
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis, and anticancer evaluation of novel coumarin/thiazole congeners as potential CDK2 inhibitors with molecular dynamics†
Samir Bondock *a,
Nada Alabbada,
Aisha Hossana,
Moaz M. Abdoub,
Ali A. Shatic,
Mohammad Y. Alfaific,
Serag E. I. Elbehairic and
Nada M. Mohamedd
*a,
Nada Alabbada,
Aisha Hossana,
Moaz M. Abdoub,
Ali A. Shatic,
Mohammad Y. Alfaific,
Serag E. I. Elbehairic and
Nada M. Mohamedd
aChemistry Department, Faculty of Science, King Khalid University, 9004 Abha, Saudi Arabia. E-mail: bondock@kku.edu.sa; bondock@mans.edu.eg
bEgyptian Petroleum Research Institute, Nasr City, 11727, Cairo, Egypt
cBiology Department, Faculty of Science, King Khalid University, 9004 Abha, Saudi Arabia
dPharmaceutical Chemistry Department, Faculty of Pharmacy, Modern University for Technology and Information, MTI, Cairo, Egypt
First published on 12th June 2024
Abstract
A series of novel coumarin–thiazoles was designed and synthesized as a possible CDK2 inhibitor with anticancer activity with low toxicity. The design relied on having hydrazine thiazole or its open-form thioamide to form H-bonds with the ATP binding site while coumarin maintained the crucial hydrophobic interactions for proper fitting. The biological evaluation revealed that the hydroxycoumarin-thiazole derivative 6c demonstrated the best inhibition with HepG2 and HCT116 IC50 2.6 and 3.5 μM, respectively. Similarly, its open thioamide chain congener 5c exhibited potent inhibition on MCF-7 and HepG2 with IC50 of 4.5 and 5.4 μM, respectively. Molecular docking simulations supported the assumption of inhibiting CDK2 by preserving the crucial interaction pattern with the hinge ATP site and the surrounding hydrophobic (HPO) side chains. Furthermore, molecular dynamics simulations of 5c and 6c established satisfactory stability and affinity within the CDK2 active site.
1. Introduction
Cancer is a multi-process disease of variable etiology that results in rapid and uncontrolled cell division with the overexpression of its relevant machinery enzymes. One of those important enzymes is the cyclin-dependent kinase family members that control the cell cycle. Cyclin-CDK2 is a member of the serine/threonine kinase family that regulates the G1/S phase of the cell cycle upon binding with the cyclin family, initiates DNA synthesis, and regulates the exit from S-phase.1–3 Nonetheless, its overexpression was monitored in several malignancies.4–6 Classically, the ATP binding site of CDK2 is located at the hinge region between the β-sheets N-terminal lobe and the α-helices C-terminal lobe.7,8 Moreover, CDK2 is activated through dimerization by binding to cyclin E/A or phosphorylating its catalytic segment.9 Binding to cyclin A facilitates the S-phase progression10 while binding to cyclin E promotes the retinoblastoma protein phosphorylation to access the G1/S-phase (Fig. 1).11 Structurally, there are four possible binding sites of CDK2 where only one emerges upon activation due to the consequent conformational changes.12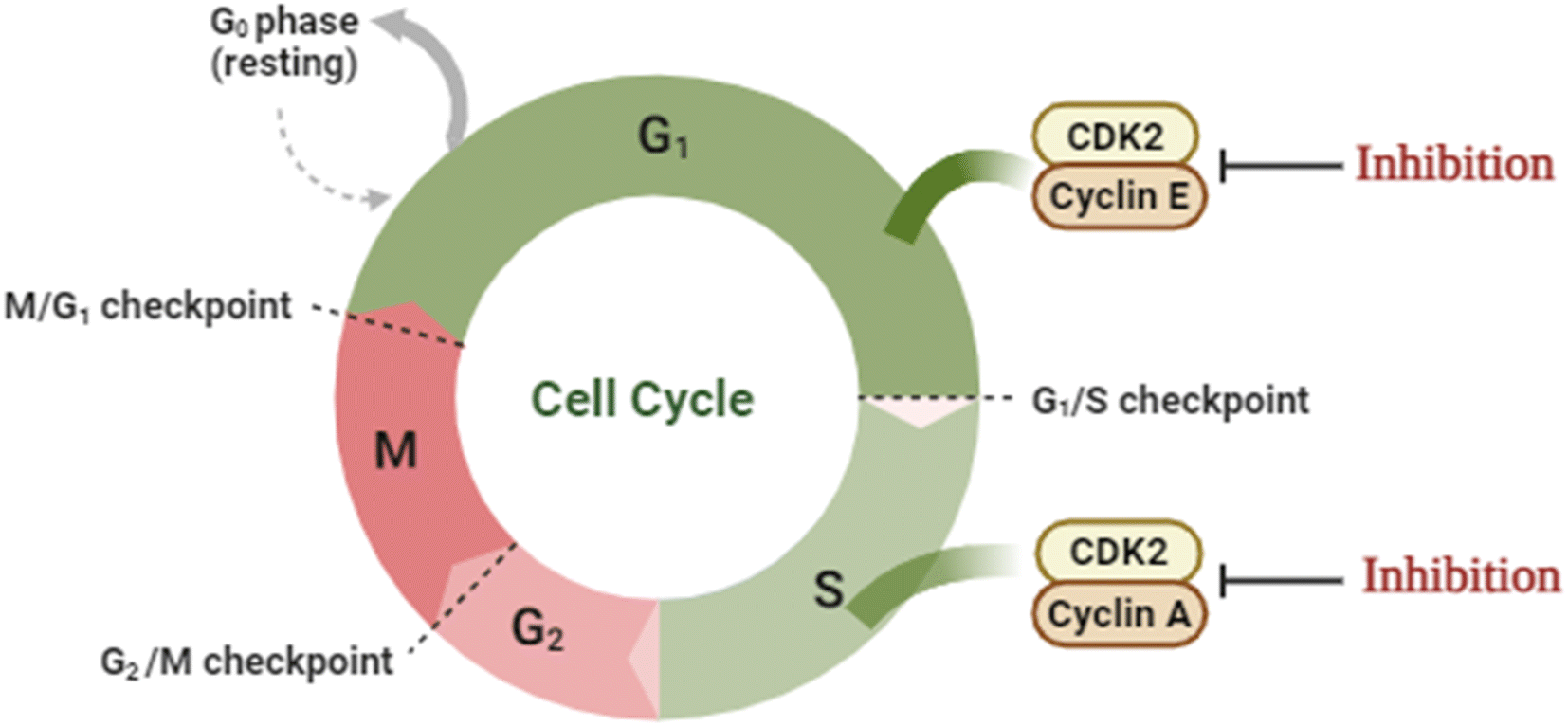 | ||
| Fig. 1 An illustrative presentation of CDK2's role in the cell cycle. | ||
Thiazole and coumarin scaffolds have proved anticancer activity alone or hybridized in one structure, especially with the presence of hydrazine or hydrazone linkage.13–15 Hydrazine and hydrazone moiety were reported to anticipate the overall structure of anticancer activity through the ability of their –NH to form hydrogen bonds with the targeted enzymes which enhanced the binding.16,17
One of the proposed mechanisms for thiazole and coumarin antiproliferative activity is through inhibition of CDK2 (Fig. 2). Compound I demonstrated CDK2 IC50 0.93 μM through binding to the ATP-binding site by its thiazole moiety in addition to forming hydrogen bonds with the crucial Glu81 and Leu83 (PDB ID: 3QTR).18 Moreover, compound II could downregulate CDK2 leading to promising anticancer effects.13 In the same context, hybridizing coumarin and thiazole with hydrazone linkage in IIIa–c resulted in CDK2 IC50 0.022–1.629 nM with antiproliferative activity IC50 0.0596–0.0091 μM against human cervical carcinoma HeLa cell line. The proposed binding pattern of IIIa–c to CDK2 through the interactions of the 2-aminothiazole moiety with the crucial leu83 backbone.19 Furthermore, the thiazole hydrazone derivative IV showed CDK2 IC50 0.39 μM.20
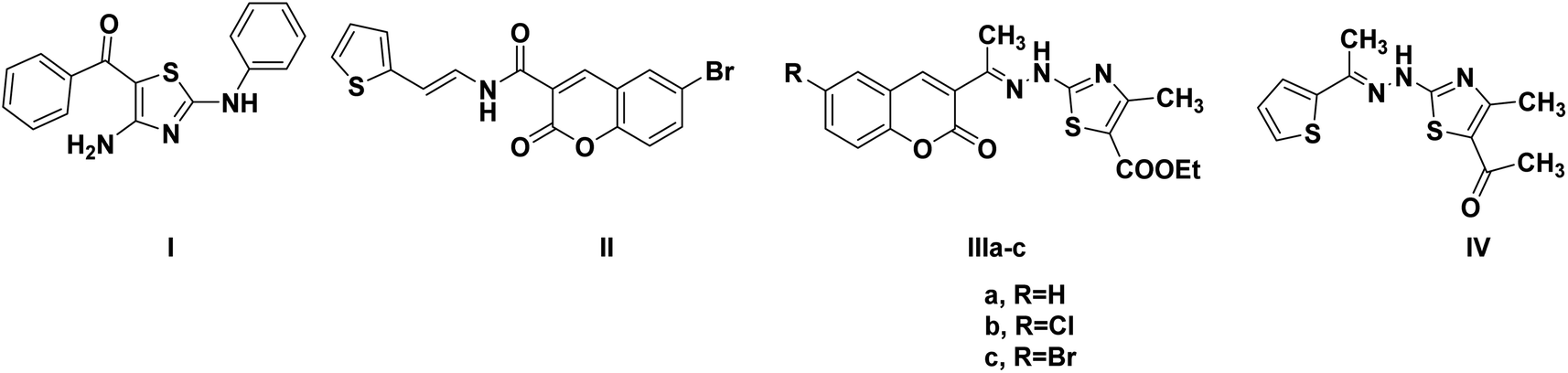 | ||
| Fig. 2 Reported thiazole and coumarin-containing derivatives with CDK2 inhibitory activity. | ||
By investigating the binding conformation of the previously reported III, its thiazole and hydrazine linker showed the essential binding to ATP-binding sites Asp145 and Leu83, respectively. In contrast, its coumarin moiety demonstrated the crucial hydrophobic interactions for proper fitting to CDK2 hydrophobic side chains.19 In this study, the effect of adding a carbonyl group to the hydrazine linker on forming extra H-bonding with the hinge ATP region was inspected. Additionally, the impact of changing the thiazole ring into its parent open thioamide on their ability to consolidate the hinge H-bonding pattern was investigated as well in strategy A (Fig. 3). Moreover, the thiazole ring was substituted with extra phenyl moiety at C4 to achieve more hydrophobic interactions with the hinge surrounding hydrophobic regions for better fitting. On the other hand, the necessity of having a coumarin ring was evaluated by its substitution with a smaller cyano group while preserving the phenyl thiazole-carbohydrazide moiety for H-bonding and the phenyl group for the hydrophobic fitting in strategy B (Fig. 3).
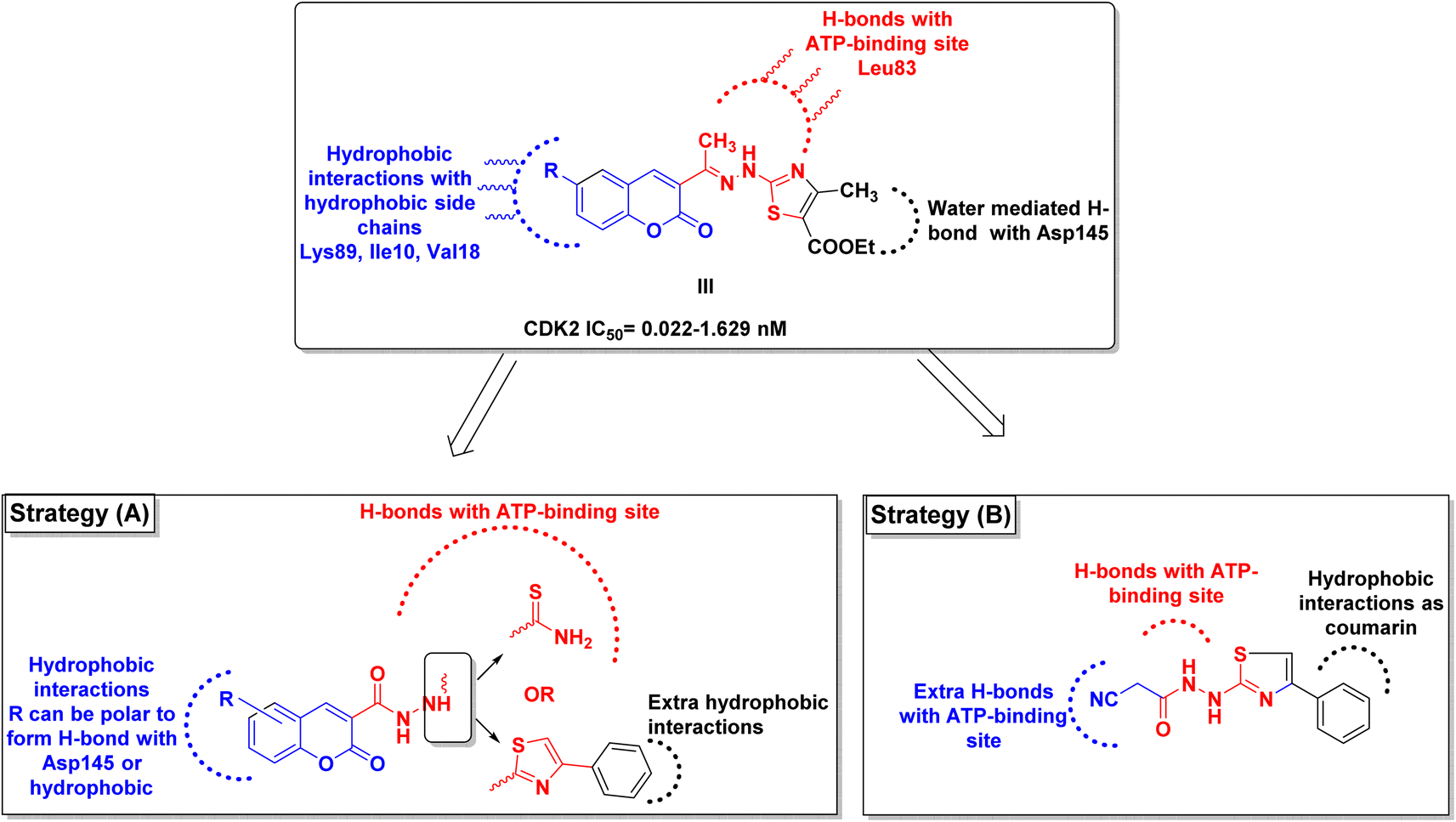 | ||
| Fig. 3 Graphical presentation of the study rationale. | ||
2. Results and discussion
2.1. Chemistry
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and CS functions at frequencies 3415, 3270, 3210, 3137, 2257, 1688, and 1294 cm−1, respectively. The 1HNMR spectrum of 3 displayed five singlet signals resonated at δ 3.63, 7.68, 7.99, 9.35, and 10.14 ppm due to methylene protons, NHa-4, NHb-4, NH-2, and NH-1 protons, respectively. The 13CNMR spectrum displayed four carbon signals at δ 24.73, 116.18, 162.64, and 182.32 ppm, characteristic of CH2, CN, CO, and CS, respectively.
O, and CS functions at frequencies 3415, 3270, 3210, 3137, 2257, 1688, and 1294 cm−1, respectively. The 1HNMR spectrum of 3 displayed five singlet signals resonated at δ 3.63, 7.68, 7.99, 9.35, and 10.14 ppm due to methylene protons, NHa-4, NHb-4, NH-2, and NH-1 protons, respectively. The 13CNMR spectrum displayed four carbon signals at δ 24.73, 116.18, 162.64, and 182.32 ppm, characteristic of CH2, CN, CO, and CS, respectively.
