
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, docking and biological evaluation of purine-5-N-isosteresas anti-inflammatory agents†
Ahmed M. El-Saghier *a,
Souhaila S. Enailiab,
Aly Abdoua,
Amany M. Hameda and
Asmaa M. Kadrya
*a,
Souhaila S. Enailiab,
Aly Abdoua,
Amany M. Hameda and
Asmaa M. Kadrya
aChemistry Department, Faculty of Science, Sohag University, 282524, Sohag, Egypt. E-mail: el.saghier@science.sohag.edu.eg
bChemistry Department, Faculty of Science, Al Zawiya University, Al Zawiya, Libya
First published on 3rd June 2024
Abstract
An operationally simple one-pot three-component and convenient synthesis method for a series of diverse purine analogues of 5-amino-7-(substituted)-N-(4-sulfamoylphenyl)-4,7-dihydro-[1,2,4]-triazolo[1,5-a][1,3,5]triazine-2-carboxamide derivatives generated in situ via the reaction of 2-hydrazinyl-N-(4-sulfamoylphenyl)-2-thioxoacetamide, cyanoguanidine and a variety of aldehydes was achieved under green conditions. This experiment was conducted to evaluate the anti-inflammatory effect of the newly synthesized compounds using indomethacin as a reference medication; all compounds were tested for in vitro anti-inflammatory activity using the inhibition of albumin denaturation, RBC hemolysis technique and COX inhibition assay. The results showed that all evaluated compounds exhibited significant in vitro anti-inflammatory efficacy leading to excellently effective RBC membrane stabilization, inhibition of protein denaturation, and inhibition of COX enzymes when compared to those of indomethacin. At concentrations of 50, 100, 200, and 300 μg ml−1, these compounds decreased COX-1 and COX-2 activities more than indomethacin and have IC50 values in the range of 40.04–87.29 μg ml−1 for COX-1 and 27.76–42.3 μg ml−1 for COX-2 while indomethacin showed IC50 = 91.57 for COX-1 and 42.66 μg ml−1 for COX-2. The anti-inflammatory findings show the need for more investigation to define the properties underlying the evaluated compounds' anti-inflammatory abilities. The enzyme cyclooxygenase-2 (COX 2) (PDB ID: 5IKT) was docked with ten synthetic substances. With docking scores (S) of −8.82, −7.82, and −7.76 kcal mol−1, 7-furan triazolo-triazine (4), 7-(2-hydroxy phenyl) triazolo-triazine (11), and 7-(4-dimethylamino phenyl) triazolo-triazine (12) had the greatest binding affinities, respectively. Therefore, these substances have COX-2 (PDB ID: 5IKT) inhibitory capabilities and hence may be investigated for COX 2 targeting development. Furthermore, both the top-ranked compounds (4 and 11) and the standard indomethacin were subjected to DFT analysis. The HOMO − LUMO energy difference (ΔE) of the mentioned compounds was found to be less than that of indomethacin.
1. Introduction
When a damaging stimulus, trauma, or infection is present, the body responds with an intricate series of reactions known as inflammation. Under physiological circumstances, this response is advantageous to the host but chronic inflammation can cause serious host damage, tissue loss, and even organ failure due to the release of pathogenic mediators.1,2 Pathologic hyper reactive inflammatory reactions are the outcome of reduced cytoprotective NRF2 cascade.3Thus, to be able to prevent the onset of a number of persistent inflammatory conditions such as Alzheimer's disease, periodontitis, colitis, rheumatoid arthritis, inflammatory bowel disease, septic shock, cardiovascular disease, and osteoporosis,4,5 it is crucial for the host to discover mechanisms for regulating, avoiding, or reducing the release of inflammatory mediators in comparison with natural products.6 Organic and medicinal chemistry efforts are ongoing to develop, synthesise, assess, and isolate substances with the potential to treat inflammation. These molecules are typically derived from heterocyclic compounds with sulfur or nitrogen atoms in their structures. Because they represent a significant class of both natural and synthetic compounds with a wide range of physicochemical characteristics and pharmacological actions, azaheterocycles are of increased interest.6,7 Triazine derivatives are a particular class of heterocycles that contain nitrogen and have a wide range of properties, including a pain reliever, an antifungal, an anticancer, an antiprotozoal, and an antitubercular. They are consequently regarded as a crucial heterocycle framework for chemical pharmaceuticals.8 The heterocycle purine, which contains nitrogen, is abundant in nature. In RNA and DNA, adenine and guanine are made of these fundamental structures. Purine nucleotides (ATP, GTP, cAMP, cGMP, NAD, and FAD) work as co-factors, substrates, or mediators in the functioning of various proteins, which is primarily what attracts biological attention to them.9,10 These proteins are thought to contain half of the targets that are most amenable to medication, primarily enzymes and receptors. For instance, protein kinases carry out the phosphorylation of ATP, while the cyclic nucleotide phosphodiesterase is involved in the hydrolysis of cAMP and cGMP.11,12 The essential enzyme xanthine oxidase is involved in the breakdown of purines, while the ubiquitous purine nucleoside phosphorylase is essential for the purine salvage route. A very promising class of G-protein-coupled receptors (GPCRs) are adenosine receptors.13–15
Purine isosteres, a structurally similar heterocyclic system, saw parallel developments in chemistry and biology as a result of improvements in the purine motif-based design and development of drugs. They are suitable for the identification of innovative medicines focusing on purine-dependent enzymes and receptors to increase molecular diversity by employing different purine isosteres. There are many well-known medications with purine isostere core structures on the market, such as xanthine, hypothanthine, theobromine, caffeine, uric acid, and isoguanine.14,15
The tubercidin deazapurines,16 toyocamycin,17 and sangivamycin18 act as metabolite inhibitors and are employed as broad-spectrum antibiotics, which is also remarkable. Purine isosteres are the subject of considerable and ongoing research and development efforts, and one of the most promising avenues focuses on employing purine isosteres based on triazine.
Azolo-triazine systems in this category can be broadly classified as purine 5-N-isosteres because they contain a nitrogen atom in position 5 of the purine ring. As a result, the basic structure of this work focuses on certain areas of the compounds' medicinal features rather than how they relate to the heterocyclic system. Research on the triazolo–triazines and the deazapurines has been increasing.19,20 In this paper, the authors highlight the significance of purine isosteres, specifically the 1,3,5-triazines, in medicinal chemistry by establishing a solid rationale for their research by explaining how these compounds target purine-binding proteins with therapeutic potential. Also, the importance of designing drugs with reduced side effects, particularly in the context of non-steroidal anti-inflammatory drugs (NSAIDs), is emphasized.
In accordance with a previous literature review, our focus in this work is synthesizing a novel purine analogue of 1,3,4-triazolo-1,3,5-triazines bearing sulfamoyl carboxamide moieties with a variety of substituted aryls at position 7 which are expected to have potential function as active pharmacophores against COX-2 enzyme because of the significance of isosteres systems as anti-inflammatory agents, and we further confirm it using a molecular docking technique.
2. Methodology
2.1. Chemistry
The melting point of every prepared target compound was calculated using the Fisher–John mechanical approach. Sigma-Aldrich provided the chemicals and solvents needed for this investigation. The synthesized compounds' infrared (ν cm−1) spectra were examined using the KBr technique, and a Bruker ADVANCE 400 MHz spectrometer was used to record the 1H NMR (δ ppm) and 13CNMR spectra (Sohag University). A Vario El Fab-Nr elemental analyzer (Cairo University) was used to record elemental analyses.2.1.1.1 General procedure. To a solution of 2-hydrazinyl-N-(4-sulfamoylphenyl)-2-thioxoacetamide (1) (0.001 mol), cyanoguanidine (2) (0.001 mol) and 0.001 mol of aldehyde derivatives (3a–j) in ethanol (15 ml) and HCl (0.001 mol, 1 ml) were added, and the reaction mixture was refluxed with stirring for about 3 h. The reaction was cooled, and the solid precipitate was collected by filtration and crystalized with ethanol.
2.1.1.2 5-Amino-7-(furan-2-yl)-N-(4-sulfamoylphenyl)-4,7-dihydro-[1,2,4]-triazolo[1,5-a][1,3,5]triazine-2-carboxamide (4). Greenish yellow solid, yield 89%. Mp. 212–214 °C. FT IR (KBr) νmax cm−1: 3347, 3248, 3224 (2NH, 2NH2), 3142 (CH-arom.), 2924, 2756 (CH-aliph.), 1694 (C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Oamide, st), 1667(CN) and 1159 (SO, st). 1H-NMR (δ ppm): 11.28 (s, 1H, NHamide, exchangeable by D2O), 10.50 (s, 1H, NHtriazine, exchangeable by D2O), 9.36 (s, 1H, CHtriazole), 7.90–7.78 (m, 6H, 4Harom + NH2), 7.29–7.23 (m, 3H, NH2 + 1CHfuran), 6.46 (s, 2H, 2CHfuran); 13CNMR (DMSO-d6), δ ppm: 158.89 (CO), 153.23 (C–O), 147.99, 144.24, 141.53 (3CN), 141.14, 139.30 (2Carom), 138.18 (CH–O), 126.92 (C–NH), 121.11, 120.50 (CHarom), 111.20, 108.23 (CHfuran) and 67.11 (CHtriazine). CF/MW: C15H14N8O4S/402; elemental analysis: calc./found: C, 44.77/44.80; H, 3.51/3.49; N, 27.85/27.79; S, 7.97/7.96%.
Oamide, st), 1667(CN) and 1159 (SO, st). 1H-NMR (δ ppm): 11.28 (s, 1H, NHamide, exchangeable by D2O), 10.50 (s, 1H, NHtriazine, exchangeable by D2O), 9.36 (s, 1H, CHtriazole), 7.90–7.78 (m, 6H, 4Harom + NH2), 7.29–7.23 (m, 3H, NH2 + 1CHfuran), 6.46 (s, 2H, 2CHfuran); 13CNMR (DMSO-d6), δ ppm: 158.89 (CO), 153.23 (C–O), 147.99, 144.24, 141.53 (3CN), 141.14, 139.30 (2Carom), 138.18 (CH–O), 126.92 (C–NH), 121.11, 120.50 (CHarom), 111.20, 108.23 (CHfuran) and 67.11 (CHtriazine). CF/MW: C15H14N8O4S/402; elemental analysis: calc./found: C, 44.77/44.80; H, 3.51/3.49; N, 27.85/27.79; S, 7.97/7.96%.
2.1.1.3 5-Amino-N-(4-sulfamoylphenyl)-7-(o-tolyl)-4,7-dihydro-[1,2,4]-triazolo[1,5-a][1,3,5]triazine-2-carboxamide (5). Yellow solid, yield 79%. Mp. 222–224 °C. FT IR (KBr) νmax cm−1: 3390, 3334, 3253 (2NH, 2NH2), 3070 (CH-arom.), 2925, 2756 (CH-aliph.), 1685 (C
Oamide, st), 1663 (CN) and 1156 (SO, st). 1H-NMR (δ ppm): 11.27 (s, 1H, NHamide), 10.49 (s, 1H, NHtriazine), 9.36 (s, 1H, CHtriazine), 7.89–7.77 (m, 6H, 4Harom + NH2.), 7.45–7.24 (m, 6H, 4Harom + NH2), 2.34 (s, 3H, CH3); 13CNMR (DMSO-d6), δ ppm: 158.87 (CO), 141.74, 141.18, 140.23 (3CN), 139.45, 139.24, 137.87, 134.74 (4Carom), 131.75, 131.14 (2CHarom), 128.85, 126.83, 120.99, 120.22 (4CHarom), 72.11 (CHtriazine) and 19.39 (CH3). CF/MW: C18H18N8O3S/426.46; elemental analysis: calc./found: C, 50.70/50.68; H, 4.25/4.20; N, 26.28/26.26; S, 7.52/7.48%.
