
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring the impact of trifluoromethyl (–CF3) functional group on the anti-cancer activity of isoxazole-based molecules: design, synthesis, biological evaluation and molecular docking analysis†
Paramita Pattanayak a,
Sripathi Nikhithab,
Debojyoti Halderb,
Balaram Ghosh*b and
Tanmay Chatterjee*a
a,
Sripathi Nikhithab,
Debojyoti Halderb,
Balaram Ghosh*b and
Tanmay Chatterjee*a
aDepartment of Chemistry, Birla Institute of Technology and Science, Pilani (BITS Pilani), Hyderabad Campus, Jawahar Nagar, Hyderabad 500078, Telangana, India. E-mail: tanmay@hyderabad.bits-pilani.ac.in
bEpigenetic Research Laboratory, Department of Pharmacy, Birla Institute of Technology and Science, Pilani (BITS Pilani), Hyderabad Campus, Jawahar Nagar, Hyderabad 500078, Telangana, India. E-mail: balaram@hyderabad.bits-pilani.ac.in
First published on 12th June 2024
Abstract
Herein we report the design and synthesis of a series of fully-substituted 4-(trifluoromethyl)isoxazoles and evaluation of their anti-cancer activities against MCF-7, 4T1 and PC-3 cell lines as a proof of concept study. 4-(Trifluoromethyl)isoxazole is a synthetically challenging class of molecules and very few synthetic methods have been developed so far and all of them suffered from several serious limitations. Recently we developed a novel, metal-free, and general synthetic strategy to access synthetically challenging 4-(trifluoromethyl)isoxazoles starting from readily available chalcones using cheap CF3SO2Na as the source of the –CF3 group and multitasking tBuONO as an oxidant as well as the source of N and O and thus we have overcome the limitations of the previous methods. Based on the structure of an isoxazole-based anti-cancer agent, 3-(3,4-dimethoxyphenyl)-5-(thiophen-2-yl)isoxazole 14, we designed a set of 4-(trifluoromethyl)isoxazoles for synthesis and further anti-cancer evaluation. Among various molecules, 3-(3,4-dimethoxyphenyl)-5-(thiophen-2-yl)-4-(trifluoromethyl)isoxazole 2g (IC50 = 2.63 μM) and 3-(thiophen-2-yl)-5-(4-(thiophen-2-yl)-1H-pyrrol-3-yl)-4-(trifluoromethyl)isoxazole 5 (IC50 = 3.09 μM) exhibited the best anti-cancer activity against the human breast cancer cell-lines (MCF-7), 2g being the lead molecule among all. Interestingly, 2g is found to be almost 8 times more active compared to its non-trifluoromethylated analogue, i.e., 3-(3,4-dimethoxyphenyl)-5-(thiophen-2-yl)isoxazole 14 (IC50 = 19.72 μM) which revealed the importance of a ‘CF3’ moiety in enhancing the anti-cancer activity of 14. Further studies such as apoptosis induction, cell cycle analysis, and nuclear staining revealed an apoptotic cell death mechanism. The in silico molecular docking, induced fit analysis, and ADME studies further supported the effect of a –CF3 moiety on the enhancement of anti-cancer activity of isoxazole-based anti-cancer molecules. Further exploration of the biodistribution and therapeutic efficacy of lead 2g in vivo holds significant promise, positioning it as a potential candidate for anticancer therapy.
Introduction
Isoxazole is indeed a significant heterocyclic framework in medicinal chemistry.1–3 Its versatility in pharmacology makes it a valuable scaffold for drug discovery and development.4 The diverse biological activities such as anticancer,5 antibacterial,6 antifungal,7 antituberculosis,8 antidepressants,9 antirheumatic, anti-inflammatory,10 immunomodulatory, antialzheimer, and antidiabetic activities highlight the importance of isoxazole as a valuable pharmacophore in drug discovery and development (Fig. 1). Researchers continue to explore and optimize isoxazole-based compounds for various therapeutic applications, making them a versatile and critical component of medicinal chemistry. | ||
| Fig. 1 Isoxazole-based pharmaceutically important molecules including drugs. | ||
On the other hand, cancer is defined as “a group of diseases characterized by the uncontrolled growth and spread of abnormal cells” and is one of the deadliest diseases globally.11 Among all, breast cancer (BCa) is one of the most frequently diagnosed cancers in women. Particularly, 70% of BCa diagnoses are estrogen receptor α positive (ERα+) making it a critical therapeutic target. As a result, the effects of ERα and ERβ subtypes of ER, on BCa cells are different. While ERα encourages cancerous activities, ERβ isoform has the opposite effects. The FDA-approved medicines, i.e., tamoxifen, toremifene, raloxifene, and fulvestrant, which bind to the estrogen binding site of the receptor, were discovered through ER-directed small molecule drug discovery for BCa. Sometime these non-selective ER-directed inhibitors cause resistance in BCa cells and raise the risk of developing endometrial cancer.12 Therefore, it is imperative to create new medications with different ERα targeting mechanisms in order to overcome the drawbacks of traditional anti-ERα treatments.
Poutiainen et al. have reported 4,5-dihydroisoxazoles as agonist of ERα and ERβ.13 In 2016, Rangappa and co-workers synthesized some 3,5-di(hetero)arylisoxazoles and evaluated their anti-cancer activity.14 They found that among several compounds, 3-(3,4-dimethoxyphenyl)-5-(thiophen-2-yl)isoxazole D (Fig. 1) exhibited the best anticancer (IC50 = 19.72 μM against MCF-7) and apoptotic activity in breast adenocarcinoma cell line by inhibiting the tumor growth of DMBA-induced mammary carcinoma tumours along with the downregulation of ERα. However, the anti-cancer activity of D was not very promising, and the synthesis required synthetically challenging starting material or multistep process starting from commercially available compounds involving hazardous and/or toxic reagents and solvent such as benzene. Hence, we intend to develop novel isoxazole-based anti-breast cancer molecules possessing superior anti-cancer activity which could be synthesized from readily available starting materials using inexpensive reagents in one step.
The incorporation of a trifluoromethyl (–CF3) group is a valuable tool in medicinal chemistry and drug discovery.15 It allows scientists to fine-tune the properties of organic molecules to optimize their therapeutic potential. Incorporating a –CF3 functional group into an organic molecule can indeed have a profound impact on its physical, chemical, and biological properties such as lipophilicity, binding selectivity, metabolic stability, and bioavailability.16 Hence, isoxazoles bearing a trifluoromethyl group are expected be more promising druggable candidates to develop better pharmaceuticals or therapeutics for the treatment of breast cancer.
