
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile and efficient transformation of thiols to disulfides via a radical pathway with N-anomeric amide†
Xiaobo Xu *,
Leyu Yan,
Weijie Huang,
Yanping Wang,
Mengya Wang,
Liming Feng,
Panpan Wang and
Shengqiang Wang*
*,
Leyu Yan,
Weijie Huang,
Yanping Wang,
Mengya Wang,
Liming Feng,
Panpan Wang and
Shengqiang Wang*
College of Chemistry and Pharmaceutical Engineering, Huanghuai University, Zhumadian 463000, People's Republic of China. E-mail: xuxb@huanghuai.edu.cn; wshq988@126.com
First published on 3rd June 2024
Abstract
Radical coupling of thiols is an attractive route for the synthesis of disulfides, but this approach should be promoted by strong oxidants and/or metal salts in combination with additives, which limits its substrate scope and application. In this work, the N-anomeric amide was first found to be able to realize the conversion of thiols to sulfur radicals with high efficiency in the absence of an oxidant or any additives for the synthesis of symmetrical disulfides. The protocol features mild reaction conditions, good functional group tolerance, and moderate to excellent yields.
Introduction
Organosulfur compounds, although usually associated with unpleasant odors, are considered non-toxic and have important functions in natural compounds,1 biological agents and drugs,2 as well as in advanced materials.3 Among compounds containing sulfur, disulfides play a dominant role in biological systems, since they are fundamental factors in protein folding and oligomerization, as they stabilize the 3D structure and affect their biological function.4 Disulfides are also important intermediates for many synthetic pathways and present various applications in agro-chemicals and pharmaceuticals.5 These findings render disulfides as moieties of great importance and synthetic interest. In recent decades, various feasible strategies have been developed for the synthesis of symmetrical disulfides, such as thiolysis,6 reductive dimerization of thiocyanates,7 nucleophilic substitution of S–H bonds,8 and reductive coupling of sulfonyl chlorides.9 However, the formation of byproducts arising from leaving groups is inevitable in these strategies. In contrast, extensive studies have indicated the direct dehydrogenation of thiols to disulfides followed by sulfur radical coupling, with H2 as the sole byproduct, and have attracted much attention due to their high atom and step-economy for the synthesis of disulfides (Scheme 1(a)).10 Oxidative coupling of thiols as a direct method avoids the pre-activation of thiols, which was promoted by strong oxidants or oxidants assisted with