
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSeparation of terbium as a first step towards high purity terbium-161 for medical applications†
Meryem Özge Arman *ab,
Angelo Mullaliua,
Bart Geboesb,
Karen Van Heckeb,
Ganghadar Dasc,
Giuliana Aquilantic,
Koen Binnemansa and
Thomas Cardinaelsab
*ab,
Angelo Mullaliua,
Bart Geboesb,
Karen Van Heckeb,
Ganghadar Dasc,
Giuliana Aquilantic,
Koen Binnemansa and
Thomas Cardinaelsab
aKU Leuven, Department of Chemistry, Celestijnenlaan 200F, P.O. 2404, B-3001 Leuven, Belgium
bBelgian Nuclear Research Centre SCK CEN, Institute for Nuclear Energy Technology, Boeretang 200, Mol, B-2400, Belgium. E-mail: bart.geboes@sckcen.be
cElettra Sincrotrone Trieste, 34149 Basovizza, Trieste, Italy
First published on 20th June 2024
Abstract
Terbium-161 is a medical radiolanthanide that has a beta decay energy and half-life similar to that of lutetium-177, which makes it a promising alternative for therapeutic purposes. The production route using an enriched gadolinium-160 target necessitates the purification of terbium-161 from the untransmuted target material as well as from its stable decay product, dysprosium-161. The separation of neighbouring lanthanides is challenging due to their similar chemical properties and prominent trivalent oxidation states. In this work, the aim is to change the oxidation state of terbium, resulting in the altering of chemical properties that ease the intragroup separation. To this end, a novel separation method is investigated, involving the electrochemical oxidation of terbium (3+) to terbium (4+) followed by anion exchange chromatography. The electrolysis conditions are set to the highest achievable conversion rate, followed by a dilution step during which the pH and electrolyte concentration are slightly lowered to obtain conditions that are compatible with the separation method. XAS analysis is done to characterize the carbonato complex of both oxidation states and to further elucidate the separation mechanism. The results show that the separation approach of combining electrochemical oxidation with anion exchange chromatography is promising for the purification of 161Tb for medical use.
Introduction
Terbium is called a ‘Swiss army knife’ for nuclear medicine,1 since it has four different radioisotopes that are of potential interest to medical applications: 149Tb (t1/2 = 4.12 h, Eα = 3.967 MeV (16.7%)), 152Tb (t1/2 = 17.5 h, Eβ+,av = 1.14 MeV (20.3%)), 155Tb (t1/2 = 5.32 d, Eγ = 86.55 keV (32%) and 105.3 keV (25%)) and 161Tb (t1/2 = 6.96 d, Eβ−,av = 0.154 MeV (100%)). These isotopes have complementary decay characteristics. 161Tb is suitable for therapeutic use due to the emission of low-energy β− particles, conversion and Auger electrons.2 161Tb has a half-life of 6.98 days, and emits low-energy β−-particles with a maximum energy of 0.59 MeV, which is very similar to clinically used 177Lu (t1/2: 6.65 d, Emaxβ: 0.49 MeV).3There are many in vitro and in vivo studies, comparing the therapeutic properties with a well-established 177Lu counterpart.3–7 Recently imaging of cancer patients has been successfully performed after administration of 161Tb-DOTATOC.8 Due to the comparable results and the fact that it is well-tolerated by the patients, the 161Tb-DOTATOC conjugate is considered to be a suitable alternative for 177Lu-DOTATOC. The efficacy of 161Tb in targeted therapy and its potential for personalized treatment approaches, make it a great candidate for medical applications.
161Tb is commonly produced through the non-carrier-added reactor production route, which involves neutron irradiation (n, γ) of an enriched 160Gd target. The neutron capture of 160Gd produces short-lived 161Gd, which decays through β−-emission into 161Tb. When a highly enriched 160Gd target material is used, radionuclidically pure 161Tb can be obtained at the end of the irradiation.9 161Tb further decays to the stable 161Dy, which can interfere with radiolabeling due to their chemical resemblance. As a result, in addition to isolating 161Tb from 160Gd, 161Tb and 161Dy must also be separated to preserve the high specific activity. The existing methods3,10,11 only deal with the separation of micro-amounts of 161Tb from a macro-amount of 161Gd target material.