Based on the structure of the anti-cancer agent D and the importance of a –CF3 moiety, we planned and designed a series of CF3-bearing isoxazoles, in particular, 4-(trifluoromethyl)isoxazoles including the trifluoromethyl analogue of D (Fig. 2) to evaluate their anti-cancer activity in search of novel isoxazole-based anti-cancer agents. However, the synthesis of 4-(trifluoromethyl)isoxazoles is challenging and only a few synthetic methods have been developed which suffered from a series of serious limitations such as (a) very limited substrate scope and poor yield of products, (b) requirement of highly expensive reagent such as Togni's reagent or Umemoto's reagent as a trifluoromethyl source, and/or rare earth transition-metal (Ir, Pd) catalyst etc.17–19 Recently, we have overcome these challenges by developing a metal-free, versatile and direct synthetic strategy for the synthesis of a wide variety of 4-(trifluoromethyl)isoxazoles strating from easily available α,β-unsaturated carbonyls including chalcones and commercially available cheap trifluoromethyl source, CF3SO2Na and tBuONO which played double role of an oxidant as well as the source of N and O.20
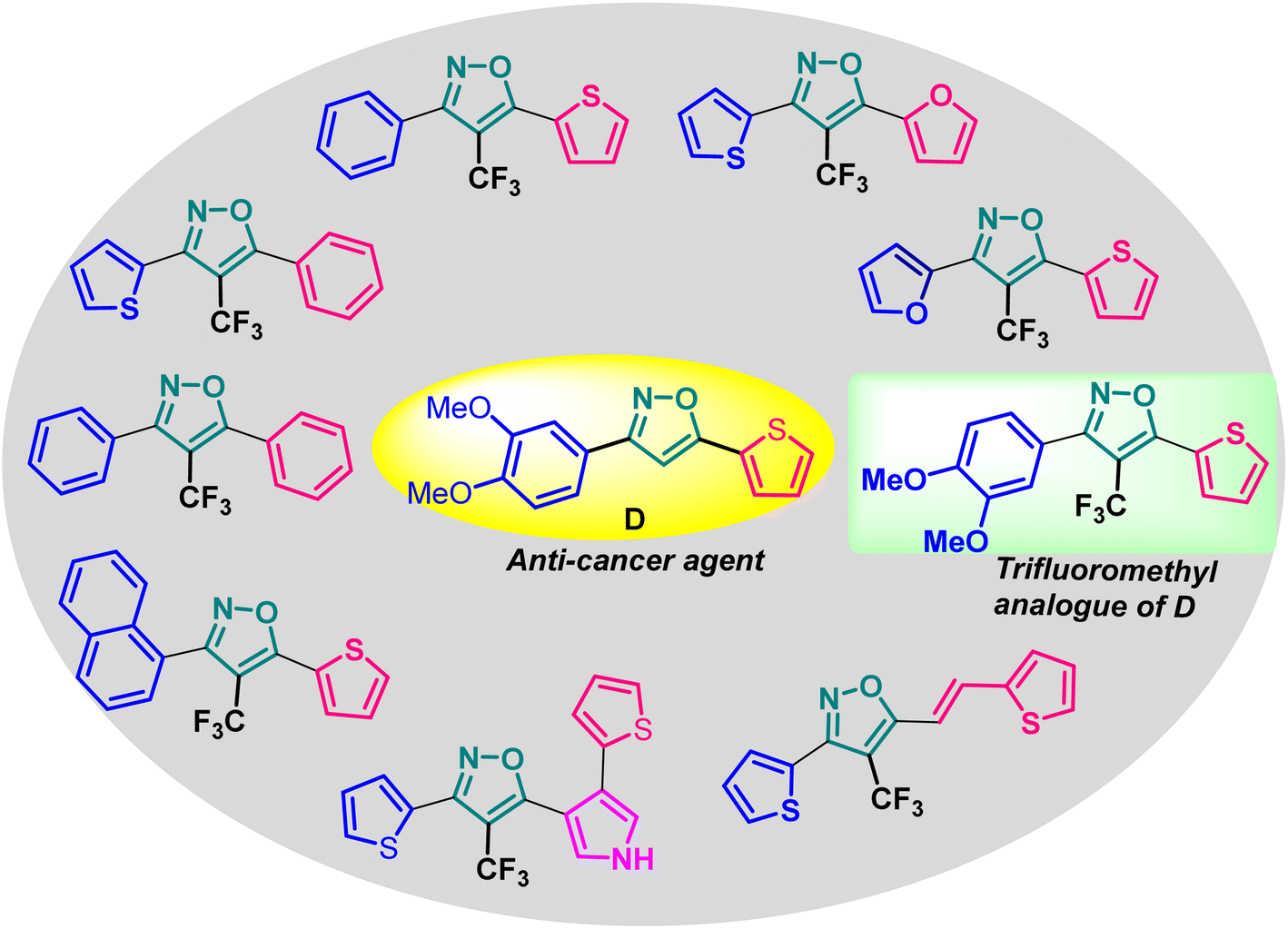 | ||
| Fig. 2 Design of 4-(trifluoromethyl)isoxazoles based on the structure of an anti-cancer agent, D. | ||
Result and discussion
Synthesis of 4-(trifluoromethyl)isoxazoles (2a–2g and 4) from chalcones (1 and 3)
The designed 4-(trifluoromethyl)isoxazoles (2a–2g, 4) bearing a thienyl and a (hetero)aryl or vinyl group at the 3rd and 5th position respectively or vice versa were synthesized by following our previously established synthetic strategy (Scheme 1).20 | ||
| Scheme 1 (A) Synthesis of 4-(trifluoromethyl)isoxazoles and (B) 4-(trifluoromethyl)-2-vinylisoxazole from respective chalcones. | ||
All the molecules were synthesised with moderate to good yield (40–70%) through a metal-free, cascade regio- and stereoselective trifluormethyloximation, cyclization, and elimination reaction with readily available chalcones (α,β-unsaturated ketones) using CF3SO2Na (3 equiv.) as the trifluoromethyl source, and tBuONO (4 equiv.) as an oxidant as well as the source of N and O. When (1E, 4E)-1,5-di(thiophen-2-yl)penta-1,4-dien-3-one (3) was employed as the starting material, compound 4 bearing a vinyl group at the 5th position was synthesised.
