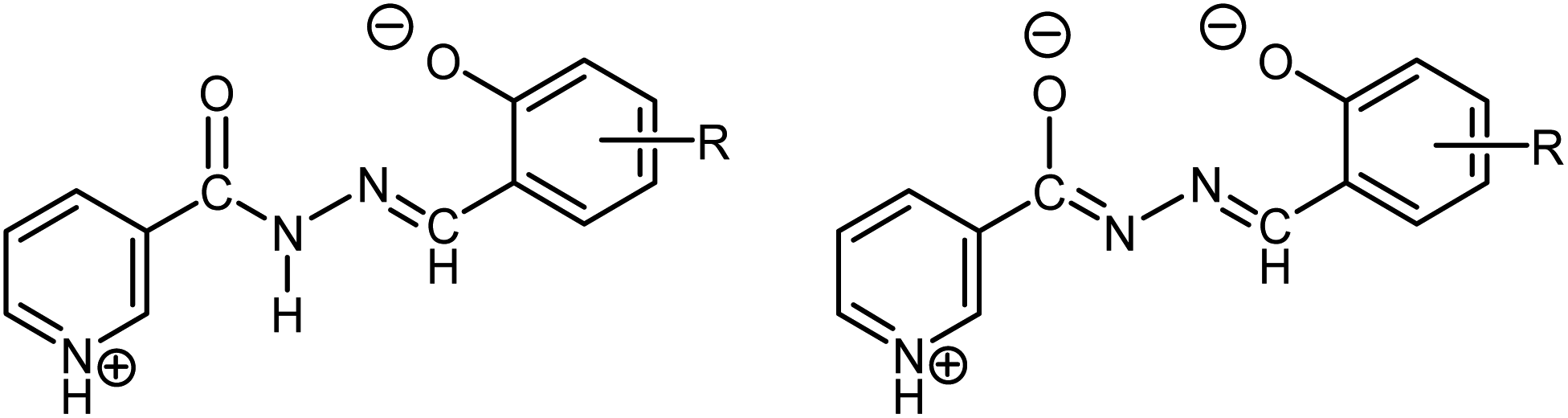 | ||
| Scheme 1 Nicotinoyl hydrazonato ligands in neutral zwitterionic and singly-deprotonated (HL−) forms bearing OMe group at positions 3 (H2L1), 4 (H2L2), and 5 (H2L3). | ||
All hybrids were characterized using the single-crystal and powder X-ray diffraction (XRD) methods, infrared (IR-ATR) spectroscopy, thermogravimetric analysis (TGA), and elemental analysis. In the case of Class II assemblies, both coordination modes through the terminal Mo = Ot and bridging μ2-O oxygen atoms to two peripheral Mo-coordination subunits were successfully obtained. The β-octamolybdate hybrid [{Mo2O4(HL2)(H2L2)}{MoO2(HL2)}Mo8O26]·2CH3CN·H2O corresponds to the first example of an asymmetrical arrangement of Mo-fragments around the POM anion.
Quantum chemical calculations were employed to support experimental results and estimate the influence of the ancillary ligand, hydrazone substituent, and POM coordination type on the hybrid-class formation.
We envisaged those POM-hybrid architectures, combining favourable properties of coordination and inorganic moieties, as promising catalysts for the epoxidation of olefins. The covalently anchored ones would be particularly appealing because the activity of Mo-complexes could be directly related to the POM bonding nature. In our research, the catalytic activity of hybrids in the selective oxidation of cyclooctene was evaluated using tert-butyl hydroperoxide (TBHP) as the oxidant (in water or decane) and with or without the addition of the organic solvent (CH3CN).
Results and discussion
Synthesis and characterization of hybrids
Ligands H2L1–3 (Scheme 1) were prepared by the reaction of isonicotinyl hydrazine with an appropriate aldehyde according to the published procedures.26,47 The synthetic pathways for POM-based hybrids and a list of all hybrids are provided in Scheme 2. Almost all assemblies accommodate lattice solvent molecules. Their formulae are consistent with analytical, spectroscopic and X-ray diffraction data. A corresponding designation of compounds can be found in Scheme 3. | ||
| Scheme 2 Summary of POM-based hybrids (Class I and Class II). Black arrows depict the syntheses carried out at room temperature in acetone or acetonitrile. Orange and blue arrows stand for the solvothermal transformations carried out in acetone and acetonitrile, respectively. | ||
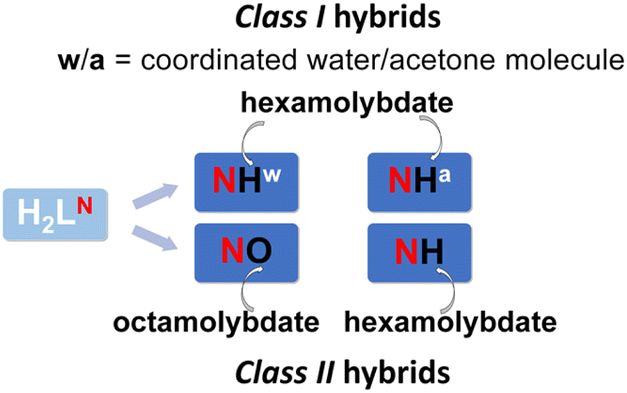 | ||
| Scheme 3 Compounds designation – N = 1, 2, and 3 correspond to hybrids with L1, L2, and L3 within the [MoO2(HL)(D)]+ cation. Hexamolybdates and octamolybdate are abbreviated as H and O, respectively, whereas a and w represent ancillary ligand D, acetone and water. | ||
Reactions of [MoO2(acac)2] (acac = acetylacetonate) and hydrazone ligands H2L1–3 in a molar ratio of 3.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in acetone resulted initially in orange suspensions, indicating the MoVI hydrazone complex formation. A significant quantity of the orange solids was separated. A comparison of the PXRD patterns identified them to be a metallosupramolecular species [MoO2(L1–3)]n, known from our previous investigation.26,31 Upon standing the filtrate at room temperature, the condensation of the remaining metal oxo-units led to the formation of the Class I assemblies [MoO2(HL1–3)(D)]2[Mo6O19]·2CH3COCH3, where D = acetone in 1Ha·2CH3COCH3, or water in 2Hw·2CH3COCH3 and 3Hw·2CH3COCH3. The red crystals deposited after about one to two weeks.
:1 in acetone resulted initially in orange suspensions, indicating the MoVI hydrazone complex formation. A significant quantity of the orange solids was separated. A comparison of the PXRD patterns identified them to be a metallosupramolecular species [MoO2(L1–3)]n, known from our previous investigation.26,31 Upon standing the filtrate at room temperature, the condensation of the remaining metal oxo-units led to the formation of the Class I assemblies [MoO2(HL1–3)(D)]2[Mo6O19]·2CH3COCH3, where D = acetone in 1Ha·2CH3COCH3, or water in 2Hw·2CH3COCH3 and 3Hw·2CH3COCH3. The red crystals deposited after about one to two weeks.
The reactions of [MoO2(acac)2] with H2L1 and H2L3 in a molar ratio of 10:1 in acetonitrile proceeded similarly. Despite the presence of a higher amount of [MoO2(acac)2], which is more favourable for the formation of octamolybdates,45 the reactions with H2L1 and H2L3 always resulted in the formation of hexamolybdate hybrids [MoO2(HL1)(H2O)]2[Mo6O19] (1Hw) and [{MoO2(HL3)}2Mo6O19]·2CH3CN (3H·2CH3CN). This could be due to the better stability of the hexamolybdate anion in acetonitrile in the presence of Mo-complexes with these two ligands. The resulting hybrids 1Hw and 3H·2CH3CN, belong to Class I and Class II, respectively. However, in the case of H2L2, the condensation always resulted in the asymmetrical Class II hybrid [{Mo2O4(HL2)(H2L2)}{MoO2(HL2)}Mo8O26]·CH3CN·H2O (2O·CH3CN·H2O). To our knowledge, this is the first example of a POM-based hybrid with octamolybdate covalently expanded by three Mo–hydrazone complex units.