2.1.1.4 (E)-5-Amino-7-styryl-N-(4-sulfamoylphenyl)-4,7-dihydro-[1,2,4]-triazolo[1,5-a][1,3,5]triazine-2-carboxamide (6). Dark yellow solid, yield 88%. Mp. > 300 °C. FT IR (KBr) νmax cm−1: 3444, 3385, 3266, 3226 (2NH, 2NH2), 3070 (CH-arom.), 2926, 2857 (CH-aliph.), 1681 (C
Oamide, st), 1664 (CN) and 1160 (SO, st). 1H-NMR (δ ppm): 11.28 (s, 1H, NHamide), 10.82 (s, 1H, NHtriazine), 10.45 (s, 1H, CHtriazine), 8.01–7.65 (m, 9H, NH2 + 5H-arom + 2CH), 7.37–7.25 (m, 6H, 4H-arom + NH2); 13CNMR (DMSO-d6), δ ppm: 157.10 (CO), 156.17, 155.57, 154.90 (3CN), 143.94, 141.25, 140.25 (3Carom), 135.94, 134.88 (CHarom), 131.67, 129.45 (2CH), 128.36, 127.04, 121.00 (5CHarom) and 68.99 (CHtriazine). CF/MW: C19H18N8O3S/438.47; elemental analysis: calc./found: C, 52.05/52.00; H, 4.14/4.11; N, 25.56/25.53; S, 7.31/7.29%.
2.1.1.5 5,5′-Diamino-N2,N2′-bis(4-sulfamoylphenyl)-4,4′,7,7′-tetrahydro-[7,7′-bi[1,2,4]triazolo[1,5-a][1,3,5]triazine]-2,2′-dicarboxamide (7). White solid, yield 84%. Mp. 228–230 °C. FT IR (KBr) νmax cm−1: 3375, 3332, 3295 (2NH, 2NH2), 3070 (CH-arom.), 2929, 2853 (CH-aliph.), 1694 (C
Oamide, st), 1663 (CN) and 1160 (SO, st). 1H-NMR (δ ppm): 11.30 (s, 1H, NHamide, exchangeable by D2O), 10.49 (s, 1H, NHtriazine, exchangeable by D2O), 8.04–7.77 (m, 6H, NH2 + 4Harom), 7.30 (s, 2H, NH2), 5.55 (s, 1H, CH); 13CNMR (DMSO-d6), δ ppm: 159.45 (CO), 158.61, 157.25, 155.22 (3CN), 142.06, 139.88 (2Carom), 127.04, 121.37 (CHarom) and 56.11 (CHtriazine). CF/MW: C22H22N16O6S2/670.64; elemental analysis: calc./found; C, 39.40/39.42; H, 3.31/3.29; N, 33.42/33.40; S, 9.56/9.55%.
2.1.1.6 5-Amino-7-(2-chlorophenyl)-N-(4-sulfamoylphenyl)-4,7-dihydro-[1,2,4]-triazolo[1,5-a][1,3,5]triazine-2-carboxamide (8). White solid, yield 84%. Mp. 220–222 °C. FT IR (KBr) νmax cm−1: 3372, 3334, 3303, 3264 (2NH, 2NH2), 3070 (CH-arom.), 2752, 2726 (CH-aliph.), 1694 (C
Oamide, st), 1666 (CN) and 1160 (SO, st). 1H-NMR (δ ppm): 11.51 (s, 1H, NH-amide), 11.30 (s, 1H, NHtriazine), 9.49 (s, 1H, CH-triazine), 8.32 (s, 2H, NH2), 8.10–7.70 (m, 8H, Harom), 7.31 (s, 2H, NH2); 13CNMR (DMSO-d6), δ ppm: 166.05 (CO), 163.66 (C–Cl), 157.11, 155.41, 150.18 (3CN), 141.20, 141.03, 140.52, 140.15 (4Carom), 137.93, 131.44 (2CHarom), 130.32, 127.05, 121.11 (CHarom) and 69.08 (CHtriazine). CF/MW: C17H15ClN8O3S/446.87; elemental analysis: calc./found: C, 45.69/45.70; H, 3.38/3.36; Cl, 7.93/7.0; N, 25.08/25.00; S, 7.17/7.15%.
2.1.1.7 5-Amino-7-(4-methoxyphenyl)-N-(4-sulfamoylphenyl)-4,7-dihydro-[1,2,4]-triazolo[1,5-a][1,3,5]triazine-2-carboxamide (9). Dark yellow solid, yield 71%. Mp. 254–256 °C. FT IR (KBr) νmax cm−1: 3351, 3264, 3169 (2NH, 2NH2), 3070 (CH-arom.), 2925, 2855(CH-aliph.), 1690 (C
Oamide, st), 1681 (CN) and 1161 (SO, st). 1H-NMR (δ ppm): 11.26 (s, 1H, NHamide), 10.45 (s, 1H, NHtriazine), 9.22 (s, 1H, CH-triazine), 7.91 (s, 2H, NH2), 7.81–7.24 (m, 8H, Harom), 6.88 (s, 2H, NH2), 3.81 (s, 3H, OCH3); 13CNMR (DMSO-d6), δ ppm: 159.16 (CO), 157.82, 155.87, 153.86 (3CN), 141.78, 141.25, 140.19, 139.18 (4Carom), 128.09, 127.03, 120.63, 120.21 (CHarom), 69.28 (CHtriazine) and 56.49 (OCH3). CF/MW: C18H18N8O4S/442.45; elemental analysis: calc./found: C, 48.86/48.84; H, 4.10/4.09; N, 25.33/25.31; S, 7.25/7.22%.
2.1.1.8 5-Amino-7-(naphthalen-1-yl)-N-(4-sulfamoylphenyl)-4,7-dihydro-[1,2,4]-triazolo[1,5-a][1,3,5]triazine-2-carboxamide (10). White solid, yield 86%. Mp. 202–204 °C. FT IR (KBr) νmax cm−1: 3442, 3398, 3378 (2NH, 2NH2), 3070 (CH-arom.), 2925, 2855 (CH-aliph.), 1690 (C
Oamide, st), 1663 (CN) and 1152 (SO, st). 1H-NMR (δ ppm): 11.28 (s, 1H, NH-amide, exchangeable by D2O), 10.54 (s, 1H, NHtriazine, exchangeable by D2O), 9.54 (s, 1H, CHtriazine), 8.22–7.24 (m, 15H, 2NH2 + 11H-arom); 13CNMR (DMSO-d6), δ ppm: 159.02 (CO), 141.74, 140.98, 139.26 (3CN), 138.58, 136.66, 134.04, 132.62, 131.19 (5Carom), 129.47, 126.93, 125.94, 121.14, 120.24 (CHarom) and 71.79 (CH-triazine). CF/MW: C21H18N8O3S/462.49; elemental analysis: calc./found: C, 54.54/54.55; H, 3.92/3.89; N, 24.23/24.20; S, 6.93/6.90%.
2.1.1.9 5-Amino-7-(2-hydroxyphenyl)-N-(4-sulfamoylphenyl)-4,7-dihydro-[1,2,4]-triazolo[1,5-a][1,3,5]triazine-2-carboxamide (11). Canary yellow solid, yield 90%. Mp. 250–252 °C. FT IR (KBr) νmax cm−1: 3468 (OH), 3351, 3264, 3168 (2NH, 2NH2), 3070 (CH-arom.), 2825, 2725 (CH-aliph.), 1692 (C
Oamide, st), 1663 (CN) and 1161 (SO, st). 1H-NMR (δ ppm): 11.27 (s, 1H, NHamide), 10.45 (s, 1H, NHtriazine), 9.93 (br, 1H, OH), 9.20 (s, 1H, CHtriazine), 8.17 (s, 2H, NH2), 8.01–7.23 (m, 8H, Harom), 6.88 (s, 2H, NH2); 13CNMR (DMSO-d6), δ ppm: 159.03 (CO), 157.15, 155.74, 153.82 (3CN), 140.82, 140.24, 139.18, 133.68 (4Carom), 127.03, 120.99, 120.23, 117.02, 115.77 (CHarom), 68.96 (CHtriazine). CF/MW: C17H16N8O4S/428.43; elemental analysis: calc./found: C, 47.66/47.60; H, 3.76/3.70; N, 26.16/26.00; S, 7.48/7.41%.
2.1.1.10 5-Amino-7-(4-(dimethyl amino) phenyl)-N-(4-sulfamoylphenyl)-4,7-dihydro-[1,2,4]-triazolo[1,5-a][1,3,5]triazine-2-carboxamide (12). Red solid, yield 88%. Mp. 200–202 °C. FT IR (KBr) νmax cm−1: 3346, 3306, 3259, 3172 (2NH, 2NH2), 3055 (CH-arom.), 2926 (CH-aliph.), 1693 (C
Oamide, st), 1640 (CN) and 1157 (SO, st). 1H-NMR (δ ppm): 11.34 (s, 1H, NHamide), 10.44 (s, 1H, NHtriazine), 9.70 (s, 1H, CH-triazine), 8.21 (s, 2H, NH2), 8.01–7.99 (m, 8H, Harom), 7.44 (s, 2H, NH2), 3.10–3.05 (s, 6H, 2CH3); 13CNMR (DMSO-d6), δ ppm: 158.97 (CO), 156.14, 154.96, 152.83 (3CN), 141.80, 140.86, 140.60, 131.99 (4Carom), 127.82, 127.05, 121.06, 120.29 (CHarom), 63.30 (CHtriazine), 18.75 and 14.40 (2CH3). CF/MW: C19H21N9O3S/455.50; elemental analysis: calc./found: C, 50.10/50.11; H, 4.65/4.55; N, 27.68/27.65; S, 7.04/7.00%.
2.1.1.11 5-Amino-7-(3-hydroxyphenyl)-N-(4-sulfamoylphenyl)-4,7-dihydro-[1,2,4]-triazolo[1,5-a][1,3,5]triazine-2-carboxamide (13). White solid, yield 74%. Mp. 252–254 °C. FT IR (KBr) νmax cm−1: 3381 (OH), 3380, 3332, 3290, 3202 (2NH, NH2), 3106 (CHarom.), 2989, 2728 (CHaliph.), 1695 (C
Oamide, st), 1664 (CN), and 1162 (SO, st). 1H-NMR (δ ppm): 11.31 (s, 1H, NHamide), 10.31 (s, 1H, NHtriazine), 9.92 (s, 1H, OH), 9.22 (s, 1H, CHtriazine), 8.06 (s, 2H, NH2), 7.99–7.84 (m, 8H, Harom), 7.30 (s, 2H, NH2); 13CNMR (DMSO-d6), δ ppm: 159.41 (CO), 158.41, 157.08, 155.33 (3CN), 141.00, 140.27, 138.40, 138.14 (4Carom), 137.93, 131.12 (2CHarom), 130.71, 127.04, 121.03, 119.70 (CHarom), and 63.22 (CH-triazine). CF/MW: C17H16N8O4S/428.43; elemental analysis: calc./found: C, 47.66/47.68; H, 3.76/3.71; N, 26.16/26.11; S, 7.48/7.50%.
2.2. Biological evaluation
Three adult male Wistar rats (age 10–12 weeks, 180–200 g) were housed in a 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) h dark/light cycle animal facility with controlled temperature (20–25°C) and humidity (40–70%). Food and water were given ad libitum throughout the study.21
h dark/light cycle animal facility with controlled temperature (20–25°C) and humidity (40–70%). Food and water were given ad libitum throughout the study.21
A common protective response to tissue damage caused by physical trauma, toxic chemicals, or microbiological agents is inflammation. It is also the localized defense mechanism of living mammalian tissue against pathogens.22 In the current work, the synthesized compounds' in vitro anti-inflammatory activity was evaluated by following methods.
| %Inhibition = ((absorbance control − absorbance sample)/(absorbance control)) × 100 |
The IC50 values of the synthesized compounds and the standard were also calculated.