Synthetic diversification of 4-(trifluoromethyl)-5-vinylisoxazole (4) to (5)
(E)-3-(Thiophen-2-yl)-5-(2-(thiophen-2-yl)vinyl)-4-(trifluoromethyl) isoxazole (4) was further synthetically diversified to a tetra-hetero aryl molecule bearing –CF3 functional group, i.e., (E)-3-(thiophen-2-yl)-5-(2-(thiophen-2-yl)vinyl)-4-(trifluoromethyl)isoxazole (5) in excellent yield (98%) via a [3 + 2] cycloaddition reaction of 4 with toluenesulfonylmethyl isocyanide (TosMIC) at room temperature (Scheme 2).20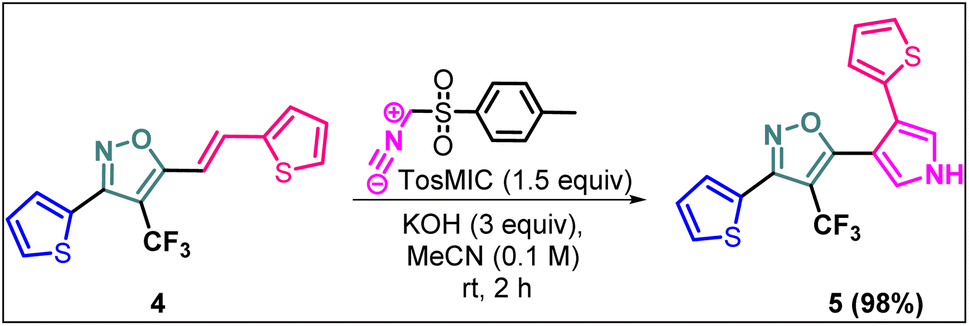 | ||
| Scheme 2 Synthesis of (E)-3-(thiophen-2-yl)-5-(2-(thiophen-2-yl)vinyl)-4-(trifluoromethyl)isoxazole 5. | ||
Synthesis of non-trifluoromethyl analogues (7 and 9) of 2a and 2c
To explore the actual effect of –CF3 functional group on the anti-cancer activity of the isoxazoles, we planned to synthesise a couple of non-trifluoromethyl analogues of some of our designed isoxazoles, i.e., 3,5-diphenylisoxazole (7) and 3-phenyl-5-(thiophen-2-yl)isoxazole (9) from the corresponding ynones 6 and 8 respectively (Scheme 3).21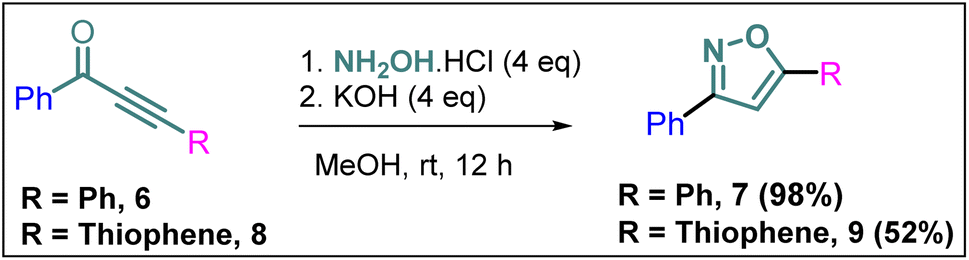 | ||
| Scheme 3 Synthesis of 7 and 9 from ynones (6 and 8). | ||
Synthesis of non-trifluoromethyl analogue (14) of 2g [compound D]
Since compound 2g is our model compound, we synthesized the non-trifluoromethyl analogue of 2g, i.e., 3-(3,4-dimethoxyphenyl)-5-(thiophen-2-yl)isoxazole (14). Compound 14 was synthesised via a multistep process starting from vanillin 10 (Scheme 4). At first, vanillin was methylated by a methylating reagent, methyl iodide to form 11 which was subjected to an oximation reaction with hydroxylamine hydrochloride to afford 12.22 Chlorination of 12 with NCS furnished 13.23 Finally, a Cu-catalysed coupling reaction of 13 with 2-ethynylthiophene afforded 3-(3,4-dimethoxyphenyl)-5-(thiophen-2-yl)isoxazole 14 or compound D in 90% yield. | ||
| Scheme 4 Synthesis of 3-(3,4-dimethoxyphenyl)-5-(thiophen-2-yl)isoxazole 14 from vanillin in four steps. | ||
Evaluation of anti-cancer activities of the synthesized isoxazoles
| Entry | Compound | Molecular structure | MCF-7 IC50 (μM) | 4T1 IC50 (μM) | PC-3 IC50 (μM) | HEK-293 IC50 (μM) |
|---|---|---|---|---|---|---|
| 1 | 2a |  |
11.34 ± 0.13 | 37.83 ± 0.20 | 16.14 ± 0.15 | 114.94 ± 0.15 |
| 2 | 2b |  |
9.76 ± 0.09 | 35.36 ± 0.19 | 13.60 ± 0.17 | 103.28 ± 0.17 |
| 3 | 2c |  |
8.19 ± 0.12 | 28.78 ± 0.19 | 11.73 ± 0.16 | 98.76 ± 0.24 |
| 4 | 2d | 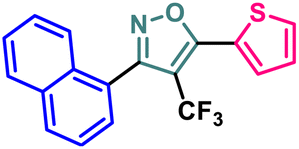 |
10.59 ± 0.19 | 37.18 ± 0.22 | 15.59 ± 0.14 | 111.83 ± 0.17 |
| 5 | 2e |  |
5.48 ± 0.14 | 23.50 ± 0.21 | 11.64 ± 0.14 | 94.55 ± 0.20 |
| 6 | 2f |  |
5.32 ± 0.19 | 18.12 ± 0.15 | 10.26 ± 0.15 | 86.98 ± 0.23 |
| 7 | 2g |  |
2.63 ± 0.15 | 8.75 ± 0.09 | 7.16 ± 0.18 | 81.20 ± 0.17 |
| 8 | 4 |  |
13.71 ± 0.13 | 44.36 ± 0.20 | 17.99 ± 0.13 | 122.03 ± 0.16 |
| 9 | 5 |  |
3.09 ± 0.11 | 14.96 ± 0.13 | 9.56 ± 0.18 | 84.86 ± 0.19 |
| 10 | 7 | 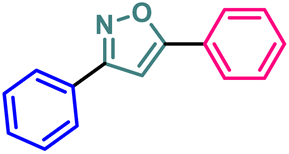 |
13.03 ± 0.13 | 41.58 ± 0.21 | 17.42 ± 0.17 | 118.72 ± 0.21 |
| 11 | 9 | 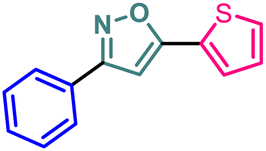 |
9.99 ± 0.10 | 36.24 ± 0.20 | 14.46 ± 0.17 | 109.28 ± 0.19 |
| 12 | 14 | 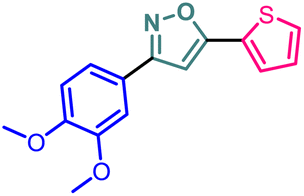 |
19.72 ± 0.16 | 48.39 ± 0.19 | 30.21 ± 0.22 | 131.50 ± 0.13 |
| 13 | BG-45 |  |
43.55 ± 0.18 | 59.14 ± 0.17 | 28.67 ± 0.14 | 144.56 ± 0.14 |
 | ||
| Fig. 3 IC50 values and dose–response curves of designed 4-(trifluoromethyl)isoxazoles along with BG-45. All compounds were tested at doses ranging from 0.195 μM to 200 μM on (A) MCF-7, (B) 4T1, (C) PC-3, and (D) HEK-293. Cell viability was assessed using the in vitro MTT assay after cells were treated with compounds for 48 hours. The data is presented as mean ± SD (n = 2) and plotted in a dose–response format using Graph Pad Prism 8.0.1 and a nonlinear regression analysis approach, the IC50 was determined. | ||
BG-45 is a class I selective histone deacetylase (HDAC) inhibitor with preferential selectivity for HDAC3 isoform. It has been reported as an anticancer molecule.37 The molecule has been used in this manuscript to show that the anti-proliferative/anticancer assay is validated with the reproducible activity of BG-45 in the assay. The anticancer potential of the newly developed molecules can also be compared with the reference compound BG-45 even though the anticancer activity of BG-45 is mechanistically different than that of the reported molecules in the manuscript. The compound BG-45 is commercially available. The molecule has been procured from Selleck, USA (cat# S7689; https://www.selleckchem.com/products/bg45.html).