Traces of moisture in the solvents are crucial for the polyoxometalate anion formation. The addition of acetic acid during the reactions increased the purity of the compounds, but there was no difference in final hybrid product formation. The powder X-ray diffraction (PXRD, Fig. S1, see ESI†) patterns of the bulk products are well-matched with the calculated ones. However, the yields depend on the quantity of the formed coordinated polymers. It should be mentioned that the reaction in acetone in a molar ratio of 10:1 yielded each time a gelatinous reaction mixture, whereas the synthesis in acetonitrile in a molar ratio of 3.3:1 gave a mixture of products.
Solvothermal treatment of the corresponding hybrid salts 1Ha·2CH3COCH3, 1Hw, 2Hw·2CH3COCH3, and 3Hw·2CH3COCH3 in dry acetonitrile resulted in the exclusion of coordinated solvent molecules (acetone or water) and POM anion immobilisation. This approach resulted in [{MoO2(HL1–3)}2Mo6O19]·xCH3CN, x = 0 or 2 (1H, 2H, and 3H·2CH3CN), Class II hybrids. The condensation under solvothermal conditions in dry acetone was successful in the transformation of 1Ha·2CH3COCH3 and 3Hw·2CH3COCH3 into 1H and 3H, respectively. In the case of 1Hw and 2Hw·2CH3COCH3, such conversion failed to proceed.
Attenuated Total Reflectance Infrared (ATR-IR) spectra for all compounds are consistent with the formation of hybrids (Fig. S2–S4, see ESI†). Whereas, the vibrations around 950 cm−1 could be assigned to the ν(Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) in the POM anion and the bands in the region of 942−886 cm−1 corresponds to the terminal ν(MoO) of the Mo–hydrazonato complex. The bands in the range of 789 cm−1 to 712 cm−1 may be assigned to the ν(Mo–O–Mo) vibrations of the hexamolybdate Mo-oxo groups, while those in the range of 689 cm−1 to 642 cm−1 are related to ν(Mo–O–Mo) vibrations of the octamolybdate cluster.48,49
O) in the POM anion and the bands in the region of 942−886 cm−1 corresponds to the terminal ν(MoO) of the Mo–hydrazonato complex. The bands in the range of 789 cm−1 to 712 cm−1 may be assigned to the ν(Mo–O–Mo) vibrations of the hexamolybdate Mo-oxo groups, while those in the range of 689 cm−1 to 642 cm−1 are related to ν(Mo–O–Mo) vibrations of the octamolybdate cluster.48,49
Crystals of solvates 2Hw·2CH3COCH3, 3Hw·2CH3COCH3, and 3H·2CH3CN exhibit excellent stability and retain their structural integrity at room temperature for long periods without decomposing. According to the thermal analysis (Fig. S2 and S3, see ESI†), they lose the coordinated and/or non-coordinated solvent molecules in one mass loss step in the range 122–161 °C (2Hw·2CH3COCH3), 158–187 °C (3Hw·2CH3COCH3), and 167–222 °C (3H·2CH3CN). In the second step, mass loss started around 290 °C.
However, in the case of 1Ha·2CH3COCH3 and 2O·CH3CN·H2O, a complete loss of lattice solvent occurred at room temperature after a few minutes. The analysis of residuals indicates that they are of the composition 1Ha and 2O. Thermogravimetric curves of 1Ha and 1Hw exhibit two separate mass loss steps (Fig. S2 and S3†). The first mass loss between 130 °C and 157 °C and between 182 °C and 213 °C corresponds to the loss of coordinated acetone and water molecules, respectively.
Solvent-free hybrids 1, 2, 2O, and 3 are very stable. As shown in Fig. S3 and S4†, the mass loss starts at 275 °C, 243 °C, 295 °C, and 317 °C, respectively, with the decomposition of the metal-oxo cluster. In all the cases, the final step mass losses agree with the calculated values, considering the product of the decomposition to be the MoO3.
Description of molecular and crystal structures
Within the structures of the ionic type one can distinguish those where the complex cation is of the [MoO2(HL)(D)]+ kind (Fig. 1), where HL− denotes a singly deprotonated form of the ligand and D represents ancillary ligand, acetone or water molecule, fulfilling the deformed octahedral coordination environment of the Mo atom (1Ha·2CH3COCH3, 3Hw·2CH3COCH3, 2Hw·2CH3COCH3 and 1Hw). In all structures of ionic type of hybrids, polyoxometalate anions are classical Lindqvist ones, Mo6O192−. | ||
| Fig. 1 Molecular structures of 1Ha·2CH3COCH3 with the atom-labeling scheme as illustrative example of ionic structures described in this work. Ellipsoids are drawn at 30% probability level. Hydrogen atoms are presented as spheres of arbitrary small radii. The O11 oxygen atom is situated on an inversion center, and for clarity, only one of the two equally occupied positions for the non-coordinated acetone molecule is shown. Dihedral angles between the planes of the pyridyl and the phenyl ring of the aldehyde residue amount to 5°. | ||
Within the class of covalently linked hybrids, one finds those in which the Lindqvist anion Mo6O192− acts a bridging unit between two [MoO2(HL)]+ fragments (1H, 2H, 3H, and 3H·2CH3CN), and the one where β-octamolybdate bridges [Mo2O4(HL)(H2L)]3+ and [MoO2(HL)]+ fragments, 2O·2CH3CN·H2O.
It should be highlighted that in all structurally characterized compounds, except the 2O·2CH3CN·H2O one, the hydrazone ligand coordinates in the monoanionic form via O1, N1 and O2 atoms, which is evident from the relevant bond distances (Scheme 1, Tables S1 and S2 in the ESI†), and also consistent with previously reported examples.45 It is also interesting to note that coordinated ligands assume in most cases an essentially planar conformation (Fig. 1, 2 and S5, see ESI†), while the largest deviations from planarity are observed for the ligands in 3H·2CH3CN and 2O·2CH3CN·H2O.
 | ||
| Fig. 2 Molecular structures of: (a) 3H·2CH3CN and (b) 2O·2CH3CN·H2O, and (c) 3H with the atom-labeling scheme. Ellipsoids are drawn at 30% probability level, whereas hydrogen atoms are shown as spheres of arbitrary small radii. In (a) O11 and in (c) O9 atoms are situated on an inversion center. For the clarity of presentation, in (b) solvent molecules are omitted. Dihedral angles between the planes of the pyridyl and the phenyl ring of the aldehyde residue for 3H·2CH3CN and 3H, respectively amount to ca. 33.3° and ca. 2.2° (for 2O·2CH3CN·H2O are discussed below). | ||
Molecular structures of ionic hybrids 1Ha·2CH3COCH3, 3Hw·2CH3COCH3, 2Hw·2CH3COCH3 and 1Hw contain Lindqvist anions, Mo6O192−, and complex cations of the type [MoO2(HL)(D)]+ (D = CH3COCH3 or H2O), in which the Mo atoms assume distorted octahedral environments (Fig. 1 and S5b–d†). As expected for octahedral coordination geometry, the longest distances within the coordination sphere of the Mo atom of the [MoO2(HL)(D)]+ units are those positioned trans to MoO functionalities, namely Mo1–O6 and Mo1–N1 bonds. The presence of additional donor molecules at the sixth coordination site of molybdenum in [MoO2(HL)(D)]+ cations along with the presence of solvent molecules in 1Ha·2CH3COCH3, 3Hw·2CH3COCH3, 2Hw·2CH3COCH3 lead to diverse packing scenarios (Fig. S6–S9, see ESI†). The crystal structure of 1Ha·2CH3COCH3 unveils strong hydrogen bonding between the protonated pyridine moiety and non-coordinated acetone molecule in a discrete D11(2) graph-set motif. In the absence of further strong hydrogen bond donors, hexamolybdate anions remain involved only in C–H⋯O interactions with the [MoO2(HL)(D)]+ units and the solvent molecules within the crystal structure (Fig. S6†). The same, evidently robust D11(2), hydrogen bond motif, established between the protonated pyridine moiety and the acetone molecule is also found in the structures of 2Hw·2CH3COCH3 and 3Hw·2CH3COCH3 (Fig. S7 and S8†).