various catalysts.11 In order to use mild conditions, catalytic oxidation was explored by adopting various metal-based catalysts12 or heterogeneous catalysts,13 on the other hand, photochemistry14 and electrochemistry15 recently have drawn extensive attention. However, these methods suffer from some serious drawbacks such as over-oxidation, very high or low reaction temperatures, use of strong basic or acidic media and tedious work-up procedures. Moreover, the use of metal catalysts is not preferred for late-stage functionalization of biologically active molecules, such as peptides or other pharmaceuticals.16 Given the importance of organic disulfides, the development of mild and effective methods remains a major objective.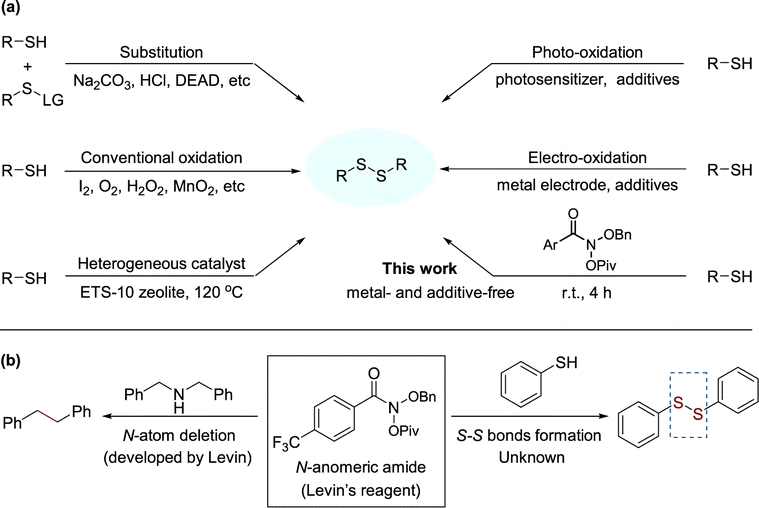 | ||
| Scheme 1 (a) Approaches for the synthesis of disulfides from thiols. (b) Structure and applications of the N-anomeric amide. | ||
In order to skeletal editing of nitrogen-containing molecules, Levin and coworkers developed the N-anomeric amide (Levin's reagent) that enables straightforward nitrogen deletion of secondary amines and deamination of primary amines (Scheme 1(b)).17 The transformation relies on the facile in situ generation of an isodiazene intermediate, which can undergo thermal homolytic N2 extrusion to forms a new C–C bond.17a Inspired by N-anomeric amide's excellent electrophilic properties17,18 and our long-term interest in organosulfur chemistry,19 we initially programmed a project aiming at the formation of S–S bonds starting from thiols. Subsequently, we have successfully detected of disulfide product when the reaction was conducted in thiol with Levin's reagent at room temperature (Scheme 1(b)). To the best of our knowledge, this is the first construction of S–S bond using N-anomeric amide beyond deaminative functionalization.20 Herein, we report our realization of a mild and efficient method for the synthesis of organic disulfides via a sulfur radical mechanism with N-anomeric amide.
Results and discussion
We mainly investigated several N-anomeric amide reagents to achieve S–S bond formation of 4-methylbenzenethiol 1a to yield disulfide 3a (Table 1). The reaction did not occur in the absence of the N-anomeric amide 2a (Table 1, entry 1). Using MeCN as a solvent at room temperature, the addition of N-anomeric amide 2a resulted in a successful formation of disulfide to give a desired product 3a in 58% yield (entry 2). The increase of 2a loading (1.2 equiv.) resulted in significantly increased yields (73%, entry 3), but the doubling of it did not give better reaction outcome (70%, entry 4). While the extension or reduction of reaction