It has been observed that the 4(trifluoromethyl)isoxazoles (2a, 2c, and 2g) possess superior anti-cancer activity against MCF-7 as compared to their corresponding non-trifluoromethyl analogues (7, 9, and 14) and the effect is most prominent for 2g. These results demonstrated the role of a “–CF3” moiety on the enhancement of anti-cancer activity of isoxazole-based molecules (Fig. 4).
 | ||
| Fig. 4 Effect of ‘–CF3’ moiety on the anticancer activity of isoxazole-based molecules. | ||
Cytotoxicity against normal HEK-293 cell lines in vitro
We also investigated their in vitro cytotoxicity against a normal human embryonic kidney (HEK-293) cell line to determine their IC50. Interestingly, all of the synthesized compounds were preferentially selective for cancer cell lines and had very less cytotoxicity against normal cell lines. The lead compound 2g demonstrated 30.8-fold selectivity for MCF-7 cells and 9.2-fold and 11.3-fold selectivity for other cancer cell lines over HEK-293 cell lines.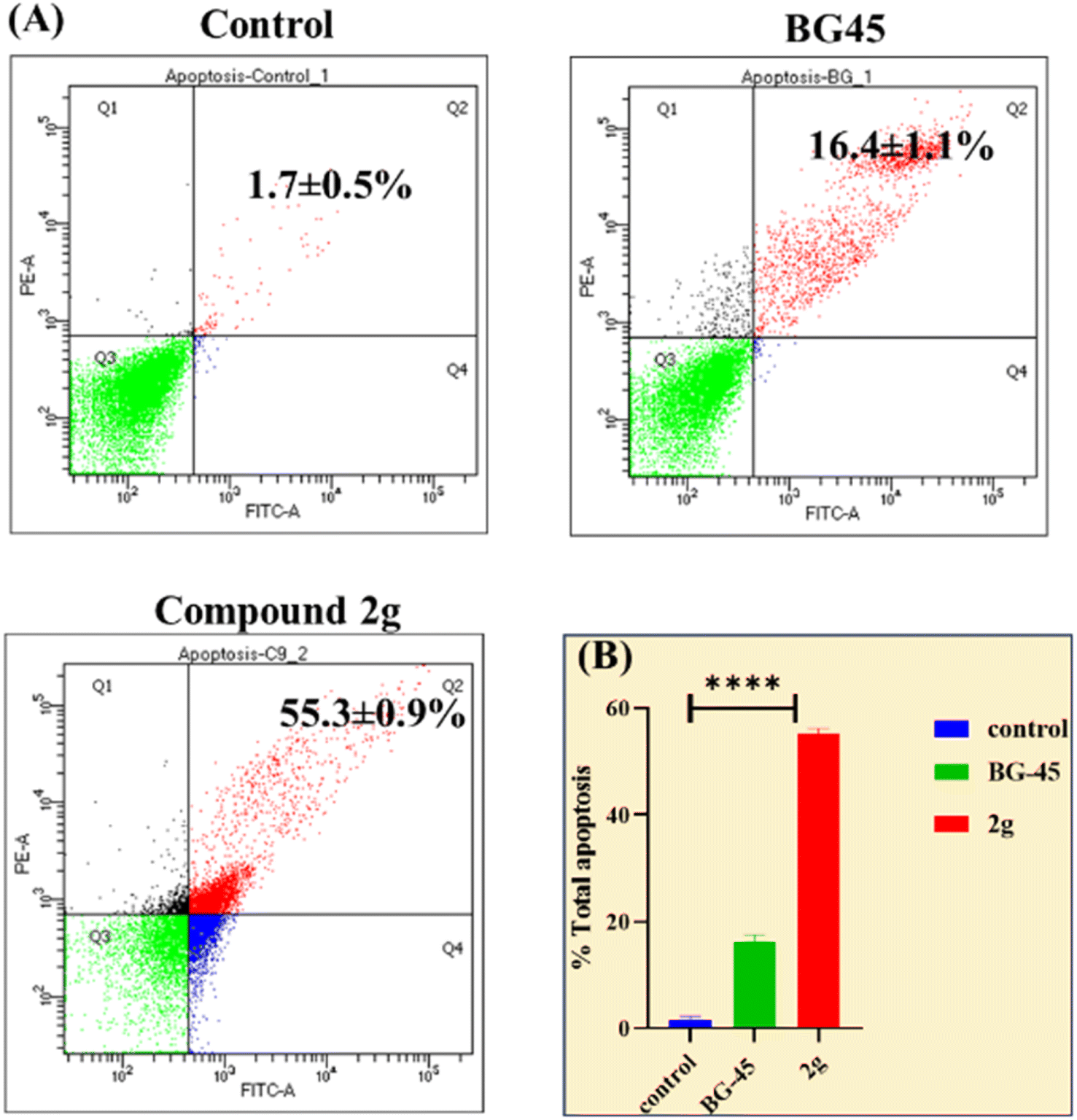 | ||
| Fig. 5 (A) Flow cytometric study of apoptosis utilizing the Annexin V/PI double staining assay. The same cultures of MCF-7 cells were treated with vehicle control, BG-45, and compound 2g for 48 hours (Q1 – necrotic cells, Q2 – late apoptosis, Q3 – live cells, Q4 – early apoptotic cells) at their respective in vitro IC50 values.(The X and Y axes show the intensities of annexin V and propidium iodide). (B) Graphical representation of overall apoptotic percentage analysis in MCF-7 cells. | ||
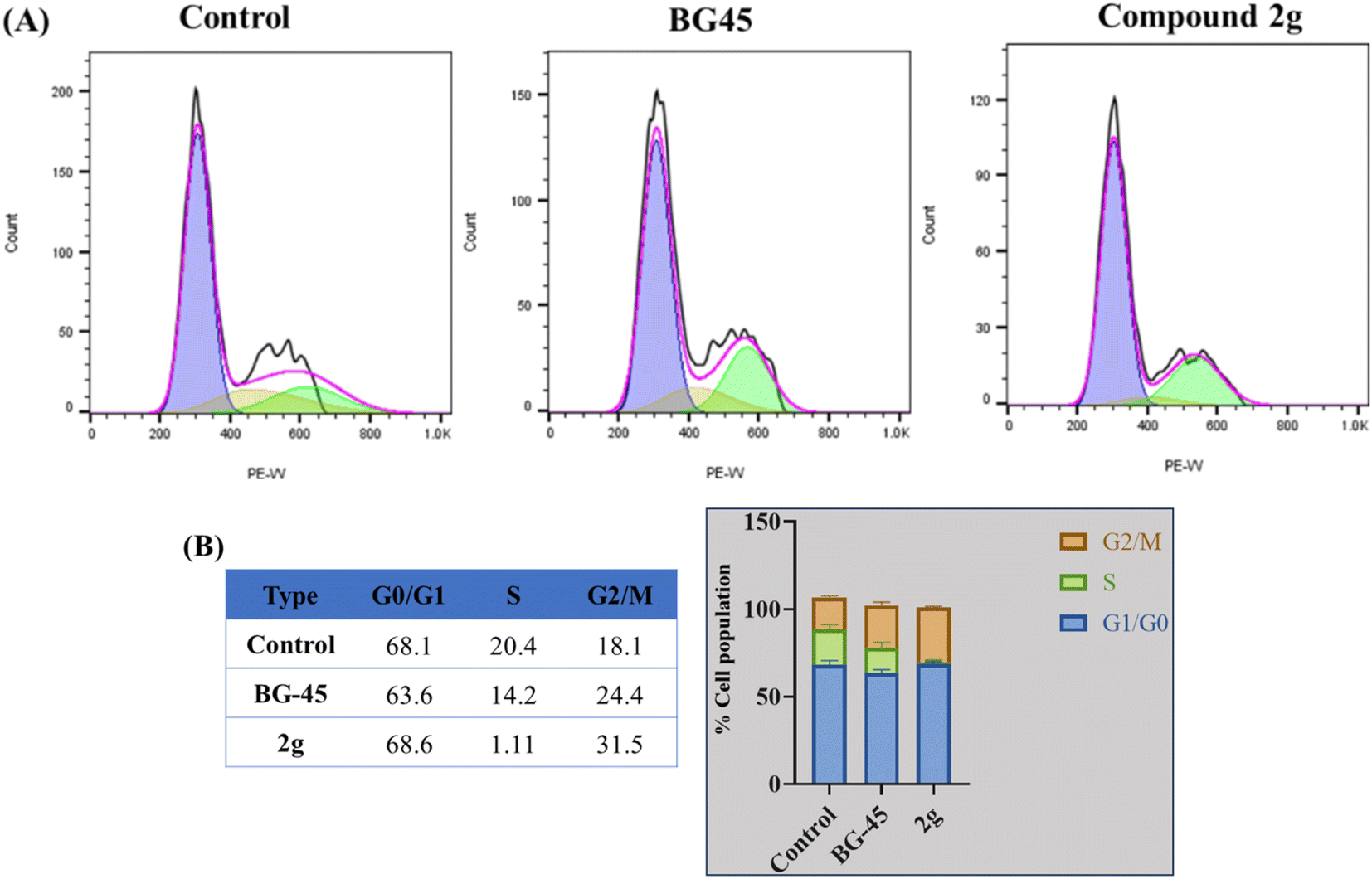 | ||
| Fig. 6 (A) Cell cycle analysis in MCF-7 cells treated for 48 hours with reference drug BG-45, and compound 2g at IC50 concentrations. Cell cycle analysis was performed after the appropriate treatment durations and was then evaluated using a flow cytometer (BD Aria III) (B). G1, S, and G2/M phases of the cell cycle in MCF-7 cells are represented graphically and in tabular form, respectively. | ||
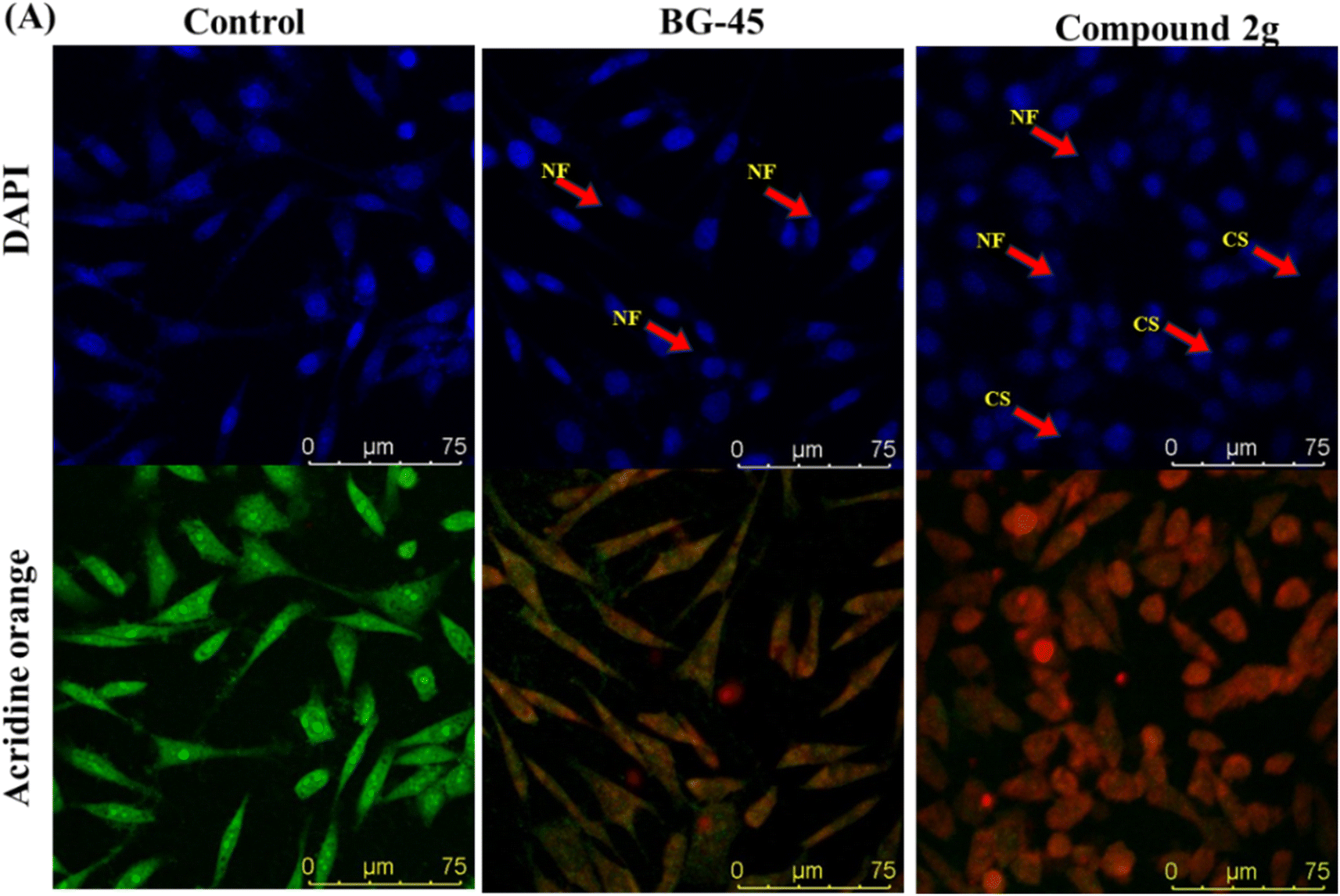 | ||
| Fig. 7 Nuclear morphology analysis by nuclear staining experiment in vitro in MCF-7 cells after treatment with IC50 concentrations of BG-45 and compound 2g along with control (A) MCF-7 cells using the staining solutions of DAPI and AO after the treatment period and NF symbolizes nuclear fragmentation, while CS represents cell shrinkage. The stained nuclei were observed using a laser scanning confocal microscope DMI8 Leica Microsystems. | ||
 | ||
