
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular simulation of CO production and adsorption in a coal–kaolinite composite gangue slit model†
Jing Zhangabcd,
Zhi Li *a,
Xuping Li*abcd,
Xiaopeng Rena,
Chenhong Zhoua and
Tianyu Lia
*a,
Xuping Li*abcd,
Xiaopeng Rena,
Chenhong Zhoua and
Tianyu Lia
aSchool of Mining and Coal, Inner Mongolia University of Science and Technology, China. E-mail: lizhi7422@qq.com; nkdlxp@163.com
bInner Mongolia Key Laboratory of Mining Engineering, China
cInner Mongolia Research Center for Coal Safety Mining and Utilization Engineering and Technology, China
dInner Mongolia Cooperative Innovation Center for Coal Green Mining and Green Utilization, China
First published on 17th June 2024
Abstract
To reveal the mechanism of CO gas generation and adsorption in coal gangue slits at the microscopic level, a new composite kaolinite–coal–kaolinite (KCK) slit model was constructed by combining the Hongqingliang (HQL) coal molecular model and the Bish kaolinite model to characterize the crack structure of the gangue. It is compared with the kaolinite model (TriK) commonly used in gangue research. Molecular dynamics was used to study the production of CO in different oxygen environments and variation in the adsorption amount, adsorption sites and diffusion coefficient in the temperature range from 293.15 K to 333.15 K. The results indicate that CO mainly comes from the decomposition of ether and phenol in organic structures, and the lower the oxygen concentration, the lesser the CO production time. The KCK model has a higher average adsorption capacity and weaker diffusion capacity mainly due to the additional adsorption sites provided by the carbon-containing structural layer, and CO is mainly adsorbed near the oxygen-containing functional groups. Although kaolinite exhibits bonding adsorption on the Al–O plane, its adsorption site is limited to the surface. The slit model with the carbon structure can better reflect the complex conditions of gas motion in the gangue, thus providing a reference to determine the spontaneous combustion conditions of the gangue hill via the index gas.
1 Introduction
Coal gangue is a solid waste produced during coal mining and washing, consisting mainly of clay ore, quartz, pyrite and residual crushed coal, among others. It has a low caloric value and exhibits flammability, and a large amount of accumulated gangue hills are prone to spontaneous combustion, posing harm to the surrounding environment.1–3 The spontaneous combustion process of coal gangue will release CO gas, which is a common index gas produced by spontaneous combustion and is of great significance for monitoring the spontaneous combustion area and oxidation of gangue hills.4 However, coal gangue also has a certain gas adsorption capacity, which may lead to the misinterpretation of monitoring results. At present, the research on coal gangue index gas focuses on macroscopic experimental analysis and molecular simulation methods that simply replace coal gangue with kaolinite, which makes it difficult to accurately analyze the mechanism of CO index gas production and adsorption at the microscopic level.5,6 Therefore, it is of great practical significance to comprehensively study the microscopic mechanism of CO production by coal gangue using a new composite macromolecular model.To study the mechanism of index gas production via the spontaneous combustion of coal gangues, many scholars have carried out experimental or simulation research. Zhang7 studied the pore and thermodynamic properties of coal gangue immersed in water at different times through adsorption, infrared spectroscopy and thermogravimetric analysis and found that soaking in water would lead to the oxidation of pyrite inside the gangue, which would significantly promote the oxidation of gangue and the expansion of internal fissures, and enhance the adsorption of O2 by the gangue. Gao8 established a coal macromolecule model through experiments and fully verified the applicability of reactive force field (ReaxFF) in simulated coal pyrolysis using thermogravimetric analysis. Zhao9 used Grand Canonical Monte Carlo (GCMC) to simulate the effect of pore structures on the adsorption of water using a low-rank coal model and found that the adsorption of water in coal is positively correlated with fugacity, porosity and other factors. Sui10 studied the simulation of adsorption of methane, CO2 and other gases by modified silica, and found that different functional groups had great differences in the priority of gas adsorption. Through molecular simulation, Wang11 found that the adsorption amount of H2 is positively correlated with the coal rank and affected by the chemical properties and pores of the coal surface. Wang12,13 used a double-layer kaolinite model instead of a coal gangue to study the adsorption behavior of the gangue. They also found that a gangue with a high moisture content has a good effect on oxygen adsorption, while a gangue with a pressure below 10 MPa has a significant change in the system energy for oxygen adsorption through gangue slits, and based on this, the process of oxygen adsorption and seepage in the gangue is divided into four stages, which shows that the spontaneous combustion of the coal gangue can be controlled by controlling the pressure.
It can be seen that scholars have made certain achievements in the study of gas generation and adsorption characteristics of coal and gangues, and the application of molecular dynamics simulation method in coal is relatively mature. However, the research results of the coal gangue are relatively simple, and most of them are macro experimental research or kaolinite simulation research, ignoring the synergistic effect of the carbon content and kaolinite on the gas adsorption and spontaneous combustion of gangue. Therefore, we propose to use molecular dynamics methods to develop a new model for coal gangue slits, in which coal molecules from the coal mine where the gangue is collected replace the carbon-containing components in the gangue. The mechanism of CO generation in the gangue combustion process was studied, and the adsorption and diffusion of CO gas under different pressure and temperature conditions were simulated to analyze the adsorption characteristics and gaps among various components, so as to explore the basic characteristics of index gas adsorption and spontaneous combustion of coal gangue.