time led to a decreasing product yields in this reaction (entries 5 and 6). Another N-anomeric amide bearing electron-deficient substituent (2b) showed outstanding reactivity and smoothly afforded the corresponding disulfide 3a in superior yield (94%) (entry 7). However, the anomeric amide bearing electron-rich substituent (2c) was used to give inferior efficiency (entry 8). Screening of different solvents revealed the initially used MeCN was the best (entries 9–11).
|
||||
|---|---|---|---|---|
| Entry | N-Anomeric amdie (equiv.) | Solvent | Time (h) | Yieldb (%) |
| a Unless otherwise noted, reactions were performed with 1a (0.2 mmol), N-anomeric amide 2, solvent (1.0 mL), room temperature, under argon atmosphere.b Yields of isolated product are shown after silica gel chromatography. | ||||
| 1 | — | MeCN | 4 | 0 |
| 2 | 2a (1.0) | MeCN | 4 | 58 |
| 3 | 2a (1.2) | MeCN | 4 | 73 |
| 4 | 2a (2.0) | MeCN | 4 | 70 |
| 5 | 2a (1.2) | MeCN | 6 | 65 |
| 6 | 2a (1.2) | MeCN | 2 | 51 |
| 7 | 2b (1.2) | MeCN | 4 | 94 |
| 8 | 2c (1.2) | MeCN | 4 | 62 |
| 9 | 2b (1.2) | DMF | 4 | 43 |
| 10 | 2b (1.2) | DMSO | 4 | 65 |
| 11 | 2b (1.2) | THF | 4 | 76 |
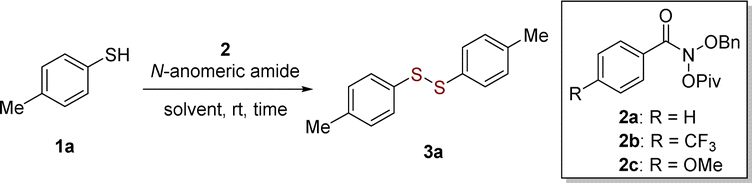
Having established the optimum reaction conditions, we then investigated the scope of substrate, such as aryl and heteroaryl thiols 3a–3o (Scheme 2). Substituted thiophenols bearing weak-electron-donating group (EDG) of para-Me and strong EDG of para-OMe reacted to yield the corresponding disulfides in excellent yields of 92–94% without obvious substituted effect (Scheme 2, 3a–3c). For meta- and para-substitutions of unprotected hydroxyl group on aromatic moiety were well tolerated, leading to products in 68–77% yields (Scheme 2, 3d and 3e). It is noteworthy that the homocoupling of functionalized aromatic thiols with amino group worked well to produces desired disulfide 3f in moderate yield. It follows that thiols have more reactivity than amines even in the presence of nitrogen-atom deletion reagent in this reaction.17b The substrates bearing electron-withdrawing groups (EWGs), such as F, Cl, Br and NO2 groups, also provided mediocre to satisfactory yields (up to 93%) (Scheme 2, 3g–3j). The bulky group ortho-CO2Me group induced a slight drop in the yield of 65% (Scheme 2, 3k). 2-Naphthyldisulfide (3l) with an extended π-framework was synthesized in 71% yield. Extending the scope beyond the aforementioned substrates, the protocol effectively facilitated the conversion of heteroaromatic (3m to 3o) thiols into their respective disulfides.
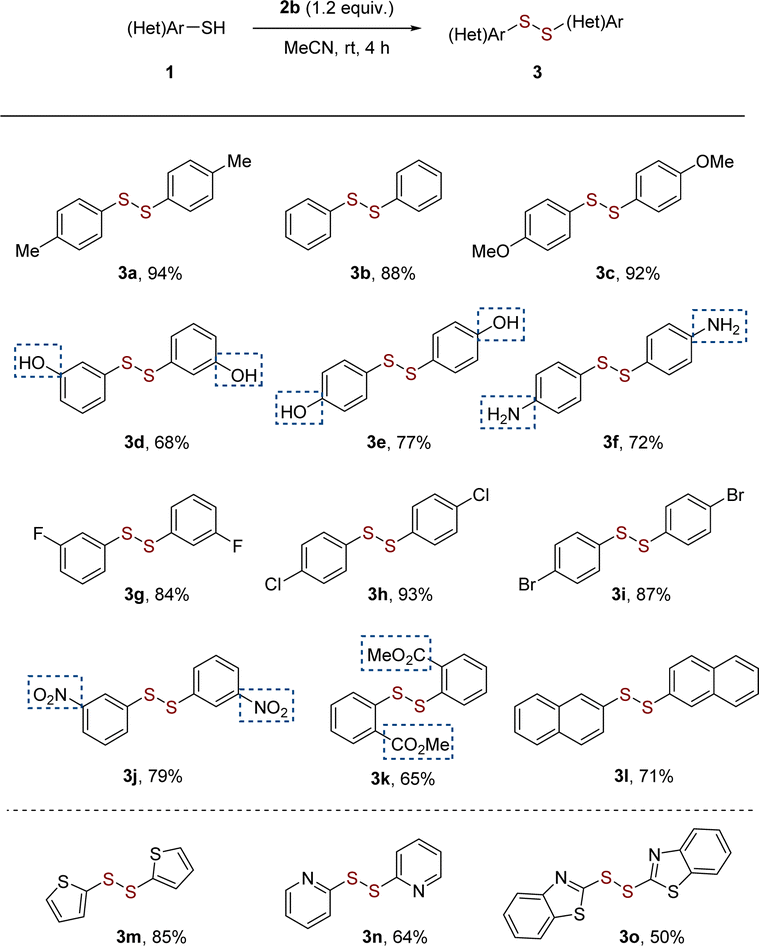 | ||