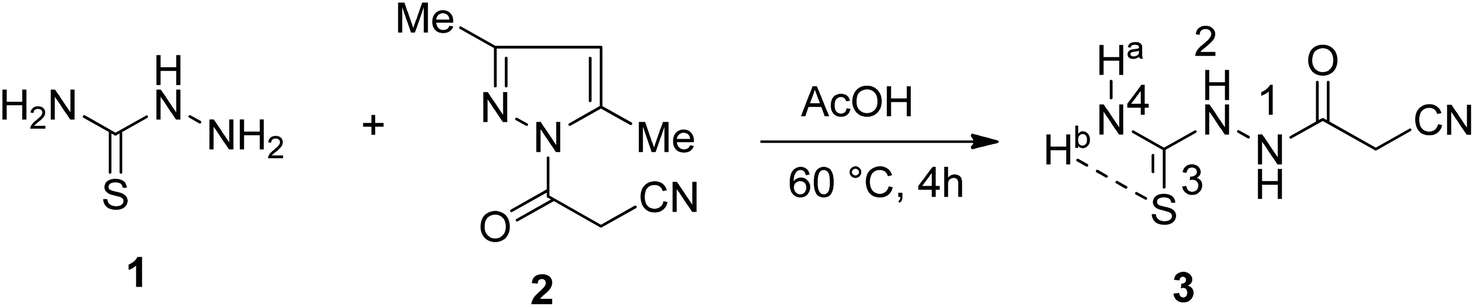 | ||
| Scheme 1 Synthesis of 1-cyanoacetylthiosemicarbazide 3. | ||
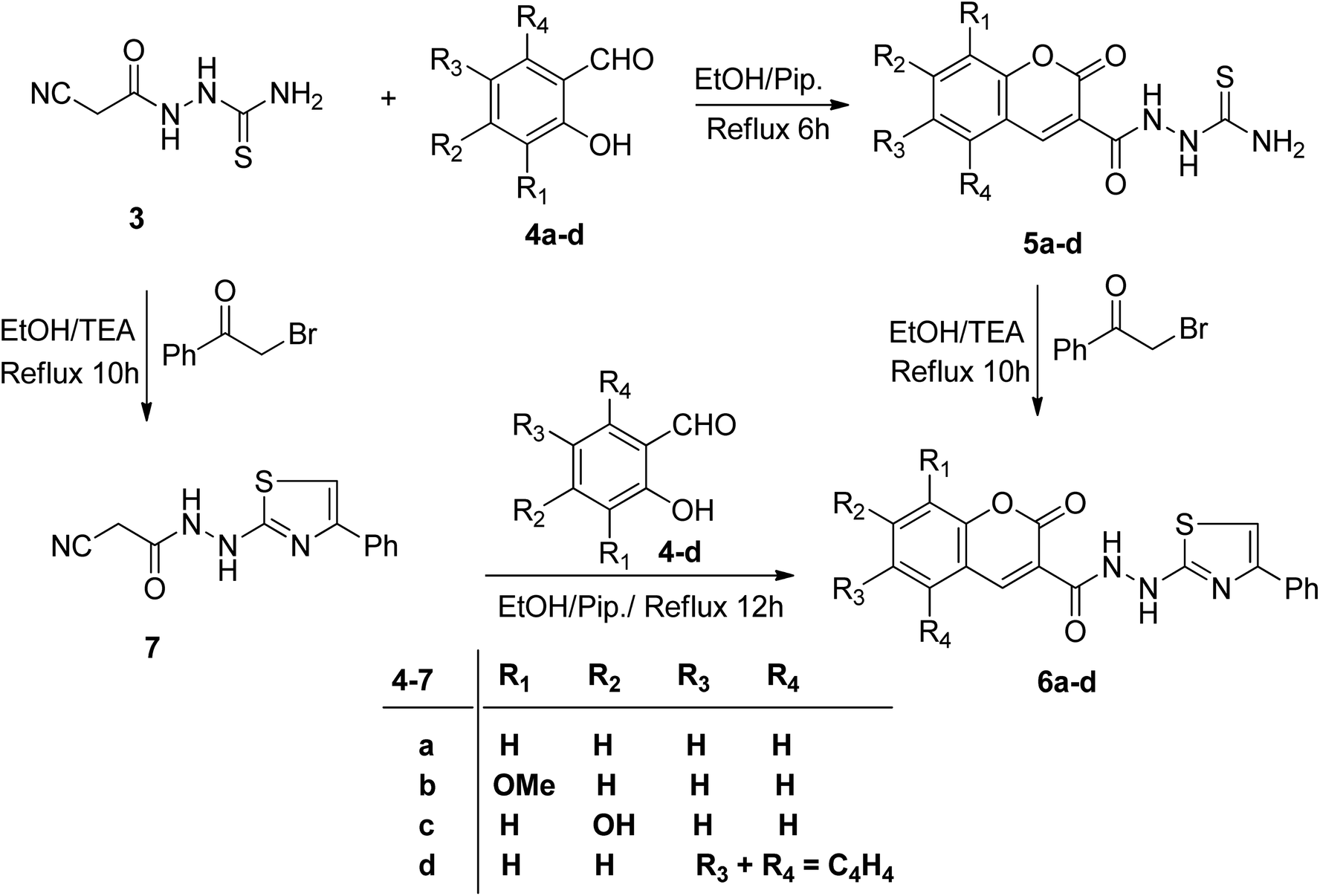
2.1.2.1. Synthesis of 1-(2-oxo-2H-chromene-3-carbonyl)thiosemicarbazide derivatives 5a–d. Merging two bioactive pharmacophores, coumarin, and thiazole, in a single molecule may lead to the discovery of new potent anticancer agents with low toxicity. In this regard, we studied the reactivity of 3 towards sets of 2-hydroxybenzaldehydes and phenacyl bromide in a basic medium to attain some novel coumarin–thiazole hybrids. Thus, the reaction of 3 with certain sets of substituted 2-hydroxybenzaldehydes 4a–d in the presence of piperidine afforded the corresponding thiosemicarbazide derivatives 5a–d (Scheme 2). The IR spectra secured the molecular structures of compounds 5a–d, revealing the absence of stretching absorption peaks of C–H sp3 and nitrile functions (ESI, Fig. S4, S8, S11, and S17†). The characteristic absorption peaks for NH2, NH, lactone C
O, amidic CO, and CS groups were seen at ranges 3404–3253, 3282–3098, 1721–1704, 1674–1651, 1273–1205 cm−1, respectively. The 1HNMR spectrum of coumarin 5a, as an example, displayed two sets of three downfield singlet signals at δ 9.11, 9.48 (coumarin-H4), 10.16, 10.67 (thioamide NH), and 11.52, 13.04 ppm (amidic NH), respectively. The four aromatic protons of the coumarin ring and the two magnetically nonequivalent protons of thioamide resonate at a range of δ 7.41–8.45 ppm. The appearance of doubling signals is ascribed to the existence of compound 5a in two rotamers, syn and anti, attributed to the restricted rotation around the C–N amide bond. Based on the integration values of the more deshielded amidic proton, it is concluded that the two rotamers exist in a ratio (76![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :24). The 13CNMR spectrum of 5a revealed nine carbon peaks. The three more deshielded carbon peaks of CS, coumarin CO, and amidic CO appeared at δ 182.12, 161.97, and 158.30 ppm (ESI, Fig. S9†). The mass spectra of 5a–d revealed molecular ion peaks, which support their molecular weights in each case (ESI, Fig. S7 and S10†). Heterocyclization of thiosemicarbazides 5a–d with 2-bromoacetophenone in the presence of triethylamine afforded coumarin–thiazole conjugates 6a–d in good yields (70–85%) (Scheme 2).
:24). The 13CNMR spectrum of 5a revealed nine carbon peaks. The three more deshielded carbon peaks of CS, coumarin CO, and amidic CO appeared at δ 182.12, 161.97, and 158.30 ppm (ESI, Fig. S9†). The mass spectra of 5a–d revealed molecular ion peaks, which support their molecular weights in each case (ESI, Fig. S7 and S10†). Heterocyclization of thiosemicarbazides 5a–d with 2-bromoacetophenone in the presence of triethylamine afforded coumarin–thiazole conjugates 6a–d in good yields (70–85%) (Scheme 2).
 | ||
| Scheme 2 Synthesis of coumarin–thiazole hybrids 6a–d. | ||
Microanalyses and spectral data confirmed the structures of 6a–d (ESI, Fig. S13–S27†). Their IR spectral data explored the absence of absorption bands for NH2 and CS functions. The characteristic absorption peaks of coumarin CO, amidic CO, and NH groups were seen at ranges 1715–1687, 1682–1635, and 3291–3113 cm−1, respectively. For example, the 1H-NMR spectrum of 6a showed the absence of an NH2 signal and a new singlet signal at δ 7.30 ppm due to the thiazole-H5 proton. Three peaks of the monosubstituted phenyl ring resonate at δ 7.85 (d, 2H), 7.40 (t, 2H), and 7.29 (t, 1H). Also, there are three downfield singlet signals characteristic of coumarin-H4, thiazole-NH, and amidic NH protons seen at δ 8.85, 9.92, and 10.68 ppm, respectively, besides the expected four aromatic signals of coumarin ring residue (ESI, Fig. S14†). The downfield shift signal of the amidic proton may be attributed to the formation of intramolecular hydrogen bonding with the CO group of the coumarin ring. Its 13CNMR spectrum displayed the seventeen carbon signals, which agree with its molecular structure. The most important two carbonyl carbon signals resonated at 161.98 (amidic-CO) and 159.91 (coumarin-CO). The thiazole carbon peaks appeared at δ 103.99, 150.94, and 171.40 ppm due to C-5, C-4, and C-2, respectively (ESI, Fig. S15†).
Conversely, the chemical structures of 6a–d were elucidated via an alternative synthetic pathway. Thus, treating 3 with ω-bromoacetophenone in ethanol containing triethylamine as a base, under reflux, afforded 2-cyano-N′-(4-phenylthiazol-2-yl)acetohydrazide 7. TLC checked the purity of compound 7 and its identity was confirmed by analytical and spectral data (MS, IR, 1HNMR, 13CNMR). The NH2 absorption band was missed in the IR spectrum of 7 and the characteristic absorption bands were displayed at 3415, 3270, and 3195 cm−1 for 2nd amines and at 2257 and 1701 cm−1 for nitrile and amidic CO functions, respectively (ESI, Fig. S28†). In the 1HNMR spectrum of compound 7 characteristic doubling of signals for CH2, thiazole-NH, and amidic NH is noted that corresponds to the existence of two conformers, syn and anti, a common feature of amide compounds because of restricted rotation about the C–N amide bond. Based on the integration values of methylene protons that appeared at δ 3.82, and 3.92 ppm in the 1HNMR spectrum of 7, the two isomers exist in a ratio (84:16) (ESI, Fig. S29†). The 13CNMR spectrum of compound 7 also showed a paring of ten carbon peaks which agree with its molecular structure and support the existence of two rotamers. The more intense characteristic carbon peaks resonate at δ 24.36, 116.24, and 163.71 ppm due to the carbons of methylene, nitrile, and amidic CO, respectively. The thiazole carbon peaks were seen at δ 103.95 (C-5), 151.42 (C-4), and 171.76 ((C-2) ppm) (ESI, Fig. S30†). Heterocyclization of the cyanoacetamide moiety of 7 with a series of 2-hydroxybenzaldehyde derivatives 4a–d in refluxing ethanol containing one drop of piperidine as a base afforded compounds that are consistent with coumarin–thiazole conjugates 6a–d in all aspects (MP, TLC, IR, NMR, MS). The mass fragmentation pattern of compound 6c (ESI, Fig. S31†). As an example of the series prepared, is depicted in Scheme 3.
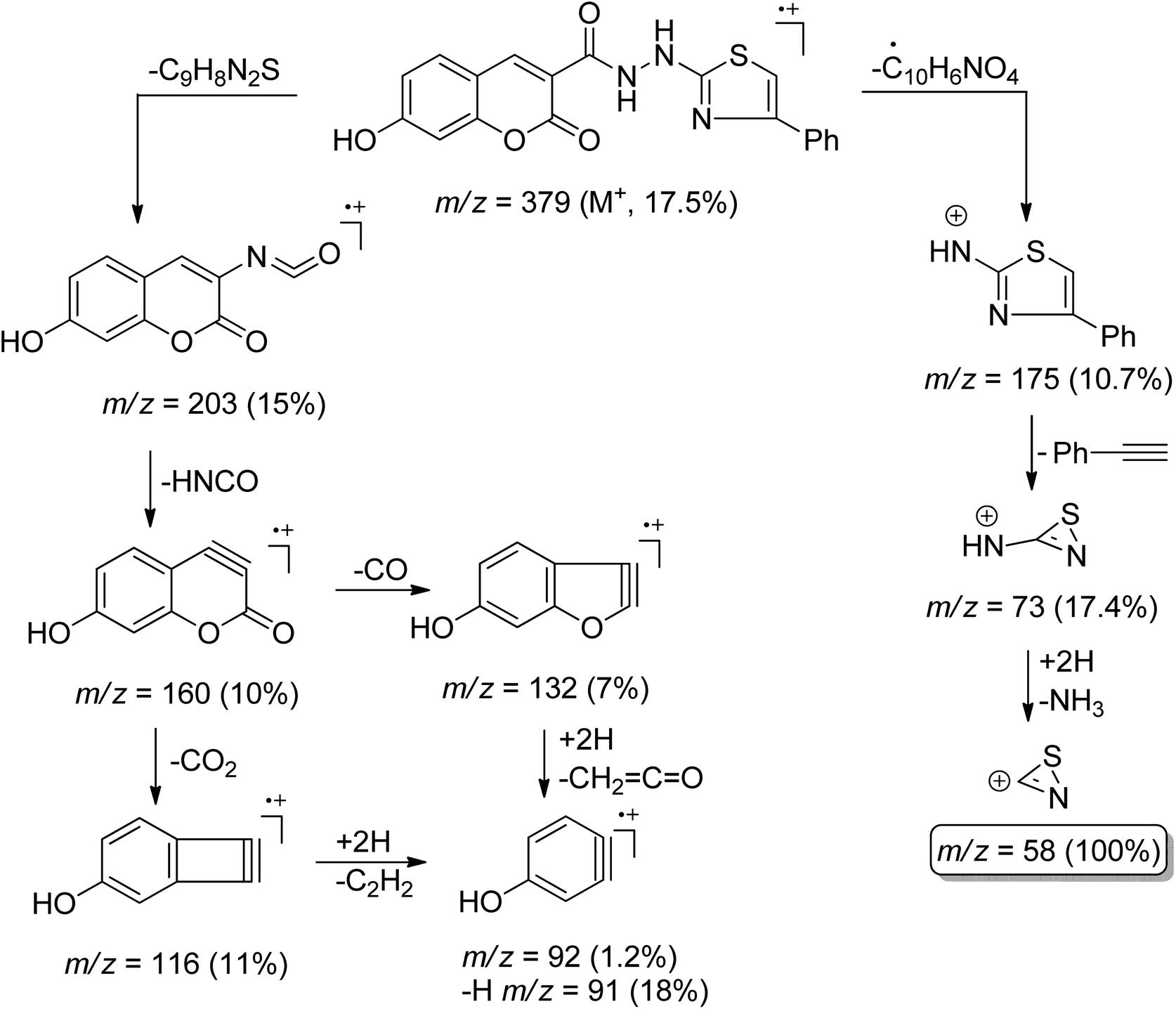 | ||
| Scheme 3 Mass fragmentation pattern of compound 6c. | ||
C bond in 2-butene, which needs about 39 kcal mol−1. In comparison, the C–N bond in methyl N,N-dimethylcarbamate necessitates approximately 15 kcal mol−1.23–25 Consequently, while amide bonds don't allow for free rotation akin to single bonds, they aren't as rigid as double bonds either. This restricted rotation around the C–N amide bond implies that the interconversion between the two conformers, syn & anti, of coumarin amides 5a–d and thiazole 7 (Fig. 4), would be slow enough to manifest as distinct sets of signals in 1HNMR and 13C-NMR spectra.