2 Methodology and theory
2.1 Simulation theory
ReaxFF, GCMC and molecular dynamics were used in the simulation. ReaxFF proposed by van Duin et al.14 is a molecular force field based on bond order, which can simulate the formation and fracture of chemical bonds on the basis of molecular dynamics, so as to simulate the reaction of macromolecular systems. Many cases have proved that this force field can well simulate the oxidation and pyrolysis reactions of organic macromolecules.15,16 The formula for calculating system potential energy under the ReaxFF force field is shown in (1):| Esystem = Ebond + Eover + Eunder + Eval + Epen + Etors + Econj + EvdWaals + ECoulomb | (1) |
Esystem is the potential energy of the system, which describes the interaction between particles. It is divided into bond potential energy and non-bond potential energy. Ebond, Eover, Eunder, Eval, Epen, Etors, Econj, EvdWalls and ECoulombis represent bond energy, overcoordination term, undercoordination term, valence angle term, penalty energy term, torsion angle term, conjugation term, van der Waals energy and coulombic interaction energy respectively.17
Monte Carlo method was proposed in the 1940s, is a numerical calculation method using random numbers to solve problems, and can be used for adsorption in porous media.18 In order to characterize the mathematical relationship between adsorption capacity and adsorption conditions and analyze the microscopic mechanism of gas adsorption, The Freundlich adsorption equation was used for characterization, and its expression is shown as eqn (2):
| y = axbx(−c) | (2) |
Other than that, to better reveal the diffusion law of CO in the coal gangue and to assess the diffusivity of CO in coal gangue slit structures, the MSD and Einstein equations have been used to calculate the self-diffusion coefficient of CO in coal gangue models. This diffusion property is closely related to the type of material, diffusion medium and applied conditions.22 Einstein's equation is shown in eqn (3):
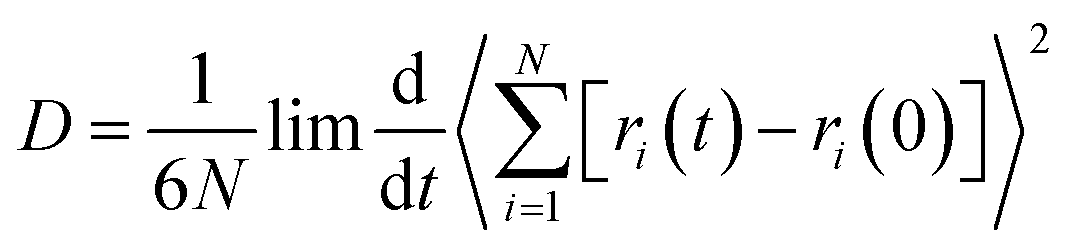 | (3) |
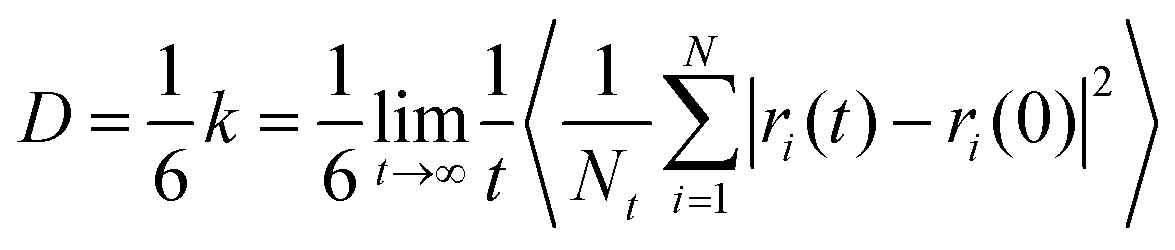 | (4) |
2.2 Establishment of the gangue slit model
The main components of coal gangue are about 70% kaolinite (clay mineral), about 15% carbon and other small amounts of impurities such as Fe2O3, CaO and so on.23 Kaolinite has a periodic crystal structure, while fixed carbon in the gangue exists in the form of crushed coal and less metamorphic carbon. In order to fully reflect the complex composition and a large number of microporous cracks in the coal gangue, and also take into account the possible adsorption difference between the two different surfaces of kaolinite crystals, Ordos HQL lignite coal molecules24 were selected to represent fixed carbon components and Bish kaolinite crystals to represent clay mineral components. The three-layer macromolecular structure of kaolinite–coal–kaolinite (KCK) was established. Because the degree of coal metamorphism is low, and the sulfur content is very low, it is relatively representative. The coal molecular formula is C140H118N2O19, and the kaolinite molecular formula is Al4[Si4O10](OH)8.Since the adsorbates in the simulations contain both organic coal macromolecules and inorganic clay mineral structures, COMPASS II was used as the simulated force field, which was proposed by Huai Sun and is a modified model of the COMPASS force field belonging to an ab initio parametric force field. After many studies, it has been proved that it is mainly applicable to the atomic simulation of organic molecules, inorganic small molecules and polymers, and is a widely applicable all-molecular general force field.25,26 The Materials Studio software was used to build the model. First, the Forcite module was used to optimize the geometry, energy, molecular dynamics, and annealing of the coal molecules, and the charge was calculated using Qeq. Then the amorphous cell module was used to construct the basic plate structure of coal, 18 coal molecules were set, the target density was 1.5 g cm−3, COMPASS II force field was selected, the temperature was 298 K, the calculation time was 100 ps, and the Ewald method (accuracy 10−4) was selected for the coulombic force calculation. The atom-based method was used to calculate the van der Waals forces, and the cutoff radius was 1.2 nm. A long slab of the coal molecular cell was constructed as a layer containing a carbon structure to ensure that the various functional groups on the coal molecules were in full contact with the environment. It was then optimized again with Forcite to ensure that it was in its lowest energy configuration. The (0 0 1) surface and 8 × 16 × 1 supercell were constructed for kaolinite crystal cells to enlarge the surface and optimize it. Finally, a new KCK slit structure was constructed using the build-layer command, so that the two sides of the carbon-containing structure layer faced the alumino–oxygen (Al–O) and silico–oxygen (Si–O) planes of kaolinites, respectively. Studies have shown that micropores below 2 nm in structures such as coal and gangue provide adsorption and storage space for most of the gas.27,28 Therefore, a 2 nm vacuum layer was set between the model layers. After these steps, a new three-layer large cell structure a = 41.192 Å, b = 142.944 Å, c = 90.659 Å, α = 90.8°, β = 96.2°, γ = 89.7° was constructed, as shown in Fig. 1. At the same time, in order to compare the adsorption capacity and diffusion capacity of CO, a triple-layer model of kaolinite (TriK) with the same pore size as the above-mentioned model was established, which is the focus of many scholars studying the coal gangue.29,30
 | ||
| Fig. 1 Coal and gangue slit model used in simulation: (a) optimized model of HQL coal, (b) kaolinite crystal model, (c) KCK slit model, and (d) TriK slit model. | ||
2.3 Simulation details
The ReaxFF module of LAMMPS was used to calculate the gas production mechanism. Since kaolinite does not contain a C element, this part mainly studies the mechanism and influence of the carbon-containing structure layer on CO gas production. In order to prevent the error caused by atomic overlap and take into account the calculation speed, the reaction kinetics simulation selects 3 HQL molecules to construct the reaction box. The air flow in the deep part of the waste heap is small, and the gangue is usually in a negative oxygen state during spontaneous combustion.31 Therefore, oxygen molecules with an equivalent ratio (which is the ratio of oxygen required for complete oxidation combustion of coal molecules to actual oxygen32) of coal to oxygen of 1–4 were added to the reaction box respectively, and the Berendsen constant temperature hot bath method was used for relaxation at 300 K. The force field file is HCONSB.ff,33 and the time step is 0.25 fs. The model density is 0.5 g cm−3. It then heats up to 5500 K in the NVT ensemble runs 90 ps, and maintains the temperature for the last 30 ps to stabilize the product.The sorption module of Materials Studio was used to calculate the adsorption. First, the energy minimization optimization of the gas molecular model was carried out, the task of fixed pressure was selected, the calculation method was Metropolis, the temperature was set at 293.15–333.15 K, and the fugacity value was calculated using the Peng–Robinson conversion equation.34 The conversion relationship between the fugacity coefficient and the pressure of CO is shown in Fig. 2. The pressure was set at 0–10 MPa, a gradient was set for every 10 K and 1 MPa, and a normal pressure of 0.1 MPa was set separately. Single-component CO gas was injected into the system, and the production time was 100 ps. The simulation force field and charge calculation, van der Waals force calculation, cutoff radius and other settings were followed when the model was constructed (Section 2.2). As the unit of adsorption obtained by the Sorption module is moles per uc, which represents the number of CO molecules in a unit cell, eqn (5) was used to convert the unit into mmol g−1 when processing the data as follows:
 | (5) |
 | ||
| Fig. 2 Relation between the fugacity coefficient and pressure of CO. | ||
In the part of the diffusion coefficient calculation, the adsorption site configurations at various temperatures and pressure points obtained from the locate task are used for the Forcite module calculation. First, geometric optimization, annealing and molecular dynamics optimization were performed for each configuration, and NVT and NVE ensembles were optimized. Finally, the output trajectory of the last 50 ps was analyzed.