| Scheme 2 Substrate scope of aryl and heteroaryl thiols 1. Reaction conditions: 1 (0.2 mmol) and 2b (0.24 mmol), MeCN (1 mL), room temperature, 4 h, under argon atmosphere. Isolated yields. | ||
Encouraged by the results, the substrate scope was further expended to benzylic and aliphatic thiols 5a–5o (Scheme 3). The reported aerobic approaches to symmetrical disulfides always suffered from limited substrate scopes or unavailable catalysts. To our delight, the present strategy showed a broad substrate scope in the synthesis of both dibenzyl and dialkyl disulfides (Scheme 3). Compared to aryl thiols, the dimerization of aliphatic thiols to disulfides was more difficult and require longer reaction time. The benzylic thiols bearing electron-donating groups or electron-withdrawing groups, such as OMe, Cl and NO2 groups, also smoothly to afford disulfides 5a–5d in high yields. The heterocycle thiols bearing furan (5e) exhibited good coupling ability with acceptable product yields (76%). The straight-chain and branched aliphatic thiols (5f–5m) were also suitable reaction partners. The reaction has good compatibility with ester group (Scheme 3, 5n). Notably, the successful intramolecular coupling of unprotected 1,4-dithiothreitol afforded trans-4,5-dihydroxy-1,2-dithiane 5o with 75% yield, which revealed the potential in chemoselective construction of disulfide bridge in nature product synthesis.
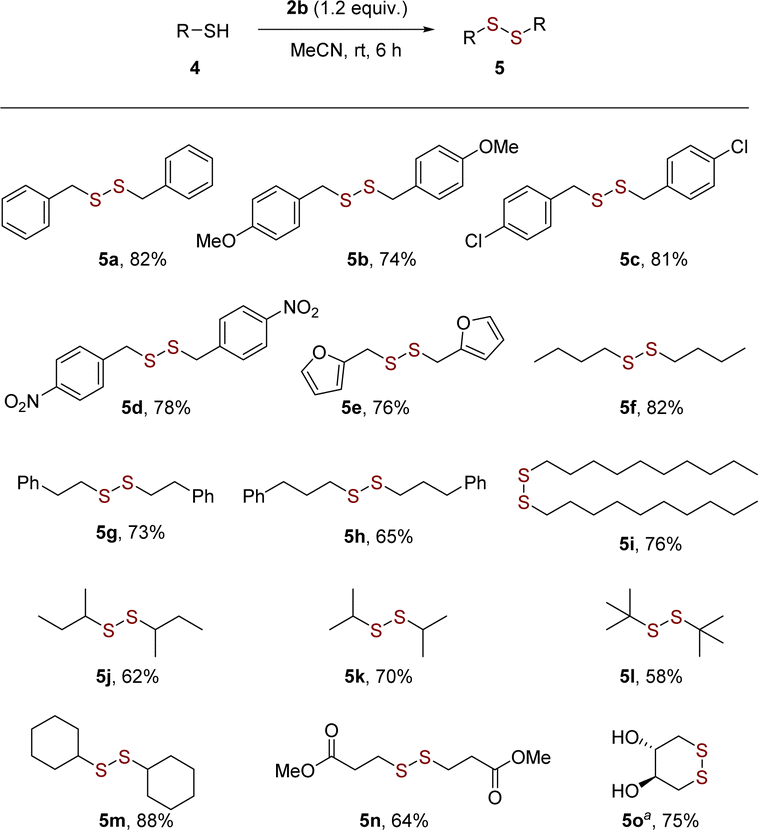 | ||
| Scheme 3 Substrate scope of benzylic and aliphatic thiols 4. Reaction conditions: 4 (0.2 mmol) and 2b (0.24 mmol), MeCN (1 mL), room temperature, 6 h, under argon atmosphere. Isolated yields. a2b (0.48 mmol), MeCN (4 mL). | ||
To gain further understanding about the reaction mechanism, control experiments were conducted. First, performing the reaction under air atmosphere instead of Ar resulted in product 3a with reduced yield, implying that the processes were partly restrained by the introduced air (Scheme 4(a)). Next, a radical capture experiment was performed and 2,2,6,6-tetramethylpiperidine N-oxide (TEMPO) was used as a radical scavenger in the reaction.21 When using 5.0 equiv. of TEMPO, the yield of 3a was decreased from 94% to 16%, the product yields were sufficiently suppressed, indicating radical intermediates were involved in the reaction. Then, we analyzed both reaction mixtures via GC-MS (Scheme 4(b)). The corresponding adduct 2,2,6,6-tetramethyl-1-((p-tolylthio)oxy)piperidine 6 of the thiyl radical with TEMPO was identified,22 illustrating that thiyl radical was produced during the reaction process. These results demonstrate our hypothesis that the N-anomeric amide under room temperature is able to generate a thiyl radical.