In contrast to 1Ha·2CH3COCH3, the presence of additional hydrogen bond donors, i.e. coordinated water molecules in [MoO2(HL)(H2O)]+, affords a complex hydrogen bonded networks in both cases. Namely, in the 2Hw·2CH3COCH3 the coordinated water molecule forms hydrogen bonds with the Lindqvist anion, and with the acetone molecule yielding thus complex supramolecular chains along the crystallographic a-axis (Fig. S8, see ESI†). On the other hand, in 3Hw·2CH3COCH3 one observes supramolecular dimers of [MoO2(HL)(H2O)]+ units, formed via O6–H6A⋯N2 hydrogen bonds, which are bridged by Mo6O192− anions. Crystal structures in both cases are stabilized by plethora of C–H⋯O interactions (Fig. S7, S8 and Table S3, see ESI†). Supramolecular architecture of 1Hw differs from the previously described ones, as there are no solvent molecules present in the structure. Here the [MoO2(HL)(H2O)]+ units associate via O–H⋯O and N–H⋯O interactions into complex assemblies which are joined by hydrogen-bond anchored Lindqvist anions (Fig. S9†). It is interesting to note that in the absence of the solvent molecules in 1Hw, i.e. acetone, the protonated pyridine moiety establishes hydrogen bond with one of the oxygen atoms of the {MoO2}2+ core of the [MoO2(HL)(H2O)]+ unit.
In the structures of neutral complexes polyoxomolybdate anions, Mo6O192− in 1H and 3H·2CH3CN and Mo8O244− in 2O·2CH3CN·H2O, act as bridging entities (Fig. 2). Polyoxoanions in these structures coordinate via their terminal oxygen atoms to molybdenum atoms of the {MoO2(HL)} fragments, thus providing the Mo atoms of the later octahedral environment.
Structure of 2O·2CH3CN·H2O is unique as the Mo8O264− unit bridges one dinuclear [Mo2O4(HL)(H2L)]3+ and one mononuclear [MoO2(HL)]+ entity (Fig. 2b). In the dinuclear unit one finds the coordinated ligand both in singly-deprotonated HL− as well as in the neutral zwitterionic form H2L (Scheme 1), whereas in mononuclear unit [MoO2(HL)]+ ligand is coordinated in its singly-deprotonated form (Scheme 1). In this structure one can observe substantial differences in the conformation of zwitterionic form of H2L with respect to singly-deprotonated ones, HL−. Namely, the angle between the pyridyl and aldehyde rings of the zwitterionic form of H2L amounts to ca. 2.5°, in the case of HL− in the [Mo2O4(HL)(H2L)]3+ unit it is ca. 35.4° and for the [MoO2(HL)]+ is ≈21.9°. It is worthwhile to note that singly-deprotonated forms of the ligand are found both with syn- and anti- oriented pyridyl moieties with respect to the coordinating ONO compartment of HL−. The crystal structure reveals a rich assortment of hydrogen bonds. Namely, hydrogen atoms of the protonated pyridyl moieties of all three coordinated ligands afford bifurcated hydrogen bonds with the octamolybdate anions. Additional hydrogen bonding involves crystal water molecules and the octamolybdate anions, as well as the hydrazinic N–H group (zwitterionic form H2L) and the oxygen atoms of the {MoO2}2+ core, forming thus a complex supramolecular network (Fig. S10†). It should be noted that acetonitrile molecules are not involved in strong intermolecular interactions, which explains the observed positional disorder, related to acetonitrile molecules, in the structure. So far, only Class I octamolybdate hybrids with coordinated solvent molecules on the {MoO2}2+ core have been studied, offering valuable insight into their structural properties.45,46
When comparing the structures of 1H and 3H·2CH3CN, one observes the difference in the engagement of the protonated pyridine moiety in the hydrogen bonding due to the presence of solvent molecules in 3H·2CH3CN (Fig. 3). Namely, in 1H hydrogen atom of the protonated pyridine moiety participates in bifurcated hydrogen bond, N3–H3⋯O4 and N3–H3⋯O15, forming thus supramolecular layers in the ab-plane (Fig. S11, see ESI†).
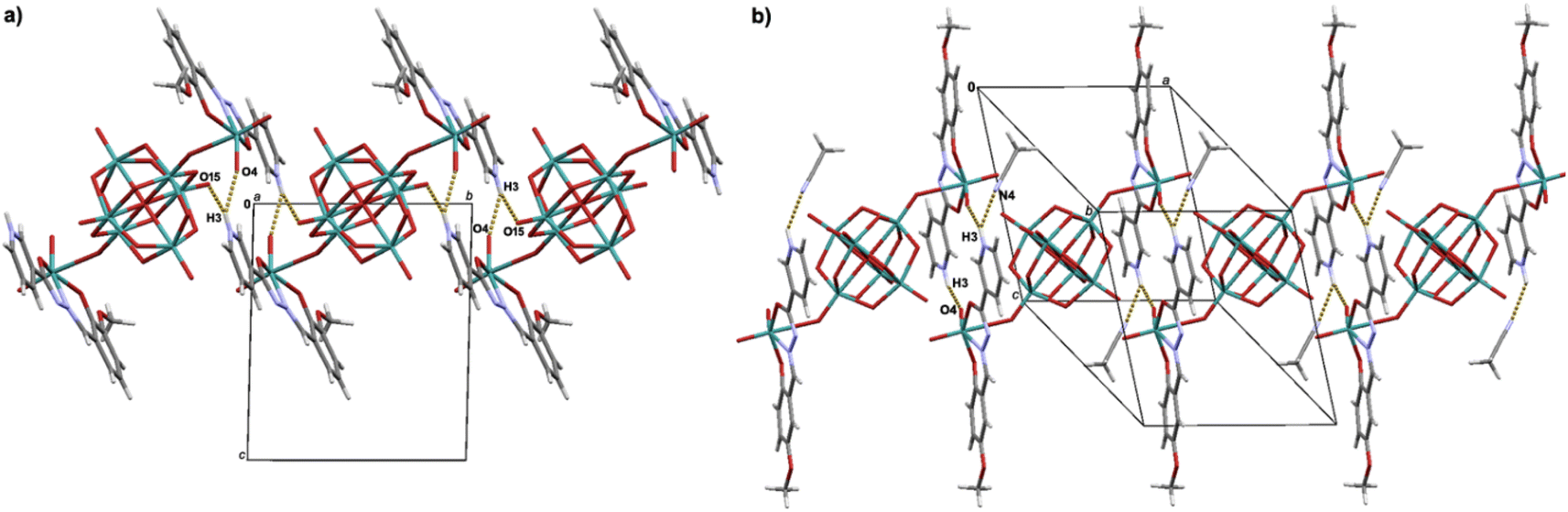 | ||
| Fig. 3 Supramolecular assemblies observed in the crystal structures of: (a) 1H, and (b) 3H·2CH3CN. In (a) N3–H3⋯O4 hydrogen bonds yield a R22(16) motif, and N3–H3⋯O15 a C11(12) motif. In (b) N3–H3⋯O4 and N3–H3⋯N4 hydrogen bonds yield R22(16) and D11(2) motifs. | ||
Molecular and crystal structures of the hybrids 2H and 3H are similar. Namely, in both cases molecular structures consist of the [MoO2(HL)]+ type of cations which are bridged by Lindqvist anions, Mo6O192−, to afford overall neutral species (Fig. 2c and S5a†). It is interesting to observe that in both 2H and 3H bridging oxygen atoms of the {Mo6O19} units coordinate to Mo1 atoms of the complex unit {MoO2(HL)}, with the corresponding distances being 2.723(5) for 2H and 2.705(6) for 3H (in both cases Mo1–O6 distances), Table S4, see ESI†.