3 Results and discussion
3.1 Mechanism analysis of CO production
After the simulation of LAMMPS, the output oxidation products were statistically analyzed, and the changes in CO products with different equivalence ratios along with the reaction time were obtained, as shown in Fig. 3. | ||
| Fig. 3 Rule of CO production under different coal–oxygen equivalent ratio conditions. | ||
It can be seen from the figure that the overall CO production increases with the increase in oxygen content under the condition of negative oxygen. The variation trend of CO during the warming process is consistent with the results of the experimental study of Zhang et al.,35 which proves the rationality of the simulation results. When the equivalent ratio of moles is 1, CO molecules are first produced at the position of 71.77 ps and the peak is 35 moles per uc, while when the equivalent ratio is 4, CO molecules are produced at 47.07 ps and the peak is 11 moles per uc. It can be seen that as the number of oxygen molecules decreases, the generation time of CO products is significantly earlier, and the peak increases continuously. In addition, the CO products at the end of the reaction were gradually reduced under the condition of high equivalence ratios.
Combining the picture and output files to analyze the product generation process, it can be found that the initial reaction is mainly chemical adsorption of oxygen, and some molecular fragments fall off, such as hydroxyl groups and carboxyl groups. However, as the temperature increases and the reaction intensifies, the carbon chain breaks over a wide range of scales, and a large number of ether and phenolic structural cracks result in increased CO production. In addition, part of the CO2 produced by early decarboxylation and methyl oxidation will bind to C and transform into CO as the temperature increases (Fig. 4). However, under conditions of high equivalent ratios, oxygen is insufficient and the carbon structure is difficult to fully oxidize to produce CO2, so the production of CO is more advanced and the amount produced is not high. Later in the reaction, when the reaction time is sufficient, CO participates in the hydrogenation reaction or is completely oxidized to CO2, resulting in a decrease in the CO product at the end of the reaction. It can be predicted that if the reaction time is prolonged, the CO product will gradually decrease at low equivalence ratios.
 | ||
| Fig. 4 Main path of CO generation in HQL coal gangue. | ||
Spontaneous combustion of the coal gangue mainly due to the combustibility of carbon-containing components and kaolinite plays a relatively minor role in the chemical reaction process that produces CO from the spontaneous combustion of the coal gangue. Kaolinite will shed its hydroxyl group at a high temperature above 1200 K,36 which slightly promotes the oxidation of coal. However, the gas adsorption capacity of kaolinite may affect the spontaneous combustion of gangues.13,37 It can be seen that the amount of CO produced by the spontaneous combustion of gangues is large, and the characteristics are obvious in the oxygen-poor state inside the gangue hill. As an indicator gas for monitoring spontaneous combustion, further studies of its adsorption and diffusion are of strong practical interest.
3.2 Adsorbing capacity
Based on the average adsorption capacity of all conformations at each temperature and pressure point, the adsorption data of CO at temperatures of 293.15 K, 303.15 K, 313.15 K, 323.15 K, and 333.15 K and pressures of 0–10 MPa were obtained in the slit models, and the Freundlich adsorption equation was used to fit. The obtained adsorption isotherm of the KCK slit model for CO adsorption at different temperatures is shown in Fig. 5(a), and the adsorption isotherm of the TriK slit model is shown in Fig. 5(b). | ||
| Fig. 5 Adsorption isotherms of CO at different temperatures: (a) adsorption capacity of the KCK model and (b) adsorption capacity of the TriK model. | ||
The simulated equivalent adsorption heat of each point is less than 42 kJ mol−1, which belongs to physical adsorption.38 It can be seen from Fig. 5(a and b) that the adsorption isotherms of the KCK model and the TriK model show very similar rules for the adsorption of CO molecules. With the increase in pressure, the adsorption capacity of CO molecules in the two models increases, and the growth rate gradually slows down, which conforms to the Freundlich law and belongs to type I isotherm.39 This is because during the initial stages of adsorption, the CO gas molecules quickly occupy the preferred adsorption sites in the adsorbent molecules, forming a stable adsorption system, and the preferentially occupied adsorption sites tend to saturate, leading to a slower adsorption rate. At the same pressure, however, the adsorption capacity decreases with the increase in temperature. Due to the intensification of Brownian motion between molecules of CO gas, the molecular kinetic energy breaks through the adsorption of the active surface, which makes it difficult to be captured and easy to desorption. Therefore, high temperatures are not conducive to adsorption. However, it can also be seen from the figure that compared with the TriK model, the KCK model has a faster growth rate of adsorption capacity at each temperature, and the adsorption capacity at each point of the latter is significantly lower and the curve is more gentle. It can be found that the adsorption capacity of the KCK model is 110.4–132.9% that of TriK model.