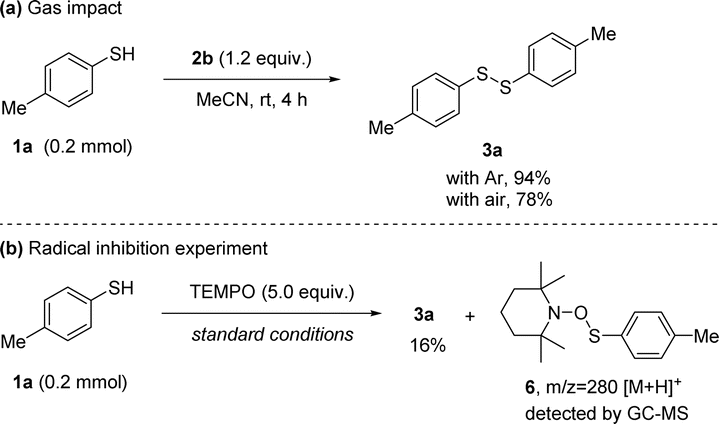 | ||
| Scheme 4 Mechanistic studies. | ||
Based on the aforementioned experimental results and preceding reports,17 a mechanistic scenario has been proposed to rationalize the observed reaction outcomes, as illustrated in Scheme 5. The reaction commences with the nucleophilic substitution of thiol 1b with Levin's reagent 2b, producing intermediate A. Subsequently, A undergoes homolysis of C–N bond to generate the p-toluenethiyl radical B and N-benzyloxy 4-(trifluoromethyl)benzoyl amide 7. Finally, the self-coupling of p-toluenethiyl radical B forms target product p-tolyl disulfide (3a).
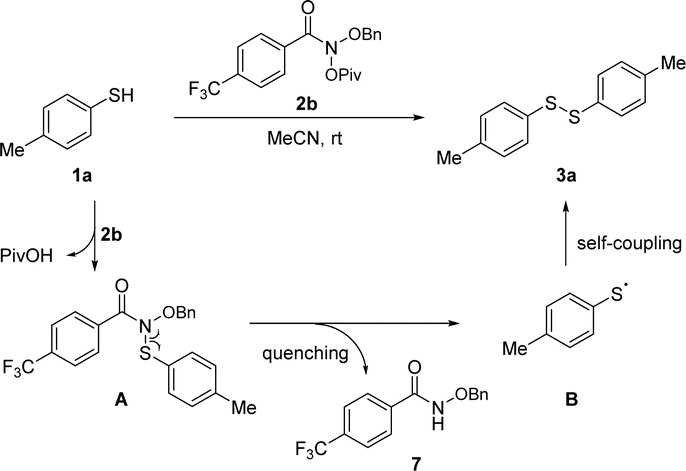 | ||
| Scheme 5 Plausible mechanism. | ||
Conclusions
In summary, we have developed a mild and convenient method for the S–S bonds homocoupling reaction of aromatic, heteroaromatic, benzylic and aliphatic thiols, offering a valuable complement to established methods for aerobic oxidation. This reaction is promoted by the readily accessible Levin's reagent in absence of metal and additive under room temperature. Significantly, this is the first application of N-anomeric amide in the synthesis of disulfides. Experimental mechanistic investigations have unveiled a radical mechanism in this reaction. Given the potential applications of the products and the synthetic practicality of the protocol, we anticipate that this method will find valuable utility in the realms of organic synthesis and medicinal chemistry.Experimental section
General procedure for the synthesis of disulfides
To a solution of thiol 1a (0.2 mmol) and N-anomeric amide 2b (0.24 mmol) in dry MeCN (1.0 mL) under argon atmosphere at room temperature. After 4 h the reaction was completed, the reaction was quenched with H2O (5 mL), and the resulting mixture was extracted with ethyl acetate (3 × 20 mL). The organic phases were combined, washed with saturated brine, dried over anhydrous Na2SO4 and then concentrated under vacuum. The residue was purified by flash chromatography (petroleum ether/ethyl acetate = 50/1–10/1) on silica gel to afford the desired product 3a. The products obtained were known compounds and were identified by NMR spectroscopy.Spectroscopy data of disulfides