Hypotonic solution-induced rat erythrocyte hemolysis: the membrane stabilizing activities of the synthesized compounds were assessed using hypotonic solution-induced rat erythrocyte hemolysis.24 The test sample consisted of a stock erythrocyte (RBC) suspension (0.5 ml) mixed with 5 ml of hypotonic solution in 10 mM sodium phosphate buffered saline (pH 7.4) containing the synthesized compound (50–300 μg ml−1) or indomethacin as a standard drug. The control sample consisted of 0.5 ml of RBC mixed with hypotonic buffered saline solution alone. The mixtures were incubated for 10 min at room temperature and centrifuged for 10 min at 3000 rpm, and the absorbance of the supernatant was measured at 540 nm. The percentage inhibition of hemolysis or membrane stabilization was calculated by the formula
| % Inhibition of hemolysis = (OD1 − OD2/OD1) × 100 |
| COX inhibition activity (%) = (1 − T/C) × 100 |
The results were expressed in terms of IC50 values and a COX-1/COX-2 selectivity index was calculated (Table 2).
2.3. In silico analysis
The in silico pharmacokinetic analysis, including the Lipinski score and BOILED egg plot, was done using SWISS ADME.26 Additionally, the in silico toxicity assessment was explored using OSIRIS Property Explorer.272.4. Molecular docking
The ten tested compounds (4–13) as ligands underwent molecular docking investigations using the MOE.25 Using the MOE program's builder interface, 3D models of the target compounds (4–13) were generated.28,29 The ligand preparation process was performed on the target compounds with the default features. The resulting database was then stored as an MDB file for the docking calculations. The atomic coordinates of the crystal structures of the studied enzyme, the structure of tolfenamic acid bound to human cyclooxygenase-2 with the Protein Data Bank (PDB) identification number 5IKT,30 as receptor were obtained from the protein databank. The target enzyme structure was prepared by adding polar hydrogen atoms and removing water molecules, native ligands, and unwanted chains that were exposed.31,32 The active site of 5IKT was docked with the ten compounds (4–13) and 30 conformations were generated for each compound.33 The London G energy scoring function was used to evaluate the ligand–receptor complexes and the lowest scoring one was selected for further analysis of the binding orientations.25The validity of the docking process was verified by a re-docking and superimposition strategy.34,35 The native ligand of the 5IKT enzyme was extracted and then re-docked into the active site. The ESI† (S.I. Docking validation) shows the details of this process.
2.5. Density functional theory (DFT) exploration
DFT analyses play an essential role in the computation of molecular orbital characteristics.36,37 In this framework, the top two compounds from the screening process (4 and 11) underwent a structure-based DFT analysis utilizing B3LYP38 and a 6–311g(d, p) basis set39 via G09w.40 Comparative investigation between the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energies was performed.413. Results and discussion
3.1. Chemistry
As part of our ongoing research into the synthesis of novel heterocycles,42–52 we prepared in this article a new series of 5-amino-7-(substituted)-N-(4-sulfamoylphenyl)-4,7-dihydro-[1,2,4]triazolo[1,5-a][1,3,5]triazine-2-carboxamidederivatives 4–13 by a new method, a smooth way, one-pot reaction, energy saving (at room temperature), low cost (without catalyst), short period (about 3 h), green solvent (EtOH) method, with high yields (79–93%) and no requirements for toxic chemicals, which follow the green synthesis rules. Thiocarbohydrazide 1 was allowed to react with cyanoguanidine 2 and various aldehydes 3a–3j, namely, furfural, 2-methylbenzaldehyde, cinnamaldehyde, glyoxal, 2-chlorobenzaldehyde, 4-methoxybenzaldehyde, 1-naphthaldehyde, 2-hydroxybenzaldehyde, 4(N,N)-dimethyl benzaldehyde, and 3-hydroxybenzaldehyde. The one pot-reaction, three component system was applied in ethanol in the presence of a few drops of conc. HCl with stirring under reflux for 3 h, and the products 4–13 were precipitated on heat and separated by filtration (Scheme 1). | ||
| Scheme 1 Synthesis of 5-amino-7-(substituted)-N-(4-sulfamoylphenyl)-4,7-dihydro-[1,2,4]triazolo[1,5-a][1,3,5]triazine-2-carboxamide derivatives (4–13). | ||
The reaction mechanism was assumed to occur via nucleophilic attack of the amino group of thiocarbohydrazide 1 at the cyano group of cyanoguanidine to afford a biguanidine intermediate, followed by nucleophilic attack of the NH of biguanidine at the carbon of thiocarbonyl (CS), with elimination of H2S (which is detected by paper dampened with lead acetate) to give 5-guanidino-N-(4-sulfamoylphenyl)-1H-1,2,4-triazole-3-carboxamide as an intermediate, and then nucleophilic attack of the NH of the triazole ring and NH of guanidine at the carbonyl group of the ketone with tautomerism to afford the desired triazolo-triazine compounds with elimination of water (Scheme 2).
 | ||
| Scheme 2 Reaction mechanism for 5-amino-7-(substituted)-N-(4-sulfamoylphenyl)-4,7-dihydro-[1,2,4]triazolo[1,5-a][1,3,5]triazine-2-carboxamidederivatives (4–13). | ||
1,3,4-Triazolo[1,3,5]triazine derivatives 4–13 were validated with physical and spectral analysis. The FT-IR spectra illustrated the appearance of the NH, NH2 stretching band at 3395–3170 cm−1. Also, the disappearance of the CS stretching band at 1287 cm−1 and the C–S–C stretching band at 745 cm−1 provided a definite sign that the triazolo-triazine system formed as a result of the reaction. In the 1H NMR spectra, signals appeared at 11.62–11.08 (s, 1H, NH-amide), 11.35–10.09 (s, NH-triazine, exchangeable by D2O), 9.54–9.20 (s, 1H, CH-triazine), 8.32–8.06 (s, 2H, NH2-triazine), 8.21–7.32 (m, CH-aromatic), and 7.73–7.06 (s, 2H, NH2, exchangeable by D2O). The formations of triazolo-triazine compounds 4–13 were proved by a clear band at 76.90–63.30 ppm in the 13C-NMR for CH-triazine. The number of aliphatic and aromatic carbons confirmed the expected structure of the title compounds in the 13C-NMR spectrum. Finally, elemental analysis spectroscopic methods obtained information about the elemental compositions of the synthesized compounds.
In this work, the formed materials 4–13 were prepared under different conditions to detect the best method to give a high product yield, short time and green methodology with operational simplicity. Initially, we selected thiocarbohydrazide (1) to react with low-cost cyanoguanidine (2) and p-methoxy-benzaldehyde (3f) as model substrates for the synthesis of 5-amino-7-(4-methoxyphenyl)-N-(4-sulfamoylphenyl)-4,7-dihydro-[1,2,4]triazolo[1,5-a][1,3,5]triazine-2-carboxamide (9) in a one pot method to optimize the reaction conditions, including the effects of type of acidic catalysis, solvent, temperature, time and yield. It is noted that the reaction rate went up when increasing the temperature; here, we recorded that no reaction happened at room temperature in the presence of AlCl3 (Table 1, entry 1). Using a different type of acidic catalysis with the same temperature and solvent enhanced compound 9's yield marginally to 43% (Table 1, entries 2–4); however, compound 9's yield marginally climbed to 44% with a shorter reaction time (Table 1, entry 5). We also assessed how different solvent types affected the product yield during the same reaction time, finding that utilizing acid catalysis—such as HCl—significantly increased the yield of 9 in each of the solvents (Table 1, entries 6–8). Accordingly, using the same amount of HCl (1 equivalent), dioxane produced greater efficiency (60%) than acetonitrile (MeCN) (49%). We refluxed the reaction mixture under a shorter reaction time in order to further prepare triazolo-triazine 9 in a high yield (72%) (Table 1, entry 9). Remarkably, a yield of 79% of 9 was achieved with 1 equivalent of HCl refluxed for 3 hours (Table 1, entry 10). However, when HCl (2 equiv.) was added at the same reaction period, the reaction yield decreased to 74% (Table 1, entry 11). Compound 9 was synthesized using a one-pot method that adheres to green chemistry guidelines. Compound 9's elemental analysis and 13C NMR and 1H NMR data all agree well with the published data.
| Entry | Acidic cat. (equiv.) | Temperature (°C) | Solvent | Time (h) | Yield (%) |
|---|---|---|---|---|---|
| 1 | AlCl3 (1.0) | RT | EtOH | 18 | Nil |
| 2 | Acetic acid (1.0) | Reflux | EtOH | 7 | 37 |
| 3 | Trifluoroacetic acid (1.0) | Reflux | EtOH | 7 | 40 |
| 4 | AlCl3 (1.0) | Reflux | EtOH | 7 | 43 |
| 5 | AlCl3 (2.0) | Reflux | EtOH | 3 | 44 |
| 6 | HCl (1.0) | RT | MeCN | 18 | 49 |
| 7 | HCl (1.0) | RT | Dioxane | 18 | 60 |
| 8 | HCl (1.0) | RT | EtOH | 18 | 64 |
| 9 | HCl (1.0) | Reflux | EtOH | 7 | 72 |
| 10 | HCl (1.0) | Reflux | EtOH | 3 | 79 |
| 11 | HCl (2.0) | Reflux | EtOH | 3 | 74 |
3.2. Biological evaluation
Many inflammatory illnesses can be treated with nonsteroidal anti-inflammatory medicines (NSAIDs, such as ibuprofen, aspirin, or naproxen), herbal supplements, such as curcumin, capsaicin, and Boswellia serrata, and corticosteroids, such as prednisone, to reduce pain and fever.53 However, 1–2% of NSAID users have been observed to have renal toxicity. Research indicates that using NSAIDs can result in renal failure, both acute and chronic.54 Because of their toxic nature, NSAIDs cannot be used to treat inflammatory conditions. Therefore, it is crucial for pharmacological researchers to find new, affordable anti-inflammatory drugs with the fewest side effects possible. This is a difficult assignment for scientists.55 Bioactive substances found in nature have long been utilized in traditional medicine to treat inflammatory conditions like pain, fever, arthritis, and migraines.56 Heterocyclic compounds are widely distributed in nature and are vital to all living organisms. Numerous natural compounds, such as hormones, pigments, antibiotics, and vitamins, include them as fundamental subunits.57 As a result, scientists have given them a lot of attention in their quest to create novel bioactive substances.58 Nitrogen-containing heterocycles are a significant class and have made significant contributions to the field of medicinal chemistry research.59Here, maximum denaturation of egg albumin was inhibited, and membrane stabilization of the RBC was achieved with synthesized compounds 4, 5, 8, 11 and 12 at 50–300 μg ml−1. In the study of membrane stabilization activity, all the newly synthesized compounds protected the erythrocyte membrane significantly in a concentration dependent manner against lysis induced by hypotonic solution. Indomethacin at the same concentrations used as standards also offered significant (p < 0.05) protection of the RBC membrane against the damaging effects induced by hypotonic solution. The membrane stabilization action and inhibitory effect of different concentrations of the synthesized compounds are presented in Fig. 1a and b. Both membrane stabilization activity and effect on protein denaturation contribute to the in vitro anti-inflammatory activity of all the synthesized compounds used in our study. Table 2 shows the IC50 values for the % inhibition of denaturing egg albumin and stabilizing synthetic compounds.