As can be seen from the Freundlich fitting parameters in Table 1, since the molecular thermal motion increases with the increase in temperature, coefficient a gradually decreases with the increase in temperature, and the decreasing trend of the KCK model is more obvious. Coefficient b also shows an upward trend. They directly reflect that the adsorption capacity and adsorption rate of the two models decrease with the increase in temperature, which hinders adsorption. The KCK model has stronger ultimate adsorption capacity and greater pressure required for saturation adsorption, so it shows a more obvious changing trend.
| Temperature K | KCK model | TriK model | ||||
|---|---|---|---|---|---|---|
| a (mL g−1) | b (MPa−1) | R2 | a (mL g−1) | b (MPa−1) | R2 | |
| 293.15 | 2.58 | 0.80 | 0.9992 | 2.20 | 0.85 | 0.9991 |
| 303.15 | 2.29 | 0.83 | 0.9990 | 1.76 | 0.94 | 0.9995 |
| 313.15 | 2.07 | 0.90 | 0.9982 | 1.52 | 0.95 | 0.9985 |
| 323.15 | 1.93 | 0.91 | 0.9989 | 1.97 | 0.90 | 0.9985 |
| 333.15 | 1.80 | 0.94 | 0.9996 | 1.55 | 1.04 | 0.9992 |
It can be seen from the above that compared with the common TriK model, the adsorption law of the KCK model does not change greatly due to the addition of carbon-containing structure layers. However, the free volume of this model is larger, and slits and pores between carbon chains bring more space for storing adsorbed gas and active adsorption sites, so the overall adsorption capacity is higher. The pressure required to achieve adsorption saturation is also greater.
In order to more obviously compare the difference in adsorption capacity between the 2 types of layers, the cleave surface method is adopted to cut several groups of locate files after adsorption according to the common method of determining the adsorption distance by 1 nm. Taking the 1 nm thickness of the gap near the layers as the cutting surface, the statistical average adsorption capacity in different regions is shown in Fig. 6.
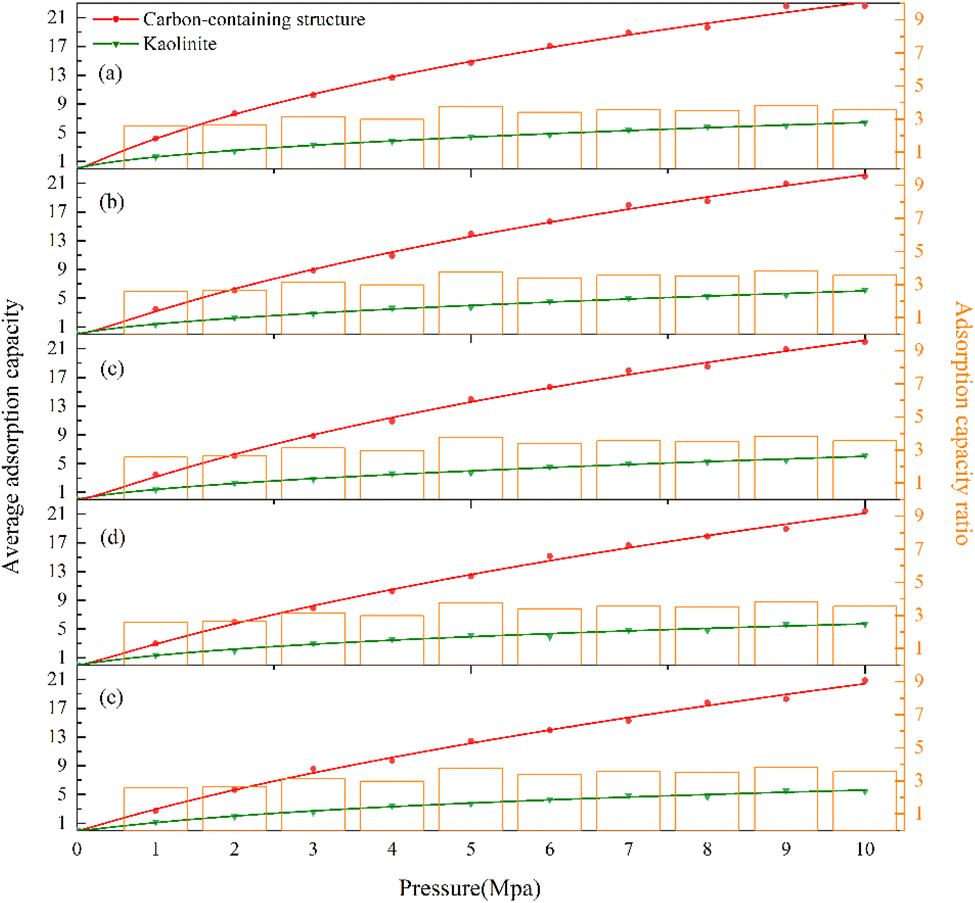 | ||
| Fig. 6 Distribution of adsorption capacity of each layer in the KCK model at different temperatures: (a) 293.15 K, (b) 303.15 K, (c) 313.15 K, (d) 323.15 K, and (e) 333.15 K. | ||
As shown in Fig. 6, the red and dark green curves are the Freundlich fitting curves for the total adsorption capacity of the carbon-containing structure layer and the kaolinite layer, respectively. It can be seen that the adsorption capacity of the carbon-containing structure layer increases significantly with pressure in all five temperature regimes, and is in the range of 20 to 23 mmol g−1 at a pressure of 10 MPa. However, the adsorption capacity of the kaolinite layer changes only slightly and is only 4 mmol g−1 at a pressure of 10 MPa. From the temperature point of view, the adsorption capacity of the carbon-containing structure layer at each temperature point decreases slightly as the temperature increases. Although the decrease in kaolinite adsorption capacity is not obvious visually in the figure, in fact, due to the low amount of kaolinite adsorption, the CO adsorption capacity of the carbon-containing structure layer at 333.15 K at 10 MPa pressure decreased by 4.8% compared with that at 293.15 K temperature, while the adsorption capacity of the kaolinite layer decreased by 16.7%.
The total CO adsorption curve of the carbon-containing structure layer and kaolinite layer conforms to the Freundlich rule, but the fitting effect of adsorption capacity of the Al–O plane and Si–O plane is poor. In addition, as can be seen in Fig. 7, the average adsorption capacity of the Al–O plane is higher than that of the Si–O plane at 293.15 K - 333.15 K, and the gap is largest at 323.15 K, reaching 0.688 mmol g−1. This indicates that the hydroxyl group can enhance the adsorption capacity of the Al–O surface.
 | ||
| Fig. 7 Average adsorption capacity of kaolinite on Al–O and Si–O surfaces at different temperature points. | ||
The ratio of the adsorption capacity between the carbon-containing structure layer and the kaolinite layer is shown by the orange histogram in Fig. 6. The adsorption capacity ratio is 258.40 to 377.25% at 293.15 K, 296.43 to 375.32% at 303.15 K, 256.49 to 383.78% at 313.15 K, 215.81% to 383.48% at 323.15 K, and 226.79% to 380.71% at 333.15 K. It can be seen that the average adsorption capacity of the carbon-containing structure layer is essentially 2–4 times larger than that of the kaolinite layer, and it can be found that the carbon-containing structure/kaolinite adsorption ratio in the low-pressure environment is lower than that in the high-pressure environment.