Lanthanides typically exist in the trivalent oxidation state, which results in a similar coordination chemistry for all the lanthanides. This gives a flexibility to select the radiolanthanide with the most suitable half-life, decay mode and decay energy for either diagnosis or therapy, from a range of possible radiolanthanides, that are all compatible with the same carrier molecule.9,12–16 On the other hand, separation of lanthanides becomes challenging, due to these similar chemical properties. The current method to obtain non-carrier-added (nca) 161Tb is using cation exchange and extraction chromatography with α-hydroxy-isobutyric acid (α-HIBA) as the complexing agent.3,11 However, these methods have only limited efficiency since the process requires long separation times and multiple separation steps. Another strategy involves substantially altering one of the lanthanide ions' chemical characteristics by changing its oxidation state, which creates possibilities for more effective separation methods.17–19
Terbium (E0 = +3.1 V vs. SHE) is, besides cerium, one of the lanthanides that can be oxidized to its tetravalent state, but there is only limited information available in the literature on the oxidation of Tb3+.20–22 In our previous work, a detailed parametric study was performed on electrochemical oxidation of Tb3+ in various aqueous media.23 Although the applied potential could be lowered to +0.9 V vs. Ag/AgCl in periodate medium, the most stable Tb4+ complex was obtained in concentrated carbonate medium. Therefore, an aqueous carbonate medium seems to be the most promising compared to the other media for developing a separation process.
Although there are many examples on rare earth separation,24–30 there are only a few examples of separation processes in general using carbonate medium. These examples consist of extraction of lanthanides and transplutonium elements from alkaline and carbonate media can be achieved mainly via liquid–liquid extraction,31–36 using various extractants such as quaternary ammonium salts,37–39 primary amines40 or alkylpyrocatechols.41 There are also just a few examples of using extraction chromatography in carbonate media.42,43 Thanks to its simplicity, ease of operation, speed, efficiency and the ease to be automated, solid phase extraction is used for the separation process.
In this study, a strategy for the separation of terbium from gadolinium is investigated based on the change of the valence state of terbium. An electrochemical oxidation is performed in highly concentrated carbonate medium, followed by a column separation using Dowex 1-X8 anion exchange resin. Prior to the separation, a dilution step is applied to lower the carbonate concentration. It is established in the work of Sherry et al.42 that at lower carbonate concentrations, higher distribution coefficients can be obtained. The Tb4+ complex was found to remain stable after dilution, which is advantageous for an anion exchange separation process. X-ray absorption spectroscopy measurements were performed to validate the change in the oxidation state as well as to gain information on the structure of the complexes.
Experimental
Chemicals
TbCl3·6H2O (99.9%), GdCl3·xH2O (99.9%) and DyCl3·6H2O (99.9%) were obtained from Strem Chemicals, Inc. (Newburyport, USA). K2CO3 (anhydrous, ≥98%) and KOH pellets (≥85%) were purchased from VWR International B. V. (Leuven, Belgium). Dowex 1-X8 (chloride form) 200–400 mesh ion exchange resin was obtained from Thermo Fisher Scientific (Merelbeke, Belgium). All chemicals were used as received without any further purification. All aqueous solutions were prepared with ultra-pure water (18.2 MΩ cm at 25 °C).Production of 161Tb