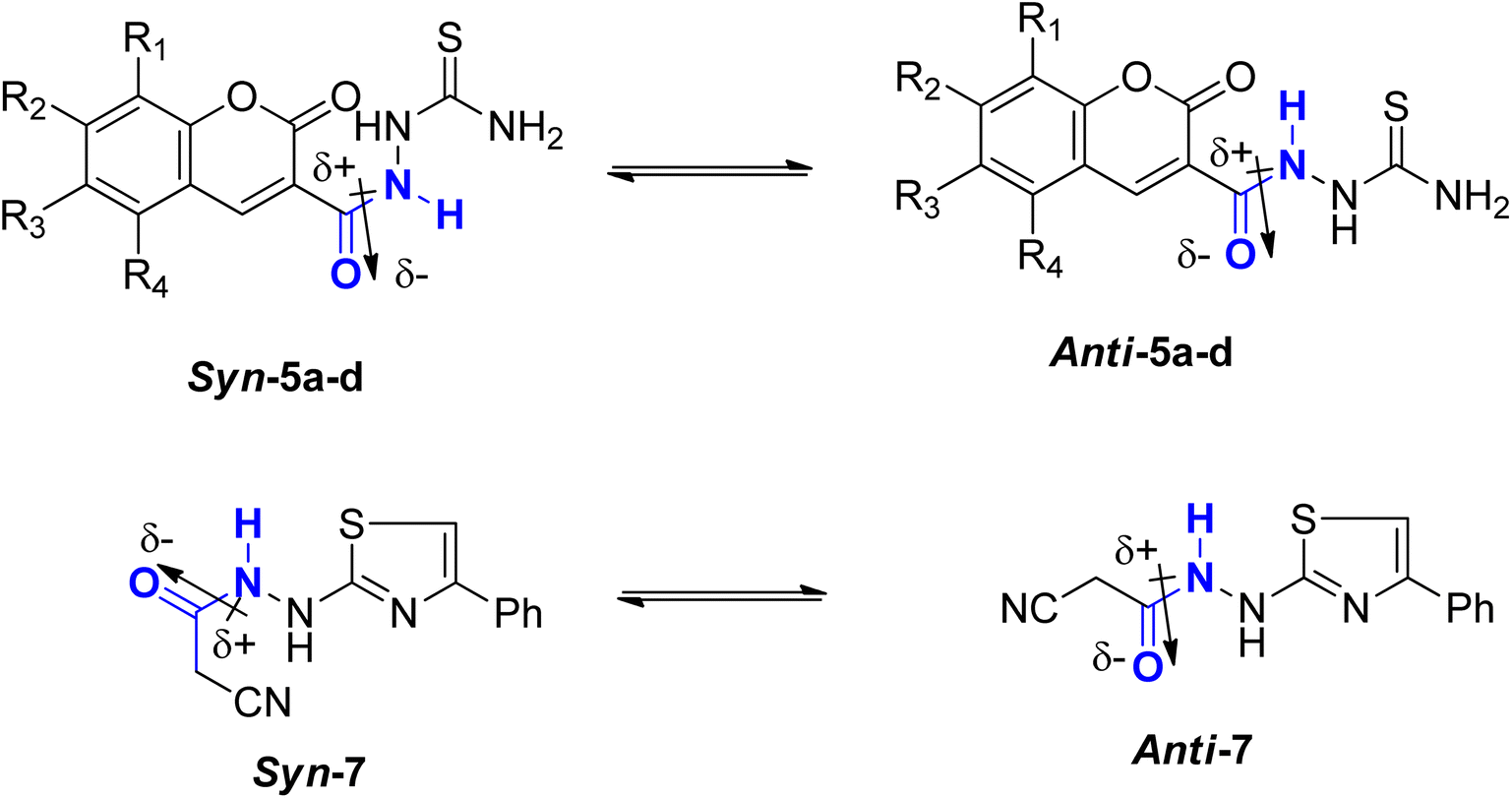 | ||
| Fig. 4 Syn & anti conformers of coumarin amides 5a–d & thiazole amide 7. | ||
The predominant conformer of coumarin and thiazole amides was anticipated to correspond to the more prominent signals, with the NH proton oriented in the anti-conformation to the CO group, while the minor signals were expected to correspond to the syn-conformation (Fig. 4). This assignment stemmed from two primary reasons. Firstly, in the syn-conformer, the proximity between the two groups leads to repulsion due to steric interactions, particularly between bulky groups, thus favoring a shift in equilibrium towards the more stable anti-conformer. Secondly, the dipole moment across the C–N amide bond26 results in a partial negative charge on one side of the bond and a partial positive charge on the opposite side. The region with partial negative charge possesses higher electron density at any given time, causing a shielding effect on surrounding protons.27
In the anti-conformation of thiazole amide 7, the amide NH proton resides directly on the positive end of the dipole. Consequently, the NH signal is expected to be less shielded, appearing more downfield (at approximately 10.60 ppm) in the 1HNMR spectrum. Conversely, in the syn-conformer of 7, the amide NH proton is distant from the positive end, resulting in the NH signal appearing more upfield (around 10.15 ppm) (Fig. 5). Similar assignments of syn and anti-conformations of amides using 1HNMR spectra have been previously documented by LaPlanche and Rogers.28
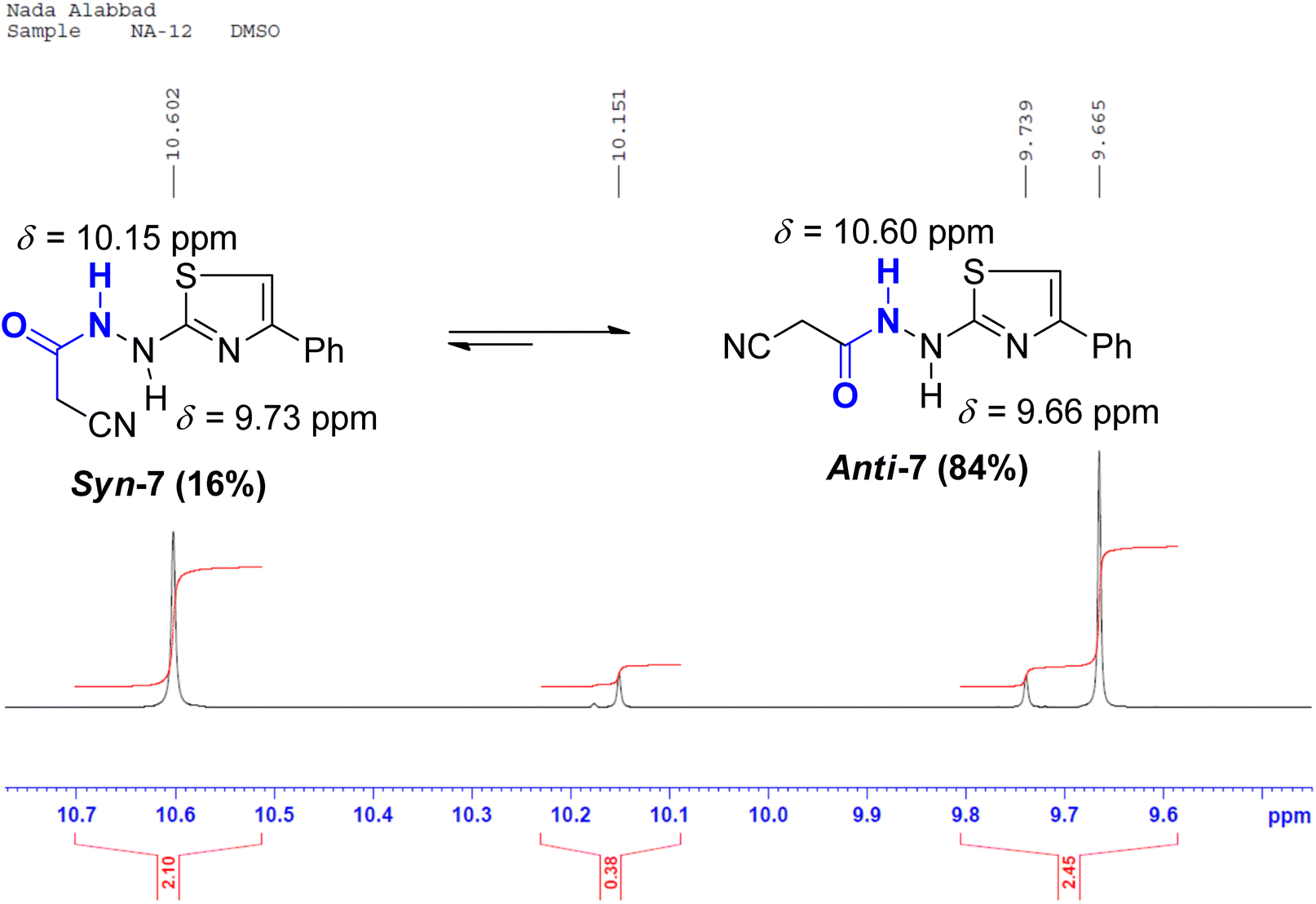 | ||
| Fig. 5 A segment of 1HNMR spectrum (500 MHz, DMSO-d6) of thiazole syn-7 & anti-7. | ||
On the contrary, the more intense signals in the 1HNMR spectrum of coumarin amide 5a were assigned to syn-conformer, and the less intense signals belonged to the minor isomer, anti-conformer. This assignment was consistent with the assumption of the deshielding effect of the dipole of a carbonyl group to the amidic NH proton in the anti-conformation. Fig. 6 shows that the amidic NH proton in the anti-isomer appears highly downfield at δ 13.04 ppm due to forming a possible intramolecular hydrogen bond with CO of the coumarin ring.
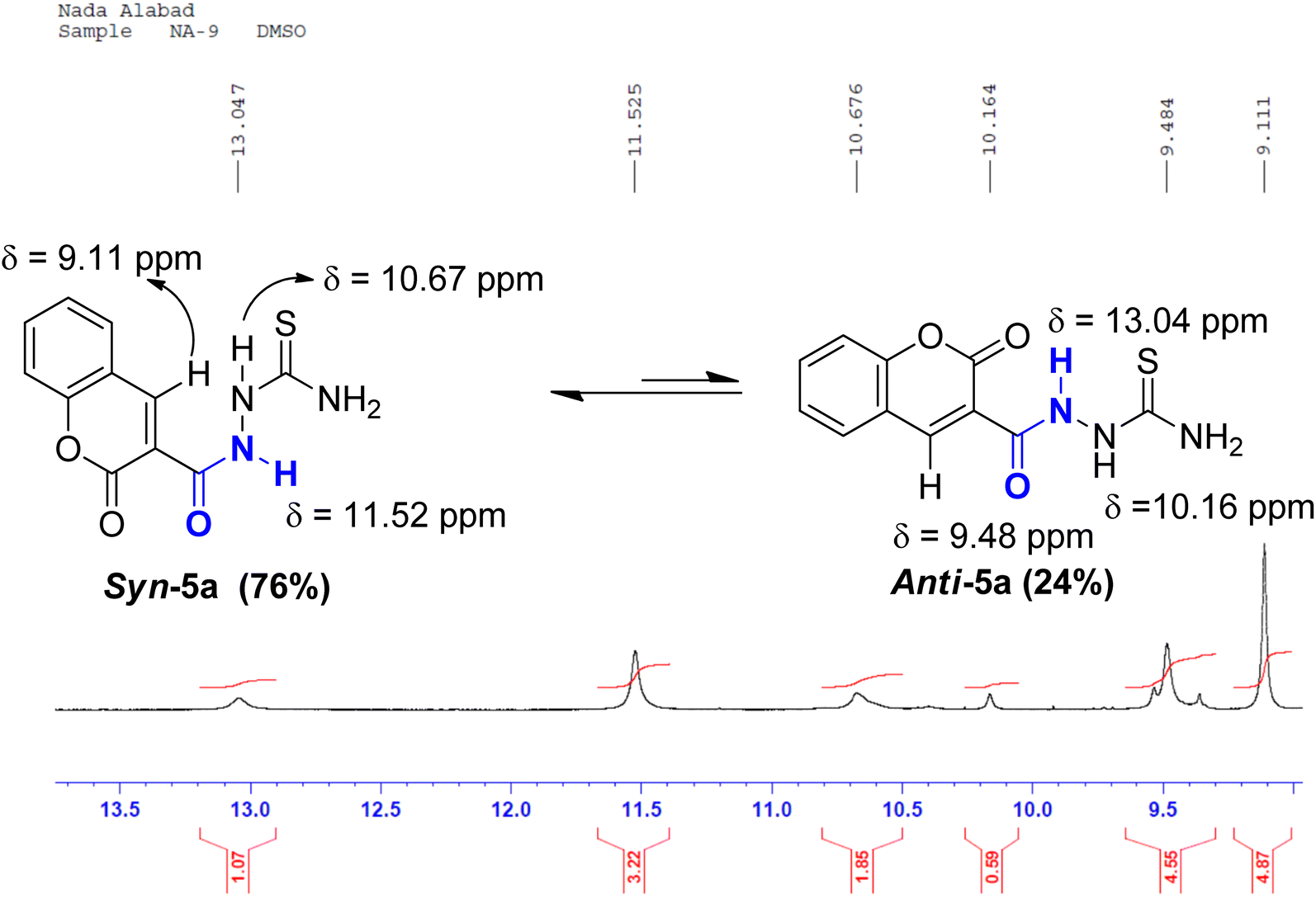 | ||
| Fig. 6 A segment of 1HNMR spectrum (500 MHz, DMSO-d6) of coumarin syn-5a & anti-5a. | ||
While in the syn-isomer, this amidic proton displays at δ 11.54 ppm due to the shielding effect of the dipole of the carbonyl group around the C–N bond. Interestingly, the coumarin-H4 also appears at two different resonances. In the minor isomer, anti-conformer, this signal resonates slightly downfield at δ 9.48 ppm because of the magnetic anisotropic deshielding effect of the amidic carbonyl group. This finding also aligns with the research reported by Gribble and Bousquet.29
2.2. Antiproliferative evaluation and SAR study
The antiproliferative activity of the synthesized derivatives 5a–d, 6a–d, and 7 were evaluated against three human cancer cell lines in addition to normal cells using doxorubicin as a reference. The human cancerous cell lines were breast MCF-7, hepatocellular carcinoma HepG2, and colorectal HCT116 where their toxicity on normal cells was assessed using the somatic hybrid cells EA.hy926. The achieved results presented in Table 1 demonstrated the privilege of the open chain thioamides 5a–d over their cyclized phenyl thiazole 6a–d congeners against the three cancerous cell lines. The superiority of the hydroxyl substituted coumarins 5c and 6c owed to their ability to form extra H-bond with the hinge Asp145 as discussed later in the molecular docking section. The open chain 5c showed an IC50 range of 4.5–7.5 μM while its thiazole congener 6c demonstrated an IC50 range of 2.6–10.0 μM against the three tested cancerous cell lines. Nonetheless, 6c exhibited a lower selectivity ratio than 5c which showed normal EA.hy926 IC50 8.4 μM compared to 26.6 μM of 5c. Moreover, coumarin substitution with the electron-donating methoxy group 5b and 6b dropped the activity by 2–5 folds among the three cell lines. On the other hand, changing coumarin into phenyl coumarin 5d improved its binding to CDK2 hydrophobic side chains, thus preserving the same activity as its hydroxy substituted congener 5c. However, further cyclization of 5d terminal thioamide into phenyl thiazole 6d diminished its cytotoxicity probably because of its larger size that did not accommodate into the CDK2 hydrophobic pocket (see the docking section). In the same context, substituting the coumarin moiety with the cyano group in 7 managed to conserve the necessary binding pattern to the CDK2 hinge and hydrophobic pocket therefore, appreciated cytotoxicity was observed against the three cancerous cell lines (IC50 = 6.5–10.3 μM) with good selectivity ratio. A summary of the derivatives structure–activity relationship is presented in Fig. 7.| Compound | MCF-7 | HepG2 | HCT116 | EA.hy926 | ||||
|---|---|---|---|---|---|---|---|---|
| IC50 (μM) ± SD | IC50 (μM) ± SD | IC50 (μM) ± SD | IC50 (μM) ± SD | |||||
| 5a | 28.6 | 0.3 | 17.7 | 0.1 | 15.2 | 0.7 | 20.0 | 0.7 |
| 5b | 21.8 | 0.8 | 10.8 | 0.5 | 36.2 | 1.6 | 25.8 | 0.4 |
| 5c | 4.5 | 0.4 | 5.4 | 0.3 | 7.5 | 0.3 | 26.6 | 0.3 |
| 5d | 9.3 | 0.6 | 6.9 | 0.2 | 3.5 | 0.2 | 31.6 | 0.4 |