It can be seen that the adsorption capacity of the carbon-containing structure in the coal gangue is much stronger than that of kaolinite, because it is a porous material, and in addition to interlayer pores, there are a large number of pores between carbon chains to provide gas adsorption sites. In addition, a large number of functional group structures can form strong adsorption of CO gas. The crystal layer spacing and surface structure of kaolinite do not allow CO molecules to be adsorbed in its bulk structure, so there is only surface adsorption and part of the gas is free in the nearby vacuum layer. It can be seen that as the pressure increases, the adsorption capacity gap between carbon-containing structure and kaolinite also increases to some extent. The adsorption capacity of the carbon-containing structure increases more in the high-pressure environment, while the effect of the high-temperature environment on the adsorption capacity of kaolinite is more significant.
3.3 CO molecular adsorption distribution
Since the adsorption capacity of the KCK model is not large in the atmospheric pressure environment, the adsorption distribution characteristics of each layer are not obvious. Therefore, the simulated adsorption conformations for each set of temperature points at 1 MPa are used as the analysis objects, and the resulting adsorption models were calculated and analyzed using the Forcite analysis module. The calculation direction was set as (0 0 1) and the Bins value as 30, and the average of five groups of final data was adopted. The relative density distribution curve of CO molecules was obtained, as shown in Fig. 8. | ||
| Fig. 8 Distribution of CO adsorption density at 313 K and 1 MPa: (a) relative density distribution curve and (b) density distribution field. | ||
It can be seen from Fig. 8(a) that the peaks near 1.2 nm and 7.6 nm represent the CO molecular layer density interacting with the Al–O and Si–O surfaces of kaolinite, where the left peak area is larger, and it contains 10 moles per uc more CO molecules than the peak on the right, while the middle peak representing the adsorption density of the carbon-containing structure layer has the largest relative area and lower peak value. In contrast to the surface adsorption of kaolinite, the adsorption sites of carbon-containing structure layers are more numerous, with stronger dispersion and adsorption capacity, in agreement with the above-mentioned conclusions. Carbon-containing structures have a large impact on the overall CO gas molecular adsorption structure.
Since the main adsorption sites do not change at each pressure and temperature point, only the amount of adsorption at each adsorption site is different. Therefore, adsorption configurations at 313 K and 1 MPa were selected for the analysis. As shown in Fig. 8(b), the adsorption density field of CO adsorbed by the KCK model in Monte Carlo steps is shown. The darker and denser the blue label color, the greater the adsorption density of CO. It can be seen that on kaolinite, CO molecules gather to form an adsorption layer roughly parallel to the wall only near the slit wall, while at a place away from the wall, CO molecules are only scattered in a free state, and the molecular kinetic diameter of CO is 0.376 nm. Due to the crystal structure of kaolinite, CO molecules are not adsorbed in the bulk structure. It can be seen that the CO molecules adsorbed by the carbon-containing structure are mostly concentrated in the tiny cavities between carbon chains and near specific functional groups, and the concentrated adsorption sites are relatively fixed.
Fig. 9 shows the adsorbed CO conformation of Al–O and Si–O planes of kaolinite, in which the adsorbent is displayed in the form of a polyhedron and the CO molecule is displayed in the form of CPK. It can be seen that the Al–O plane in Fig. 9(a) adsorbs CO more closely, while the Si–O plane in Fig. 9(b) adsorbs CO more loosely. This is due to the presence of hydroxyl groups on the Al–O surface, which form hydrogen bonds with CO in addition to electrostatic forces, while the Si–O surface is adsorbed by electrostatic forces. In addition, the CO molecules in Fig. 9(a) are mostly adsorbed above the holes surrounded by the hydroxyl group on the Al–O surface, while the CO molecules in Fig. 9(b) are mostly C atoms pointing to the center of the Si–O tetrahedral ring, while O atoms are far away from the wall. With the exception of a small number of CO molecules that are free in the vacuum layer, the rest of the adsorbed layer molecules on the kaolinite wall essentially obey this rule, which is due to the low adsorption energy and the stable structure at this adsorption site. The electronegativity of O in CO is greater, and the negatively charged wall of kaolinite is more likely to attract C atoms, while repelling O atoms with high electronegativity.40–42
 | ||
| Fig. 9 CO molecular adsorption sites in the kaolinite layer: (a) Al–O plane and (b) Si–O plane. | ||
Fig. 10 shows the adsorption sites of a portion of the carbon-containing structure layer. It can be seen that except for free CO in the slit space, CO adsorbed by HQL molecules is basically near the oxygen-containing functional groups, especially the functional groups such as –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, –C–O, and –OH. There is also a very small amount of adsorbed CO molecules near the aliphatic hydrocarbon –CH2. This is because CO is a weak quadrupole linear polar fluid molecule, and polar molecules are easy to interact with strong polar oxygen-containing functional groups by dipole force, and the differences in the polarity and relative content of each oxygen-containing functional group will cause differences in the adsorption capacity of CO.43,44 In addition, measure/change was used to measure the distance between CO molecules on the stable adsorption and adjacent functional groups, and it was found that the minimum adsorption distance was 3.21 Å, all of which were greater than 3 Å, indicating that CO did not form hydrogen bonds in the adsorption of carbon-containing structure layer, but still dominated by intermolecular forces.
O, –C–O, and –OH. There is also a very small amount of adsorbed CO molecules near the aliphatic hydrocarbon –CH2. This is because CO is a weak quadrupole linear polar fluid molecule, and polar molecules are easy to interact with strong polar oxygen-containing functional groups by dipole force, and the differences in the polarity and relative content of each oxygen-containing functional group will cause differences in the adsorption capacity of CO.43,44 In addition, measure/change was used to measure the distance between CO molecules on the stable adsorption and adjacent functional groups, and it was found that the minimum adsorption distance was 3.21 Å, all of which were greater than 3 Å, indicating that CO did not form hydrogen bonds in the adsorption of carbon-containing structure layer, but still dominated by intermolecular forces.