[161Tb]TbCl3 was produced by following an adapted literature procedure.11 Highly enriched 160Gd2O3 (1.0 mg, 98.2%, Isoflex USA) was loaded as a nitrate salt into a quartz ampoule, sealed inside an aluminum capsule. The irradiation was performed in the BR2 Reactor at the Belgian Nuclear Research Centre (SCK CEN) at a thermal neutron flux of 3.0 × 1014 n cm−2 s−1. After 10 days of irradiation, the target was removed from the reactor and cooled for 5 days. The irradiated material was then dissolved in trace-metal grade water and the 161Tb was purified using high-pressure ion chromatography (HPIC, Shimadzu), with a strong cation exchange column (ϕ: 6 mm, l: 50 mm, Shodex IC R-621) by elution with α-hydroxyisobutyric acid. The collected fractions containing [161Tb] were further concentrated by loading them onto an LN3 column (ϕ: 2.1 mm, l: 30 mm, TrisKem International) and eluted with 0.05 mol L−1 hydrochloric acid. The isolated solutions of [161Tb]TbCl3 had a radionuclidical purity of 99.998%. An activity of about 2.5 MBq 161Tb was added to a solution containing 4 mol L−1 K2CO3 and 0.07 mol L−1 TbCl3·6H2O. The pH was then increased to 14 by adding KOH. This step is crucial for the electrolysis to be successful as well as for the stability of the Tb4+ complex in carbonate medium.20,23 The final volume was set to 10 mL to ensure the compatibility with the electrolysis cell volume.Electrochemical oxidation of terbium
The electrochemical oxidation of terbium was performed in an undivided three-electrode voltammetry cell (BASI MF-1052 VC-2). A platinum-mesh electrode was used as working electrode, and a platinum wire (ϕ: 0.5 mm, l: 65 mm) served as auxiliary electrode and BASi MF-2056 Ag/AgCl in a 3 mol L−1 KCl solution as reference electrode. A Metrohm Autolab PGSTAT302N potentiostat, operated by Nova 2.1.5 software was used in potentiostatic mode to apply constant a potential of +1.3 V vs. Ag/AgCl for 3 hours.UV-Vis spectroscopy
During the electrolysis, a colour change was observed, starting from the electrode surface, and dissipating into the solution, from colourless to dark brown, indicating the Tb4+ complex formation. This change in the oxidation state was characterized with UV-Vis spectroscopy, by a broad charge transfer band with an absorption peak centred at 365 nm.20,23Absorption spectra were recorded in the 230–1000 nm range using a Shimadzu 1900i spectrophotometer, operated by LabSolutions UV-Vis software. The light source changeover occurred at 340.8 nm. A quartz cuvette with 1 mm path length was used. Spectra were collected at a fast-scanning rate of 300 nm min−1 with steps of 0.5 nm. Baseline correction was done with the blank solutions of carbonate solutions without terbium.
Extraction chromatography
Experiments were conducted in a column separation setup using Triskem 2 mL bed volume cartridges with 1 cm inner diameter and a total length of 2.5 cm. An ISMATEC IPC 8-channel peristaltic pump was used to control the flow rate of the mobile phase. The Dowex 1-X8 resin in chloride form was packed into the column/cartridge as a slurry to ensure uniform packing, and to avoid cracks or holes that might end up in channel formation, hence impact the separation capability and column performance. The packed column was preconditioned with 4 mol L−1 K2CO3 solution, and thus converted to carbonate form. The feed solution was prepared by diluting the initial carbonate concentration from 4 mol L−1 to 1 mol L−1 (pH ≈ 12), and thus the Tb3+ concentration decreases to 0.018 mol L−1. 0.4 mL of the feed solution was loaded onto the column with a flow rate of 0.3 mL min−1 by use of a peristaltic pump. Afterwards, the column was first eluted with water for the elution of Tb4+. The mobile phase was switched to 2 mol L−1 K2CO3 after collecting 9 fractions of 0.5 mL. The fractions were measured by gamma spectrometry for 161Tb. All experiments were performed at room temperature.Gamma spectrometry
Gamma spectrometric analysis of 161Tb (74.57 keV) and 175Yb (396.33 keV) was done using a Canberra High Purity Germanium detector (model: GC2520) with a DSA-1000 Multi-Channel Analyzer (Canberra Semiconductors N. V., Olen, Belgium). Genie 200 gamma analysis software was used (Mirion Technologies). The relevant peaks showed at least 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 counts with the uncertainty of less than 1%, used for the quantitative analysis.
000 counts with the uncertainty of less than 1%, used for the quantitative analysis.