| 6a | 72.9 | 0.8 | 36.5 | 1.8 | 82.5 | 0.5 | 31.9 | 1.2 |
| 6b | 186.6 | 5.6 | 116.3 | 3.7 | 192.6 | 3.2 | 89.0 | 3.5 |
| 6c | 10.0 | 1.0 | 2.6 | 0.1 | 3.5 | 0.1 | 8.4 | 0.3 |
| 6d | 104.9 | 2.7 | 73.5 | 1.4 | 76.9 | 1.4 | 96.6 | 0.5 |
| 7 | 10.3 | 0.6 | 9.9 | 0.2 | 6.5 | 0.4 | 11.6 | 0.4 |
| Doxorubicin | 2.1 | 0.5 | 1.6 | 0.4 | 1.9 | 0.3 | 1.7 | 0.4 |
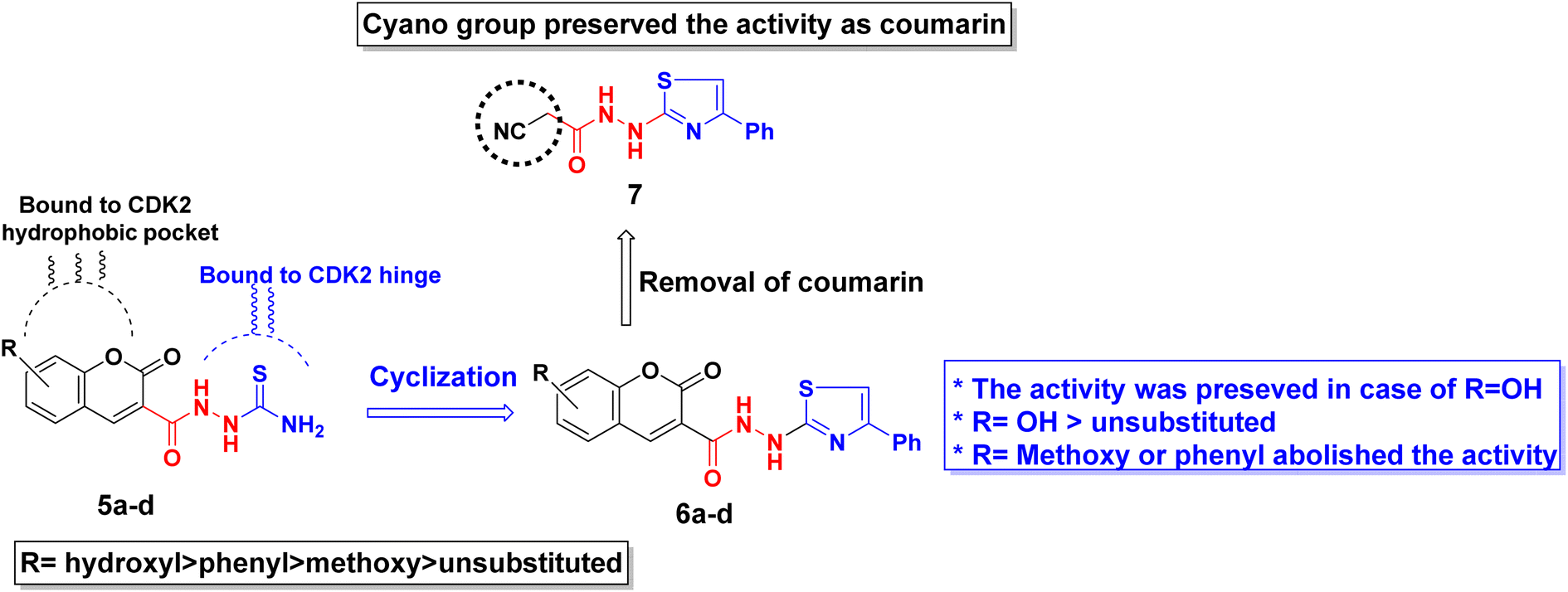 | ||
| Fig. 7 Graphical presentation of the synthesized derivatives structure–activity relationship. | ||
2.3. Molecular docking simulations
To assess the assumption of inhibiting CDK2, the synthesized derivatives were subjected to molecular docking simulations using the deposited crystal structure PDB ID: 3QTR, 1.85 Å.30,31 The docking protocol was validated before commencing the actual simulation through self-docking of the co-crystallized ligand X36 giving RMSD of 1.98 Å difference between the original and re-docked pose (ESI, Fig. S32†) using the open source DockRMSD.32–34 Moreover, during derivatives preparation at the default docking forcefield MMFF94x, the carbohydrazide linkage was automatically changed into carbodiazene at the default temperature and pH. The obtained results (Table 2, Fig. 8–11) revealed that most of the interactions occurred in the ATP-binding site with residues Lys33, Leu83, Glu81 and the gatekeeper Phe80 suggesting a competitive mechanism of inhibition.34–36 All tested derivatives except 6a and 7 showed H-bond with the hinge Leu83 at an average distance of 2.50 Å through their diazene nitrogen or thioamide terminus resembling the purine ring of ATP. In a similar way, all derivatives expect 5a and 5d showed either H-bond or electrostatic attraction with the crucial Asp86 imitating X36 (Table 2 and Fig. 8a). Furthermore, the coumarin scaffold was better oriented inside the hydrophobic pocket of CDK2 than the smaller benzoyl moiety of X36 developing several π-sigma, π-alkyl and alkyl interactions with Ile10, Val18, Ala31, Leu134 and Ala144 (Table 2, Fig. 8–11).| Compound | Binding energy in kcal mol−1 | Interaction type | Interacting residues | Distance in Å | H-bond angle |
|---|---|---|---|---|---|
| X36 | −9.20 | H-donor | Asp86 | 2.41 | 121.30 |
| HPO amide-π | A: GLN85: C,O; ASP86: N | 5.04 | |||
| HPO π-alkyl | ILe10 | 5.34 | |||
| HPO π-alkyl | Va18 | 4.66 | |||
| HPO π-alkyl | Va18 | 5.47 | |||
| HPO π-alkyl | Ala31 | 4.60 | |||
| HPO π -alkyl | Val64 | 5.21 | |||
| HPO π-alkyl | Leu134 | 5.30 | |||
| HPO π-alkyl | Ala144 | 3.88 | |||
| HPO π-alkyl | Ile10 | 4.12 | |||
| 5a | −8.50 | H-acceptor | Leu83 | 2.00 | 158.83 |
| H-donor | Leu83 | 2.49 | 128.20 | ||
| Electrostatic π-cation | Phe82 | 4.27 | |||
| HPO π-sigma | Val18 | 2.55 | |||
| π-Sulfur | Phe82 | 5.24 | |||
| HPO π-alkyl | Ile10 | 5.21 | |||
| HPO π-alkyl | Val18 | 5.12 | |||
| HPO π-alkyl | Ala31 | 4.29 | |||
| HPO π-alkyl | Leu134 | 4.30 | |||
| HPO π-alkyl | Ala31 | 4.82 | |||
| HPO π-alkyl | Leu134 | 5.44 | |||
| 5b | −8.90 | H-donor | Asp86 | 2.28 | 1.53.293 |
| Electrostatic | Asp86 | 3.76 | |||
| H-donor | Leu83 | 2.52 | 108.32 | ||
| HPO π-sigma | Phe80 | 3.61 | |||
| HPO alkyl | Ala144 | 3.41 | |||
| HPO alkyl | Val64 | 4.11 | |||
| HPO π-alkyl | Ile10 | 5.33 | |||
| HPO π-alkyl | Val18 | 4.28 | |||
| HPO π-alkyl | Ala31 | 4.98 | |||
| HPO π-alkyl | Leu134 | 4.86 | |||
| HPO π-alkyl | Val18 | 3.81 | |||
| 5c | −9.80 | Electrostatic | Asp86 | 4.11 | |
| Electrostatic | Asp86 | 3.97 | |||
| H-donor | Leu83 | 2.36 | 166.32 | ||
| H-donor | Asp145 | 2.38 | 118.21 | ||
| HPO π-sigma | Val18 | 2.80 | |||
| HPO π-alkyl | Ile10 | 5.43 | |||
| HPO π-alkyl | Ala31 | 4.62 | |||
| HPO π-alkyl | Leu134 | 4.93 | |||
| HPO π-alkyl | Val18 | 4.70 | |||
| HPO π-alkyl | Ala31 | 4.72 | |||
| HPO π-alkyl | Ala144 | 4.41 | |||
| 5d | −10.20 | H-acceptor | Leu83 | 1.92 | 162.22 |
| H-donor | Leu83 | 2.87 | 121.27 | ||
| H-donor | His84 | 2.43 | 150.70 | ||
| Electrostatic π-cation | Phe82 | 4.29 | |||
| HPO π-sigma | Val18 | 2.50 | |||
| HPO π-alkyl | Ile10 | 5.25 | |||
| HPO π-alkyl | Val18 | 5.11 | |||
| HPO π-alkyl | Ala31 | 4.16 | |||
| HPO π-alkyl | Leu134 | 4.34 | |||
| HPO π-alkyl | Ala31 | 4.65 | |||
| HPO π-alkyl | Leu134 | 5.50 | |||
| HPO π-alkyl | Val18 | 3.76 | |||
| 6a | −7.40 | H-donor | Asp86 | 2.53 | 156.72 |
| Electrostatic | Asp86 | 4.13 | |||
| H-donor | Ile10 | 2.26 | 115.92 | ||
| HPO amide-π | A: GLN85: C, O; ASP86: N | 5.03 | |||
| HPO π-alkyl | Ile10 | 4.30 | |||
| HPO π-alkyl | Val18 | 3.65 | |||
| HPO π-alkyl | Val18 | 3.95 | |||
| HPO π-alkyl | Ala31 | 5.30 | |||
| HPO π-alkyl | Lys33 | 4.97 | |||
| 6b | −7.50 | Electrostatic | Asp86 | 4.74 | |
| Electrostatic | Asp86 | 5.03 | |||
| H-donor | Leu83 | 2.68 | 120.30 | ||
| Electrostatic | Leu298 | 4.94 | |||
| HPO alkyl | Val18 | 3.76 | |||
| HPO alkyl | Lys33 | 4.06 | |||
| HPO π-alkyl | Ala31 | 4.76 | |||
| 6c | −10.30 | Electrostatic | Asp86 | 4.90 | |
| Electrostatic | Asp86 | 5.07 | |||
| H-acceptor | Lys89 | 3.09 | 126.25 | ||
| H-donor | Leu83 | 2.48 | 127.87 | ||
| H-donor | Asp145 | 2.50 | 109.37 | ||
| H-acceptor | Asp145 | 2.70 | 120.02 | ||
| HPO π-sigma | Val18 | 2.80 | |||
| HPO π-sigma | Gln85 | 2.93 | |||
| HPO amide-π | A: HIS84: C, O; GLN85: N amide | 4.01 | |||
| HPO π-alkyl | Ala31 | 4.35 | |||