Similarity in molecular structures of 2H and 3H is also reflected in their packing arrangements. In both cases, such complexes associate via N–H⋯O hydrogen bonds involving both {[MoO2(HL)]} and {Mo6O19} fragments (Fig. 4), which are further associated through C–H⋯O interactions (Fig. S12, S13 and Table S4, see ESI†).
 | ||
| Fig. 4 Supramolecular assembly observed in the crystal structure of 2H. {[MoO2(HL)]} units of the 2H structure is engaged in N3–H3⋯O4 hydrogen bonds forming C11(8) chains (along the b-axis), while the {Mo6O19} fragments participate in N3–H3⋯O11 hydrogen bonds shaping C11(10) motif. | ||
Finally, it is interesting to observe that in the Lindqvist containing hybrids, 2H and 3H have their pyridyl moieties-oriented anti with respect to the coordinating ONO compartment of the HL−, whereas in the structures of (1Ha·2CH3COCH3, 3Hw·2CH3COCH3, 2Hw·2CH3COCH3, 1Hw, 1H and 3H·2CH3CN) one observes both syn and anti-arrangements.
Quantum chemical calculations
In order to explain possible paths of crystallization, structures of [MoO2(HL1–3)(D)]2[Mo6O19] and [{MoO2(HL1–3)}2Mo6O19] were built starting from the crystallographically determined structures, and their geometries were optimized. For each optimized geometry, calculations of harmonic frequencies were performed and thermodynamic properties were computed. Relative differences in standard Gibbs energies of binding (Tables 1 and 2) were calculated using the calculated standard Gibbs energies of formation.
(Tables 1 and 2) were calculated using the calculated standard Gibbs energies of formation.
|
||||||
|---|---|---|---|---|---|---|
| [MoO2(HL1–3)(D)]+ syn-orientation | [MoO2(HL1–3)(D)]+ anti-orientation | |||||
| D | HL1 | HL2 | HL3 | HL1 | HL2 | HL3 |
| CH3CN | 9.22 | 3.31 | 4.24 | 3.46 | 4.17 | 3.75 |
| H2O | 4.86 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| CH3OH | 0.00 | 5.05 | 5.15 | 8.76 | 2.77 | 4.90 |
| CH3COCH3 | 14.28 | 6.80 | 7.58 | 12.26 | 7.46 | 6.99 |
| Experimental | 1Hw | 2Hw·2CH3COCH3 | 1Ha·2CH3COCH3 | 3Hw·2CH3COCH3 | ||

|
||||||
|---|---|---|---|---|---|---|
| [{MoO2(HL1–3)}2(Mo6O19)] syn-orientation | [MoO2(HL1–3)(Mo6O19)] anti-orientation | |||||
| POM donor atom | HL1 | HL2 | HL3 | HL1 | HL2 | HL3 |
| μ2-OPOM | 555.21 | 507.65 | 496.88 | 521.87 | 508.11 | 514.77 |
| Ot-POM (terminal) | 535.94 | 530.90 | 527.98 | 536.42 | 527.50 | 530.88 |
| Experimental | 1H | 3H·2CH3CN | 2H | 3H | ||
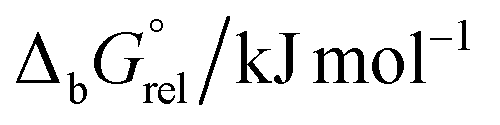
Analysis of calculated structures for [MoO2(HL1)(D)]2[Mo6O19] revealed that the most stable complex should be the one where the metal ion is coordinated with MeOH. This complex has the lowest relative standard Gibbs energy of binding  . However, for other complexes [MoO2(HL2,3)(D)]2[Mo6O19] the complexes with the lowest
. However, for other complexes [MoO2(HL2,3)(D)]2[Mo6O19] the complexes with the lowest  were the ones with coordinated H2O molecule.
were the ones with coordinated H2O molecule.
Depending on the orientation of the pyridyl moieties of the HL− ligand, being oriented anti or syn (Fig. 5), the hybrids have different Gibbs binding energies. By considering the Gibbs binding energy for the structures having syn-orientation, Fig. 5a, in the case of Mo complex containing the ligand HL1, coordination of POM via the terminal oxygen atom is preferred over the bridging one, while in the case of ligands HL2 and HL3, the coordination via the bridging oxygen atom is more stable. Those differences in Gibbs energies amount to 31.1 kJ mol−1. Even so, hybrid 3H·2CH3CN (coordinated through Ot) crystallized depending on the reaction conditions. This could probably be due to the stabilization through the hydrogen bonding implied by the presence of solvent molecules in 3H·2CH3CN and sterical hindrance directly related to the size of the POM. Nevertheless, in the case of the nicotinic hydrazone moiety anti-orientation, shown in Fig. 5b, the POM coordination via the bridging oxygen atom is more favourable. The results are in line with the experimental results for 1H, 2H, and 3H. The strong hydrogen bonds identified in those structures involve terminal oxygen atoms.