X-ray absorption spectroscopy
XAS spectra were collected at room temperature at the XAFS Beamline in Elettra Synchrotron Trieste (Italy).44 The storage ring was operating at 2 GeV with an optimal current around 300 mA. The energy of the beam was tuned by using a fixed exit monochromator equipped with a pair of Si(111) crystals. An internal reference of cobalt foil is used for the energy calibration. Data were recorded at ambient pressure at Tb L3-edge in dual transmission and fluorescence mode due to the low absorption of the material. A sample holder with Kapton® windows and a Teflon™ spacer with a thickness of 0.1 mm were used as sample holders. The spectra were recorded by scanning through the terbium L3 absorption edge (ca. 7514 eV) in energy interval of 7305–7715 eV. XAS primary data analysis was performed with the Athena program. The ab initio simulation of the XANES spectra was performed using the FDMNES software.45 The Tb L3-edge was calculated in the photoelectron energy range −10 < E < 80 eV with respect to the Fermi energy level using the Muffin-Tin approximation. The Hedin–Lundqvist complex potential46 was used to calculate the excited states. The absorption cross-section was calculated within the dipolar approximation. Clusters of 5 Å built around each non-equivalent absorbing atom were considered.Results and discussion
Stability of Tb4+ complex
Dowex 1-X8 strong anion exchange resin was used in its carbonate form. There are multiple examples of this resin being used with alkaline media, specifically carbonate/bicarbonate systems.42,43,47–50 The resin shows good physical and chemical stability in the pH range of 0–14.51 The resin was prepared with washing 4 mol L−1 K2CO3 and 0.08 mol L−1 KOH mixture before introducing the feed solution. Thus, the resin was expected to convert from Cl− form to CO32− form rather than OH− form due to the high concentration of carbonate ions in the solution. The selectivity towards Tb4+ complex rather than the Tb3+ complex, so that the former is retained on the column, could be explained by the difference in the total charge of these complexes. As the first eluent is water, the concentration of carbonate would drastically drop and preferentially elute the Tb3+ complex. The Tb4+ complex is then eluted by switching back to the carbonate solution, where the carbonate ions might be exchanging with the Tb4+ anionic complex.Although the stability of Tb4+ in carbonate medium was found to be high enough to follow up with a separation process, the possibility of back-reduction due to the resin itself can result in lower separation factors. To investigate this effect, UV-Vis absorption spectra were collected after elution of different volumes of feed solution with water and measuring the related Tb4+ fractions. The UV-Vis spectra shown in Fig. 1, are for the feed solution after the electrolysis, and for the first fractions collected after 0.5 mL and 6 mL feed solution were introduced to the column respectively. In Fig. 2, it can be observed that the absorbance value only changes by 0.01 at 365 nm for the feed and fraction collected from 6 mL of feed, showing that the back reduction due to the column material can be considered as negligible. When a small volume of feed solution (0.5 mL) was introduced, due to the dilution effect by the eluent, the absorbance obviously decreased.