| Fig. 8 Intracellular ROS generation by DCFH-DA in MCF-7 cells after 48 h of treatment with IC50 doses of BG-45 and compound 2g, as well as a control. (A) MCF-7 cells stained with DCFH-DA dye after treatment time, (B) table reflecting the fluorescence intensity values quantified by Image J program, and (C) plot of fluorescence intensity obtained. The ROS generation was visualized using a Leica microscope at 60× magnification and a laser scanning confocal microscope DMI8 (Leica Microsystems, Germany). Scale bars are 25 μm long. The acquired results are the mean standard deviation (n = 2); ***p 0.0001. ImageJ software was used to calculate the fluorescence intensity. The significance was determined using one-way ANOVA, and the graph was created in GraphPad Prism 8.0.1. | ||
In silico analysis of 2g and 14
| Compounds | Structure | Docking scores (kcal mol−1) | Interactions | MMGBSA ΔG (kcal mol−1) |
|---|---|---|---|---|
| a ALA alanine, ASP aspartic acid, GLU glutamic acid, HID histidine, LEU leucine, MET methionine, PHE phenylalanine, THR threonine, TRP tryptophan, VAL valine. | ||||
| 2g |  |
−7.773 | Hydrophobic: MET343, LEU346, ALA350, LEU354, TRP383, LEU384, LEU387, MET388, LEU391, LEU402, PHE404, LEU428, PHE425, ILE424, MET421, LEU525, MET528, polar: THR347, HID524, negative charged: ASP351 | −15.03 |
| 14 | 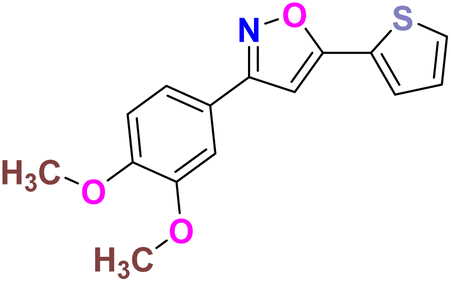 |
−6.264 | Hydrophobic: MET343, LEU346, LEU349, ALA350, TRP383, LEU384, LEU387, MET388, LEU391, LEU402, PHE404, LEU428, PHE425, ILE424, MET421, VAL418, LEU525, MET528, polar: THR347, HID524, negative charged: ASP351, GLU353 | −4.98 |
 | ||
| Fig. 9 Receptor-ligand docking analysis (2D) of compounds – (A) 2g, and (B) 14 with HERα (PDB: 3ERT). | ||
 | ||
| Fig. 10 Induced fit docking (3D interaction diagram) of compounds – (A) 2g [IFD score: −531.87], and (B) 14 [IFD score: −529.41] with HERα (PDB: 3ERT). | ||
| Compound | Molecular weight | Hydrogen bond donor | Hydrogen bond acceptor | QPlogPo/w | PSA | Percent human oral absorption | Rule of three | Rule of five |
|---|---|---|---|---|---|---|---|---|
| 2g | 355.33 | 0 | 3 | 4.62 | 41.18 | 100 | 0 | 0 |
| 14 | 287.33 | 0 | 3 | 3.92 | 40.68 | 100 | 0 | 0 |
| Compound | QPlogS | QPlogHERG | QPPCaco | QPlogBB | QPPMDCK | QPlogKhsa |
|---|---|---|---|---|---|---|
| a QPlogS: predicted aqueous solubility, QPlogHERG: predicted IC50 value for blockage of HERG K+ channels, QPPCaco: predicted apparent Caco-2 cell permeability in nm s−1, QPlogBB: predicted brain/blood partition co-efficient, QPPMDCK: predicted apparent MDCK cell permeability in nm s−1, QPlogKhsa: prediction of binding to human serum albumin. | ||||||
| 2g | −5.47 | −5.00 | 3888.86 | 0.352 | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
0.557 |
| 14 | −4.70 | −5.39 | 4063.03 | 0.163 | 3865.74 | 0.353 |
| Acceptable range | −6.5–0.5 | > −5 | Excellent > 500 | −3.0–1.2 | Excellent > 500 | −1.5–1.5 |
HERα than compound 14, throughout the receptor-ligand docking, free binding energy MMGBSA analysis, and induced fit docking at the catalytic binding site with amino acid residues of leucine with excellent fit at the antagonist binding pocket, which provide significant evidence of the anticancer activity of the molecules.