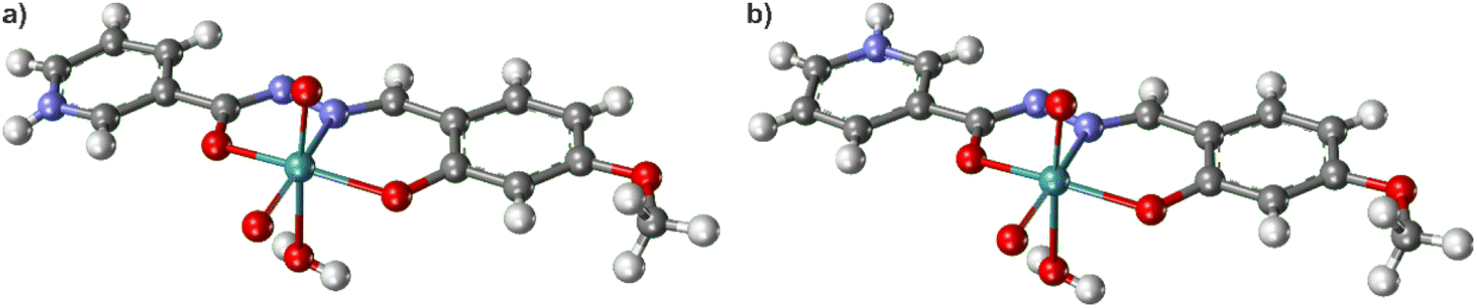 | ||
| Fig. 5 Representation of (a) syn- and (b) anti-orientation of pyridyl moieties relative to coordinating ONO compartment of HL− in the complex cation. | ||
Catalytic studies
Those hybrid species are interesting since they bear two active parts, one with the cationic [MoO2(HL)]+ complex and the other one with the anionic [Mo6O19]2−. The different interaction between both entities could potentially influence their reactivity. To test the catalytic activity of the prepared catalysts in the cyclooctene epoxidation reactions, the following parameters were studied: (i) influence of the oxidant (TBHP in water vs. TBHP in decane) and (ii) addition of the organic solvent (CH3CN) to the reaction media. The purpose of parameter variations was to evaluate the impact of green chemistry principles, based on the use of ecologically more appropriate oxidant (TBHP in water) and no addition of organic solvent to the reaction media, on the catalytic activity of the tested catalysts.Relevant catalytic parameters are summarized in Table 3. As expected, the catalytic results provided by the use of TBHP in decane are the best ones and classify all the prepared catalysts as very active, conversion of cyclooctene >93%, and highly selective towards epoxide, >82% (Table 3 and Fig. S5, see ESI†). This is following the fact that solubility in the organic phase of the MoO2(LH) moiety is favoured and the expected reactivity goes mainly through the transition metal complex. It is worth noting that epoxide can undergo further transformation into the corresponding diol, which is predominantly found in the water phase but also partially in the organic phase. Our analysis via GC primarily targets the organic layer of the reaction mixture, where the substrate, oxidizing agent, and desired product naturally reside. Given that TBHP is utilized in the water phase, the diol would be distributed across both phases, potentially affecting the accuracy of our determinations. Hence, considering the various oxidizing agents tested, our research was tailored specifically towards the epoxide.
| Catalyst | Conversiona/% | Selectivityb (%) | TOFc | TONd |
|---|---|---|---|---|
| a Conversion of cyclooctene = n(cyclooctene converted)/n(cyclooctene engaged) calculated after 6 h.b n(Cyclooctene oxide formed)/n(cyclooctene converted) after 6 h.c TOF = n(cyclooctene transformed)/3 × n(catalyst)/time at 20 minutes. All potential “active species” were considered, i.e. three potential active “sites” per species. The concept of three potential sites was adopted from our earlier published research, which considers two active sites originating from two mononuclear coordination complexes, along with one active site arising from POM.51d TON is defined at the end of the reaction n(cyclooctene transformed)/3 × n(catalyst). | ||||
| Oxidant: TBHP in decane, no organic solvent added | ||||
| 1Ha | 93 | 89 | 952 | 1233 |
| 1Hw | 97 | 87 | 1090 | 1292 |
| 1H | 99 | 82 | 1605 | 1332 |
| 2Hw·2CH3COCH3 | 99 | 85 | 876 | 1325 |
| 2H | 99 | 83 | 1256 | 1329 |
| 2O | 98 | 86 | 850 | 1300 |
| 3Hw·2CH3COCH3 | 99 | 84 | 1441 | 1313 |
| 3H·2CH3CN | 99 | 84 | 6546 | 1329 |
| 3H | 99 | 65 | 1520 | 1321 |
|
||||
| Oxidant: TBHP in water, no organic solvent added | ||||
| 1Ha· | 45 | 54 | 413 | 582 |
| 1Hw | 78 | 95 | 245 | 1032 |
| 1H | 78 | 68 | 800 | 1034 |
| 2Hw·2CH3COCH3 | 87 | 95 | 570 | 1164 |
| 2H | 85 | 75 | 570 | 1121 |
| 2O | 61 | 87 | 408 | 706 |
| 3Hw·2CH3COCH3 | 87 | 84 | 492 | 1157 |
| 3H·2CH3CN | 98 | 96 | 587 | 1309 |
| 3H | 97 | 80 | 494 | 1268 |
|
||||
| Oxidant: TBHP in water, CH3CN added | ||||
| 1Ha· | 41 | 44 | 410 | 548 |
| 1Hw | 69 | 71 | 646 | 926 |
| 1H | 80 | 71 | 264 | 1067 |
| 2Hw·2CH3COCH3 | 63 | 97 | 966 | 836 |
| 2H | 53 | 87 | 568 | 710 |
| 2O | 52 | 59 | 503 | 705 |
| 3Hw·2CH3COCH3 | 65 | 95 | 500 | 879 |
| 3H·2CH3CN | 84 | 84 | 560 | 1119 |
| 3H | 96 | 56 | 512 | 1098 |
The activity of the complex 3H·2CH3CN should be highlighted, Fig. 6 and 7. TOF20min is extremely high (value of 6546), implying very fast and abrupt activation of the complex towards active species. The observation correlates with the results obtained for the theoretical calculations above, where 3H·2CH3CN is less stable than 3H, faster transforming into catalytically active species. For all the catalysts the first 60 min of the reaction were carefully followed and different trends can be observed. For the reactions with the complex 2Hw·2CH3COCH3 induction time could be distinguished, implying slower activation than in the case of other studied catalysts. This could be due to the presence of the coordinated water molecule to molybdenum. The same type of phenomenon was seen in the case of 1Hw, with the slowest activity in the first 20 min of the reaction (Table 3).
 | ||
| Fig. 6 Kinetic profile for cyclooctene conversion when using TBHP in decane as oxidant with the catalysts 3H·2CH3CN (red), 3Hw (blue) and 3H (black). | ||
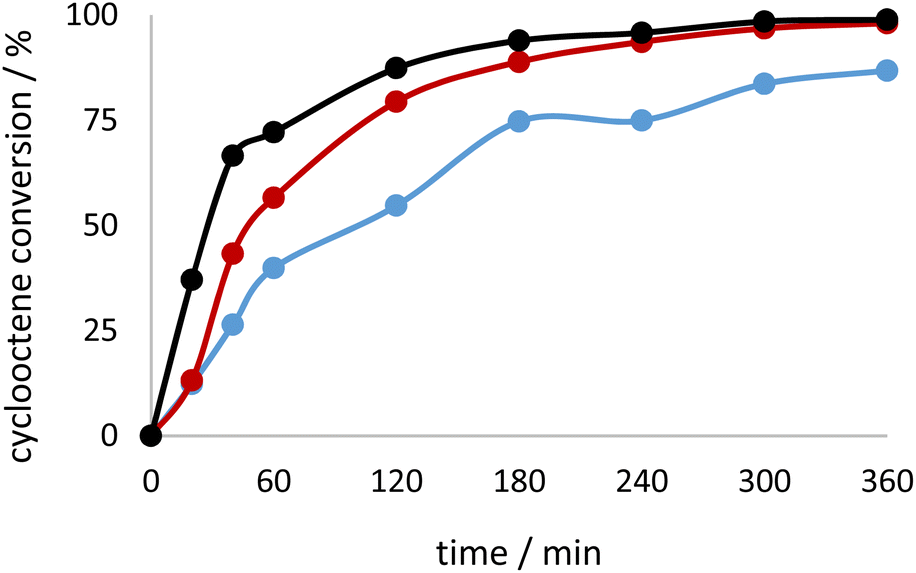 | ||
| Fig. 7 Kinetic profile for cyclooctene conversion when using TBHP in water as oxidant with the catalysts 3H·2CH3CN (red), 3Hw (blue) and 3H (black). | ||
In general, the use of TBHP in water, as environmentally friendly media, resulted in very good activity and high selectivity, for most of the tested catalysts, justifying the green chemistry concept pursued in this research (Fig. 7). The complex 3H, obtained from H2L3 ligand, operates the best under eco-friendly conditions. It can be assumed that the water is promoting epoxide ring opening and the formation of the diol that was not quantified through GC measurements since only the organic layer was analysed. On the other hand, the addition of CH3CN surprisingly inhibited the catalytic activity, but the selectivity towards epoxide did not seem strongly affected. The reverse observation was noted for the tungsten metal complexes coordinated by a similar class of ligands.50
Having in mind that the self-assembly of organic–inorganic POM-based salts in the presence of hydrazone ligands is rare, we compared the catalytic performance of the investigated compounds with the one previously reported, [MoO2(HL)]2Mo6O19·2CH3CN, where HL presents mono-deprotonated ligand obtained by the condensation of pyridoxal and nicotinic acid hydrazide.51 The compound [MoO2(HL)]2Mo6O19·2CH3CN provides the cyclooctene conversion of 72% with selectivity towards epoxide of 58% when using TBHP in water, without adding organic solvent. For the presented investigation, no clear distinction and correlation could be concluded for the nature of ligands (position of OMe group) influencing the catalytic performance of the tested catalysts. Further, all the tested complexes in this research show much better activity and selectivity than the previously tested “referent” system (Bu4N)2Mo6O19,51 justifying the role of [MoO2L] species (conversion parameter 30%, selectivity towards epoxide 53%, TOF 42, TON 123, under the same reaction conditions).