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the National Natural Science Foundation of China (No. 22101096), the Programs for Science and Technology Development of Henan Province (No. 212102310329, 222102310559 and 222102310321), the Key Scientific Research Projects of Universities in Henan Province for financial support (No. 19A150033), and the National Project Cultivation Foundation of Huanghuai University (No. XKPY-2019006).References
- (a) R. J. Cremlyn, An Introduction to Organosulfur Chemistry, John Wiley & Sons, Chichester, 1996 Search PubMed; (b) M. Wang and X. Jiang, ACS Sustain. Chem. Eng., 2022, 10, 671–677 CrossRef CAS; (c) L. Melguizo-Rodríguez, E. García-Recio, C. Ruiz, E. De Luna-Bertos, R. Illescas-Montes and V. J. Costela-Ruiz, Food Funct., 2022, 13, 2415–2426 RSC.
- (a) N. Martins, S. Petropoulos and I. C. F. R. Ferreira, Food Chem., 2016, 211, 41–50 CrossRef CAS PubMed; (b) M. Mustafa and J. Y. Winum, Expet Opin. Drug Discov., 2022, 17, 501–512 CrossRef CAS PubMed.
- P. Sang, Q. Chen, D.-Y. Wang, W. Guo and Y. Fu, Chem. Rev., 2023, 123, 1262–1326 CrossRef CAS PubMed.
- (a) E. Gross, C. S. Sevier, A. Vala, C. A. Kaiser and D. Fass, Nat. Struct. Mol. Biol., 2002, 9, 61–67 CrossRef CAS PubMed; (b) J. R. Winther and C. Thorpe, Biochim. Biophys. Acta, 2014, 1840, 838–846 CrossRef CAS PubMed; (c) M. Gongora-Bernitez, J. Tulla-Puche and F. Albericio, Chem. Rev., 2014, 114, 901–926 CrossRef PubMed.
- (a) J.-X. Gong, X. Shen, L.-G. Yao, H. Jiang, K. Krohn and Y.-W. Guo, Org. Lett., 2007, 9, 1715–1716 CrossRef CAS PubMed; (b) B. Mandal and B. Basu, RSC Adv., 2014, 4, 13854–13881 RSC; (c) V. Pace, A. Pelosi, D. Antermite, O. Rosati, M. Curini and W. Holzer, Chem. Commun., 2016, 52, 2639–2642 RSC; (d) L. Lelo, V. Pillari, N. Gajic, W. Holzer and V. Pace, Chem. Commun., 2020, 56, 12395–12398 RSC.