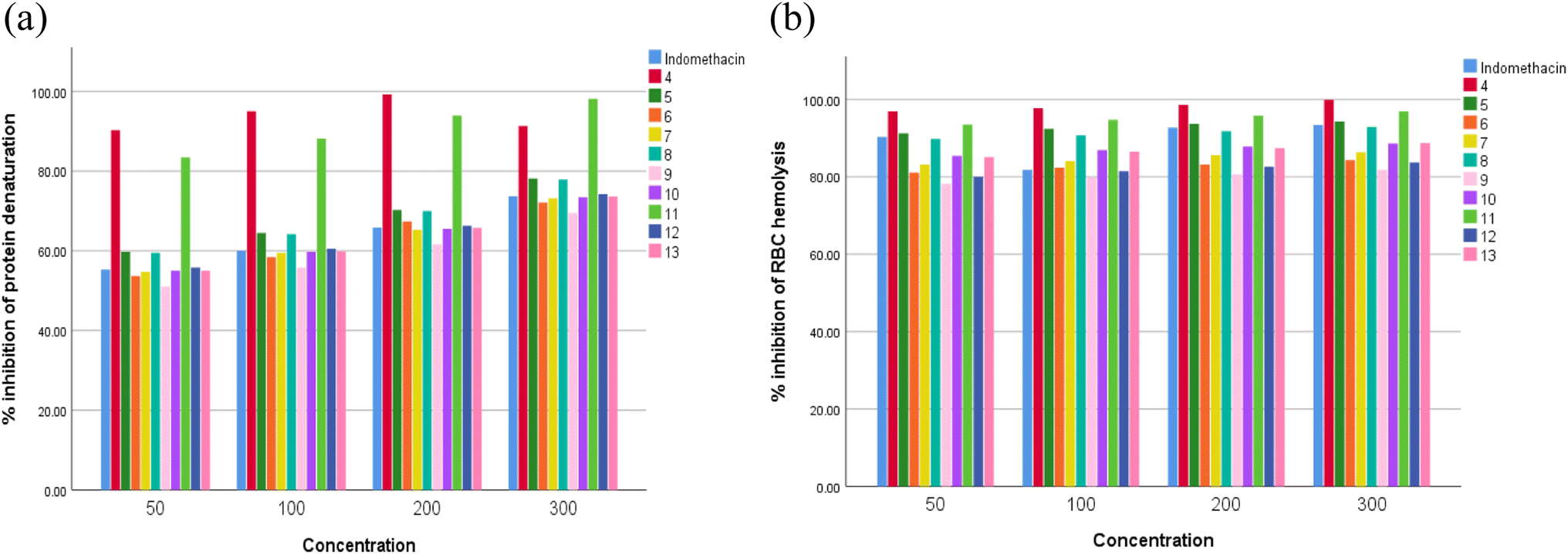 | ||
| Fig. 1 In vitro anti-inflammatory activity by (a) protein denaturation and (b) membrane stabilization. | ||
| Compound code | Conc. (μg ml−1) | Inhibition of protein denaturation (%) | IC50 (μg ml−1) for protein denaturation | Inhibition of RBC hemolysis (%) | IC50 (μg ml−1) for RBC hemolysis |
|---|---|---|---|---|---|
| a IC50 value is the compound concentration required to produce 50% inhibition. All the values are significant when compared to indomethacin (p < 0.05). | |||||
| Indomethacin | 50 | 55.26 | 45.2 | 90.3 | 27.6 |
| 100 | 60 | 91.8 | |||
| 200 | 65.78 | 92.7 | |||
| 300 | 73.68 | 93.4 | |||
| 4 | 50 | 90.26 | 27.6 | 96.9 | 25.7 |
| 100 | 95 | 97.7 | |||
| 200 | 99.2 | 98.6 | |||
| 300 | 91.3 | 99.9 | |||
| 5 | 50 | 59.73 | 41.8 | 91.2 | 27.4 |
| 100 | 64.47 | 92.4 | |||
| 200 | 70.26 | 93.7 | |||
| 300 | 78.15 | 94.3 | |||
| 6 | 50 | 53.68 | 46.5 | 81.1 | 30.8 |
| 100 | 58.42 | 82.4 | |||
| 200 | 67.36 | 83.2 | |||
| 300 | 72.1 | 84.3 | |||
| 7 | 50 | 54.73 | 45.6 | 83.2 | 30 |
| 100 | 59.47 | 84.1 | |||
| 200 | 65.26 | 85.6 | |||
| 300 | 73.15 | 86.3 | |||
| 8 | 50 | 59.47 | 63.3 | 89.8 | 27.8 |
| 100 | 64.21 | 90.7 | |||
| 200 | 70 | 91.8 | |||
| 300 | 77.89 | 92.9 | |||
| 9 | 50 | 51.05 | 42 | 78.2 | 31.9 |
| 100 | 55.78 | 79.8 | |||
| 200 | 61.57 | 80.6 | |||
| 300 | 69.47 | 81.8 | |||
| 10 | 50 | 55 | 45.4 | 85.4 | 29.2 |
| 100 | 59.73 | 86.9 | |||
| 200 | 65.52 | 87.8 | |||
| 300 | 73.42 | 88.6 | |||
| 11 | 50 | 83.42 | 29.9 | 93.5 | 26.7 |
| 100 | 88.15 | 94.7 | |||
| 200 | 93.94 | 95.8 | |||
| 300 | 98.15 | 96.9 | |||
| 12 | 50 | 55.78 | 44.8 | 80 | 27.2 |
| 100 | 60.52 | 81.4 | |||
| 200 | 66.31 | 82.6 | |||
| 300 | 74.21 | 83.7 | |||
| 13 | 50 | 55 | 45.4 | 85.1 | 29.3 |
| 100 | 59.83 | 86.5 | |||
| 200 | 65.72 | 87.4 | |||
| 300 | 73.62 | 88.7 | |||
We evaluated the capacities of the synthesized compounds to inhibit COX-1 and COX-2 activities by an in vitro test and compared them to indomethacin (reference NSAID). As shown in Fig. 2a and b, the synthesized compounds 4, 5, 8, 11 and 12 induced a strong and dose-dependent inhibition. At 50, 100, and 150 μg ml−1, compounds 4, 5, 8, 11 and 12 decreased COX-1 and COX-2 activities more than indomethacin. The results demonstrated that the newly synthesized compounds 4, 5, 8, 11 and 12 have IC50 values in the range of 40.04–87.29 μg ml−1 for COX-1 and 27.76–42.3 μg ml−1 for COX-2, while the positive control indomethacin showed an IC50 of 91.57 for COX-1 and 42.66 μg ml−1 for COX-2. It was noteworthy that compounds 4, 5, 8, 11 and 12 showed higher inhibition for COX activities than indomethacin, with excellent selectivity indexes in the 1.44–2.07 range for the synthesized compounds (Table 3).
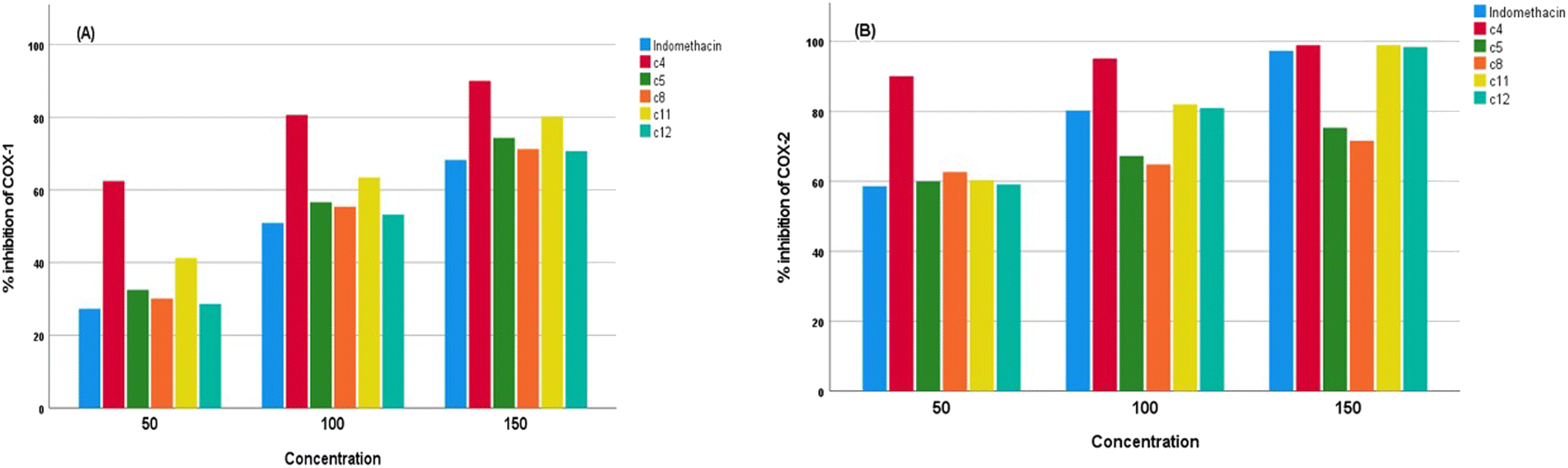 | ||
| Fig. 2 Effect of synthesized compounds on (a) COX-1 activity and (b) COX-2 activity. | ||
| Compound code | Concentration (μg ml−1) | Inhibition of COX-1 (%) | Inhibition of COX-2 (%) | COX-1 IC50 (μg ml−1) | COX-2 IC50 (μg ml−1) | Selectivity index |
|---|---|---|---|---|---|---|
| Indomethacin | 50 | 27.3 ± 0.08 | 58.6 ± 0.04 | 91.57 | 42.66 | 2.14 |
| 100 | 50.88 ± 0.1 | 80.2 ± 0.05 | ||||
| 150 | 68.24 ± 0.2 | 97.3 ± 0.03 | ||||
| 4 | 50 | 62.43 ± 0.01 | 90.03 ± 0.02 | 40.04 | 27.76 | 1.44 |
| 100 | 80.64 ± 0.01 | 95.11 ± 0.01 | ||||
| 150 | 90.02 ± 0.02 | 98.92 ± 0.02 | ||||
| 5 | 50 | 32.51 ± 0.03 | 60.01 ± 0.05 | 76.89 | 41.65 | 1.84 |
| 100 | 56.64 ± 0.01 | 67.25 ± 0.02 | ||||
| 150 | 74.32 ± 0.03 | 75.31 ± 0.01 | ||||
| 8 | 50 | 30.12 ± 0.02 | 62.64 ± 0.01 | 83 | 39.91 | 2.07 |
| 100 | 55.41 ± 0.05 | 64.78 ± 0.01 | ||||
| 150 | 71.23 ± 0.05 | 71.61 ± 0.01 | ||||
| 11 | 50 | 41.28 ± 0.04 | 60.3 ± 0.05 | 60.56 | 41.45 | 1.46 |
| 100 | 63.43 ± 0.04 | 81.99 ± 0.01 | ||||
| 150 | 80.12 ± 0.03 | 98.97 ± 0.06 | ||||
| 12 | 50 | 28.64 ± 0.04 | 59.1 ± 0.01 | 87.29 | 42.3 | 2.06 |
| 100 | 53.24 ± 0.03 | 80.98 ± 0.02 | ||||
| 150 | 70.68 ± 0.04 | 98.4 ± 0.02 |
Because it is simple and accurate, the heat-induced egg albumin denaturation test is a commonly used method. This test was chosen because albumin protein denaturation produces antigens that set off type III hypersensitivity reactions that result in inflammation.60 According to the findings of the current research, these synthetic compounds have a more potent anti-inflammatory impact than indomethacin. All the synthetic compounds inhibited the denaturation of egg albumin at 50, 100, 200, and 300 μg ml−1. The maximum activities were found in 4, 5, 8, 11, and 12 compared to the other studied compounds and indomethacin as a standard drugs.