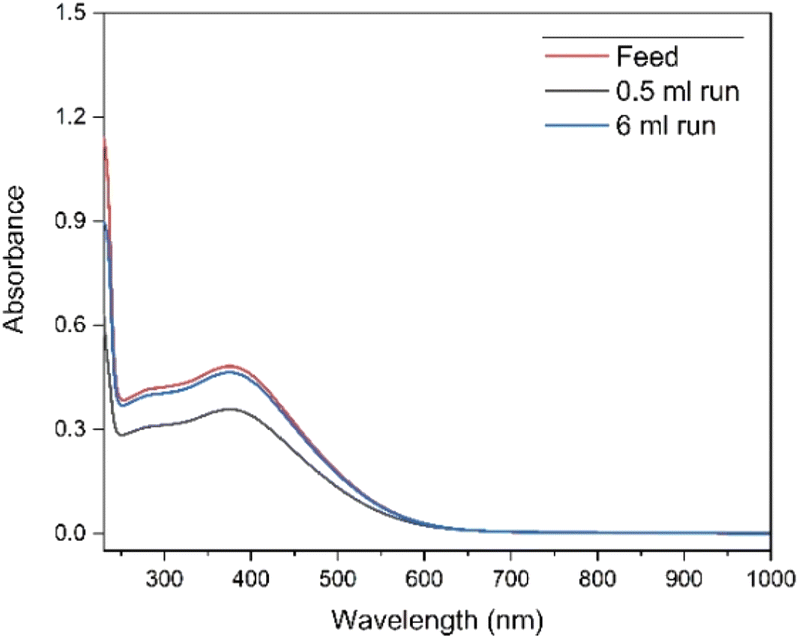 | ||
| Fig. 1 UV-Vis absorption spectra of the fractions collected at different feed volumes of 0.07 mol L−1 Tb4+ in 4 mol L−1 K2CO3 solution at pH = 14. Electrolysis was performed at constant potential +1.3 V vs. Ag/AgCl for 3 h. The formation of Tb(IV) was followed by the absorbance peak at 365 nm. Blank carbonate solution was used as the reference sample for baseline correction. The cell path length was 1 mm. | ||
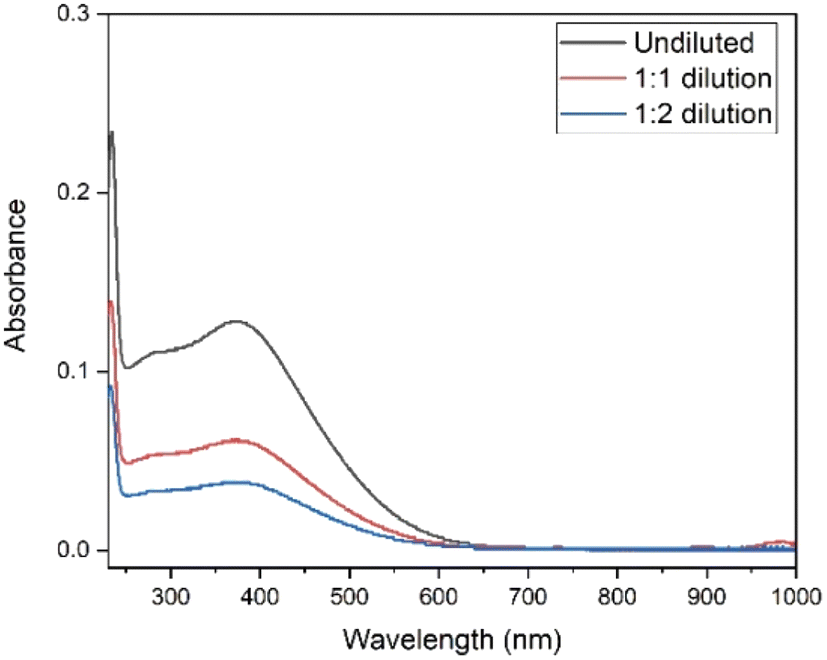 | ||
| Fig. 2 UV–Vis absorption spectra of the undiluted 0.07 mol L−1 Tb4+ in 4 mol L−1 K2CO3 solution at pH = 14, 1:1 (v/v) dilution and 1:2 (v/v) dilution. Electrolysis was performed on the undiluted sample at constant potential +1.3 V vs. Ag/AgCl during 30 min. Blank carbonate solution was used as the reference sample for baseline correction. The path length of the quartz cuvette was 1 mm. | ||
In the studies of Sherry et al., carbonate and bicarbonate complexes of lanthanides were investigated in a K2CO3 concentration range of 0.3 to 3 mol L−1.42,43 These authors also showed that distribution coefficients of the rare earth carbonate complexes were drastically dropping with increasing carbonate concentration.42,43 Therefore, to optimize the separation process, a dilution step was added after the electrolysis. The electrolysed solution was diluted from 4 mol L−1 to 2 mol L−1 and 1.3 mol L−1 K2CO3. To check the stability of Tb4+ complex after the dilution step, UV-Vis spectra were recorded at various time intervals (ESI Fig. S1 and S2†). Absorbance was measured immediately after the electrolysis and any necessary dilutions are prepared swiftly in order to measure both diluted and undiluted samples in the same time frame. As can be seen in Fig. 2, the absorbance values, for the undiluted, 1:1 and 1:2 diluted solutions are 0.127, 0.061 and 0.038 at 365 nm respectively. These values agree well with the dilution factors and these results show that the Tb4+ complex remains stable under dilution, and the influence of back-reduction is negligible (ESI Fig. S1 and S2†).