Materials and methods
General information for chemistry
All the required chemicals were purchased from various companies and used without purification. The products were characterized by 1H and 13C NMR. NMR spectra were recorded on a Bruker 400 MHz instrument (400 MHz for 1H NMR and 100 MHz for 13C NMR). Copies of 1H and 13C NMR spectra can be found at the end of the ESI.† 1H NMR experiments are reported in units, parts per million (ppm), and were measured relative to residual chloroform (7.26 ppm) in the deuterated solvent. 13C NMR spectra are reported in ppm relative to deuterochloroform (77.00 ppm), and all were obtained with 1H decoupling. Coupling constants were reported in Hz. Reactions were monitored by thin layer chromatography (TLC) and 1H-NMR of the crude reaction mixture using 1,3,5- trimethoxybenzene as the internal standard. Mass spectral data of unknown compounds were obtained on a high-resolution mass spectrometer, HRMS. Melting points of unknown compounds were recorded on a KRUSS Optronic M3000 apparatus.
Compound (5) were synthesized by following our previously reported synthetic method.20 The synthesized compounds were characterized by 1H NMR which matched with the literature.

Step-1. PdCl2(PPh3)2 (140 mg, 0.200 mmol) and CuI (76.2 mg, 0.400 mmol) were charged into a two-necked round flask and the flask was refilled with N2. THF (17.0 mL) was added to the flask. Benzoyl chloride (703 mg, 5.00 mmol), phenyl acetylene (613 mg, 6.00 mmol), and triethylamine (506 mg, 5.00 mmol) were added to the mixture at room temperature. The reaction mixture was stirred at room temperature overnight. Then the saturated NH4Cl solution was added to the mixture and the resulting aqueous phase was extracted with EtOAc. The combined organic phase was washed with brine, dried over MgSO4. After removal of the solvent, the resulting crude mixture was purified by silica gel column chromatography (hexane/EtOAc = 45/1) to give 1,3-diphenylprop-2-yn-1-one (6) as a pale yellow solid (1.02 g, 4.95 mmol, 88% yield).

Step-2. The treatment of 6 (0.1 g, 0.5 mmol) with NH2OH·HCl (0.1 g, 2 mmol, 4 equiv.) in the presence of KOH (0.112 g, 2 mmol, 4 equiv.) at room temperature in a mixture solvent of MeOH/H2O (9:1) gave oxazole derivative 7 (0.108 g, 0.5 mmol) in 98% yield. The products of step-1 and step-2 were characterized by 1H NMR which matched with the literature.21
Compound 9 was also synthesized following the same protocol as mentioned above and characterized by 1H NMR which matched with the literature.24
Step-1. Vanillin (12.172 g, 8 mmol) was added into 130 mL of acetone, and the mixture was warmed to 60 °C to dissolve the vanillin. Then 11.222 g (8 mmol, 4 equiv.) of K2CO3 and 8 mL (12 mmol, 2 equiv.) of MeI were added, and the mixture was stirred at 60–70 °C overnight (17 hours) under reflux. The reaction mixture was filtered, and the solid was washed with acetone, then the filtrate was concentrated under vacuum to yield the desired product, which was further purified by flash column chromatography, to give 13.2 g (99.3% yield) of pure product.
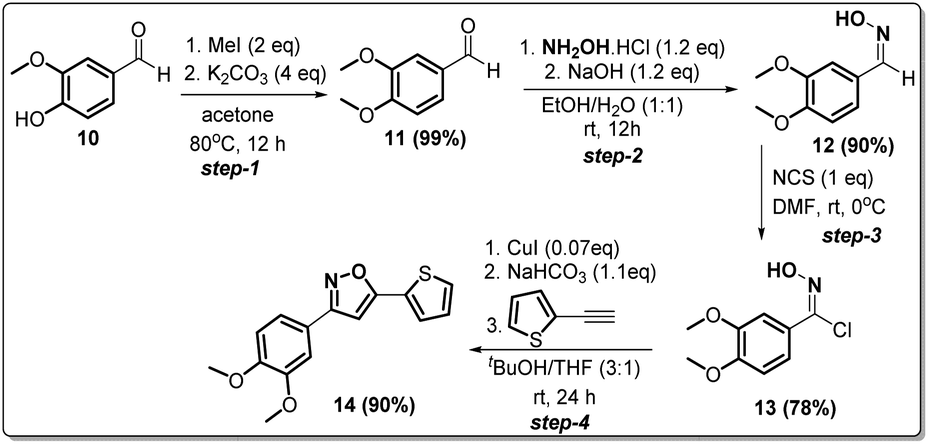
Step-2. To a 50 mL round-bottomed flask, 3,4-dimethoxybenzaldehyde 11 (1 g, 6 mmol), hydroxylamine chloride (0.238 g, 7.22 mmol, 1.2 equiv.) and ethanol/water 1:1 (10 mL) was added and the mixture stirred for 10 min., after that it was added slowly to a solution of NaOH (0.288 g, 7.22 mmol, 1.2 equiv.). The mixture was stirred at room temperature for 3 h, forming a white suspension. The reaction was acidified to pH 6.0. The reaction mixture was extracted with dichloromethane (3 × 20 mL). The organic layer was dried over Na2SO4, evaporated, and recrystallized from n-heptane to obtain 0.985 g of 12 in 90% yield.