In other ways, the in vitro membrane stabilization method indicated more effective, flexible, and useful. The study found that the synthesized compounds were more resistant to RBC lysis than the common drug indomethacin. Compounds 4, 5, 8, 11 and 12 were able to prevent lysis more successfully than indomethacin because they have superior membrane stability and strong anti-inflammatory properties, while substances 6, 7, 9, 10 and 13 are less effective at reducing inflammation than indomethacin. As a result of the relatively significant anti-inflammatory capabilities of synthesised compounds 4, 5, 8, 11 and 12, they displayed good potency against COX-1 and COX-2 enzymes in the COX inhibition assay. In comparison to indomethacin, the other investigated substances (6, 7, 9, 10 and 13) demonstrated less inhibition against these enzymes. The results of the current investigation indicated that compounds 4, 5, 8, 11 and 12 were the most effective at inhibiting both COX-1 and COX-2 at concentrations of 50, 100, and 150 μg ml−1. These derivatives may prove to be effective therapeutic molecules for inflammatory illnesses if further laboratory research is conducted on them.
3.3. In silico analysis
P (partition coefficient) less than 5, no more than 5 hydrogen bond donors (H donor), and no more than 10 hydrogen bond acceptors (H acceptor), along with fewer than 10 rotatable bonds. Among the compounds tested, compounds 4, 5, 6, 8, 9, 10, 11, 12, and 13 all adhere to Lipinski's rule, exhibiting properties conducive to good oral bioavailability. However, compound 7 fails to meet Lipinski's criteria, with violations in three areas: excessive molecular weight (670 g mol−1), significantly negative logP (−2.153), and an excess of hydrogen bond acceptors (18) and donors (8). These results suggest that, while most of the compounds show promise for oral bioavailability, compound 7 may face challenges in this regard due to its physicochemical properties.
| MW (g mol−1) | LogP (<5) |
Rotatable bonds | H acceptor (<10) | H donor (<5) | Lipinski's rule | |
|---|---|---|---|---|---|---|
| 4 | 402 | 0.058 | 4 | 4 | 10 | Yes (1 violation) |
| 5 | 426 | 0.773 | 4 | 9 | 4 | Yes (0 violation) |
| 6 | 438 | 1.130 | 5 | 9 | 4 | Yes (0 violation) |
| 7 | 670 | −2.153 | 7 | 18 | 8 | No (3 violations) |
| 8 | 446 | 1.118 | 4 | 9 | 4 | Yes (0 violation) |
| 9 | 442 | 0.474 | 5 | 10 | 4 | Yes (0 violation) |
| 10 | 462 | 1.162 | 4 | 9 | 4 | Yes (0 violation) |
| 11 | 428 | 0.171 | 4 | 10 | 5 | Yes (0 violation) |
| 12 | 455 | 0.531 | 5 | 10 | 4 | Yes (0 violation) |
| 13 | 428 | 0.171 | 4 | 10 | 5 | Yes (0 violation) |
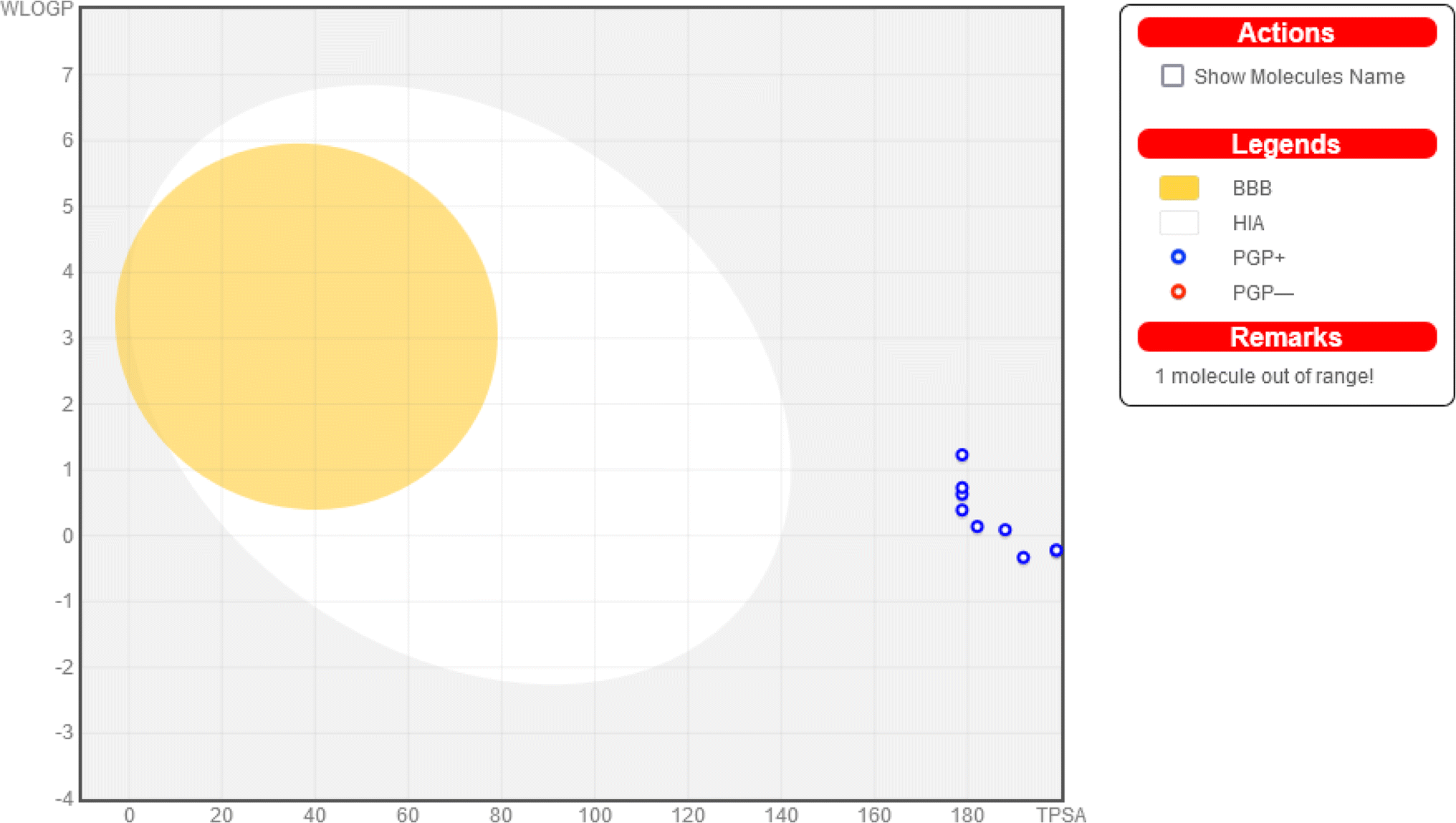 | ||
| Fig. 3 Combined BOILED egg representation (created using Swiss ADME) of the studied compounds. | ||
| Pharmacodynamics | Pharmacokinetics | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Mutagenic | Tumorigenic | Eye and skin irritation | Reproductive effect | Solubility | Absorption | TPSA | Drug-likeliness | Drug score | |
| 4 | No | No | No | Medium | −4.76 | −0.27 | 191.9 | 7.34 | 0.55 |
| 5 | No | No | No | Medium | −5.42 | 0.89 | 178.7 | 7.15 | 0.47 |
| 6 | No | No | No | Medium | −5.09 | 0.94 | 178.7 | 6.23 | 0.49 |
| 7 | No | No | No | Medium | −6.11 | −2.63 | 357.5 | 7.22 | 0.28 |
| 8 | No | No | No | Medium | −5.81 | 1.15 | 178.7 | 7.64 | 0.43 |
| 9 | No | No | No | Medium | −5.09 | 0.47 | 187.9 | 7.43 | 0.49 |
| 10 | No | High | No | Medium | −6.68 | 1.74 | 178.7 | 7.03 | 0.22 |
| 11 | No | No | No | Medium | −4.78 | 0.2 | 198.9 | 7.26 | 0.53 |
| 12 | No | High | No | Medium | −5.11 | 0.44 | 182.0 | 6.25 | 0.29 |
| 13 | No | No | No | Medium | −4.78 | 0.2 | 198.9 | 7.41 | 0.53 |
3.4. Molecular docking
The process of molecular docking involves the prediction of the optimal arrangement and strength of binding between a ligand (herein, the ten tested compounds 4–13) and a receptor (herein, the 5IKT molecule), resulting in the formation of a stable complex.63 One of the receptors that has received a great deal of notice in recent years is 5IKT. The 5IKT receptor is the structure of tolfenamic acid bound to human cyclooxygenase2, which is an enzyme that catalyzes the synthesis of prostaglandins that are associated with inflammation and pain. Tolfenamic acid is a kind of nonsteroidal anti-inflammatory medication (NSAID) that prevents the enzyme cyclooxygenase from gaining accessibility to its target arachidonic acid.64,65 Molecular docking may be used to create unique medicines targeting cyclooxygenase-2 in a substrate-selective manner, which means that they can selectively block the oxygenation of endocannabinoids, which are another class of substrates for cyclooxygenase-2, without affecting the oxygenation of arachidonic acid. This may have consequences for the design of new cyclooxygenase-2-targeting anti-inflammatory and analgesic medications. Therefore, the 5IKT receptor is important in molecular docking because it can provide structural information for designing new drugs that modulate the activity of cyclooxygenase-2 and its substrates. Overall, molecular docking is considered a powerful tool in exploring, discovering, and designing new drugs that can specifically target a certain enzyme.66Here, the docking process was checked by re-docking and superimposition.51 The re-docking procedure used the same procedures that had been used before. The re-docked 5IKT native ligand was found to be wholly overlaid on the native co-crystallized ligand (Fig. S1†). The re-docking and superimposition protocol gave good evidence for docking protocol validation.