Further comparison was made considering Mo tetranuclear and polynuclear coordination compounds obtained from H2L1–3 ligands (Table S7, see ESI†).26,31 With TBHP in decane conversion was 74% and selectivity towards epoxide 85%, while with TBHP in water, the investigated parameters were much lower (27% and 56%, respectively). In this case, it seems that the presence of polyoxomolybdate anion positively influences the catalytic performance of the tested catalyst, supporting the fact that the Lindqvist-type anion is one active site of the catalyst.
Experimental section
Materials and instruments
The commercially available acetylacetone, nicotinic acid hydrazide, 2-hydroxy-3-methoxybenzaldehyde, 2-hydroxy-4-methoxybenzaldehyde, 2-hydroxy-5-methoxybenzaldehyde, and organic solvents were purchased from Alfa Aesar or Aldrich. Unless otherwise mentioned, all solvents were used without further purification. [MoO2(acac)2] (acac = acetylacetonate),52 and ligands26,47 were synthesised following the previously published procedures. Dry solvents were used for the solvothermal reactions. Acetonitrile was dried by the use of phosphorus pentoxide and then distilled. Acetone was dried with anhydrous potassium carbonate for at least 48 hours and then distilled prior to use. Light was excluded from all reactions using foil to avoid Mo reduction.Attenuated Total Reflectance Infrared (ATR-IR) spectra were recorded at room temperature on a PerkinElmer Spectrum One spectrometer fitted with an ATR reflectance attachment. All data were processed in OMNIC software. NMR data for organic compounds was recorded on a Bruker Avance 400 MHz at room temperature.
Thermal stability was examined by conducting thermogravimetric analysis (TGA) on a Mettler-Toledo TGA/SDTA851 at a heating rate or 5 °C min−1. The samples were heated from 25 °C to 600 °C in an oxygen atmosphere (flow rate: 20 mL min−1) using Al2O3 crucibles. The results were developed by applying the Mettler STARe evaluation software. Elemental analysis (C, H, and N) was determined by the Analytical Services Laboratory of the Ruđer Bošković Institute, Zagreb.
X-ray crystallography
The powder X-ray diffraction data were collected using a Malvern Panalytical Aeris powder diffractometer in the Bragg–Brentano geometry with a PIXcel1D detector, using CuKα radiation (λ = 1.5418 Å). Samples were contained on a Si sample holder. Powder patterns were collected at room temperature (5 < 2Θ < 40°). The data were collected and visualized utilizing the Malvern Panalytical HighScore Software Suite.53High-quality single crystals of 1Ha·2CH3COCH3, 2Hw·2CH3COCH3, 3Hw·2CH3COCH3, 1Hw, 1H, 2H, 2O·2CH3CN·H2O, 3H, 3H·2CH3CN were grown by crystallization from the corresponding solutions. Diffracted intensities were collected on Rigaku XtaLAB Synergy diffractometer equipped with Dualflex source using CuKα radiation (λ = 1.54184 Å) for 1Ha·2CH3COCH3, 3Hw·2CH3COCH3, 2H, 2O·2CH3CN·H2O, 3H and 3H·2CH3CN, as well as MoKα (λ = 0.71073) for 2Hw·2CH3COCH3, 1Hw, 1H and HyPix detector via ω-scans. The crystals were kept at 150 K or 170 K during data collection (see Table S1 in the ESI†). Data were processed using the CrysAlis program package. A summary of general and crystal data, intensity data collection and final refinement parameters are presented in Table S5 and S6 (see ESI†). The structures were solved with dual space methods using SHELXT. The refinement procedure by full-matrix least-squares methods based on F2 values against all reflections included anisotropic displacement parameters for all non-H atoms. Hydrogen atoms bound to carbon atoms were placed in geometrically idealized positions and refined by the use of the riding model with Uiso = 1.2Ueq of the connected carbon atom or as ideal CH3 groups with Uiso = 1.5Ueq. Acetonitrile molecules in 2O·2CH3CN·H2O are heavily disordered and for one of them hydrogen atoms of the methyl group could not be satisfactory modelled and were thus omitted form the model. In the structure of 1Ha·2CH3COCH3 acetone molecule in the crystal lattice was disordered over two positions with equal occupancies 0.5. Hydrogen atoms attached to heteroatoms were located in the difference Fourier maps at the final stages of the refinement procedure except for one of the coordinated ligands in 2O·2CH3CN·H2O, where hydrogen atom on the N2 was ascribed based on geometrical considerations and the charge, while considering also the most reasonable hydrogen bond geometry. All refinements were performed using SHELXL, which was operated within the Olex2 suite. Geometrical calculations and molecular graphics were done with Mercury and Platon.54,55
Catalytic studies
The Agilent 7820A chromatograph, outfitted with a Flame Ionization Detector (FID) and an HP5-MS capillary column (dimensions: 30 m × 0.32 mm × 0.25 μm), was utilized for chromatogram production. Parameters for the gas chromatography (GC) analysis were established through the examination of genuine reactant and product samples. The analysis focused on quantifying the transformation of olefins into their respective epoxides, employing calibration curves for precise measurement. Acetophenone served as the internal standard throughout this process, ensuring accuracy in quantification.Quantum chemical calculations
Optimizations of geometries for all computed complexes were performed using the hybrid functional B3LYP56 with the D3 version of Grimme's dispersion57 in combination with the Def2TZVP58,59 basis set. Initial geometries were taken from crystallographically determined structures. To confirm that the obtained geometries were local minima, harmonic frequency calculations were performed.60,61 The standard Gibbs energies of formation were calculated at T = 298.15 K and p = 101325 Pa. Strengths of the intramolecular interactions were estimated by the calculation of relative differences in the standard Gibbs energies of binding  according to the formula:
according to the formula: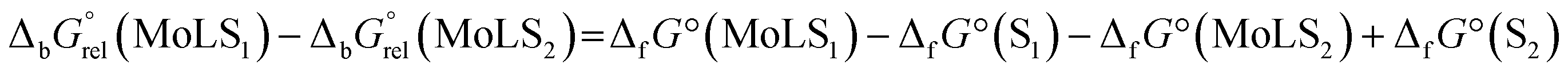
All quantum chemical calculations were carried out using the Gaussian 16 program package.62