| HPO π-alkyl | Leu134 | 4.88 | |||
| HPO π-alkyl | Ala144 | 5.26 | |||
| HPO π-alkyl | Val18 | 4.87 | |||
| HPO π-alkyl | Ala31 | 4.71 | |||
| HPO π-alkyl | Ala144 | 4.33 | |||
| 6d | −7.60 | H-acceptor | Leu83 | 2.95 | 140.57 |
| H-acceptor | Asp86 | 2.70 | 110.25 | ||
| Electrostatic | Asp145 | 3.49 | |||
| HPO π-sigma | Val18 | 2.66 | |||
| HPO π-alkyl | Ile10 | 4.71 | |||
| HPO π-alkyl | Ala31 | 4.92 | |||
| HPO π-alkyl | Val18 | 5.38 | |||
| 7 | −8.30 | H-donor | Asp86 | 2.10 | 170.08 |
| Electrostatic | Asp86 | 4.00 | |||
| H-acceptor | Glu12 | 2.96 | 100.95 | ||
| H-acceptor | Lys89 | 2.65 | 171.64 | ||
| H-acceptor | Gly13 | 2.97 | 142.60 | ||
| HPO π–π | Phe80 | 5.14 | |||
| HPO π-alkyl | Ile10 | 5.03 | |||
| HPO π-alkyl | Leu134 | 4.65 | |||
| HPO π-alkyl | Val18 | 5.18 | |||
| HPO π-alkyl | Ala31 | 4.65 | |||
| HPO π-alkyl | Leu134 | 5.07 | |||
| HPO π-alkyl | Ala144 | 3.96 |
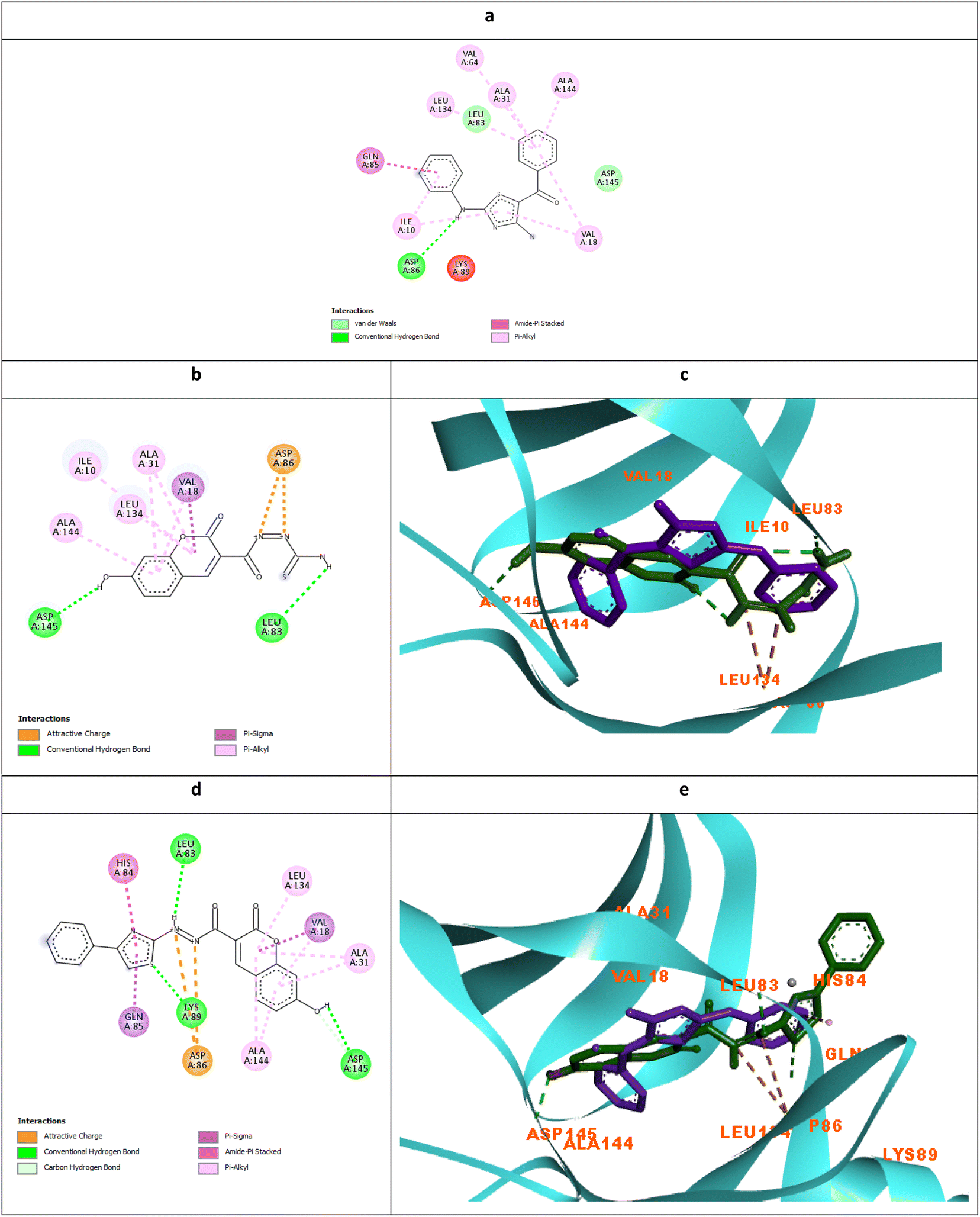 | ||
| Fig. 8 Molecular docking simulations of the co-crystallized ligand X36 (a), 5c (b and c) and 6c (d and e) using PDB ID: 3QTR, 1.85 Å. The evaluated derivatives appeared as a green stick model relative to the magenta-colored X36 showing the interaction bonds as dotted lines. | ||
It was observed that the most potent derivatives 5c, 6c, and 5d showed better binding energy to CDK2 than X36 demonstrating −9.40, −10.30, −10.20, and −9.20 kcal mol−1, respectively (Table 2). Besides achieving better binding energy, cyclizing the thioamide terminus of 5c into a thiazole ring in 6c exposed two additional amide-π stacked interactions with His84 and Glu85 while preserving the H-bond with Leu83 and electrostatic attraction with Asp86 through its diazene linkage (Fig. 8b and d). These extra interactions further supported the orientation of 6c inside the ATP-binding site better than 5c and X36 achieving double the antiproliferative effect of 5c in the case of HepG2 and HCT116 (Fig. 8c and e). On the other hand, cyclizing the thioamide terminus of the benzo-coumarin 5d into a thiazole ring in 6d dropped the activity by 10 folds which was justified by their docking orientation. The smaller 5d was better positioned inside the hinge region showing the usual H-bonds with leu83 and His84 in addition to its proper positioning in the neighboring hydrophobic side chain resulting in eight hydrophobic interactions with Ile18, Val18, Ala31, and leu134 (Fig. 9a and b). However, increasing the size of the compound by cyclizing the thioamide into a phenyl thiazole moiety exposed the large benzo-coumarin outside the hydrophobic pocket losing the aforementioned hydrophobic interactions except with Ile10 (Fig. 9c). Similarly, the cyclization of 5a and 5b into 6a and 6b, respectively resulted in abolishing their antiproliferative activity over the tested three cell lines probably by the same steric clashes with CDK2 (Fig. 10a–d).19
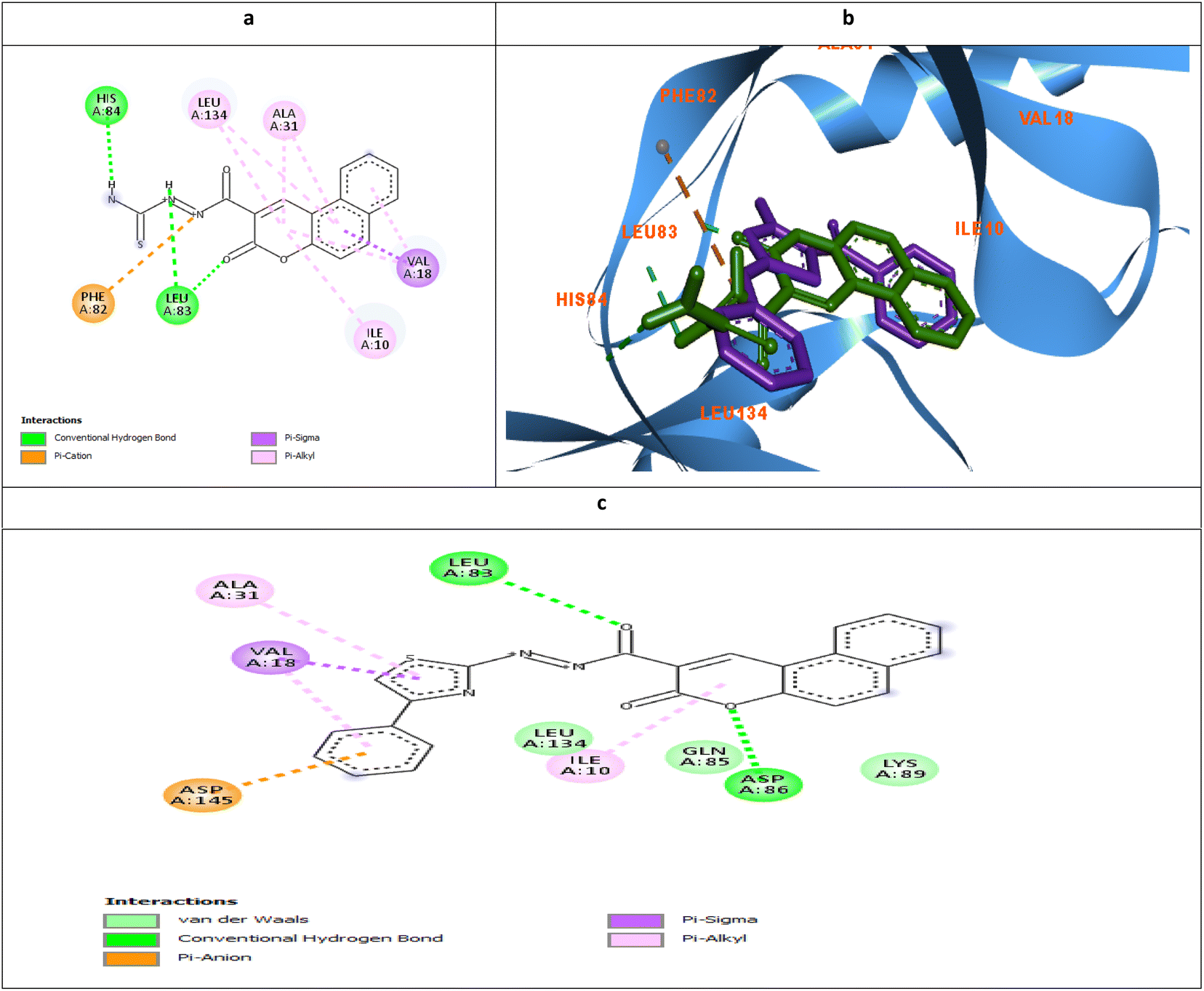 | ||
| Fig. 9 Molecular docking simulations of 5d (a and b) and 6d (c) using PDB ID: 3QTR, 1.85 Å. The evaluated derivatives appeared as a green stick model relative to the magenta-colored X36 showing the interaction bonds as dotted lines. | ||
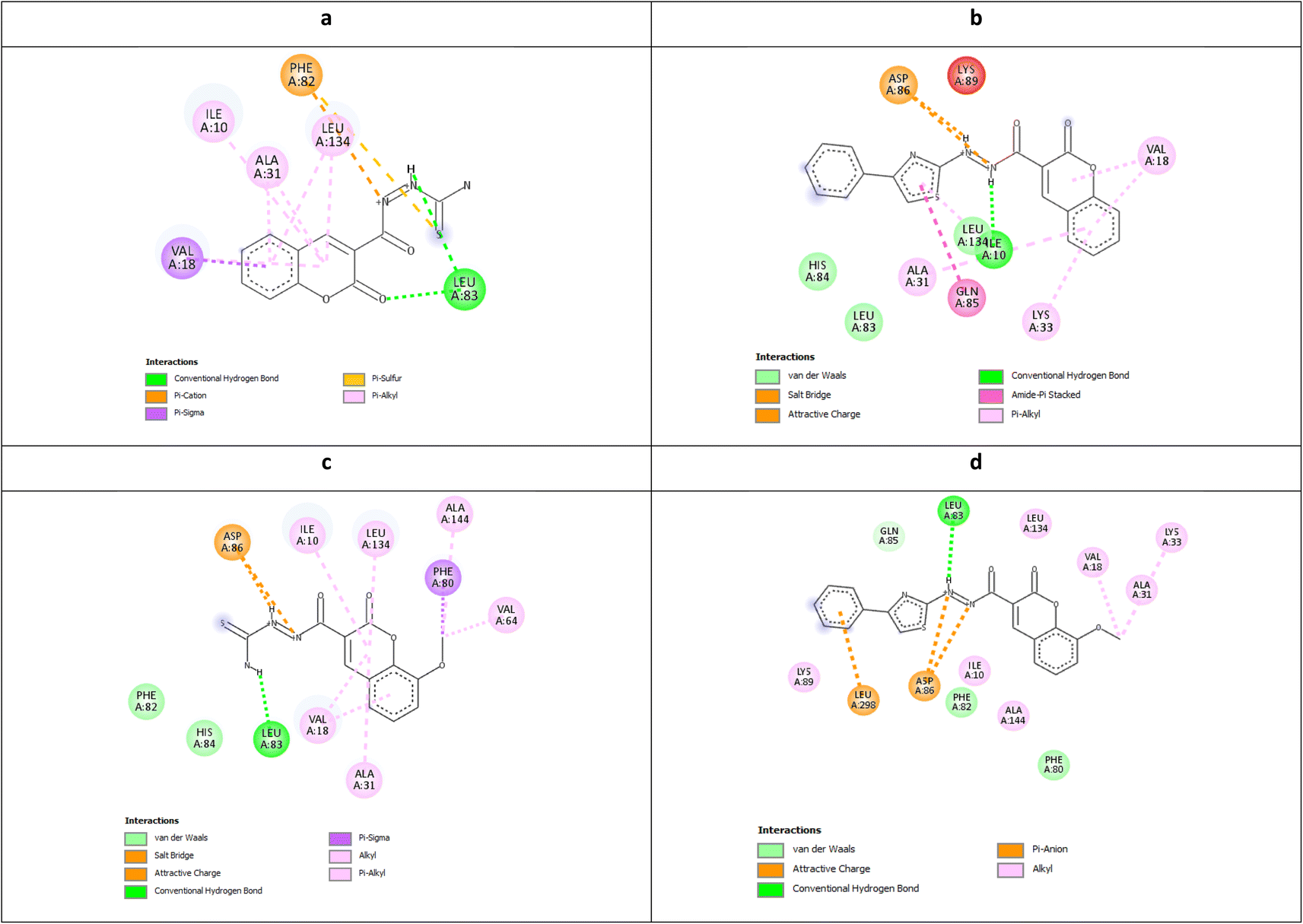 | ||
| Fig. 10 Molecular docking simulations of 5a (a), 6a (b), 5b (c), and 6b (d) using PDB ID: 3QTR, 1.85 Å. | ||
The ability of the phenyl thiazole moiety to imitate the coumarin binding pattern within the hydrophobic side chain was investigated in 7 where the coumarin was substituted with the open chain diazene acquiring a cyano group. Fig. 11a and b demonstrated the capability of the phenyl thiazole to form many hydrophobic interactions with the hydrophobic pocket residues in addition to the unique π–π interaction with the gatekeeper Phe80. Moreover, the cyano group formed a H-bond with the crucial Glu12 and Gly13 with an approximate distance of 2.96 Å which declared IC50 10.3, 9.9, and 6.5 μM against MCF-7, HepG2, and HCT116, respectively.
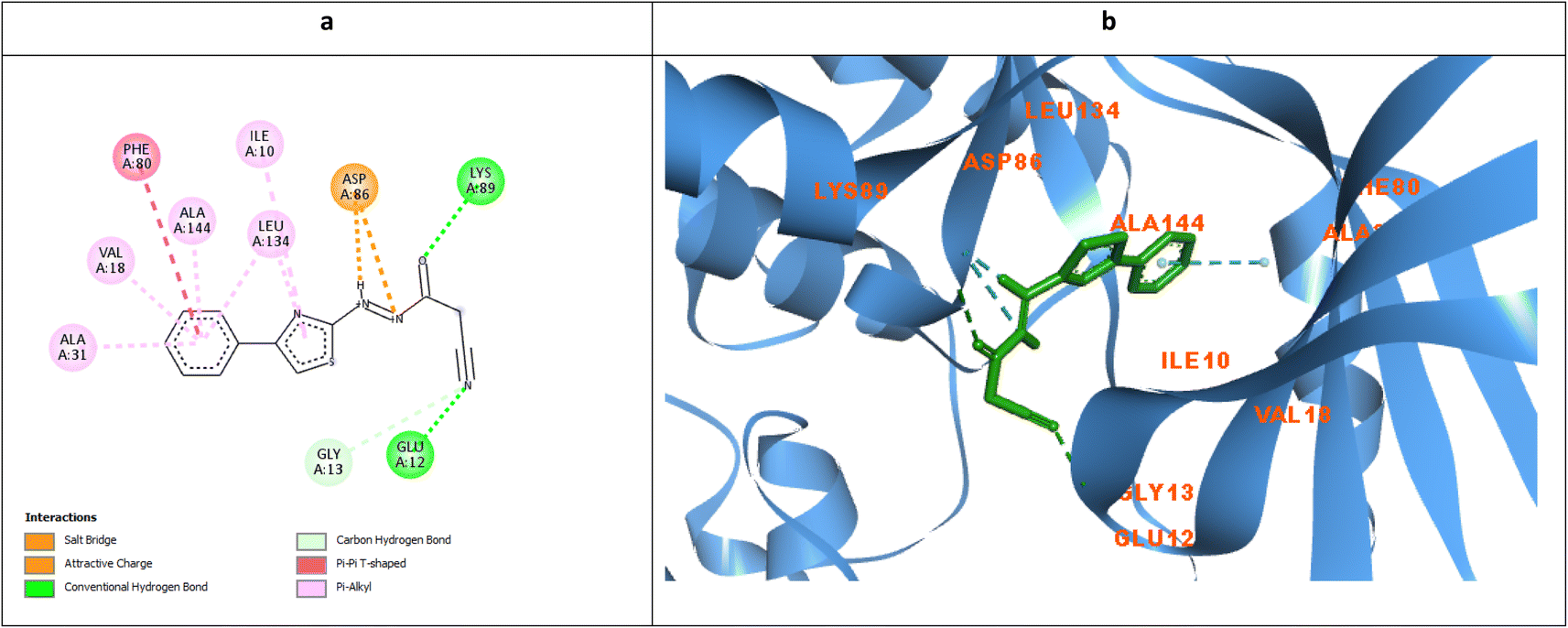 | ||
| Fig. 11 Molecular docking simulations of 7 (a and b) using PDB ID: 3QTR, 1.85 Å. The evaluated derivatives appeared as a green stick model showing the interaction bonds as dotted lines. | ||
2.4. Molecular dynamics simulation
The stability and binding affinity of compounds 5c and 6c were rigorously evaluated through molecular dynamics (MD) simulations extending for 100 nanoseconds. The analysis revealed that both derivatives maintained a commendable degree of stability throughout the simulation period. Notably, compound 5c exhibited superior stability, as evidenced by its root mean square deviation (RMSD) profiles, with an average RMSD of 2.09 Å for compound 5c compared to 3.27 Å for compound 6c (Fig. 12A).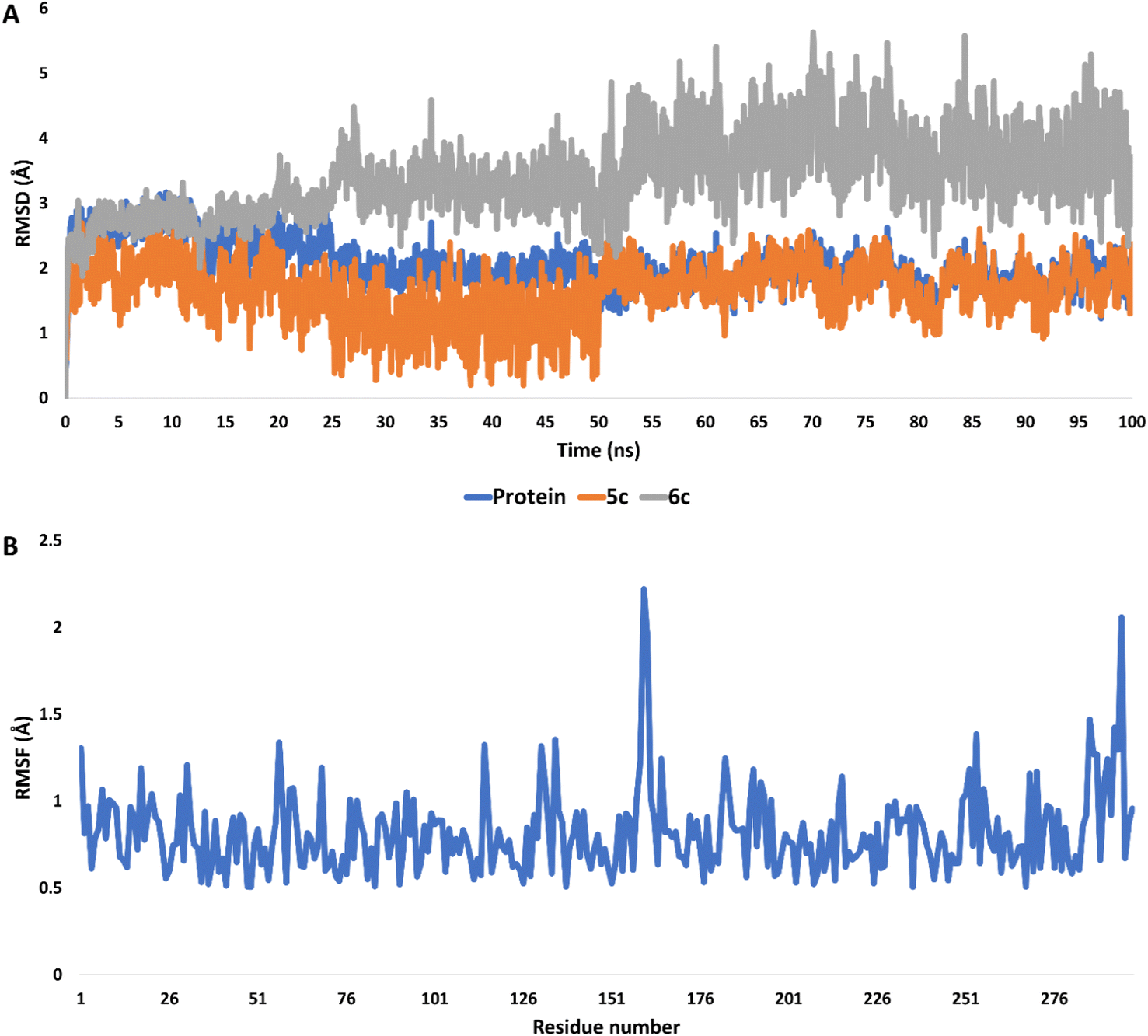 | ||
| Fig. 12 (A) RMSDs of compounds 5c and 6c inside the active site of CDK2 (PDB ID: 3QTR) along with that of the unliganded protein throughout 100 ns-long MD simulation. (B) RMSF profile of the unliganded CDK2 throughout 100 ns-long MD simulation. | ||
In the case of the unliganded protein, its structural integrity remained significantly stable, demonstrating minimal fluctuations throughout the simulation. This was quantitatively supported by an average RMSD of 2.12 Å and a root mean square fluctuation (RMSF) of 1.41 Å (Fig. 12B).