Table 6 displays the docking scores of the examined compounds (4–13) against the 5IKT enzyme. The docking scores (S) exhibited a range between −8.82 kcal mol−1 (for 4) and −6.99 kcal mol−1 (for 9).
| Ligand | Receptor | Interaction | Distance | E (kcal mol−1) | S (kcal mol−1) | |
|---|---|---|---|---|---|---|
| 4 | C5 | HIS 388 | H-donor | 3.45 | −1.70 | −8.82 |
| N8 | HIS 388 | H-donor | 3.44 | −2.40 | ||
| O14 | THR 212 | H-acceptor | 3.58 | −0.70 | ||
| 6-ring | HIS 207 | Pi-cation | 3.59 | −0.60 | ||
| 5 | N8 | HIS 388 | H-donor | 3.44 | −0.70 | −7.67 |
| N20 | GLN 454 | H-acceptor | 3.24 | −2.50 | ||
| 6-ring | HIS 386 | Pi-cation | 3.37 | −0.60 | ||
| 6 | N23 | GLN 454 | H-donor | 2.99 | −1.60 | −7.10 |
| O11 | HIS 207 | H-acceptor | 3.25 | −0.90 | ||
| N16 | HIS 207 | H-acceptor | 3.01 | −6.90 | ||
| N20 | ASN 382 | H-acceptor | 3.48 | −1.20 | ||
| 6-ring | LEU 294 | Pi-H | 4.13 | −0.70 | ||
| 7 | N12 | ASP 213 | H-donor | 2.93 | −5.30 | −7.32 |
| O10 | GLN 289 | H-acceptor | 3.51 | −1.00 | ||
| O13 | LYS 215 | H-acceptor | 2.97 | −5.90 | ||
| O33 | GLN 203 | H-acceptor | 3.21 | −2.00 | ||
| N43 | HIS 214 | H-acceptor | 3.20 | −2.90 | ||
| 5-ring | HIS 207 | Pi-cation | 4.08 | −0.80 | ||
| 8 | C5 | HIS 388 | H-donor | 3.53 | −1.30 | −7.46 |
| N8 | HIS 388 | H-donor | 3.41 | −8.70 | ||
| N13 | ASN 382 | H-donor | 3.06 | −0.90 | ||
| O14 | THR 212 | H-acceptor | 3.49 | −1.10 | ||
| 6-ring | HIS 207 | Pi-cation | 3.83 | −0.70 | ||
| 5-ring | LEU 294 | Pi-H | 4.22 | −0.70 | ||
| 6-ring | ILE 408 | Pi-H | 4.21 | −0.60 | ||
| 9 | N8 | HIS 388 | H-donor | 3.04 | −15.00 | −6.99 |
| 5-ring | HIS 207 | Pi-cation | 3.99 | −0.90 | ||
| 6-ring | THR 212 | Pi-H | 3.62 | −1.00 | ||
| 6-ring | LEU 294 | Pi-H | 4.15 | −0.80 | ||
| 10 | N7 | PHE 210 | H-donor | 3.42 | −1.30 | −7.43 |
| N23 | TYR 385 | H-donor | 3.15 | −0.70 | ||
| 11 | N13 | GLU 290 | H-donor | 3.08 | −1.10 | −7.82 |
| N23 | GLN 454 | H-donor | 3.17 | −1.00 | ||
| O30 | HIS 388 | H-donor | 2.94 | −11.80 | ||
| O11 | THR 212 | H-acceptor | 2.95 | −2.70 | ||
| O14 | ASN 222 | H-acceptor | 3.03 | −0.80 | ||
| 12 | N13 | ALA 443 | H-donor | 3.16 | −0.80 | −7.76 |
| N23 | ASN 382 | H-donor | 2.99 | −1.00 | ||
| 5-ring | HIS 207 | Pi-cation | 3.22 | −0.80 | ||
| 6-ring | THR 212 | Pi-H | 4.11 | −0.70 | ||
| 13 | N13 | TRP 387 | H-donor | 2.88 | −1.30 | −7.37 |
| O11 | GLN 203 | H-acceptor | 3.24 | −1.50 | ||
| 6-ring | GLN 203 | Pi-H | 3.63 | −0.60 | ||
| 5-ring | HIS 207 | Pi-cation | 3.45 | −2.30 |
Compound 4 exhibited the highest activity with the most negative score among the compounds, with a value of −8.82 kcal mol−1. Following this, compound 12 displayed a score of −7.76 kcal mol−1, compound 5 had a score of −7.67 kcal mol−1, compound 8 had a score of −7.46 kcal mol−1, compound 10 had a score of −7.43 kcal mol−1, compound 13 had a score of −7.37 kcal mol−1, compound 7 had a score of −7.32 kcal mol−1, compound 6 had a score of −7.10 kcal mol−1, and finally compound 9 had a score of −6.99 kcal mol−1. Accordingly, compound 9 exhibited the lowest activity with the least negative score of −6.99 kcal mol−1. Fig. 4 illustrates the precise binding positions of the compounds under investigation inside the active site of the 5IKT interaction in both three-dimensional (3D) and two-dimensional (2D) representations. Additionally, Table 6 presents the docking data associated with these compounds.
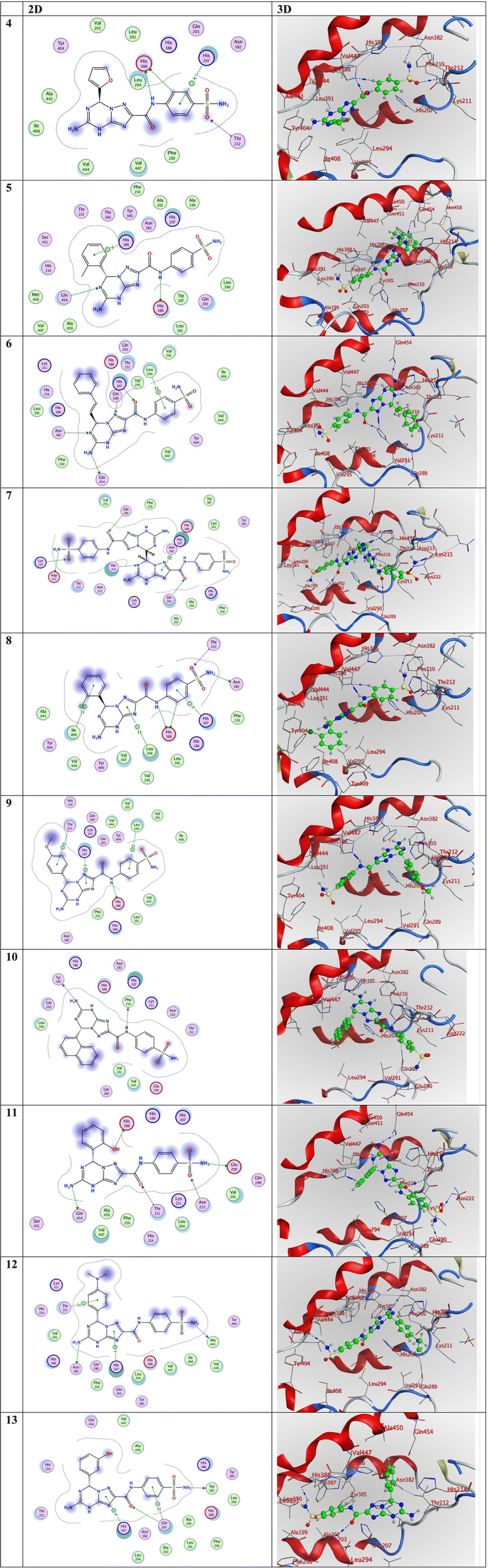 | ||
| Fig. 4 3D and 2D representations of the molecular interactions of the investigated compounds (from compound 4 to compound 13) against the PDB ID: 5IKT. | ||
In the examination of the molecular interactions in compound 4, it is shown that two H-donor and one H-acceptor bonds are established between C5 with HIS388, N8 with HIS388, and O14 with THR212, respectively, at distances of 3.45, 3.44, and 3.58 Å, respectively. Furthermore, it is seen that one pi-cation interaction was established between 6-ring with HIS207 at distance of 3.59 Å. These findings are listed in Table 6. In the instance of compound 11, three H-donors and two H-acceptor bonds are established between N13 with GLU290, N23 with GLN454, O30 with HIS388, O11 with THR212, and O14 with ASN222, respectively, at distances of 3.08, 3.17, 2.94, 2.95, and 3.03 Å, respectively. In compound 12, two hydrogen-donor bonds were observed, with distances of 3.16 and 2.99 Å, between N13 with ALA443 and N23 with ASN382, respectively (Table 6). Also, one pi-cation and one pi-H interactions were established between 5-ring with HIS207 and 6-ring with THR212 at 3.22 and 4.11 Å, respectively, as shown in Table 6.
3.5. Density functional theory (DFT) exploration
DFT calculations were performed to correlate the calculated HOMO–LUMO energy differences with the in vitro biological activity results, looking for patterns or trends that may help explain the observed experimental outcomes.67 Indomethacin was selected as it was used as the standard in the in vitro biological activity experiments. Considering the structural variations between the target compounds and indomethacin, we assess how these structural differences influence the electronic properties and reactivity and whether they can be linked to the biological activity.The DFT investigation was explored via the B3LYP functional with the 6–311g(d,p) basis set. Fig. 5 displays the DFT assessments of the two highest-ranking compounds (4 and 11) obtained from the screening method and those of the reference drug indomethacin. The results of the DFT analysis demonstrated that the HOMO–LUMO energy differences (ΔE) of compounds 4 (2.43 eV) and 11 (2.18 eV) were lower than that of indomethacin (2.95 eV). This finding indicates the possibility and significance of molecular charge transfer.68–70 Furthermore, the hardness (η, eV), and softness (σ, eV−1) values of the two highest-ranking compounds (4 and 11) identified by the screening approach, together with those of the reference drug indomethacin, were determined by evaluating the energies of their HOMO and LUMO orbitals using the Parr and Pearson interpretation. Interestingly, compounds 4 and 5 had the highest chemical softness values (0.82 eV−1 and 0.92 eV−1, respectively) and lowest chemical hardness values (1.22 eV and 1.09 eV, respectively) compared to those of indomethacin (0.68 eV−1 and 1.47 eV, respectively), which might be responsible for the increased chemical reactivity compared to indomethacin.
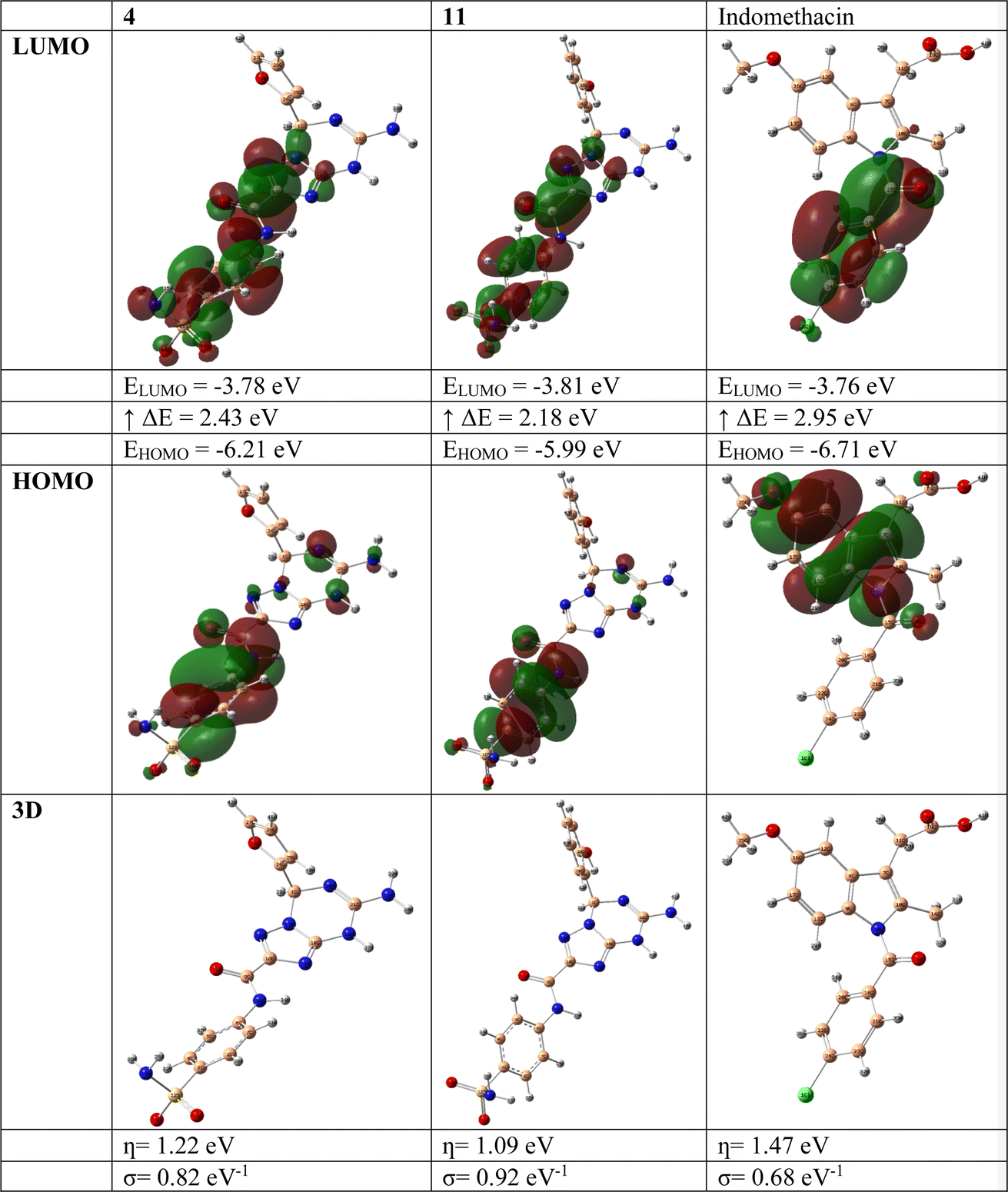 | ||
| Fig. 5 DFT exploration of compounds 4 and 11 and reference drug indomethacin. | ||
4. Conclusion
The synthesis and characterization of new fused heterocyclic compounds based on purine analogues made from thiocarbohydrazide are reported in this study. The preparation method involved stirring conventionally in ethanol with drops of concentrated HCl. This is a simple process that provides high purities, high yields of triazolo-triazines with a sulfamoyl moiety, and the anticipated biological activity of the compounds. The molecular structures of the synthesized compounds were verified using spectroscopic methods and the compounds demonstrated highly significant anti-inflammatory activity. Particularly, compounds 4, 5, 8, 11, and 12 exhibited excellently effective RBC membrane stabilization, inhibition of protein denaturation, and inhibition of COX enzymes compared to those of indomethacin. According to the in vitro anti-inflammatory activity, compounds 4, 5, 8, 11 and 12 are promising as therapeutic candidates for treating inflammatory diseases. Careful and systematic plans could lead to further research employing innovative techniques and animal models. The COX inhibition study showed that these compounds exhibit remarkable COX-1/COX-2 inhibition, comparable with conventional indomethacin. COX 2 (PDB ID: 5IKT) was docked with the ten synthetic compounds (4–13). Compounds 4, 11 and 12 had the highest binding affinity. The results suggest that these compounds inhibit COX 2 (PDB ID: 5IKT) and could be further studied for COX 2 targeting. DFT analysis was performed on the two most favorable compounds (4 and 11) identified through the screening methodology and the reference pharmaceutical agent indomethacin. The DFT analysis revealed that compounds 4 and 11 exhibited a reduced HOMO–LUMO energy difference in comparison to that of indomethacin. This discovery elucidates the potential importance of intermolecular charge transfer in this system.Animal rights
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Sohag University and experiments were approved by the Animal Ethics Committee of “Committee for Scientific Research Ethics (CSRE) of Faculty of Science, Sohag University, Sohag, Egypt (protocol no. CSRE-26-23)”.Conflicts of interest
There are no conflicts to declare.References
- N. Fujiwara and K. Kobayashi, Macrophages in inflammation, Curr. Drug Targets: Inflammation Allergy, 2005, 4(3), 281–286 CAS.