 | ||
| Fig. 10 Adsorption sites of the CO molecule in the carbon-containing structure layer. | ||
3.4 Diffusivity analysis
As mentioned in the above section, MSD was calculated under representative conditions, and the diffusion coefficients and fitting curves of CO under various conditions of 313 K and 1 MPa were obtained, as shown in Fig. 11.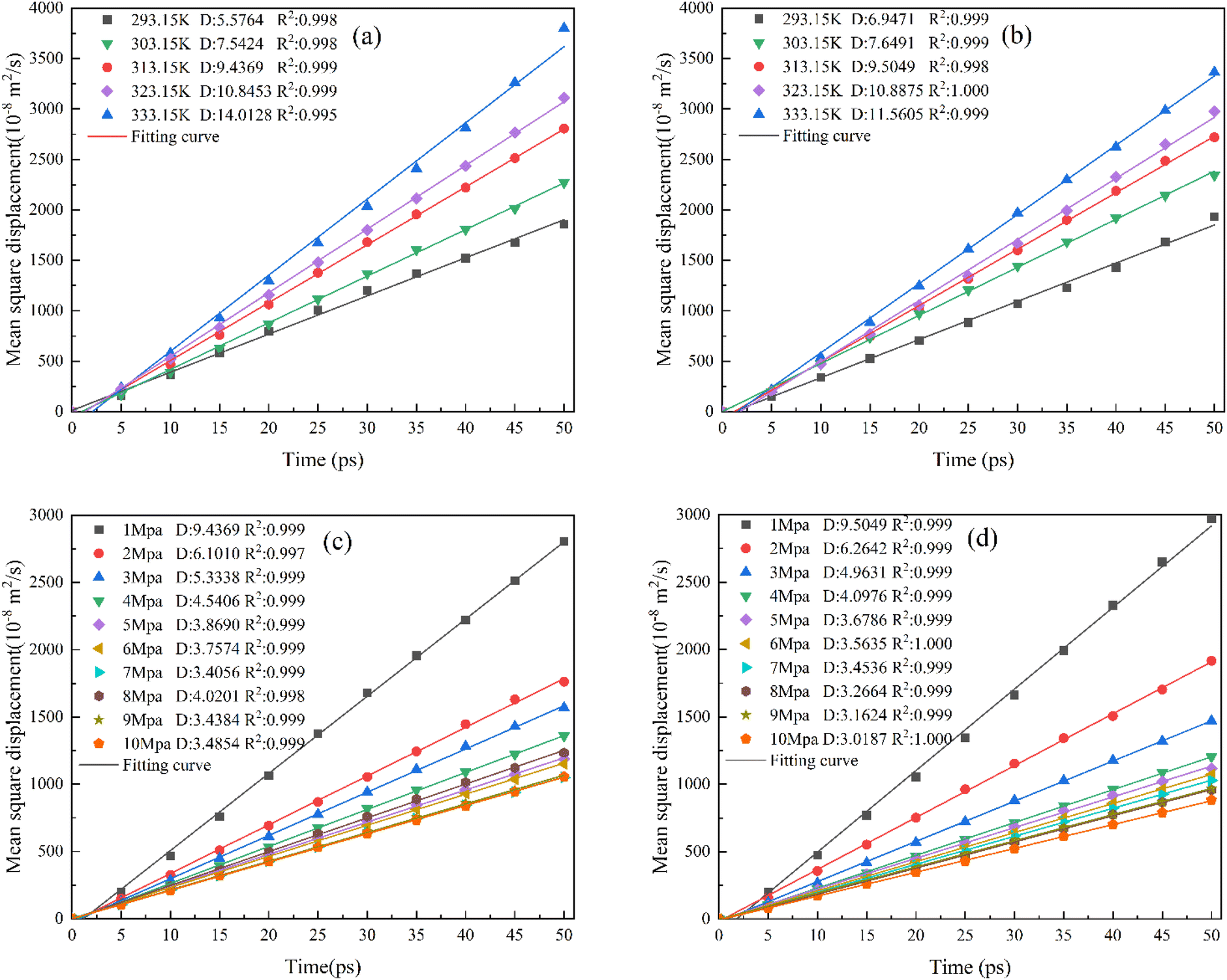 | ||
| Fig. 11 Linear regression curve of CO diffusion. (a) The KCK model at 1 MPa, (b) TriK model at 1 MPa, (c) KCK model at 313.15 K, and (d) TriK model at 313.15 K. | ||
It can be seen from Fig. 11(a) that the diffusion coefficient of CO in the KCK model is (5.58–14.01) × 10−8 m2 s−1 under a fixed pressure of 1 MPa. As the temperature increases, the CO diffusion coefficient increases because the internal energy of the molecule increases. According to the law of conservation of energy, it can be inferred that its internal energy is converted into kinetic energy, which increases the kinetic energy of the CO molecule in the high temperature regime, so that the molecular motion gradually intensifies and the diffusion coefficient increases. As shown in Fig. 11(c), the diffusion coefficient at a fixed temperature of 313.15 K is (3.49–9.44) × 10−8 m2 s−1. With the increase in pressure, the diffusion coefficient shows a downward trend on the whole, but it stabilizes around 3.4 × 10−8 m2 s−1 after 6 MPa, and the downward trend is less obvious. This is because the increase in pressure causes the gap to be filled with a large number of crowded gas molecules, and the adsorption capacity of the structure is also improved, hence the strengthening of the intermolecular effect reduces the overall diffusion capacity of CO gas molecules, but when the pressure increases to 4 MPa, this trend gradually weakens as the gas molecules get closer and closer to adsorption saturation.
It can be seen from Fig. 11(b) that the CO diffusion coefficient in the TriK model is in the range of (6.94–11.56) × 10−8 m2 s−1 under a fixed pressure, and its variation trend is basically the same as that in the KCK slit (a). In Fig. 11(d), the diffusion coefficient of kaolinite structures at a fixed temperature is in the range of (3.02–10.89) × 10−8 m2 s−1, and the variation trend is the same as that shown in Fig. 11(c). At a higher pressure, the diffusion coefficient is basically concentrated at 3.1 × 10−8 m2 s−1, while the difference between the CO diffusion coefficients of the two structures under the same conditions is relatively small. This suggests that the addition of a carbon-containing structure layer does not significantly change the CO diffusion trend in coal gangues, and that temperature is the largest factor affecting the diffusion coefficient of CO molecules in such structures.
The diffusion process of CO is an activation process. In order to further study the diffusion properties and influence conditions of CO gas molecules, the Arrhenius equation was used to calculate its diffusion activation energy.45 Arrhenius's equation is shown in eqn (6):
 | (6) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D and 1/T was finally fitted, and the diffusion activation energy was determined, as shown in Fig. 11.
D and 1/T was finally fitted, and the diffusion activation energy was determined, as shown in Fig. 11.