Moreover, the interaction energies of compounds 5c and 6c were evaluated, with mean values estimated at −65.38 kcal mol−1 and −52.44 kcal mol−1, respectively, as shown in Fig. 13. Additionally, the binding free energies of these compounds, calculated using the MM-PBSA method, were determined to be −13.31 kcal mol−1 for compound 5c and −6.20 kcal mol−1 for compound 6c, indicating their potential binding affinities (Table 3).
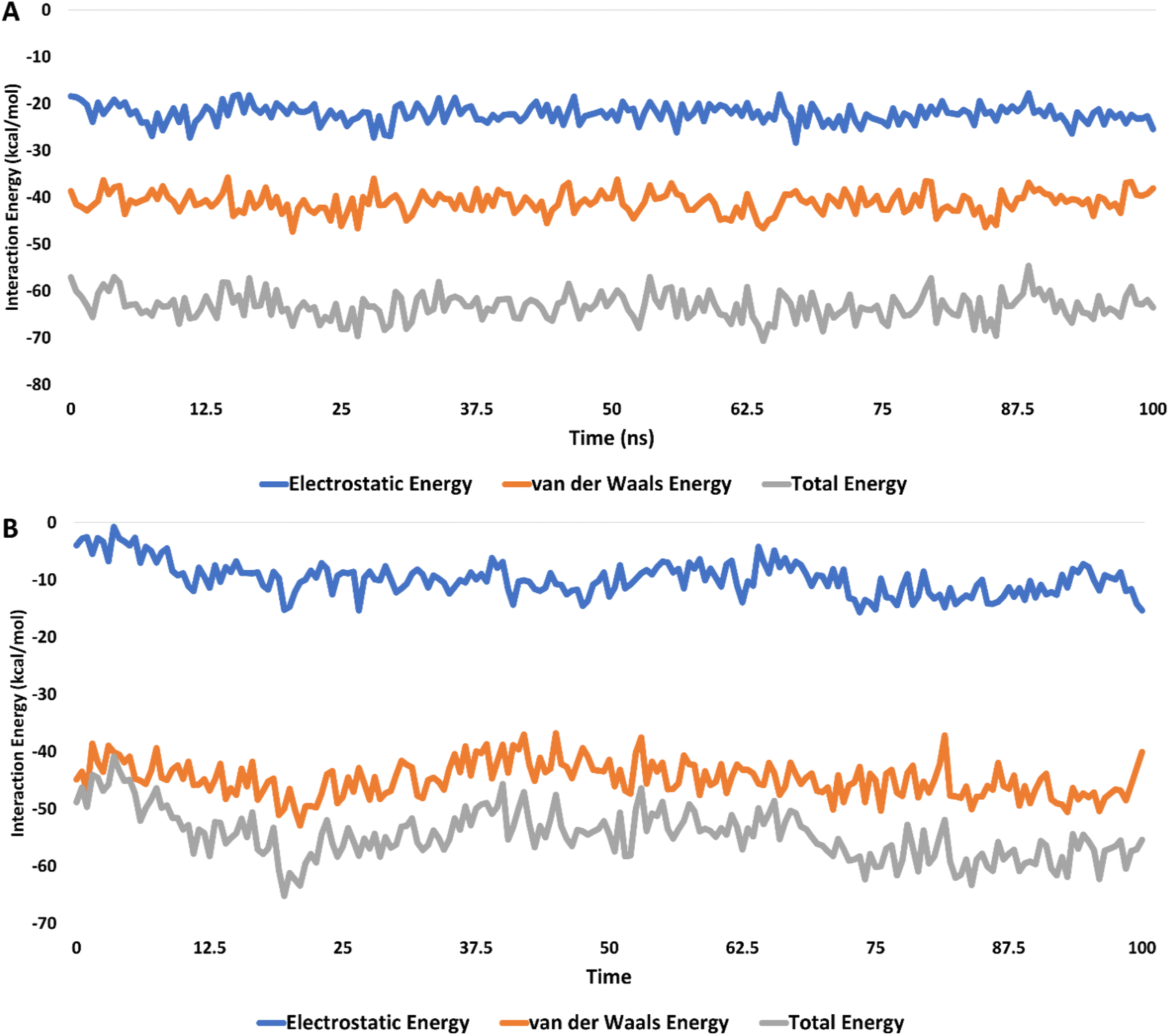 | ||
| Fig. 13 The interaction energies of compounds 5c (A) and 6c (B) inside the active site of CDK2 (PDB ID: 3QTR) for 100 ns-long MD simulation. | ||
| Energy component | 5c | 6e |
|---|---|---|
| ΔGgas | −24.6539 | −16.3099 |
| ΔGsolv | 11.3398 | 10.1038 |
| ΔGtotal | −13.3141 | −6.2061 |
The investigation also highlighted the formation of stable hydrophilic contacts, especially hydrogen bonds, with both compounds forming between 1 and 3 hydrogen bonds during the simulation (ESI, Fig. S35†).
In summation, the findings suggested that compounds 5c and 6c exhibited satisfactory to moderate levels of binding stability and affinity within the active site of the target CDK2. This observation pointed towards their potential as CDK2 inhibitors, underlining their significance in further drug development efforts.
2.5. Physicochemical and pharmacokinetics prediction
The physicochemical descriptors and pharmacokinetics properties of the synthesized derivatives were calculated using the SwissADME web tool from the Swiss Institute of Bioinformatics (SIB).37,38 The obtained prediction was summarized in ESI, Table S1† demonstrated compliance with Lipinski's rule of five giving a bioavailability score of 0.55.39,40 The predicted values of molecular weight, rotatable bonds, H-bond donor, and acceptors were within the acceptable limit of being less than 500 g per mole, 10, 5, and 10, respectively. Moreover, the topological polar surface area of the derivatives was less than 170 Å2 complying with Veber's rule of bioavailability which translated into high GI absorption except for the relatively more polar 5c.41–43 In addition, the in-house logPo/w evaluation showed values less than 5 among all tested derivatives which accounted for their promising bioavailability.44,45 They were expected to not cross the blood–brain barrier and not be affected by the efflux pump P-glycoprotein.46,47 However, some could affect the level of cytochrome P-450 isoenzymes which needs more cautious during further implementation.
3. Experimental section
3.1. Synthesis and spectroscopic characterization
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), 1688 (CO), 1294 (CS); 1H-NMR (850 MHz, DMSO-d6): δppm = 3.63 (s, 2H, CH2), 7.68 (s, 1H, NHa-4); 7.99 (s, 1H, NHb-4), 9.35 (s, 1H, NH-2), 10.14 (s, 1H, amidic-NH); 13C-NMR (212.5 MHz, DMSO-d6): δppm = 24.73 (CH2), 116.18 (CN), 162.64 (CO), 182.32 (CS); anal. calcd. For C4H6N4OS (158.18): C, 30.37; H, 3.82; N, 35.42%; found: C, 30.36; H, 3.84; N, 35.40%.
N), 1688 (CO), 1294 (CS); 1H-NMR (850 MHz, DMSO-d6): δppm = 3.63 (s, 2H, CH2), 7.68 (s, 1H, NHa-4); 7.99 (s, 1H, NHb-4), 9.35 (s, 1H, NH-2), 10.14 (s, 1H, amidic-NH); 13C-NMR (212.5 MHz, DMSO-d6): δppm = 24.73 (CH2), 116.18 (CN), 162.64 (CO), 182.32 (CS); anal. calcd. For C4H6N4OS (158.18): C, 30.37; H, 3.82; N, 35.42%; found: C, 30.36; H, 3.84; N, 35.40%.3.1.2.1. 2-(2-Oxo-2H-chromene-3-carbonyl) hydrazine-1-carbothioamide (5a). This substance was obtained from 3 and salicylaldehyde (6 mmol, 0.73 g) by heating for 4 h. Orange powder; yield: 40%; MP = 200–202 °C; IR (KBr) νmax/cm−1 = 3404, 3253 (NH2) 3282, 3098 (NH), 2964 (CH-sp3), 1721 (C
O), 1674 (amidic CO), 1273 (CS); 1H-NMR (850 MHz, DMSO-d6): δppm = 7.24 (d, J = 8.5 Hz, 2H, coumarin-H5,8)**, 7.27 (t, J = 7.65 Hz, 1H, coumarin-H6)**, 7.41–8.42 (m, 9H, coumarin-H5,6,7,8 + NH2), 9.11 (s, 1H, coumarin-H4)**, 9.48 (s, 1H, coumarin-H4)*, 10.16 (s, 1H, NH)*, 10.67 (s, 1H, NH)**; 11.52 (s, 1H, amidic-NH)**, 13.04 (s, 1H, amidic-NH)*; 13C-NMR (212.5 MHz, DMSO-d6): δppm = 115.50 (coumarin-C3), 116.71 (coumarin-C8)*, 116.80 (coumarin-C8)**, 118.75 (coumarin-C4a), 119.41 (coumarin-C4)*, 119.90 (coumarin-C4)**, 124.70 (coumarin-C6)**, 125.76 (coumarin-C6)*, 130.37 (coumarin-C5)**, 130.74 (coumarin-C5)*, 133.62 (coumarin-C7)**, 134.82 (coumarin-C7)*, 153.82 (coumarin-C8a)**, 154.33 (coumarin-C8a)*, 158.30 (amidic-CO), 161.97 (coumarin-CO), 182.12 (CS); MS m/z (%): 263 (M+, 39.82), 234 (100), 171 (31.65), 112 (27.86), 60 (26.93); anal. calcd. For C11H9N3O3S (263.27): C, 50.18; H, 3.45; N, 15.96%; found: C, 50.15; H, 3.47; N, 15.95%.Signals are ascribed to * anti and ** to syn stereoisomers in this spectrum.
3.1.2.2. 2-(8-Methoxy-2-oxo-2H-chromene-3-carbonyl)hydrazine-1-carbo-thioamide (5b). This substance was obtained from 3 and o-vanillin (6 mmol, 0.91 g) by heating for 8 h. Orange powder; yield 70%; MP = 160–162 °C; IR (KBr) νmax/cm−1 = 3404, 3341 (NH2), 3282, 3177 (NH), 2968 (CH-sp3), 1704 (C
O), 1651 (amidic CO), 1273 (CS), 1H-NMR (850 MHz, DMSO-d6): δppm = 3.80 (s, 3H, OCH3), 7.00 (s, 2H, NH2); 7.34–7.42 (m, 2H, coumarin-H6,7); 7.66 (d, J = 7.5 Hz, 1H, coumarin-H5); 8.58 (s, 1H, coumarin-H4)*, 8.89 (s, 1H, coumarin-H4)**, 9.96 (s, 1H, NH), 13.47 (s, 1H, amidic-NH)*, 13.84 (s, 1H, amidic-NH)**; 13C-NMR (212.5 MHz, DMSO-d6): δppm = 56.62 (CH3)**, 56.70 (CH3)*, 114.21 (coumarin-C3), 115.43 (coumarin-C7)**, 116.19 (coumarin-C7)*, 119.21 (coumarin-C4)**, 119.86 (coumarin-C4)*, 120.79 (coumarin-C5)*, 121.00 (coumarin-C5)**, 125.41 (coumarin-C4a)*, 125.59 (coumarin-C4a)**, 143.02 (coumarin-C6)*, 143.28 (coumarin-C6)**, 144.07 (coumarin-C8a)*, 144.35 (coumarin-C8a)**, 146.69 (coumarin-C8)**, 146.85 (coumarin-C8)*, 157.36 (amidic CO)*, 160.02 (amidic CO)**, 167.32 (coumarin CO), 177.03 (CS); MS m/z (%): 293 (M+, 17.47), 263 (30.77), 174 (12.89), 146 (14.96), 119 (17.09), 92 (43.03), 44 (100); anal. calcd. For C12H11N3O4S (293.30) C, 49.14; H, 3.78; N, 14.33%; found: C, 49.11; H, 3.77; N, 14.35%.Signals are ascribed to * anti and ** to syn stereoisomers in this spectrum.