- D. A. Hume, The many alternative faces of macrophage activation, Front. Immunol., 2015, 6, 153172 CrossRef PubMed.
- R. Kin, J. Notarte, T. Quimque, K. Macaranas, P. Adriel, V. Oliver, P. Hans, H. Arturo, I. V. Pilapil, M. Sophia, D. Tan, D. Q. Wei, W. Arlette, T. Deniz, Y. Chia-Hung, Ji. Seon, K. Gi-Young, C. Yung and M. Allan, ACS Omega, 2023, 8(6), 5377–5392, DOI:10.1021/acsomega.2c06451.
- N. Ansari, F. Khodagholi, M. Ramin, M. Amini, H. Irannejad, L. Dargahi and A. D. Amirabad, Inhibition of LPS-induced apoptosis in differentiated-PC12 cells by new triazine derivatives through NF-κB-mediated suppression of COX-2, Neurochem. Int., 2010, 57(8), 958–968 CrossRef CAS PubMed.
- Q. Cao, X. Wang, L. Jia, A. K. Mondal, A. Diallo, G. A. Hawkins, S. K. Das, J. S. Parks, L. Yu and H. Shi, Inhibiting DNA methylation by 5-Aza-2′-deoxycytidine ameliorates atherosclerosis through suppressing macrophage inflammation, Endocrinology, 2014, 155(12), 4925–4938 CrossRef PubMed.
- L. Winand, S. Angela and N. Markus, Bioengineering of Antiinflammatory Natural Products, ChemMedChem, 2021, 16(5), 767–776 CrossRef CAS PubMed.
- R. Kumar, T. Sirohi, H. Singh, R. Yadav, R. Roy, A. Chaudhary and S. Pandeya, 1, 2, 4-triazine analogs as novel class of therapeutic agents, Mini-Rev. Med. Chem., 2014, 14(2), 168–207 CrossRef CAS PubMed.
- O. A. Abd Allah, A. M. El-Saghier, A. M. Kadry and A. A. Seleem, Synthesis and Evaluation of Some Novel Curcumin Derivatives as Anti-inflammatory Agents, Int. J. Pharm. Sci. Rev. Res., 2015, 32(1), 87–92 Search PubMed.
- A. Tsar’kov, S. Smurov, E. Maevsky, I. Sedova, L. Bogdanova, V. Volsky and M. Kozhurin, Some Innovative Approaches To Maintaining The Adaptive Response Of The Organism Under Stress, Mar. Med., 2018, 4(1), 85–95 CrossRef.
- M. Legraverend and D. S. Grierson, The purines: Potent and versatile small molecule inhibitors and modulators of key biological targets, Biorg. Med. Chem., 2006, 14(12), 3987–4006 CrossRef CAS PubMed.
- M. J. Perry and G. A. Higgs, Chemotherapeutic potential of phosphodiesterase inhibitors, Curr. Opin. Chem. Biol., 1998, 2(4), 472–481 CrossRef CAS PubMed.
- G. L. Russo, M. Russo and P. Ungaro, AMP-activated protein kinase: a target for old drugs against diabetes and cancer, Biochem. Pharmacol., 2013, 86(3), 339–350 CrossRef CAS PubMed.
- B. B. Fredholm, A. P. IJzerman, K. A. Jacobson, J. Linden and C. E. Müller, International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors—an update, Pharmacol. Rev., 2011, 63(1), 1–34 CrossRef CAS PubMed.
- A. Bzowska, E. Kulikowska and D. Shugar, Purine nucleoside phosphorylases: properties, functions, and clinical aspects, Pharmacol. Ther., 2000, 88(3), 349–425 CrossRef CAS PubMed.
- A. K. Werner and C.-P. Witte, The biochemistry of nitrogen mobilization: purine ring catabolism, Trends Plant Sci., 2011, 16(7), 381–387 CrossRef CAS PubMed.
- A. Bloch, R. J. Leonard and C. A. Nichol, On the mode of action of 7-deaza-adenosine (tubercidin), Biochim. Biophys. Acta, Nucleic Acids Protein Synth., 1967, 138(1), 10–25 CrossRef CAS PubMed.
- R. M. McCarty and V. Bandarian, Deciphering deazapurine biosynthesis: pathway for pyrrolopyrimidine nucleosides toyocamycin and sangivamycin, Chem. Biol., 2008, 15(8), 790–798 CrossRef CAS PubMed.
- A. V. Dolzhenko, A. V. Dolzhenko and W. K. Chui, 1, 2, 4-triazolo [1, 5-a][1, 3, 5] triazines (5-azapurines): synthesis and biological activity, Heterocycles, 2006, 68(8), 1723–1759 CrossRef CAS.
- R. Chovatiya and R. Medzhitov, Stress, inflammation, and defense of homeostasis, Mol. Cell, 2014, 54(2), 281–288 CrossRef CAS PubMed.
- A. Dolzhenko, A. Dolzhenko and W. Chui, Pyrazolo [1, 5-a][1, 3, 5] triazines (5-aza-9-deazapurines): synthesis and biological activity, Heterocycles, 2008, 75(7), 1575–1622 CrossRef CAS.
- S. R. Al-Ayash, T. H. Al-Noor and A. Abdou, Synthesis and Characterization of Metals Complexes with Uracil and Uracil Derivatives (A Review), Russ. J. Gen. Chem., 2023, 93(4), 987–995, DOI:10.1134/S107036322304028X.
- U. H. Hasan, A. M. Uttra and S. Rasool, Evaluation of in vitro and in vivo anti-arthritic potential of Berberis calliobotrys, Bangladesh Journal of Pharmacology, 2015, 10(4), 807–819 CrossRef.
- U. Shinde, A. Phadke, A. Nair, A. Mungantiwar, V. Dikshit and M. Saraf, Membrane stabilizing activity—a possible mechanism of action for the anti-inflammatory activity of Cedrus deodara wood oil, Fitoterapia, 1999, 70(3), 251–257 CrossRef CAS.
- M. Murias, N. Handler, T. Erker, K. Pleban, G. Ecker, P. Saiko, T. Szekeres and W. Jäger, Resveratrol analogues as selective cyclooxygenase-2 inhibitors: synthesis and structure–activity relationship, Biorg. Med. Chem., 2004, 12(21), 5571–5578 CrossRef CAS PubMed.
- C. Scholz, S. Knorr, K. Hamacher and B. Schmidt, DOCKTITE A Highly Versatile Step-by-Step Workflow for Covalent Docking and Virtual Screening in the Molecular Operating Environment, J. Chem. Inf. Model., 2015, 55(2), 398–406 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules, Sci. Rep., 2017, 7(1), 42717 CrossRef PubMed.
- N. Z. Ismail, N. Annamalai, N. N. M. Zain and H. Arsad, J. Biol. Sci. Opin., 2020, 8, 4–11 CrossRef CAS.
- A. M. Najar, A. Eswayah, M. B. Moftah, M. K. R. Omar, E. Bobtaina, M. Najwa, T. A. Elhisadi, A. Tahani, S. M. Tawati and A. M. M. Khalifa, et al., Rigidity and Flexibility of Pyrazole, s-Triazole, and v-Triazole Derivative of Chloroquine as Potential Therapeutic against COVID-19, J. Med. Chem. Sci., 2023, 6(9), 2056–2084, DOI:10.26655/jmchemsci.2023.9.14.
- S. Shaaban, Y. S. Al-Faiyz, G. M. Alsulaim, M. Alaasar, N. Amri, H. Ba-Ghazal, A. A. Al-Karmalawy and A. Abdou, Synthesis of new organoselenium-based succinanilic and maleanilic derivatives and in silico studies as possible SARS-CoV-2 main protease inhibitors, Inorganics, 2023, 11(8), 321 CrossRef CAS.
- B. J. Orlando and M. G. Malkowski, Substrate-selective inhibition of cyclooxygeanse-2 by fenamic acid derivatives is dependent on peroxide tone, J. Biol. Chem., 2016, 291(29), 15069–15081 CrossRef CAS PubMed.
- T. Murugan, R. Venkatesh, K. Geetha and A. Abdou, Synthesis, spectral investigation, DFT, antibacterial, antifungal and molecular docking studies of Ni (II), Zn (II), Cd (II) complexes of tetradentate Schiff-base ligand, Asian J. Chem., 2023, 35, 1509–1517 CAS.
- H. M. Abd El-Lateef, M. M. Khalaf, A. A. Amer, M. Kandeel, A. A. Abdelhamid and A. Abdou, Synthesis, characterization, antimicrobial, density functional theory, and molecular docking studies of novel Mn (II), Fe (III), and Cr (III) complexes incorporating 4-(2-hydroxyphenyl azo)-1-naphthol (Az), ACS Omega, 2023, 8(29), 25877–25891 CrossRef CAS PubMed.