In the proposed separation method (Fig. 3), Tb3+ and Tb4+ complexes show different affinities to the column, based on the difference in the charge of these complexes. Because of this, a selective separation can be achieved. Misumi et al. described the separation mechanism for tetravalent metal ion complexes in 0.2–0.6 mol L−1 carbonate medium as follows:48
| M4+ + xCO32− ↔ (MCO3)x4−2x | (1) |
| (MCO3)x4−2x + (y/2)R2CO3 ↔ RyM(CO3)x + (y/2)CO32− | (2) |
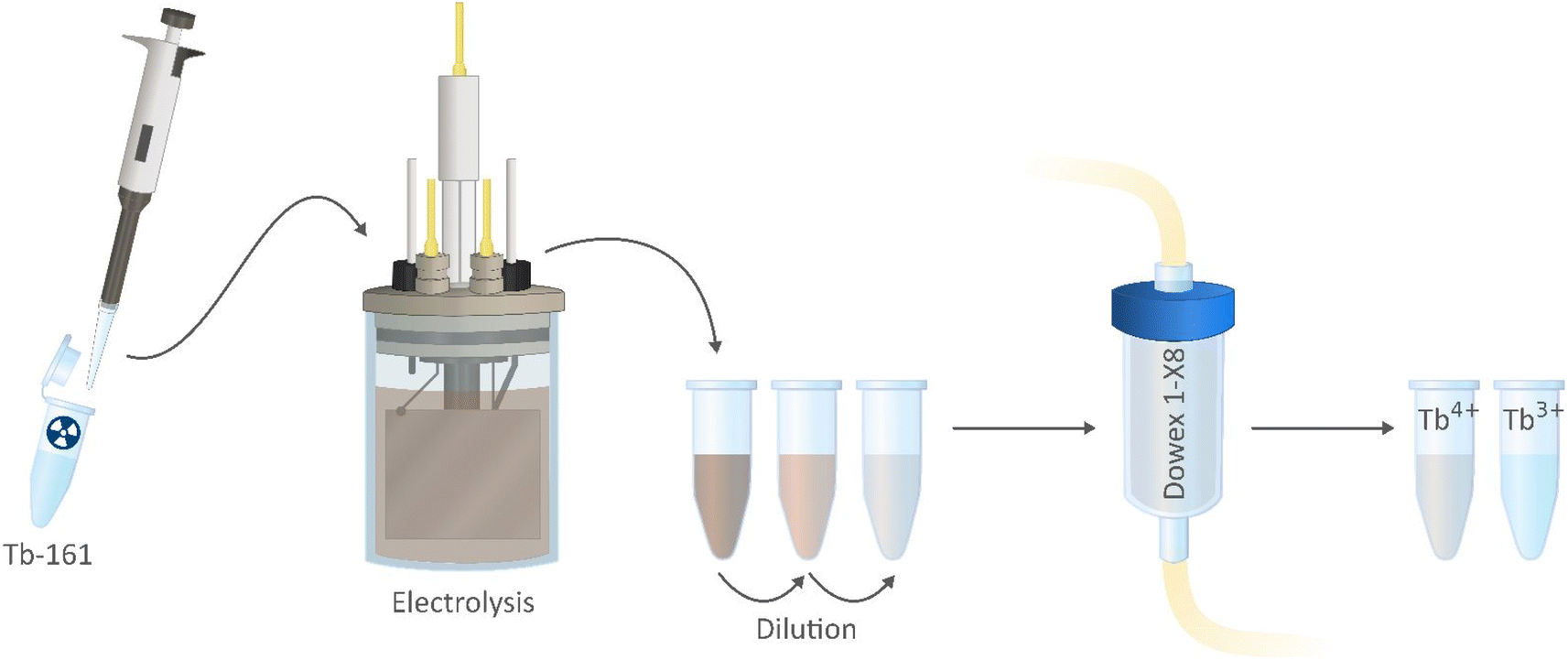 | ||
| Fig. 3 Schematic of the proposed purification method using Dowex 1-X8 resin packed column. | ||
Based on these equations, y was equal to 2x − 4 to maintain the electrical neutrality of the resin. This was based on the assumption that the tetravalent metal ion forms a single complex with the carbonate ion, neglecting the presence of hydroxide ions, especially at higher concentrations of carbonate. In the study of Varlashkin et al.,21 several stoichiometries of solid state Tb4+ carbonato–hydroxo complexes were determined based on IR spectra, and it was assumed that the Tb4+ solutions could contain complexes similar to these containing a mixture of carbonate and hydroxide groups. Similarly, in the works of Sherry et al.,42,43 rare earth carbonate systems are investigated by anion exchange column chromatography where the slope of plotted logarithmic functions of molality of the ligand and distribution coefficients, the ligand number of these complexes was predicted at lower carbonate concentrations. With increasing carbonate concentrations, the presence of hydroxide groups in the complex was assumed here as well. However, to the best of our knowledge, no further studies on Tb3+ and Tb4+ complexes in highly concentrated carbonate solutions have been reported.
Column separation using Dowex 1X-8 resin