3.1.2.3. 2-(7-Hydroxy-2-oxo-2H-chromene-3-carbonyl)hydrazine-1-carbo-thioamide (5c). This substance was obtained from 3 and 2,4-dihydroxybenzaldehyde (6 mmol, 0.83 g) by heating for 8 h. Yellow powder; yield: 56%; MP = 292–294 °C; IR (KBr) νmax/cm−1 = 3269, 3214 (2NH), 3108, 3066 (C–H sp2), 1714 (coumarin C
O), 1683 (amidic CO), 1277 (CS); 1H-NMR (850 MHz, DMSO-d6): δppm = 4.80 (s, 1H, coumarin-H8), 6.92 (d, J = 8.5 Hz, 1H, coumarin-H6), 7.20 (s, 2H, NH2), 7.67 (d, J = 8.5 Hz, 1H, coumarin-H5), 8.20 (s, 1H, NH), 8.59 (s, 1H, coumarin-H4), 10.83 (s, 1H, OH), 13.86 (s, 1H, amidic NH); 13C-NMR (212.5 MHz, DMSO-d6): δppm = 105.50 (coumarin-C8), 113.30 (coumarin-C3), 136.10 (coumarin-C5), 156.50 (coumarin-CO), 168.43 (amidic-CO), 181.12 (CS); anal. calcd. For C11H9N3O4S (279.27): C, 47.31; H, 3.25; N, 15.05%; found: C, 47.33; H, 3.27; N, 15.03%.
3.1.2.4. 2-(3-Oxo-3H-benzo[f]chromene-2-carbonyl)hydrazine-1-carbothio-amide (5d). This substance was obtained from 3 and 2-hydroxy-1-naphthaldehyde (6 mmol, 1.03 g) by heating for 8 h. Orange powder; yield: 78%; MP = 186–187 °C; IR (KBr) νmax/cm−1 = 3312, 3259 (NH2), 3163 (NH), 2980, 2970 (CH sp3), 1716 (coumarin C
O), 1675 (amidic CO), 1273 (CS); 1H-NMR (850 MHz, DMSO-d6): δppm = 7.20 (s, 2H, NH2), 7.62 (t, J = 6.8 Hz, 1H, coumarin-H8), 7.76 (t, J = 7.65 Hz, 1H, coumarin-H9), 8.15 (d, J = 8.5 Hz, 2H, coumarin-H10); 8.27 (d, J = 8.5 Hz, 1H, coumarin-H7), 8.68 (d, J = 8.5 Hz, 1H, coumarin-H8), 9.35 (s, 1H, coumarin-H4), 9.76 (s, 1H, NH), 10.70 (s, 1H, amidic-NH); 13C-NMR (212.5 MHz, DMSO-d6): δppm = 113.07 (coumarin-C3), 116.97 (coumarin-C4a), 118.01 (coumarin-C10), 122.89 (coumarin-C5), 126.07 (coumarin-C7), 127.15 (coumarin-C6), 128.03 (coumarin-C8), 129.48 (coumarin-C8a), 135.04 (coumarin-C5a), 136.57 (coumarin-C9), 155.11 (coumarin C4), 159.75 (amidic CO), 163.15 (coumarin-CO), 183.12 (CS); anal. calcd. For C15H11N3O3S (313.33): C, 57.50; H, 3.54; N, 13.41%; found: C, 57.53; H, 3.52; N, 13.43%.
3.1.3.1. 2-Oxo-N′-(4-phenylthiazol-2-yl)-2H-chromene-3-carbohydrazide (6a). This substance was obtained from heating coumarin 5a (4 mmol, 1.05 g) and phenacyl bromide (4 mmol, 0.78 g) for 6 h and was crystallized from EtOH. Orange powder; yield: 61%; MP = 205–207 °C; IR (KBr) νmax/cm−1 = 3291, 3113 (NH), 3078 (CH-sp2), 2980, 2971 (CH-sp3), 1715 (coumarin-C
O), 1682 (amidic CO); 1H-NMR (850 MHz, DMSO-d6): δppm = 7.29 (t, J = 6.8 Hz, 1H, Ar-H4), 7.30 (s, 1H, thiazole-H5), 7.40 (t, J = 7.65 Hz, 2H, Ar-H3,5), 7.46 (t, J = 7.65 Hz, 1H, coumarin-H6), 7.53 (d, J = 8.5 Hz, 1H, coumarin-H8), 7.77 (t, J = 7.65 Hz, 1H, coumarin-H7), 7.85 (d, J = 7.65 Hz, 2H, Ar-H2,6); 8.00 (d, J = 7.65 Hz, 1H, coumarin-H5), 8.85 (s, 1H, coumarin-H4), 9.92 (s, 1H, thiazole-NH), 10.68 (s, 1H, amidic-NH); 13C-NMR (212.5 MHz, DMSO-d6): δppm = 103.99 (thiazole-C5), 116.69 (coumarin-C3), 118.72 (coumarin-C8), 119.39 (coumarin-C4a), 125.63 (coumarin-C4,6), 126.06 (Ar-C2,6), 128.02 (coumarin-C5), 129.06 (Ar-C3,5), 130.79 (coumarin-C7), 134.87 (Ar-C4), 148.26 (Ar-C1), 150.94 (thiazole-C4), 154.47 (coumarin-C8a), 159.91 (coumarin-CO), 161.98 (amidic-CO), 171.40 (thiazole-C2); MS m/z (%): 363 (M+, 10.88), 324 (38.92), 227 (10.08), 196 (18.17), 181 (6.75), 136 (19.04), 110 (9.88), 82 (26.88), 69 (100), 58 (37.24), 44 (18.43).
3.1.3.2 8-Methoxy-2-oxo-N'-(4-phenylthiazol-2-yl)-2H-chromene-3-carbo-hydrazide (6b). This substance was obtained from heating coumarin 5b (4 mmol, 1.17 g) and phenacyl bromide (4 mmol, 0.78 g) for 7 h and was crystallized from a mixture of EtOH and CHCl3. Brown powder; yield: 69%; MP = 229–231 °C; IR (KBr) νmax/cm−1 = 3291, 3265 (NH), 3113, 3055 (
C–H sp2), 2968 (–C–H sp3); 1682 (coumarin CO), 1635 (amidic CO), 1H-NMR (500 MHz, DMSO-d6): δppm = 2.51 (s, 3H, OCH3), 6.83 (s, 1H, thiazole-H5), 7.37–7.47 (m, 4H, Ar-H3,4,5 + coumarin-H6), 7.84–7.91 (m, 2H, coumarin-H5,7), 7.95 (s, 1H, NH), 7.97 (d, J = 5 Hz, 2H, Ar-H2,6)*, 8.04 (d, J = 5 Hz, 2H, Ar-H2,6)**, 8.19 (s, 2H, coumarin-H4 + amidic-NH); 13C-NMR (125 MHz, DMSO-d6): δppm = 55.54 (OCH3), 109.41 (thiazole-C5), 110.48 (coumarin-C3), 112.56 (coumarin-C4a), 125.61 (coumarin-C4,7), 126.45 (Ar-C2,6), 128.82 (coumarin-C6), 128.96 (coumarin-C5), 129.28 (Ar-CH3,5), 129.43 (Ar-CH4), 133.59 (Ar-C1)*, 133.84 (Ar-C1)**, 134.41 (coumarin-C8a), 139.49 (coumarin-C8), 142.72 (Thiazole-C4), 151.89 (coumarin CO)**, 152.16 (coumarin-CO)*, 160.61 (amidic CO)*, 160.97 (amidic CO)**, 161.69 (thiazole-C2)**, 162.84 (thiazole-C2)*; MS m/z (%): 393 (M+, 22.67), 317 (3.21), 234 (12.13), 204 (3.12), 174 (4.67), 132 (7.11), 119 (37.94), 90 (4.86), 66 (24.64), 40 (100).Signals are ascribed to * anti and ** to syn stereoisomers in this spectrum.
3.1.3.3. 7-Hydroxy-2-oxo-N′-(4-phenylthiazol-2-yl)-2H-chromene-3-carbohy-drazide (6c). This substance was obtained from heating coumarin 5c (4 mmol, 1.12 g) and phenacyl bromide (4 mmol, 0.78 g) for 10 h and was crystallized from EtOH. White powder; yield: 31%; MP = 258–260 °C; IR (KBr) νmax/cm−1 = 3241 (NH), 3065 (C–H sp2), 2973, 2925 (C–H sp3), 1710 (coumarin C
O), 1682 (amidic CO); 1H-NMR (850 MHz, DMSO-d6): δppm = 4.85 (s, 2H, NH + coumarin-H8), 6.80 (s, 1H, thiazole-H5), 6.86 (d, J = 8.5 Hz, 1H, coumarin-H6), 7.57 (t, J = 7.65 Hz, 2H, Ar-H3,5), 7.69 (t, J = 7.65 Hz, 1H, Ar-H4), 7.72 (d, J = 8.5 Hz, 1H, coumarin-H5), 8.05 (d, J = 7.65 Hz, 2H, Ar-H2,6), 8.63 (s, 1H, coumarin-H4), 10.97 (s, 1H, OH), 13.93 (s, 1H, amidic NH); 13C-NMR (212.5 MHz, DMSO-d6): δppm = 102.52 (coumarin-C8), 111.62 (thiazole-C5), 115.32 (coumarin-C3), 128.88 (Ar-C2,6 + coumarin-C4a,4), 129.29 (Ar-C3,5 + coumarin-C6,7,8a), 131.71 (Ar-C4), 134.06 (Ar-C1), 136.15 (coumarin-C5), 143.57 (thiazole-C4), 156.03 (coumarin-CO), 162.52 (amidic-CO), 168.43 (thiazole-C2); MS m/z (%): 379 (M+, 17.57), 203 (15), 175 (10.7), 160 (10), 132 (7%), 116 (11%), 92 (1.2), 91 (18), 73 (17.4), 58 (100).
3.1.3.4. 2-Oxo-N′-(4-phenylthiazol-2-yl)-2H-benzo[h]chromene-3-carbohydr-azide (6d). This substance was obtained by heating coumarin 5d (4 mmol, 1.25 g) and phenacyl bromide (4 mmol, 0.78 g) for 9 h and was purified from EtOH. Brown powder; yield: 36%; MP = 214–215 °C; IR (KBr) νmax/cm−1 = 3247, 3191 (NH), 3058, 3025 (C–H sp2), 1715 (coumarin C
O), 1642 (amidic CO); 1H-NMR (850 MHz, DMSO-d6): δppm = 7.29 (m, 2H, Ar-H4 + thiazole-H5), 7.39 (t, J = 7.65 Hz, 2H, Ar-H3,5), 7.68 (t, J = 7.65 Hz, 3H, NH + Ar-H2,6), 7.80 (t, J = 6.8 Hz, 1H, coumarin-H8), 7.86 (t, J = 7.65 Hz, 1H, coumarin-H9), 8.12 (d, J = 8.5 Hz, 2H, coumarin-H10); 8.37 (d, J = 8.5 Hz, 1H, coumarin-H7), 8.64 (d, J = 8.5 Hz, 1H, coumarin-H8), 9.47 (s, 1H, coumarin-H4), 9.96 (s, 1H, NH), 10.74 (s, 1H, amidic-NH); 13C-NMR (212.5 MHz, DMSO-d6): δppm = 104.03 (thiazole-C5), 113.07 (coumarin-C3), 116.97 (coumarin-C4a), 118.01 (coumarin-C10), 122.89 (coumarin-C5), 126.07 (coumarin-C7), 127.15 (coumarin-C6), 128.03 (coumarin-C8), 129.07 (Ar-C2,6), 129.34 (coumarin-C8a), 129.48 (Ar-C5), 129.58 (Ar-C3), 129.65 (Ar-C4), 130.48 (Ar-C1), 135.04 (coumarin-C5a), 136.57 (coumarin-C9), 143.72 (coumarin C4), 150.97 (thiazole-C4), 155.11 (coumarin C10a), 159.85 (coumarin-CO), 162.15 (amidic CO), 171.52 (thiazole-C2); MS m/z (%): 413 (M+, 18.62), 337 (17.21), 295 (16.70), 272 (19.69), 188 (100), 157 (13.03), 104 (41.57), 53 (30.16).
O); 1H-NMR (850 MHz, DMSO-d6): δppm = 3.82 (s, 2H, CH2)**, 3.92 (s, 2H, CH2)*, 7.28 (t, J = 7.4 Hz, 1H, Ar-H4), 7.30 (s, 1H, thiazole-H5), 7.38 (t, J = 7.65 Hz, 1H, Ar-H3,5), 7.82 (d, J = 7.65 Hz, 1H, Ar-H2,6), 9.66 (s, 1H, thiazole-NH)**, 9.74 (s, 1H, thiazole-NH)*, 10.15 (s, 1H, amidic-NH)*, 10.60 (s, 1H, amidic-NH)**; 13C-NMR (212.5 MHz, DMSO-d6): δppm = 23.83 (CH2)*, 24.36 (CH2)**, 103.95 (thiazole-C5)**, 104.94 (thiazole-C5)*, 115.97 (CN)**, 116.24 (CN)*, 126.06 (Ar-C3,5), 128.06 (Ar-C4)**, 128.20 (Ar-C4)*, 129.06 (Ar-C2,6)**, 129.11 (Ar-C2,6)*, 134.82 (Ar-C1)*, 134.99 (Ar-C1)**, 151.06 (thiazole-C4)**, 151.43 (thiazole-C4)*, 163.18 (amidic-CO)**, 168.16 (amidic CO)*, 171.58 (thiazole-C2)*, 171.77 (thiazole-C2)**; MS m/z (%): 258 (M+, 14.05), 190 (13.52), 112 (23.25), 83 (41.90), 77 (100).Signals are ascribed to * syn and ** to anti stereoisomers in this spectrum.
3.2. Antiproliferative evaluation
Three human tumor cells (MCF-7, HCT-116, and HepG2) and EA. hy926 as a normal human cell line was used for evaluation using sulforhodamine B (SRB) assay.48–51 The cell lines were purchased from ATCC and cultured in RPMI-1640 with penicillin (100 U mL−1)–streptomycin (100 μg mL−1) and heat-inactivated fetal bovine serum (10% v/v) at 37 °C and 5% (v/v) CO2. The growing cells were trypsinized and cultured in a 96-well tissue culture plate for 24 h before being manipulated. Cells treated with compounds (0.01, 0.1, 1, 10, and 100 μg mL−1) were mixed with the untreated (control) cells. Following 72 h of dosage exposure, the cells were fixed with 10% w/v TCA for an hour at 4 °C. Following many washes, cells were stained with a 0.4% (w/v) SRB solution for ten minutes in the dark. Glacial acetic acid, 1% (v/v), was used to eliminate any leftover discoloration residue. SRB-stained cells were dissolved in Tris–HCl and allowed to dry overnight before being submitted to a microplate reader to determine the color intensity at 540 nm.3.3. Molecular docking simulations
The molecular docking simulation utilized the open-source PyRx (https://pyrx.sourceforge.io/) embedded Vina tool. The X-ray protein crystal of CDK2 was retrieved from the Protein Data Bank (RCSB: PDB) as PDB ID 3QTR.46 The protein structure was prepared using the embedded DockPrep tool of Chimera17.1. This preparation tool includes the removal of unnecessary water, ions, and the co-crystallized ligand followed by adding hydrogen atoms and assigning charges to protein atoms. The tested derivatives were drawn and energy minimized using OSIRIS DataWarrior and then saved in sdf format (https://openmolecules.org/datawarrior/). The docking simulation utilized a grid box that contained the co-crystallized X36 binding site using the default MMFF94x forcefield. The grid box dimensions were x = 17.4734 Å, y = 17.1954 Å and z = 15.9998 Å. The output binding conformations were visualized by BIOVIA Discovery Studio Visualizer 2021 (https://discover.3ds.com/discovery-studio-visualizer-download).3.4. Molecular dynamics simulations
NAMD 3.0.0 was used for MD simulations applying Charmm-36 forcefield.52,53 The protein chains were constructed using the QwikMD toolkit of the VMD software53,54 and then checked for any missing hydrogens. The co-crystallized water molecules were eliminated and the protonation statuses of the amino acid residues were adjusted to pH = 7.4. After that, the entire structure was immersed in an orthorhombic TIP3P water box together with 0.15 M Na+ and Cl− ions in a solvent buffer with a 20 Å thickness. The systems were then energy-minimized and equilibrated for five nanoseconds. The simulation was started for protein-ligand complexes using the top-scoring docked conformations of 5c and 6c. ForceField Toolkit (ffTK), was used to generate the compounds' topologies and properties. The binding free energy of the docked complex was calculated as reported using 100 frames according to the following equation.55| ΔGbinding = ΔGcomplex − ΔGreceptor − ΔGinhibitor |
Each of the aforementioned terms requires the calculation of multiple energy components, including van der Waals energy, electrostatic energy, internal energy from molecular mechanics, and polar contribution to solvation energy.