- (a) M. Arisawa and M. Yamaguchi, J. Am. Chem. Soc., 2013, 125, 6624–6625 CrossRef PubMed; (b) K. Tanaka and K. Ajiki, Tetrahedron Lett., 2004, 45, 5677–5679 CrossRef CAS; (c) R. Hunter, M. Caira and N. Stellenboom, J. Org. Chem., 2006, 71, 8268–8271 CrossRef CAS PubMed; (d) R. Hunter, N. Stellenboom and M. R. Caira, Synlett, 2008, 252–254 CrossRef CAS; (e) M. Han, J. T. Lee and H. G. Hahn, Tetrahedron Lett., 2011, 52, 236–239 CrossRef CAS; (f) D. R. Boyd, N. D. Sharma, S. D. Shepherd, S. G. Allenmark and C. C. R. Allen, RSC Adv., 2014, 4, 27607–27619 RSC; (g) M. Musiejuk, T. Klucznik, J. Rachon and D. Witt, RSC Adv., 2015, 5, 31347–31351 RSC; (h) K. V. Ruddraraju, Z. D. Parsons, C. D. Lewis and K. S. Gates, J. Org. Chem., 2017, 82, 776–780 CrossRef PubMed.
- (a) C. J. Bums, L. D. Field, J. Morgan, D. D. Ridley and V. Vignevich, Tetrahedron Lett., 1999, 40, 6489–6492 CrossRef; (b) X. S. Jia and Y. M. Zhang, Chin. J. Chem., 2005, 23, 303–304 CrossRef CAS.
- M. Bao and M. Shimizu, Tetrahedron, 2003, 59, 9655–9659 CrossRef CAS.
- (a) H. Firouzabadi and B. Karimi, Synthesis, 1999, 500–502 CrossRef CAS; (b) H. Firouzabadi and A. Jamalian, Phosphorus Sulfur, 2001, 170, 211–220 CrossRef CAS; (c) N. Iranpoor, H. Firouzabadi and A. Jamalian, Synlett, 2005, 1447–1449 CrossRef CAS; (d) G. W. Kabalka, M. S. Reddy and M. L. Yao, Tetrahedron Lett., 2009, 50, 7340–7342 CrossRef CAS.
- (a) J. B. Arterburn, M. C. Perry, S. L. Nelson, B. R. Dible and M. S. Holguin, J. Am. Chem. Soc., 1997, 119, 9309–9310 CrossRef CAS; (b) A. R. Hajipour, S. E. Mallakpour and H. Adibi, J. Org. Chem., 2002, 67, 8666–8668 CrossRef CAS PubMed; (c) J. K. Vandavasi, W. P. Hu, C. Y. Chen and J. J. Wang, Tetrahedron, 2011, 67, 8895–8901 CrossRef CAS; (d) H. M. Wang, Q. Q. Lu, C. H. Qian, C. Liu, W. Liu, K. Chen and A. W. Lei, Angew. Chem., Int. Ed., 2016, 55, 1094–1097 CrossRef CAS PubMed; (e) H. H. Cui, W. Wei, D. S. Yang, Y. L. Zhang, H. J. Zhao, L. L. Wang and H. Wang, Green Chem., 2017, 19, 3520–3524 RSC; (f) X. Qiu, X. Yang, Y. Zhang, S. Song and N. Jiao, Org. Chem. Front., 2019, 6, 2220–2225 RSC.
- (a) A. K. Misra and G. Agnihotri, Synth. Commun., 2004, 34, 1079–1085 CrossRef CAS; (b) J. K. Vandavasi, W.-P. Hu, C.-Y. Chen and J.-J. Wang, Tetrahedron, 2011, 67, 8895–8901 CrossRef CAS; (c) L. Wang, L. Chen, Z. Qin, K. Ni, X. Li, Z. Yu, Z. Kuang, X. Qin, H. Duan and J. An, Molecules, 2023, 28, 6789 CrossRef CAS PubMed.
- (a) P. J. Chai, Y. S. Li and C. X. Tan, Chin. Chem. Lett., 2011, 22, 1403–1406 CrossRef CAS; (b) Y. Dou, X. Huang, H. Wang, L. Yang, H. Li, B. Yuan and G. Yang, Green Chem., 2017, 19, 2491–2495 RSC; (c) L. Song, W. Li, W. Duan, J. An, S. Tang, L. Li and G. Yang, Green Chem., 2019, 21, 1432–1438 RSC.
- (a) D. R. Dreyer, H. P. Jia, A. D. Todd, J. Geng and C. W. Bielawski, Org. Biomol. Chem., 2011, 9, 7292–7295 RSC; (b) C. Zhu, D. Wu, H. Liu, C. Meng and T. Tang, Green Chem., 2022, 24, 9033–9039 RSC.