759, 730 (MoOb), 600 (δ(ObMoOt)).Mo–O), 734, 689, 664, 649, 642 (MoOb), 555 (δ(ObMoOt)).Mo–O), 782754, 731 (MoOb), 577 (δ(ObMoOt)).Mo–O), 761, 729 (MoOb), 558 (δ(ObMoOt)).Mo–O), 764, 722 (MoOb), 573 (δ(ObMoOt)).General procedure for the epoxidation of cyclooctene using TBHP (in water or decane)
In a typical reaction, cyclooctene (2.76 mL, 20 mmol), aqueous TBHP (70% w/w, 40 mmol) or TBHP in decane (5.5 mol dm−3, 40 mmol) and acetophenone (0.1 mL) and molybdenum (pre)catalyst, 0.005 mmol, were used, and the reaction mixture was stirred for 5 h at 80 °C. The reaction was monitored by withdrawing small aliquots at certain time intervals and analyzing them by GC.General procedure for the epoxidation of cyclooctene using TBHP in water with the addition of CH3CN
The procedure was the same as without CH3CN, except the 2 mL of CH3CN was added to the reaction mixture before the catalyst was added.Conclusions
Mo-POM organic–inorganic hybrids were successfully prepared by conventional and solvothermal methods. In the Class I hybrids [MoO2(HL1–3)(D)]2[Mo6O19], POM anions tend to associate with cationic Mo-complexes via multiple non-covalent interactions. However, in Class II architectures [{MoO2(HL1–3)}2Mo6O19], POM serves as a sterically hindered ligand and completes the coordination sphere of two Mo atoms from peripheral coordination subunits either through the terminal or bridging oxygen atoms. 2H and 3H are the first examples of polyoxometalates that incorporate bridging POM coordinated architectures. The preference of Mo-complex cations for coordination with solvent and terminal or bridging oxygen groups on the POM moiety was estimated by calculation of complexes' relative standard Gibbs energies of binding. Density functional theory was utilized for the calculation of standard Gibbs energies of formation, which were used for the estimation of binding energies. In most of the complexes, there is a clear preference for coordination with water molecules, whereas the bridging oxygen group of the POM is preferred in coordination with POM moiety.The assembly process is additionally sensitive to the ligand's methoxy group position. Thus, the condensation with H2L2 resulted in the unsymmetrical octamolybdate-based hybrid [{MoO2(H2L2)}{MoO2(HL2)}Mo8O26{MoO2(HL2)}], covalently expanded by three Mo–hydrazone complex units. All Class II hybrids have long bonds Mo–OPOM due to sterical hindrance.
In this study, the catalytic activity of all compounds was investigated and compared. With TBHP in decane as an oxidant, all the prepared catalysts showed excellent catalytic activity towards the cyclooctene oxidation, where >90% conversion and >80% selectivity could be obtained. Notably, 3H·2CH3CN catalyst operates the best under eco-friendly conditions. The addition of CH3CN surprisingly inhibited the catalytic activity when TBHP in water is used as oxidant, but the selectivity towards epoxide remained almost the same.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for 1Ha·2CH3COCH3, 1Hw, 1H, 2Hw·2CH3COCH3, 2H, 3Hw·2CH3COCH3, 2O·CH3CN·H2O, 3H, 3H·2CH3CN has been deposited at the Cambridge Structural Data base under with deposition numbers CCDC 2335829–2335837.Author contributions
Jana Pisk: investigation, supervision, formal analysis, writing – original draft preparation. Mirna Mandarić: investigation and formal analysis. Tomica Hrenar: quantum chemical calculations, data curation, and writing – original draft, visualization. Dominique Agustin: supervision, review and editing. Mirta Rubčić: investigation, writing – original draft preparation, review and editing. Višnja Vrdoljak: conceptualization, supervision, investigation, writing – original draft preparation, visualization, review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been fully supported by the Croatian Science Foundation under the project (IP-2016-06-4221). We acknowledge the support of project CIuK co-financed by the Croatian Government and the European Union through the European Regional Development Fund–Competitiveness and Cohesion Operational Programme (Grant KK.01.1.1.02.0016). LCC CNRS and Chemistry Dept of IUT Paul Sabatier are acknowledged for providing the equipment for catalysis experiments. We thank Matea Pajski and Silvija Mrkonja for their technical help with catalytic studies.Notes and references
- J. J. Walsh, A. M. Bond, R. J. Forster and T. E. Keyes, Coord. Chem. Rev., 2016, 306, 217 CrossRef CAS.
- Y. Dong, Y. Yang, L. Qiu, G. Dong, D. Xia, X. Liu, M. Li and R. Fan, ACS Appl. Energy Mater., 2019, 2, 4224 CrossRef CAS.
- A. Bijelic, M. Aureliano and A. Rompel, Angew. Chem., Int. Ed., 2019, 58, 2980 CrossRef CAS PubMed.
- L. S. van Rompuy and T. N. Parac-Vogt, Curr. Opin. Biotechnol., 2019, 58, 92 CrossRef CAS PubMed.
- E. Coronado, C. Gimenez-Saiz, C. J. Gomez-Garcia and S. C. Capelli, Angew. Chem., Int. Ed., 2004, 43, 3022 CrossRef CAS PubMed.
- S.-S. Wang and G.-Y. Yang, Chem. Rev., 2019, 115, 4893 CrossRef PubMed.
- S. Roy, V. Vemuri, S. Maiti, K. S. Manoj, U. Subbarao and S. C. Peter, Inorg. Chem., 2018, 57, 12078 CrossRef CAS PubMed.
- K. Kamata, K. Yonehara, Y. Sumida, K. Yamaguchi, S. Hikichi and N. Mizuno, Science, 2003, 300, 964 CrossRef CAS PubMed.
- N. Mizuno, K. Yamaguchi and K. Kamata, Coord. Chem. Rev., 2005, 249, 1944 CrossRef CAS.
- L. Salles, J.-Y. Piquemal, R. Thouvenot, C. Minot and J.-M. Brégeault, J. Mol. Catal. A: Chem., 1997, 117, 375 CrossRef CAS.
- L. Jing, J. Shi, F. Zhang, Y. Zhong and W. Zhu, Ind. Eng. Chem. Res., 2013, 52, 10095 CrossRef CAS.
- D. Sloboda-Rozner, P. L. Alsters and R. Neumann, J. Am. Chem. Soc., 2003, 125, 5280 CrossRef PubMed.
- N. Mizuno and M. Misono, Chem. Rev., 1998, 98, 199 CrossRef CAS PubMed.
- Y. M. A. Yamada, H. Tabata, M. Ichinohe, H. Takahashi and S. Ikegami, Tetrahedron, 2004, 60, 4087 CrossRef CAS.
- D. Schaming, C. Allain, R. Farha, M. Goldmann, S. Lobstein, A. Giraudeau, B. Hasenknopf and L. Ruhlmann, Langmuir, 2010, 26, 5101 CrossRef CAS PubMed.
- Y. Wang, X. Liu, W. Xu, Y. Yue, B. Li and L. Wu, Inorg. Chem., 2017, 56, 7019 CrossRef CAS PubMed.
- H. Karoui and C. Ritchie, New J. Chem., 2018, 42, 25 RSC.
- D. Schaming, C. Costa-Coquelard, I. Lampre, S. Sorgues, M. Erard, X. Liu, J. Liu, L. Sun, J. Canny, R. Thouvenot and L. Ruhlmann, Inorg. Chim. Acta, 2010, 363, 2185 CrossRef CAS.