Step-3. A mixture of 3,4-dimethoxybenzaldehyde oxime (0.9 g, 4.97 mmol) and DMF (7 mL) was cooled to 0 °C. Under stirring was added slowly N-chlorosuccinimide (0.673 g, 4.97 mmol, 1 equiv.). The reaction mixture was stirred at room temperature for 2 h. After that, the complete reaction mixture was poured into a mixture of water/ice (20 mL) and it was extracted with ethyl acetate (3 × 30 mL). The organic layer was washed with water (2 × 50 mL), brine (50 mL), and then dried over Na2SO4 and finally the solvent was evaporated to give a yellow solid 13 (0.843 g, 78%).
Step-4. Compound 13 was used without further purification. To a 50 mL round-bottomed flask 2-ethynylthiophene (0.4 g, 3.7 mmol, 1 equiv.), N-hydroxy-3,4-dimethoxybenzimidoyl chloride 13 (0.8 g, 3.7 mmol), were suspended in 20.0 mL of a 3:1 t-butanol/THF. Catalytic portion of CuI (50 mg, 0.25 mmol) was added, and the solution was stirred for 10 min. After that, KHCO3 (0.342 g, 4 mmol) was added and the reaction mixture was stirred at room temperature for 24 h. The progress of the reaction was monitored by TLC. The crude product was purified by column chromatography to afford pure 3-(3,4-dimethoxyphenyl)-5-(thiophen-2-yl)isoxazole 14 (0.97 gm 90% yield).
General information for biology
The mouse triple-negative breast cancer cell line (4T1), a human luminal A breast cancer cell line (MCF-7), a human prostate cancer cell line (PC-3), and a normal human embryonic kidney cell line (HEK-293) were procured from the National Center for Cell Science (NCCS, Pune, India). Dulbecco's Modified Eagle Medium (DMEM) (Himedia Laboratories Pvt Ltd, Mumbai, India) was used to cultivate MCF-7, 4T1, PC-3, and HEK-293 cells. All the mentioned cell lines were cultured in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (Himedia Laboratories Pvt Ltd, Mumbai, India) and 1% antibiotic (Pen strep: A001), Himedia Laboratories Pvt Ltd, Mumbai, India. Cells were grown in a humidified environment with 5% CO2 at 37 °C. The experiment was performed using the yellow dye MTT [3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide].Chemicals and reagents used for the biological evaluation
For subsequent investigations, 100 mM stock solutions of each compound were made in DMSO and kept at 4 °C. Sigma-Aldrich supplied DAPI, Acridine Orange, propidium iodide, and RNase. TACs Annexin-V/FITC – PI assay kit was acquired from Bio-legend and utilized according to the protocol included in the kit.Studies on cytotoxicity in vitro
The Maestro module utilize grid-based methodology for docking analysis with extra precision (XP) efficacy. Glide application helps in determining the receptor binding analysis in the flexible way. Hence, the ‘Ligand docking’ methodology was employed with the ligands – 2g and 14, in the developed grid of HERα to get the binding pose, and docking score of the ligand-protein binding at the catalytic site in which the antagonist (4 hydroxy tamoxifen) was present.32,33 Furthermore, for the calculation of free binding energy analysis, MMGBSA was carried out using the earlier protocol,30 with the VSGB solvation model.34 It uses the generalized born approximation for the calculation of ligand – receptor binding strength. The Prime module analysis of MMGBSA provides crucial information about the ligand receptor affinity.35
Conclusions
Although 3-(3,4-dimethoxyphenyl)-5-(thiophen-2-yl)isoxazole 14 exhibited anticancer and apoptotic activity in breast adenocarcinoma cell line by inhibiting the tumor growth of DMBA-induced mammary carcinoma tumours along with the downregulation of Erα14 the anti-cancer activity (IC50 = 19.72 μM) was not very promising. Moreover, the synthesis of 14 is tedious involving multistep process and hazardous reagents and solvent such as benzene. As a proof of concept, we designed and synthesized a series of novel 4-(trifluoromethyl)isoxazoles including the CF3 analogue of 14, i.e., 2g from easily available chalcones in one step and evaluated their anti-cancer activities against several cancer cell lines such as MCF-7, 4T1, and PC-3. In the series of designed and synthesized compounds, 3-(3,4-dimethoxyphenyl)-5-(thiophen-2-yl)-4-(trifluoromethyl)isoxazole 2g (IC50 = 2.63 μM) and 3-(thiophen-2-yl)-5-(4-(thiophen-2-yl)-1H-pyrrol-3-yl)-4-(trifluoromethyl)isoxazole 5 (IC50 = 3.09 μM) showed excellent anti-cancer activity against the human breast cancer cell-lines (MCF-7), 2g being the lead molecule. Notably the trifluoromethyl analogues of isoxazoles such as 2a (IC50 = 11.34 μM), 2c (IC50 = 8.19 μM) and 2g (IC50 = 2.63 μM) were all found to possess superior anti-cancer activity with respect to their corresponding non-trifluoromethyl counterpart, i.e., 7 (IC50 = 13.03 μM), 9 (IC50 = 9.99 μM) and 14 (IC50 = 19.72 μM) respectively. These results revealed the effect of the trifluoromethyl (–CF3) group on the anti-cancer activity of isoxazole-based molecules. Further studies such as apoptosis induction, cell cycle analysis, and nuclear staining with the lead molecule 2g revealed an apoptotic cell death mechanism. The molecular docking studies and free binding energy MMGBSA ΔG of compound 2g was −7.773 kcal mol−1 and −15.03 kcal mol−1, respectively which represented a superior result than that of its non-trifluoromethyl analogue, i.e., compound 14 (docking score: −6.264 kcal mol−1, and MMGBSA ΔG: −4.98 kcal mol−1). Following the induced fit docking using a combined Prime and Glide protocol, the IFD score of compounds 2g was reported as 531.87, and compound 14 was 529.41. Furthermore, the drug-likeness, Lipinski rule of five, and other ADME properties of compounds 2g and compound 14 were evaluated, and it was observed that compound 2g exhibited better ADME properties and drug-likeness than compound 14. This result further supported the importance of a –CF3 moiety on the enhancement of the anti-cancer activity of isoxazole-based anti-cancer molecules. Further design and synthesis of the derivative of 2g and evaluation of their anti-cancer activities is underway in our laboratories in search of more potent anti-cancer agents. We believe that this study will motivate medicinal chemists to design and synthesize various trifluoromethyl analogues of known molecules and evaluate their biological activities in search of superior lead molecules with better therapeutic efficacy.Author contributions
Paramita Pattanayak: methodology, investigation, data curation and writing – original draft preparation for the chemistry part of the manuscript. Sripathi Nikhitha: methodology, investigation, data curation, and writing – original draft preparation for the biology part of the manuscript. Debojyoti Halder: software, validation. Balaram Ghosh: supervision, visualization, funding acquisition and writing – review & editing for the biology part of the manuscript. Tanmay Chatterjee: conceptualization, supervision, visualization, project administration, funding acquisition, and writing – review & editing for the chemistry part of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr Tanmay Chatterjee gratefully acknowledges the funding (File No. 02(0390)/21/EMR-II) from the Council of Scientific and Industrial Reasearch (CSIR), Govt. of India for this work and also thanks Science and Engineering Research Board (SERB), Govt. India for the financial support as core research grant (SERB-CRG) (File No. CRG/2023/003045). This research has also been supported by the research fund from the Department of Health Research (DHR-Indian Council of Medical Research) (File No. 11013_33_2021-GIA HR), Govt. of India and Indian Council of Medical Research (File No. 5/4-4/3/MH/2022-NCD-II) Govt. of India, provided to Dr Balaram Ghosh. The instrumentational facilities, at central analytical laboratory (CAL) of BITS-Pilani, Hyderabad campus, are also gratefully acknowledged.Notes and references
- J. Zhu, J. Mo, H. zhi Lin, Y. Chen and H. peng Sun, Bioorg. Med. Chem., 2018, 26, 3065–3075 CrossRef CAS PubMed.