- (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (b) D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed; (c) A. Talla, B. Driessen, N. J. W. Straathof, L. G. Milroy, L. Brunsveld, V. Hessel and T. Noël, Adv. Synth. Catal., 2015, 357, 2180–2186 CrossRef CAS; (d) Q. Liu and L.-Z. Wu, Natl. Sci. Rev., 2017, 4, 359–380 CrossRef CAS; (e) D. H. Dethe, A. Srivastava, B. D. Dherange and B. V. Kumar, Adv. Synth. Catal., 2018, 360, 3020–3025 CrossRef CAS; (f) L. Buzzetti, G. E. M. Crisenza and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 3730–3747 CrossRef CAS PubMed; (g) R. Cannalire, S. Pelliccia, L. Sancineto, E. Novellino, G. C. Tron and M. Giustiniano, Chem. Soc. Rev., 2021, 50, 766–897 RSC; (h) N. Spiliopoulou and C. G. Kokotos, Green Chem., 2021, 23, 546–551 RSC; (i) X. Li, J. Fan, D. Cui, H. Yan, S. Shan, Y. Lu, X. Cheng and T. P. Loh, Eur. J. Org Chem., 2022, 2022, e202200340 CrossRef CAS.
- P. H. Huang, P. Wang, S. Tang, Z. J. Fu and A. W. Lei, Angew. Chem., Int. Ed., 2018, 27, 8247–8251 CrossRef.
- (a) C. E. Garrett and K. Prasad, Adv. Synth. Catal., 2004, 346, 889–900 CrossRef CAS; (b) X. Zhao, J. Liu, J. Fan, H. Chao and X. Peng, Chem. Soc. Rev., 2021, 50, 4185–4219 RSC.
- (a) S. H. Kennedy, B. D. Dherange, K. J. Berger and M. D. Levin, Nature, 2021, 593, 223–227 CrossRef CAS PubMed; (b) K. J. Berger, J. L. Driscoll, M. Yuan, B. D. Dherange, O. Gutierrez and M. D. Levin, J. Am. Chem. Soc., 2021, 143, 17366–17373 CrossRef CAS PubMed; (c) B. D. Dherange, M. Yuan, C. B. Kelly, C. A. Reiher, C. Grosanu, K. J. Berger, O. Gutierrez and M. D. Levin, J. Am. Chem. Soc., 2023, 145, 17–24 CrossRef CAS PubMed.
- (a) J. H. Xue, Y. Li, Y. Liu, Q. Li and H. Wang, Angew. Chem., Int. Ed., 2024, 63, e202319030 CrossRef CAS PubMed; (b) J.-H. Xue, Y. Li, D.-H. Tan, F.-H. Tu, Y. Liu, Q. Li and H. Wang, iScience, 2023, 26, 106255 CrossRef CAS PubMed.
- (a) X. Xu, L. Yan, S. Wang, P. Wang, A. X. Yang, X. Li, H. Lu and Z. Y. Cao, Org. Biomol. Chem., 2021, 19, 8691–8695 RSC; (b) X. Xu, L. Yan, Z. K. Zhang, B. Lu, Z. Guo, M. Chen and Z. Y. Cao, Molecules, 2022, 27, 4675 CrossRef CAS PubMed.
- C. Zippel, J. Seibert and S. Brase, Angew. Chem., Int. Ed., 2021, 60, 19522–19524 CrossRef CAS PubMed.
- Y. Lin, Y. Liu, Y. Zheng, R. Nie, L. Guo and Y. Wu, ACS Sustain. Chem. Eng., 2018, 6, 13644–13649 CrossRef CAS.
- L. Liu, Q. Wang, Y. Li, M. Liu, B. Liu, Q. Li and K. Feng, Org. Lett., 2024, 26, 2271–2275 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra03545c |
| This journal is © The Royal Society of Chemistry 2024 |