- K. R. Valasani, J. R. Vangavaragu, V. W. Day and S. S. Yan, Structure based design, synthesis, pharmacophore modeling, virtual screening, and molecular docking studies for identification of novel cyclophilin D inhibitors, J. Chem. Inf. Model., 2014, 54(3), 902–912 CrossRef CAS PubMed.
- E. M. Terefe and A. Ghosh, Molecular docking, validation, dynamics simulations, and pharmacokinetic prediction of phytochemicals isolated from Croton dichogamus against the HIV-1 reverse transcriptase, Bioinf. Biol. Insights, 2022, 16, 11779322221125605 Search PubMed.
- K. E. Hevener, W. Zhao, D. M. Ball, K. Babaoglu, J. Qi, S. W. White and R. E. Lee, Validation of molecular docking programs for virtual screening against dihydropteroate synthase, J. Chem. Inf. Model., 2009, 49(2), 444–460 CrossRef CAS PubMed.
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38(6), 3098 CrossRef CAS PubMed.
- M. A. E. A. A. A. El-Remaily, O. Elhady, A. Abdou, D. Alhashmialameer, T. N. A. Eskander and A. M. Abu-Dief, Development of new 2-(Benzothiazol-2-ylimino)-2,3-dihydro-1H-imidazole-4-ol complexes as a robust catalysts for synthesis of thiazole 6-carbonitrile derivatives supported by DFT studies, J. Mol. Struct., 2023, 1292, 136188, DOI:10.1016/j.molstruc.2023.136188.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37(2), 785 CrossRef CAS PubMed.
- J. A. Plumley and J. Dannenberg, A comparison of the behavior of functional/basis set combinations for hydrogen-bonding in the water dimer with emphasis on basis set superposition error, J. Comput. Chem., 2011, 32(8), 1519–1527 CrossRef CAS PubMed.
- A. Tomberg, Gaussian 09w tutorial, An Introduction to Computational Chemistry Using G09W and Avogadro Software, 2013, pp. 1–36 Search PubMed.
- A. Abdou, O. A. Omran, J. H. Al-Fahemi, R. S. Jassas, M. M. Al-Rooqi, E. M. Hussein, Z. Moussa and S. A. Ahmed, Lower rim thiacalixarenes derivatives incorporating multiple coordinating carbonyl groups: Synthesis, characterization, ion-responsive ability and DFT computational analysis, J. Mol. Struct., 2023, 1293, 136264, DOI:10.1016/j.molstruc.2023.136264.
- N. A. Elkanzi, A. M. Kadry, R. M. Ryad, R. B. Bakr, M. A. E. A. A. Ali El-Remaily and A. M. Ali, Efficient and recoverable bio-organic catalyst cysteine for synthesis, docking study, and antifungal activity of new bio-active 3, 4-dihydropyrimidin-2 (1 H)-ones/thiones under microwave irradiation, ACS Omega, 2022, 7(26), 22839–22849 CrossRef CAS PubMed.
- A. M. El-Saghier, A. Abdul-Baset, O. M. El-Hady and A. M. Kadry, Synthesis of some new thiadiazole/thiadiazine derivatives as potent biologically active compounds, Sohag J. Sci., 2023, 8(3), 371–375 Search PubMed.
- A. M. El-Saghier, S. S. Enaili, A. Abdou, A. M. Hamed and A. M. Kadry, An operationally simple, one-pot, convenient synthesis, and in vitro anti-inflammatory activity of some new spirotriazolotriazine derivatives, J. Heterocycl. Chem., 2024, 61(1), 146–162 CrossRef CAS.
- O. A. Abd Allah, A. M. El-Saghier and A. M. Kadry, Synthesis, structural stability calculation, and antibacterial evaluation of novel 3, 5-diphenylcyclohex-2-en-1-one derivatives, Synth. Commun., 2015, 45(8), 944–957 CrossRef CAS.
- A. H. Abdelmonsef, A. M. El-Saghier and A. M. Kadry, Ultrasound-assisted green synthesis of triazole-based azomethine/thiazolidin-4-one hybrid inhibitors for cancer therapy through targeting dysregulation signatures of some Rab proteins, Green Chem. Lett. Rev., 2023, 16(1), 2150394 CrossRef.
- A. M. El-Saghier, M. A. Mohamed, O. A. Abdalla and A. M. Kadry, Utility of amino acid coupled 1,2,4-triazoles in organic synthesis: synthesis of some new antileishmainal agents, Bull. Chem. Soc. Ethiop., 2018, 32(3), 559–570, DOI:10.4314/bcse.v32i3.14.
- M. A. Mohamed, A. M. Kadry, S. A. Bekhit, M. A. S. Abourehab, K. Amagase, T. M. Ibrahim and A. A. Bekhit, Spiro heterocycles bearing piperidine moiety as potential scaffold for antileishmanial activity: synthesis, biological evaluation, and in silico studies, J. Enzyme Inhib. Med. Chem., 2023, 38(1), 330–342, DOI:10.1080/14756366.2022.215076.
- A. M. El-Saghier, S. S. Enaili, A. Abdou and A. M. Kadry, An efficient eco-friendly, simple, and green synthesis of some new spiro-N-(4-sulfamoyl-phenyl)-1, 3, 4-thiadiazole-2-carboxamide derivatives as potential inhibitors of SARS-CoV-2 proteases: drug-likeness, pharmacophore, molecular docking, and DFT exploration, Mol. Diversity, 2023, 1–22 Search PubMed.
- A. M. El-Saghier, A. Abdou, M. A. Mohamed, H. M. Abd El-Lateef and A. M. Kadry, Novel 2-acetamido-2-ylidene-4-imidazole derivatives (El-Saghier reaction): Green synthesis, biological assessment, and molecular docking, ACS Omega, 2023, 8(33), 30519–30531 CrossRef CAS PubMed.
- A. M. El-Saghier, S. S. Enaili, A. M. Kadry, A. Abdou and M. A. Gad, Green synthesis, biological and molecular docking of some novel sulfonamide thiadiazole derivatives as potential insecticidal against Spodoptera littoralis, Sci. Rep., 2023, 13(1), 19142 CrossRef CAS PubMed.
- M. A. Mohamed, A. A. Bekhit, O. A. Abd Allah, A. M. Kadry, T. M. Ibrahim, S. A. Bekhit, A. Kikuko and A. M. El-Saghier, Synthesis and antimicrobial activity of some novel 1,2-dihydro-[1,2,4]triazolo[1,5-a]pyrimidines bearing amino acid moiety, RSC Adv., 2021, 11, 2905–2916 RSC.
- J. C. Maroon, J. W. Bost and A. Maroon, Natural anti-inflammatory agents for pain relief, Surg. Neurol. Int., 2010, 1, 80–87 CrossRef PubMed.
- S. Harirforoosh, W. Asghar and F. Jamali, Adverse effects of nonsteroidal antiinflammatory drugs: an update of gastrointestinal, cardiovascular and renal complications, J. Pharm. Pharm. Sci., 2013, 16(5), 821–847 Search PubMed.
- H. ur Rashid, Y. Xu, N. Ahmad, Y. Muhammad and L. Wang, Promising anti-inflammatory effects of chalcones via inhibition of cyclooxygenase, prostaglandin E2, inducible NO synthase and nuclear factor κb activities, Bioorg. Chem., 2019, 87, 335–365 CrossRef CAS PubMed.
- G. Yuan, M. L. Wahlqvist, G. He, M. Yang and D. Li, Natural products and anti-inflammatory activity, Asia Pac. J. Clin. Nutr., 2006, 15(2), 143–152 CAS.
- Y. Ju and R. S. Varma, Aqueous N-heterocyclization of primary amines and hydrazines with dihalides: microwave-assisted syntheses of N-azacycloalkanes, isoindole, pyrazole, pyrazolidine, and phthalazine derivatives, J. Org. Chem., 2006, 71(1), 135–141 CrossRef CAS PubMed.
- L. Winand, A. Sester and M. Nett, Bioengineering of anti-inflammatory natural products, ChemMedChem, 2021, 16(5), 767–776 CrossRef CAS PubMed.
- M. García-Valverde and T. Torroba, Sulfur-nitrogen Heterocycles, 2005, vol. 10, pp. 318–320 Search PubMed.
- X.-Y. Meng, H.-X. Zhang, M. Mezei and M. Cui, Molecular docking: a powerful approach for structure-based drug discovery, Curr. Comput.-Aided Drug Des., 2011, 7(2), 146–157 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Delivery Rev., 1997, 23(1–3), 3–25 CrossRef CAS.
- A. Daina and V. Zoete, A boiled-egg to predict gastrointestinal absorption and brain penetration of small molecules, ChemMedChem, 2016, 11(11), 1117–1121 CrossRef CAS PubMed.
- H. M. Abd El-Lateef, M. M. Khalaf, M. Kandeel and A. Abdou, Synthesis, Characterization, DFT, Biological and Molecular Docking of Mixed Ligand Complexes of Ni(II), Co(II), and Cu(II) Based on Ciprofloxacin and 2-(1H-benzimidazol-2-yl)phenol, Inorg. Chem. Commun., 2023, 155, 111087, DOI:10.1016/j.inoche.2023.111087.
- L. Pinzi and G. Rastelli, Molecular docking: shifting paradigms in drug discovery, Int. J. Mol. Sci., 2019, 20(18), 4331 CrossRef CAS PubMed.
- H. M. Abd El-Lateef, M. M. Khalaf, F. El-Taib Heakal and A. Abdou, Fe(III), Ni(II), and Cu(II)-moxifloxacin-tri-substituted imidazole mixed ligand complexes: Synthesis, structural, DFT, biological, and protein-binding analysis, Inorg. Chem. Commun., 2023, 158, 111486, DOI:10.1016/j.inoche.2023.111486.
- H. M. Abd El-Lateef, A. M. Ali, M. M. Khalaf and A. Abdou, New iron (III), cobalt (II), nickel (II), copper (II), zinc (II) mixed-ligand complexes: Synthesis, structural, DFT, molecular docking and antimicrobial analysis, Bull. Chem. Soc. Ethiop., 2024, 38(1), 147–166 CrossRef CAS.
- R. K. Mohapatra, A. Mahal, A. Ansari, M. Kumar, J. P. Guru, A. K. Sarangi, A. Abdou, S. Mishra, M. Aljeldah and B. M. AlShehail, et al., Comparison of the binding energies of approved mpox drugs and phytochemicals through molecular docking, molecular dynamics simulation, and ADMET studies: An in silico approach, J. Biosaf. Biosecur., 2023, 5(3), 118–132, DOI:10.1016/j.jobb.2023.09.001.
- R. G. Pearson, The HSAB principle—more quantitative aspects, Inorg. Chim. Acta, 1995, 240(1–2), 93–98 CrossRef CAS.
- A. M. El-Saghier, H. F. Abd El-Halim, L. H. Abdel-Rahman and A. Kadry, Green Synthesis of new Trizole Based Heterocyclic Amino Acids Ligands and their Transition Metal Complexes. Characterization, Kinetics, Antimicrobial and Docking Studies, Appl. Organomet. Chem., 2019, 33, e4641, DOI:10.1002/aoc.464118.
- M. A. E. A. A. A. El-Remaily, T. El-Dabea, R. M. El-Khatib, A. Abdou, M. A. El Hamd and A. M. Abu-Dief, Efficiency and development of guanidine chelate catalysts for rapid and green synthesis of 7-amino-4,5-dihydro-tetrazolo[1,5-a]pyrimidine-6-carbonitrile derivatives supported by density functional theory (DFT) studies, Appl. Organomet. Chem., 2023, 37(11), e7262, DOI:10.1002/aoc.7262.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra02970d |
| This journal is © The Royal Society of Chemistry 2024 |