It can be seen from the fitting curve in Fig. 12 that there is a good linear relationship between lnD and 1/T, and the fitting parameters (R2) are 0.99 and 0.97, indicating that the trend of diffusion coefficient lnD changing with the temperature is in good agreement with the Arrhenius equation, and the diffusion of CO molecules in the two structures conforms to the one-dimensional unsteady diffusion model.46 The resulting diffusion activation energy equation is shown in eqn (7) and (8):
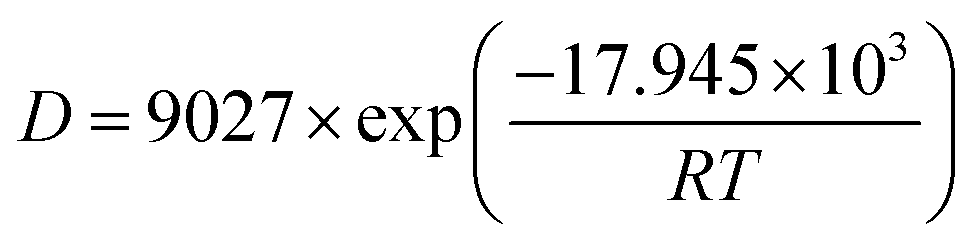 | (7) |
 | (8) |
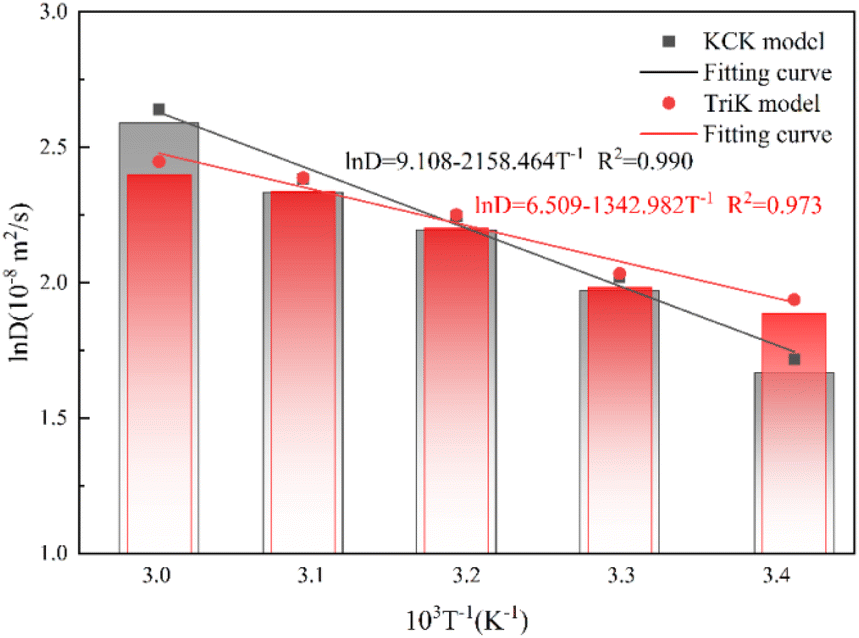 | ||
| Fig. 12 Fitting curve of the activation energy of lnD and 1/T at 1 MPa. | ||
It follows that the CO diffusion activation energy at 1 MPa is 17.945 kJ mol−1 for the KCK model and 11.165 kJ mol−1 for the TriK model. The diffusion activation energy increases with the temperature. The adsorption capacity of the TriK slit model is weak, and hence, the CO molecule diffusion reaction is easier to achieve in it.
4 Conclusions
In this research, the mechanism of CO gas generation, adsorption and diffusion in gangue slit models was studied by molecular dynamics simulation, and the following conclusions were drawn:(1) The CO characteristic gas produced by the spontaneous combustion of the HQL coal gangue mainly comes from the fracture of oxygen-containing organic structures such as ether and phenol in the carbon-containing structure and the conversion of CO2. With the reduction in oxygen molecules, the CO production time gradually advances and the output decreases.
(2) The CO adsorption by the KCK slit and TriK slit in the coal gangue under the conditions of 293.15–333.15 K and 0–10 MPa belongs to physical adsorption, which is in line with the Freundlich adsorption model and belongs to type I isotherm. Increasing the temperature or decreasing the pressure does not favor its adsorption, and the adsorption capacity of the KCK slit model is 110.4–132.9% that of the TriK model.
(3) The average adsorption capacity of the carbon-containing structure is 2.2 to 3.8 times that of kaolinite, and the difference slightly increases with pressure and temperature, exerting a stronger effect on the adsorption capacity of the carbon-containing structure than kaolinite. This indicates that the carbon-containing composition contributes significantly to the adsorption of CO by the coal gangue, which cannot be neglected in adsorption studies. The adsorption sites of carbon-containing structures are much more than that of kaolinite, and they are concentrated near oxygen-containing functional groups. The adsorption sites of kaolinite are relatively fixed, with slightly stronger adsorption in the Al–O plane, and there are both bonding adsorption and electrostatic adsorption, while the Si–O plane is dominated by electrostatic adsorption.
(4) The diffusion coefficient of CO in the KCK slit model is in the range of (3.49–14.01) × 10−8 m2 s−1, and the diffusion activation energy at 1 MPa and 313.15 K is 17.945 kJ mol−1. Under the same conditions, the CO diffusion coefficient of the TriK slit model is in the range of (3.02–11.56) × 10−8 m2 s−1, and the diffusion activation energy is 11.165 kJ mol−1. The effect of temperature on the diffusion coefficient is larger and the CO gas is more difficult to diffuse in the KCK slit model. In the actual project of the gangue hill, the influence of CO adsorption and diffusion behavior on the carbon structure such as crushed coal on spontaneous combustion gas monitoring should be fully considered, and the appropriate index gas and sampling point should be selected.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work is supported by the Fundamental Research Funds for Inner Mongolia University of Science & Technology (Grant No. 2024QNJS121).References
- Y. Li, X. Ren, Y. Zhang, Y. Zhang, X. Shi and S. Ren, Energy, 2024, 288, 129781 CrossRef CAS.
- S. Shao, B. Ma, C. Wang and Y. Chen, Fuel, 2023, 331, 125927 CrossRef CAS.
- G. Ke, H. Jiang and Z. Li, Constr. Build. Mater., 2024, 414, 135061 CrossRef CAS.
- X. Jiang, S. Yang, B. Zhou, W. Song, J. Cai, Q. Xu, Q. Zhou and K. Yang, Fire Mater., 2022, 46, 549–559 CrossRef CAS.
- Y. Wu, X. Yu, S. Hu, H. Shao, Q. Liao and Y. Fan, Process Saf. Environ. Prot., 2019, 123, 39–47 CrossRef CAS.
- Y. Liu, X. Qi, D. Luo, Y. Zhang and J. Qin, ACS Omega, 2023, 8, 47690–47700 CrossRef CAS PubMed.