In Fig. 4, the elution curve of a Tb3+ solution spiked with 161Tb that was not treated by electrolysis is shown. With water as the mobile phase, the Tb3+ carbonate complex was completely retained on the column. After the mobile phase was switched to 2 mol L−1 K2CO3, Tb3+ is completely eluted in the collected fractions. The radioactivity of the column material was checked after all fractions were collected, showing no presence of Tb161 tracers. In addition, when the activity of 0.4 mL of the feed solution (178.73 ± 2.54 kBq) is compared to the total activity collected in the fractions (175.46 ± 2.47 kBq), values are in good agreement. When the remainder of the feed solution was electrolysed during 3 h, at constant potential of +1.3 V vs. Ag/AgCl, an immediate colour change was observed. In a second experiment, the same volume (0.4 mL) of feed solution was introduced onto the column. The elution curve of Tb4+ solution can be seen in Fig. 5. In this elution curve, it is observed that the Tb4+ carbonate complex is not retained by the anion exchange resin, in contrast to the Tb3+ carbonate complex. In our previous study,23 an in-depth qualitative analysis was performed on the electrochemical oxidation of Tb3+ to Tb4+, but a quantitative electrochemical analysis could not be performed due to the limitation of the high formal reduction potential (E0 = +3.1 V vs. Ag/AgCl). In the current study, with the column chromatography experiments, full separation of Tb4+ and Tb3+ is achieved. An estimation of the conversion of Tb3+ to Tb4+ by electrochemical reduction can be done based on the assumption that back-oxidation is negligible during the dilution and separation, which was shown by the UV-Vis absorption spectroscopy results. Based on this, only ca. 11.2% of the mixture consisted of Tb4+ whereas majority (88.8%) was still Tb3+ after 2 hours of electrolysis.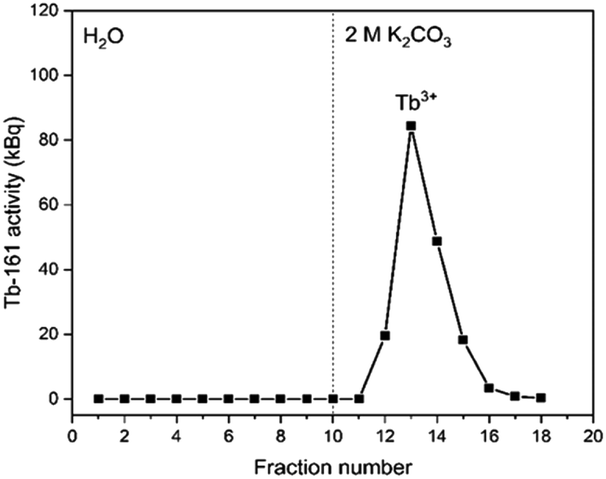 | ||
| Fig. 4 Elution curve of tracer spiked Tb3+ solution using a Dowex 1-X8 resin filled cartridge (ϕ: 1 cm, h: 2.5 cm, BV: 2 mL). Feed: 0.4 mL of a solution containing 0.018 mol L−1 Tb3+ in 1 mol L−1 K2CO3 solution (pH ≈ 12). The vertical dashed line denotes the change of mobile phase from water to 2 mol L−1 K2CO3. Flow rate: 0.3 mL min−1. | ||
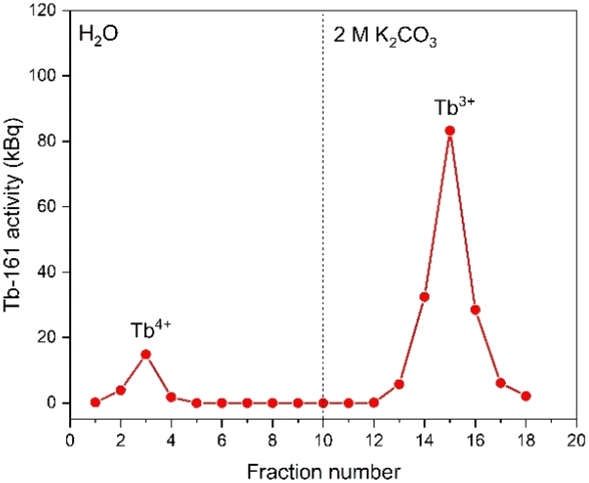 | ||