- H. Kwen, V. G. Young Junior and E. A. Maatta, Angew. Chem., Int. Ed., 1999, 38, 1145 CrossRef CAS PubMed.
- S. Schönweiz, M. Heiland, M. Anjass, T. Jacob, S. Rau and C. Streb, Eur. J. Chem., 2017, 23, 15370 CrossRef PubMed.
- N. K. Mishra, D. Bansal and S. Supriya, ACS Omega, 2022, 7(35), 31403 CrossRef CAS PubMed.
- C. Yao, L.-K. Yan, W. Guan, C.-G. Liu, P. Song and Z.-M. Su, Dalton Trans., 2010, 39, 7645 RSC.
- R. M. Smith, I. Colliard, M. Amiri, M. A. Galindo and M. Nyman, Cryst. Growth Des., 2022, 22, 2294 CrossRef CAS.
- A. Yokoyama, T. Kojima, K. Ohkubo, M. Shiro and S. Fukuzumi, J. Phys. Chem. A, 2011, 115, 986 CrossRef CAS PubMed.
- J.-H. Li, X.-L. Wang, G. Song, H.-Y. Lin, X. Wang and G.-C. Liu, Dalton Trans., 2020, 49, 1265 RSC.
- M. Mandarić, E. Topić, D. Agustin, J. Pisk and V. Vrdoljak, Int. J. Mol. Sci., 2024, 25(3), 1503 CrossRef PubMed.
- J. Pisk, D. Agustin, V. Vrdoljak and R. Poli, Adv. Synth. Catal., 2011, 353, 2910 CrossRef CAS.
- R. Bikas, V. Lippolis, N. Noshiranzadeh, H. Farzaneh-Bonab, A. J. Blake, M. Siczek, H. Hosseini-Monfared and T. Lis, Eur. J. Inorg. Chem., 2017, 6, 999 CrossRef.
- D. Biswal, N. R. Pramanik, S. Chakrabati, M. G. B. Drew, B. Sarkar, M. R. Maurya, S. K. Mukherjee and P. Chowdhury, New J. Chem., 2017, 41, 4116 RSC.
- M. R. Maurya, L. Rana and F. Avecilla, Inorganica Chim. Acta, 2015, 429, 138 CrossRef CAS.
- V. Vrdoljak, M. Mandarić, T. Hrenar, I. Đilović, J. Pisk, G. Pavlović, M. Cindrić and D. Agustin, Cryst. Growth Des., 2019, 19, 3000 CrossRef CAS.
- D. Cvijanović, J. Pisk, G. Pavlović, D. Šišak-Jung, D. Matković-Čalogović, M. Cindrić, D. Agustin and V. Vrdoljak, New J. Chem., 2019, 43, 1791 RSC.
- J. Pisk, M. Rubčić, D. Kuzman, M. Cindrić, D. Agustin and V. Vrdoljak, New J. Chem., 2019, 43, 5531 RSC.
- J. Pisk, D. Agustin and V. Vrdoljak, Catal. Commun., 2020, 142, 106027 CrossRef CAS.
- V. Vrdoljak, B. Prugovečki, D. Matković-Čalogović and J. Pisk, CrystEngComm, 2011, 13, 4382 RSC.
- V. Vrdoljak, B. Prugovečki, D. Matković-Čalogović, R. Dreos, P. Siega and C. Tavagnacco, Cryst. Growth Des., 2010, 10, 1373 CrossRef CAS.
- A. Dolbecq, A. Guirauden, M. Fourmigué, K. Boubekeur, P. Batail, M.-M. Rohmer, M. Bénard, C. Coulon, M. Sallé and P. Blanchard, J. Chem. Soc., Dalton Trans., 1999, 1241 RSC.
- B. Gai, H. He, Y. Zhao, Z. Mao and H. Fu, Chem. Res. Chin. Univ., 2016, 32, 527 CrossRef CAS.
- S. Wang, Z. Sun, C. Zhang, L. Ni, C. Wang, Y. Gao, L. Lv, J. Chang and W. Hao, Inorg. Chem. Commun., 2014, 41, 47 CrossRef CAS.
- Y. Wang, Z. Zhang, E. Wang, Y. Qi and S. Chang, Aust. J. Chem., 2008, 61, 874 CrossRef CAS.
- M. Yuan, Y. Li, E. Wang, C. Tian, L. Wang, C. Hu, N. Hu and H. Jia, Inorg. Chem., 2003, 42, 3670 CrossRef CAS PubMed.
- H. Jin, Y. Qi, E. Wang, Y. Li, X. Wang, C. Qin and S. Chang, Cryst. Growth Des., 2006, 6, 2693 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, The Cambridge Structural Database, Acta Crystallogr., 2016, B72, 171 Search PubMed.
- E. Topić, I. Landripet, M. Duguin, J. Pisk, I. Đilović, V. Vrdoljak and M. Rubčić, New J. Chem., 2020, 44, 13357 RSC.
- N. Bebić, E. Topić, M. Mandarić, T. Hrenar and V. Vrdoljak, CrystEngComm, 2021, 23, 6349 RSC.
- M. R. Maurya, S. Dhaka and F. Avecilla, Polyhedron, 2014, 67, 145–215 CrossRef CAS.
- J. Pisk, T. Hrenar, M. Rubčić, G. Pavlović, V. Damjanović, J. Lovrić, M. Cindrić and V. Vrdoljak, CrystEngComm, 2018, 20, 1804 RSC.
- A. J. Bridgeman and G. Cavigliasso, Chem. Phys., 2002, 279, 143 CrossRef CAS.
- B. Modec, J. V. Brenčič and J. Zubieta, J. Chem. Soc., Dalton Trans., 2002, 1500 RSC.
- V. Vrdoljak, J. Pisk, B. Prugovečki, D. Agustin, P. Novak and D. Matković- Čalogović, RSC Adv., 2016, 6, 36384 RSC.
- J. Pisk, B. Prugovečki, D. Matković-Čalogović, T. Jednačak, P. Novak, D. Agustin and V. Vrdoljak, RSC Adv., 2014, 4, 39000 RSC.
- J.-J. Chen, J. W. McDonald and W. E. Newton, Inorg. Chem., 1976, 15, 2612 CrossRef.
- T. Degen, M. Sadki, E. Bron, U. König and G. Nénert, The HighScore Suite, Powder Diffr., 2014, 29, S13 CrossRef CAS.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226 CrossRef CAS PubMed.
- A. L. Spek, Acta Crystallogr., 2009, D65, 148 CrossRef PubMed.
- (a) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 37, 785 CrossRef PubMed; (b) R. Peverati and D. G. Truhlar, Phys. Chem. Chem. Phys., 2021, 14, 16187 RSC.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC.
- T. Hrenar, I. Primožič, D. Fijan and M. Majerić Elenkov, Phys. Chem. Chem. Phys., 2017, 19, 31706 RSC.
- I. Primožič, T. Hrenar, K. Baumann, L. Krišto, I. Križić and S. Tomić, Croat. Chem. Acta, 2014, 87, 153 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: (1) Powder diffraction patterns, (2) ATR-IR spectra, (3) TGA curves, (3) additional figures for compounds, (4) tables of selected bond distances and angles and of hydrogen bonds parameters. Crystallographic data sets for the structures 1Ha·2CH3COCH3, 1Hw, 1H, 2Hw·2CH3COCH3, 2H, 3Hw·2CH3COCH3, 2O·CH3CN·H2O, 3H, 3H·2CH3CN. CCDC 2335829–2335837. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra03563a |
| This journal is © The Royal Society of Chemistry 2024 |