4. Conclusion
In the present work, a series of novel thiazolyl–coumarin derivatives were synthesized, starting with 1-cyanoacetylthiosemicarbazide and characterized by spectral studies. The biological results of the synthesized derivatives revealed the privilege of the open-chain thioamides 5a–d over their cyclized phenyl thiazole 6a–d against the three cancerous cell lines. The molecular docking simulation explained the superiority of the coumarins 5c and 6c due to their ability to form extra H-bond with the hinge Asp145. The open chain 5c showed an IC50 range of 4.5–7.5 μM while its thiazole congener 6c demonstrated an IC50 range of 2.6–10.0 μM against the three tested cancerous cell lines. Nonetheless, 6c exhibited a lower selectivity ratio than 5c which showed normal EA.hy926 IC50 8.4 μM compared to 26.6 μM of 5c. In the same context, substituting the coumarin moiety with the cyano group in 7 managed to conserve the necessary binding pattern to the CDK2 hinge and hydrophobic pocket therefore, appreciated cytotoxicity was observed against the three cancerous cell lines (IC50 = 6.5–10.3 μM) with good selectivity ratio. Molecular dynamics simulations showed that 5c exhibited superior stability with an average RMSD of 2.09 Å relative to 3.27 Å of 6c. Moreover, the interaction energies of both 5c and 6c were evaluated, with mean values estimated at −65.38 kcal mol−1 and −52.44 kcal mol−1, respectively. The investigation also highlighted the formation of stable hydrophilic contacts, especially hydrogen bonds, with both compounds forming between 1 and 3 hydrogen bonds during the simulation.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors extend their appreciation to the Deanship of Research and Graduate Studies at King Khalid University for funding this work through small group research under grant number RGP1/53/45.References
- R. Gerosa, R. De Sanctis, F. Jacobs, C. Benvenuti, M. Gaudio, G. Saltalamacchia, R. Torrisi, G. Masci, C. Miggiano, F. Agustoni, P. Pedrazzoli, A. Santoro and A. Zambelli, Crit. Rev. Oncol. Hematol., 2024, 196, 104324 CrossRef PubMed.
- K. L. Ferguson, S. M. Callaghan, M. J. O’Hare, D. S. Park and R. S. Slack, The Rb-CDK4/6 signaling pathway is critical in neural precursor cell cycle regulation, J. Biol. Chem., 2000, 275(43), 33593–33600 CrossRef CAS PubMed.
- L. Cen, B. L. Carlson, M. A. Schroeder, J. L. Ostrem, G. J. Kitange, A. C. Mladek, S. R. Fink, P. A. Decker, W. Wu, J.-S. Kim, T. Waldman, R. B. Jenkins and J. N. Sarkaria, p16-Cdk4-Rb axis controls sensitivity to a cyclin-dependent kinase inhibitor PD0332991 in glioblastoma xenograft cells, Neuro-Oncology, 2012, 14(7), 870–881 CrossRef CAS PubMed.
- T. Chohan, H. Qian, Y. Pan and J.-Z. Chen, Cyclin-dependent kinase-2 as a target for cancer therapy: progress in the development of CDK2 inhibitors as anti-cancer agents, Curr. Med. Chem., 2015, 22, 237–263 CrossRef CAS PubMed.
- S. Tadesse, E. C. Caldon, W. Tilley and S. Wang, Cyclin-Dependent Kinase 2 Inhibitors in Cancer Therapy: An Update, J. Med. Chem., 2019, 62(9), 4233–4251 CrossRef CAS PubMed.
- C. Sánchez-Martínez, L. M. Gelbert, M. J. Lallena and A. De Dios, Bioorg. Med. Chem. Lett., 2015, 25, 3420–3435 CrossRef PubMed.
- A. Huwe, R. Mazitschek and A. Giannis, Small molecules as inhibitors of cyclin-dependent kinases, Angew. Chem., Int. Ed., 2003, 42(19), 2122–2138 CrossRef CAS PubMed.
- K. Parang and G. Sun, Protein kinase inhibitors in drug discovery, Drug Discovery Handbook, ed. S. C. Gad, John Wiley & Sons Inc, 2005 Search PubMed.
- G. Lolli and L. N. Johnson, CAK-Cyclin-dependent activating kinase: a key kinase in cell cycle control and a target for drugs?, Cell Cycle, 2005, 4, 572–577 CrossRef CAS PubMed.
- C. J. Sherr, G1 phase progression: cycling on cue, Cell, 1994, 79(4), 551–555 CrossRef CAS PubMed.
- J. Pines and T. Hunter, Cyclin-dependent kinases: a new cell cycle motif?, Trends Cell Biol., 1991, 1(5), 117–121 CrossRef CAS PubMed.
- Y. Li, J. Zhang, W. Gao, L. Zhang, Y. Pan, S. Zhang and Y. Wang, Insights on structural characteristics and ligand binding mechanisms of CDK2, Int. J. Mol. Sci., 2015, 16, 9314–9340 CrossRef CAS PubMed.
- T. Nasr, S. Bondock and M. Youns, Anticancer activity of new coumarin substituted hydrazide-hydrazone derivatives, Eur. J. Med. Chem., 2014, 76, 539–548 CrossRef CAS PubMed.
- M. Riveiro, N. De Kimpe, A. Moglioni, R. Vazquez, F. Monczor, C. Shayo and C. Davio, Coumarins: old compounds with novel promising therapeutic perspectives, Curr. Med. Chem., 2010, 17, 1325–1338 CrossRef CAS PubMed.
- K. S. Kim, S. D. Kimball, R. N. Misra, D. B. Rawlins, J. T. Hunt, H.-Y. Xiao, S. Lu, L. Qian, W.-C. Han, W. Shan, T. Mitt, Z.-W. Cai, M. A. Poss, H. Zhu, J. S. Sack, J. S. Tokarski, C. Y. Chang, N. Pavletich, A. Kamath, W. G. Humphreys, P. Marathe, I. Bursuker, K. A. Kellar, U. Roongta, R. Batorsky, J. G. Mulheron, D. Bol, C. R. Fairchild, F. Y. Lee and K. R. Webster, Discovery of aminothiazole inhibitors of cyclin-dependent kinase 2: synthesis, X-ray crystallographic analysis, and biological activities, J. Med. Chem., 2002, 45, 3905–3927 CrossRef CAS PubMed.
- L.-Y. Li, J.-D. Peng, W. Zhou, H. Qiao, X. Deng, Z.-H. Li, J.-D. Li, Y.-D. Fu, S. Li, K. Sun, H.-M. Liu and W. Zhao, Potent hydrazone derivatives targeting esophageal cancer cells, Eur. J. Med. Chem., 2018, 148, 359–371 CrossRef CAS PubMed.
- A. Jashari, F. Imeri, L. Ballazhi, A. Shabani, B. Mikhova, G. Dräger, E. Popovski and A. Huwiler, Synthesis and cellular characterization of novel isoxazolo- and thiazolohydrazinylidene-chroman-2,4-diones on cancer and non-cancer cell growth and death, Bioorg. Med. Chem., 2014, 22, 2655–2661 CrossRef CAS PubMed.
- E. Schonbrunn and S. B. R. Alam, Development of highly potent and selective diaminothiazole inhibitors of cyclin-dependent kinases, J. Med. Chem., 2013, 56, 3768–3782 CrossRef CAS PubMed.
- S. S. A. El-Karim, Y. M. Syam, A. M. E. Kerdawy and T. M. Abdelghany, New thiazol-hydrazono-coumarin hybrids targeting human cervical cancer cells: Synthesis, CDK2 inhibition, QSAR and molecular docking studies, Bioorg. Chem., 2019, 86, 80–96 CrossRef PubMed.
- A. M. El-Naggar, M. A. El-Hashash and E. B. Elkaeed, Eco-friendly sequential one-pot synthesis, molecular docking, and anticancer evaluation of arylidene–hydrazinyl–thiazole derivatives as CDK2 inhibitors, Bioorg. Chem., 2021, 108, 104615 CrossRef CAS PubMed.
- R. Balicki and P. Nantka-Namirski, Cancerostatics. Part V. Synthesis of some acyl hydrazine derivatives, Acta Pol. Pharm., 1988, 45(1), 1–7 CAS.
- S. Bondock, N. Alabbad, A. Hossana and M. M. Abdou, 1-Cyanoacetylthiosemicarbazides: Recent advances in their synthesis, reactivity in heterocyclization, and bio-applications, Mini-Rev. Org. Chem., 2024, 21, 409–423 CrossRef CAS.
- H. Eyring, D. M. Grant and H. Hecht, The rotational barrier in ethane, J. Chem. Educ., 1962, 39(9), 466–468 CrossRef CAS.
- P. R. Rablen, Computational analysis of the solvent effect on the barrier to rotation about the conjugated C–N bond in methyl N, N-dimethylcarbamate, J. Org. Chem., 2000, 65(23), 7930–7937 CrossRef CAS PubMed.
- S. E. Barrows and T. H. Eberlein, Understanding rotation about a CC double bond, J. Chem. Educ., 2005, 82(9), 1329–1333 CrossRef CAS.
- V. Kubyshkin and N. Budisa, Amide rotation trajectories probed by symmetry, Org. Biomol. Chem., 2017, 15(32), 6764–6772 RSC.
- H. Friebolin, Basic One-And Two-Dimensional NMR Spectroscopy, John Wiley & Sons, 2010 Search PubMed.
- L. A. LaPlanche and M. T. Rogers, Cis and trans configurations of the peptide bond in N-monosubstituted amides by nuclear magnetic resonance, J. Am. Chem. Soc., 1964, 86(3), 337–341 CrossRef CAS.
- G. W. Gribble and F. P. Bousquet, Deshielding effects in the NMR spectra of ortho-substituted anilides and thioanilides, Tetrahedron, 1971, 27(16), 3785–3794 CrossRef CAS.
- S. Betzi, R. A. Alam and H. Han, CDK2 in Complex with Inhibitor RC-1-148, 2012, RCSB: Protein Data Bank: PDB Search PubMed.
- E. Schonbrunn, S. Betzi, R. Alam, M. P. Martin, A. Becker, H. Han, R. Francis, R. Chakrasali, S. Jakkaraj, A. Kazi, S. M. Sebti, C. L. Cubitt, A. W. Gebhard, L. A. Hazlehurst, J. S. Tash and G. I. Georg, Development of highly potent and selective diaminothiazole inhibitors of cyclin-dependent kinases, J. Med. Chem., 2013, 56, 3768–3782 CrossRef CAS PubMed.
- E. W. Bell and Y. Zhang, DockRMSD: an open-source tool for atom mapping and RMSD calculation of symmetric molecules through graph isomorphism, J. Cheminf., 2019, 11, 40 Search PubMed.
- Y. Zhang, DockRMSD online, available from: https://zhanggroup.org/DockRMSD/.
- Y. Li, J. Zhang, W. Gao, L. Zhang, Y. Pan, S. Zhang and Y. Wang, Insights on Structural Characteristics and Ligand Binding Mechanisms of CDK2, Int. J. Mol. Sci., 2015, 16, 9314–9340 CrossRef CAS PubMed.
- T. M. Sielecki, T. L. Johnson, J. Liu, J. K. Muckelbauer, R. H. Grafstrom, S. Cox, J. Boylan, C. R. Burton, H. Chen, A. Smallwood, C.-H. Chang, M. Boisclair, P. A. Benfield, G. L. Trainor and S. P. Seitz, Quinazolines as Cyclin Dependent Kinase Inhibitors, Bioorg. Med. Chem. Lett., 2001, 11, 1157–1160 CrossRef CAS PubMed.
- A. A. Russo, P. D. Jeffrey and N. P. Pavletich, Structural basis of cyclin-dependent kinase activation by phosphorylation, Nat. Struct. Biol., 1996, 3, 696–700 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
- Available from: https://www.swissadme.ch/index.php.
- C. Lipinski, Lead- and Drug Like Compounds: The Rule of Five Revolution, Drug Discovery Today: Technol., 2004, 1, 337–341 CrossRef CAS PubMed.
- Y. C. Martin, A bioavailability score, J. Med. Chem., 2005, 48(9), 3164–3170 CrossRef CAS PubMed.
- D. F. Veber, S. R. Johnson, H.-Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, Molecular Properties That Influence the Oral Bioavailability of Drug Candidates, J. Med. Chem., 2002, 45(12), 2615–2623 CrossRef CAS PubMed.
- A. Daina and V. Zoete, A BOILED-Egg to predict gastrointestinal absorption and brain penetration of small molecules, ChemMedChem, 2016, 11, 1117–1121 CrossRef CAS PubMed.
- W. J. Egan, K. M. Merz and J. Baldwin, Prediction of Drug Absorption Using Multivariate Statistics, J. Med. Chem., 2000, 43, 3867–3877 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, iLOGP: a simple, robust, and efficient description of n-octanol/water partition coefficient for drug design using the GB/SA approach, J. Chem. Inf. Model., 2014, 54(12), 3284–3301 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Deliv. Rev., 2001, 23, 3–25 CrossRef.
- J. H. Lin and M. Yamazaki, Role of P-Glycoprotein in Pharmacokinetics: Clinical Implications, Clin. Pharmacokinet., 2003, 42, 59–98 CrossRef CAS PubMed.
- F. J. Sharom, ABC multidrug transporters: structure, function and role in chemoresistance, Pharmacogenomics, 2008, 9, 105–127 CrossRef CAS PubMed.
- S. Bondock, T. Albarqi, M. M. Abdou and N. M. Mohamed, Computational insights into novel benzenesulfonamide-1,3,4-thiadiazole hybrids as a possible VEGFR-2 inhibitor: design, synthesis and anticancer evaluation with molecular dynamics studies, New J. Chem., 2023, 47, 20602–20618 RSC.
- S. Bondock, T. Albarqi, T. Nasr, N. M. Mohamed and M. M. Abdou, Design, synthesis, cytotoxic evaluation and molecular docking of novel 1,3,4-thiadiazole sulfonamides with azene and coumarin moieties as carbonic anhydrase inhibitors, Arabian J. Chem., 2023, 16, 104956 CrossRef CAS.
- S. Bondock, T. Albarqi, I. A. Shaaban and M. M. Abdou, Novel asymmetrical azines appending 1,3,4-thiadiazole sulfonamide: synthesis, molecular structure analyses, in silico ADME, and cytotoxic effect, RSC Adv., 2023, 13, 10353–10366 RSC.
- S. Bondock, T. Albarqi, M. Abboud, T. Nasr, N. M. Mohamed and M. M. Abdou, Tail-approach based design, synthesis, and cytotoxic evaluation of novel disubstituted and trisubstituted 1,3-thiazole benzenesulfonamide derivatives with suggested carbonic anhydrase IX inhibition mechanism, RSC Adv., 2023, 13, 24003–24022 RSC.
- J. C. Phillips, R. Braun, W. Wang, J. C. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. V. Kalé and K. Schulten, Scalable molecular dynamics with NAMD, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- J. V. Ribeiro, R. C. Bernardi, T. Rudack, K. Schulten and E. Tajkhorshid, QwikMD-Gateway for Easy Simulation with VMD and NAMD, Biophys. J., 2018, 114(3), 673a–674a CrossRef.
- W. Humphrey, A. Dalke and K. Schulten, VMD: visual molecular dynamics, J. Mol. Graphics, 1996, 14(1), 33–38 CrossRef CAS PubMed.
- B. R. Miller, T. D. McGee, J. M. Swails, N. Homeyer, H. Gohlke and A. E. Roitberg, MMPBSA. py: an efficient program for end-state free energy calculations, J. Chem. Theory Comput., 2012, 8, 3314–3321 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra02456g |
| This journal is © The Royal Society of Chemistry 2024 |