- N. Agrawal and P. Mishra, Med. Chem. Res., 2018, 27, 1309–1344 CrossRef CAS PubMed.
- A. Sysak and B. Obmińska-Mrukowicz, Eur. J. Med. Chem., 2017, 137, 292–309 CrossRef CAS PubMed.
- K. V. Chikkula and R. Sundararajan, Int. J. Pharm. Pharm. Sci., 2017, 9, 13 CrossRef CAS.
- R. M. Kumbhare, U. B. Kosurkar, M. Janaki Ramaiah, T. L. Dadmal, S. N. C. V. L. Pushpavalli and M. Pal-Bhadra, Bioorg. Med. Chem. Lett., 2012, 22, 5424–5427 CrossRef CAS PubMed.
- M. B. Bommagani, J. R. Yerrabelly, M. Chitneni, G. Thalari, N. R. Vadiyala, S. K. Boda and P. R. Chitneni, Chem. Data Collect., 2021, 31, 100629 CrossRef CAS.
- F. Xie, T. Ni, Z. Ding, Y. Hao, R. Wang, R. Wang, T. Wang, X. Chai, S. Yu, Y. Jin, Y. Jiang and D. Zhang, Bioorg. Chem., 2020, 101, 103982 CrossRef CAS PubMed.
- P. Palanisamy, S. J. Jenniefer, P. T. Muthiah and S. Kumaresan, RSC Adv., 2013, 3, 19300–19310 RSC.
- K. Singh, R. Pal, S. A. Khan, B. Kumar and M. J. Akhtar, J. Mol. Struct., 2021, 1237, 130369 CrossRef CAS.
- E. K. A. Abdelall, Bioorg. Chem., 2020, 94, 103441 CrossRef CAS PubMed.
- V. Ruiz-Torres, J. A. Encinar, M. Herranz-López, A. Pérez-Sánchez, V. Galiano, E. Barrajón-Catalán and V. Micol, Molecules, 2021, 1237, 130369 Search PubMed.
- D. Bafna, F. Ban, P. S. Rennie, K. Singh and A. Cherkasov, Int. J. Mol. Sci., 2020, 21, 1–49 Search PubMed.
- P. K. Poutiainen, T. A. Venäläinen, M. Peräkylä, J. M. Matilainen, S. Väisänen, P. Honkakoski, R. Laatikainen and J. T. Pulkkinen, Bioorg. Med. Chem., 2010, 18, 3437–3447 CrossRef CAS PubMed.
- H. Ananda, K. S. S. Kumar, M. Hegde and K. S. Rangappa, Mol. Cell. Biochem., 2016, 420, 141–150 CrossRef CAS PubMed.
- T. Chatterjee, N. Iqbal, Y. You and E. J. Cho, Acc. Chem. Res., 2016, 49, 2284–2294 CrossRef CAS PubMed.
- Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Acenã, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef CAS PubMed.
- M. A. Schmidt, K. Katipally, A. Ramirez, O. Soltani, X. Hou, H. Zhang, B. C. Chen, X. Qian and R. P. Deshpande, Tetrahedron Lett., 2012, 53, 3994–3997 CrossRef CAS.
- Y. R. Malpani, B. K. Biswas, H. S. Han, Y. S. Jung and S. B. Han, Org. Lett., 2018, 20, 1693–1697 CrossRef CAS PubMed.
- Y. Xiong, M. Wu, X. Zhang, C. Ma, L. Huang, L. Zhao and B. Tan, Org. Lett., 2014, 16, 1000–1003 CrossRef CAS PubMed.
- P. Pattanayak and T. Chatterjee, J. Org. Chem., 2023, 88, 5420–5430 CrossRef CAS PubMed.
- W. Xiong, B. Wu, B. Zhu, X. Tan, L. Wang, W. Wu, C. Qi and H. Jiang, ChemCatChem, 2021, 13, 2843–2851 CrossRef CAS.
- K. D. Parghi, J. R. Satam and R. V. Jayaram, Green Chem. Lett. Rev., 2011, 4, 143–149 CrossRef CAS.
- N. C. Tran, H. Dhondt, M. Flipo, B. Deprez and N. Willand, Tetrahedron Lett., 2015, 56, 4119–4123 CrossRef CAS.
- R. Harigae, K. Moriyama and H. Togo, J. Org. Chem., 2014, 79, 2049–2058 CrossRef CAS PubMed.
- Schrödinger Release 2023-3, LigPrep, Schrödinger, LLC, New York, NY, 2023 Search PubMed.
- D. Halder, S. Das, R. Aiswarya and R. S. Jeyaprakash, RSC Adv., 2022, 12, 21452–21467 RSC.
- A. K. Shiau, D. Barstad, P. M. Loria, L. Cheng, P. J. Kushner, D. A. Agard and G. L. Greene, Cell, 1998, 95, 927–937 CrossRef CAS PubMed.
- Schrödinger Release 2022-3, Epik, Schrödinger, LLC, New York, NY, 2021 Search PubMed.
- C. Lu, C. Wu, D. Ghoreishi, W. Chen, L. Wang, W. Damm, G. A. Ross, M. K. Dahlgren, E. Russell, C. D. Von Bargen, R. Abel, R. A. Friesner and E. D. Harder, J. Chem. Theory Comput., 2021, 17, 4291–4300 CrossRef CAS PubMed.
- D. Halder, S. Das and R. S. Jeyaprakash, Mol. Diversity, 2023, 166, 107481 Search PubMed.
- D. Halder, S. Das, A. Joseph and R. S. Jeyaprakash, J. Biomol. Struct. Dyn., 2022, 1–14 Search PubMed.
- Schrödinger Release 2023-3: Induced Fit Docking Protocol, Glide, Schrödinger, LLC, New York, NY, 2023 Search PubMed.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley, J. K. Perry, D. E. Shaw, P. Francis and P. S. Shenkin, J. Med. Chem., 2004, 47, 1739–1749 CrossRef CAS PubMed.
- J. Li, R. Abel, K. Zhu, Y. Cao, S. Zhao and R. A. Friesner, Proteins: Struct., Funct., Bioinf., 2011, 79, 2794–2812 CrossRef CAS PubMed.
- Schrödinger Release 2021-4: Protein Preparation Wizard, Epik, Schrödinger, LLC, New York, NY, 2021 Search PubMed.
- Schrödinger Release 2023-3: QikProp, Schrödinger, LLC, New York, NY, 2023 Search PubMed.
- J. Minami, R. Suzuki, R. Mazitschek, G. Gorgun, B. Ghosh, D. Cirstea, Y. Hu, N. Mimura, H. Ohguchi, F. Cottini, J. Jakubikova, N. C. Munshi, S. J. Haggarty, P. G. Richardson, T. Hideshima and K. C. Anderson, Leukemia, 2014, 28, 680–689 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C spectra, HRMS data of the synthesised compounds, docking data of the lead molecule, 2g. See DOI: https://doi.org/10.1039/d4ra02856b |
| This journal is © The Royal Society of Chemistry 2024 |