- Y. Zhang, X. Qi, J. Zou, Y. Rao, L. Chen, L. Zhang, Y. Ji and Z. Liang, Fuel, 2023, 346, 128273 CrossRef CAS.
- M. Gao, X. Li, C. Ren, Z. Wang, Y. Pan and L. Guo, Energy Fuels, 2019, 33, 2848–2858 CrossRef CAS.
- D. Zhao and X. Liu, Arabian J. Chem., 2024, 17, 105697 CrossRef CAS.
- H. Sui, F. Zhang, L. Zhang, D. Wang, Y. Wang, Y. Yang and J. Yao, Sci. Total Environ., 2024, 908, 168356 CrossRef CAS PubMed.
- G. Wang and W. Chen, Int. J. Hydrogen Energy, 2024, 51, 10–20 CrossRef CAS.
- W. Wang, P. Wang, Z. Cao, Y. Cao, Z. Wu and B. Su, Mater. Today Commun., 2023, 36, 106817 CrossRef CAS.
- W. Wang, P. Wang, Z. Cao, Y. Cao and Y. Ma, Int. J. Coal Prep. Util., 2023, 43, 2046–2064 CrossRef CAS.
- F. Castro-Marcano and A. C. T. van Duin, Combust. Flame, 2013, 160, 766–775 CrossRef CAS.
- X. Zhang, B. Lu, L. Qiao and C. Ding, Energy, 2023, 285, 129553 CrossRef CAS.
- X. Zhang, B. Lu, J. Zhang, X. Fu, H. Deng, L. Qiao, C. Ding and F. Gao, Fuel, 2023, 340, 127501 CrossRef CAS.
- Y. Chen, Z. Wang, B. Li, K. Yu, H. Wang, J. Wang, Y. Huo and J. Wang, Langmuir, 2023, 39, 18581–18593 CrossRef CAS PubMed.
- R. L. C. Akkermans, N. A. Spenley and S. H. Robertson, Mol. Simul., 2013, 39, 1153–1164 CrossRef CAS.
- R. Xu, A. Yiannikouris, U. K. K. Shandilya and N. A. A. Karrow, Toxins, 2023, 15(2), 104 CrossRef CAS PubMed.
- C. Wu, J. Li, F. Zhou and B. Shi, Int. J. Hydrogen Energy, 2024, 59, 924–936 CrossRef CAS.
- Z. Li, C. Ding, W. Wang, B. Lu and D. Gao, Energy Sources, Part A, 2022, 44, 3709–3719 CrossRef CAS.
- X. Liu, X. He, N. Qiu, X. Yang, Z. Tian, M. Li and Y. Xue, Appl. Surf. Sci., 2016, 389, 894–905 CrossRef CAS.
- J. Li and J. Wang, J. Cleaner Prod., 2019, 239, 117946 CrossRef.
- J. Zhang, J. Wang, Z. Li, J. Zhu and B. Lu, ACS Omega, 2022, 7, 11190–11199 CrossRef CAS.
- H. Sun, Z. Jin, C. Yang, R. L. C. Akkermans, S. H. Robertson, N. A. Spenley, S. Miller and S. M. Todd, J. Mol. Model., 2016, 22, 47 CrossRef PubMed.
- H. Sun, P. Ren and J. R. Fried, Comput. Theor. Polym. Sci., 1998, 8, 229–246 CrossRef CAS.
- Y. Cheng and B. Hu, Coal Sci. Soc., 2023, 48, 212–225 Search PubMed.
- L. Hong, W. Wang, D. Gao and W. Liu, PLoS One, 2022, 17(3), e0264225 CrossRef CAS.
- B. Li, J. Guo, B. Albijanic, S. Liu, L. Zhang and X. Sun, Miner. Eng., 2020, 148, 106226 CrossRef CAS.
- J. Chen, F. Min, L. Liu and C. Liu, Appl. Surf. Sci., 2019, 476, 6–15 CrossRef CAS.
- X. Jiang, S. Yang, B. Zhou and J. Cai, Combust. Sci. Technol., 2023, 195, 713–727 CrossRef CAS.
- M. Chen, Y. Zhao, L. Zhang, C. Xing, L. Liu, P. Qiu and S. Sun, Int. J. Hydrogen Energy, 2024, 49, 1268–1277 CrossRef CAS.
- F. Castro-Marcano, A. M. Kamat, M. F. Russo, A. C. T. van Duin and J. P. Mathews, Combust. Flame, 2012, 159, 1272–1285 CrossRef CAS.
- J. Li, Y. Wang, Z. Chen and S. S. Rahman, Langmuir, 2021, 37, 12732–12745 CrossRef CAS PubMed.
- Y. Zhang, X. Shi, Y. Li and Y. Liu, Can. J. Chem. Eng., 2018, 96, 1752–1761 CrossRef CAS.
- W. Ge, H. Mao, J. Chen, F. Min, H. Liu and S. Song, Appl. Clay Sci., 2024, 251, 107313 CrossRef CAS.
- J. Zhao, M. He, X. Hu and W. Gao, Chin. Phys. B, 2017, 26, 079101 CrossRef.
- J. Zhang, J. Wang, C. Zhang, Z. Li, J. Zhu and B. Lu, Sci. Rep., 2021, 11, 11706 CrossRef CAS PubMed.
- M. M. Majd, V. Kordzadeh-Kermani, V. Ghalandari, A. Askari and M. Sillanpaa, Sci. Total Environ., 2022, 812, 151334 CrossRef.
- X. Du, D. Pang, Y. Zhao, Z. Hou, H. Wang and Y. Cheng, Arabian J. Chem., 2022, 15, 103665 CrossRef CAS.
- Y. Miao, H. Yan, B. Hong, X. Zhou, L. Tong, Y. Xiao, S. Qiu, B. Yang, Q. Long, Y. Li, Y. Xia and T. Qiu, J. Mol. Liq., 2022, 368, 120819 CrossRef CAS.
- D. Richard and N. M. Rendtorff, Appl. Clay Sci., 2019, 169, 67–73 CrossRef CAS.
- B. Hu, Y. Cheng, X. He, Z. Wang, Z. Jiang, C. Wang, W. Li and L. Wang, Fuel, 2020, 262, 116675 CrossRef CAS.
- Y. Sun, L. Wang, R. Wang, S. Zheng, X. Liao, Z. Zhu and Y. Zhao, Fuel, 2022, 330, 125715 CrossRef CAS.
- J. Kohout, Molecules, 2021, 26(23), 7162 CrossRef CAS PubMed.
- H. Hu, L. Du, Y. Xing and X. Li, Fuel, 2017, 187, 220–228 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra03151b |
| This journal is © The Royal Society of Chemistry 2024 |