| Fig. 5 Elution curve of tracer spiked and electrolysed Tb4+ solution using a Dowex 1-X8 resin filled cartridge (ϕ: 1 cm, h: 2.5 cm, BV: 2 mL). Feed: 0.4 mL of a solution containing 0.018 mol L−1 Tb3+/Tb4+ in 1 mol L−1 K2CO3 solution (pH ≈ 12). The vertical dashed line denotes the change of mobile phase from water to 2 mol L−1 K2CO3. Flow rate: 0.3 mL min−1. | ||
As a next step, electrolysis was performed in a mixture of lanthanides gadolinium, terbium and dysprosium, with a concentration of each of 0.05 mol L−1 in 4 mol L−1 K2CO3 at pH = 14. After 2 hours of electrolysis at +1.3 V vs. Ag/AgCl, UV-Vis absorption spectra were recorded, as can be seen in Fig. 6. The spectra – recorded before and after the electrolysis – showed that, Tb3+ can be successfully oxidized in presence of other lanthanides without any inhibition. The concentration was chosen based on the solubility limit of the lanthanides in the carbonate solution, in which increasing the lanthanide concentration to more than 0.15 mol L−1 resulted in precipitation. In the previous work, it was established that when terbium concentrations lower than 0.04 mol L−1 were used, oxidation was no longer observed.23 Furthermore, maximum Tb3+ to Tb4+ conversion was not achieved at 3 hours of electrolysis for terbium concentrations higher than 0.15 mol L−1. This was explained by the strong complexation hindering the oxidation reaction at these concentrations.23 Thus, in these tests equimolar lanthanide mixtures were used with the total concentration of 0.15 mol L−1. After the electrolysis, the same protocol was followed for the separation process, except the feed volume was decreased to 0.2 mL instead of 0.4 mL due to the higher lanthanide concentration. In addition to 161Tb, ≈200 ± 18.2 kBq 175Yb was also added. While 161Tb tracers are expected to oxidize to tetravalent state, 165Yb tracers are added in the mixture as a representative for the trivalent lanthanides gadolinium and dysprosium. 165Yb is deemed more of a convenience for the gamma spectrometry due to its availability and relatively short half-life (4.16 d). The elution curves for 175Yb and 161Tb is shown in Fig. 7. The trivalent lanthanides, Gd, Dy and Tb were retained on the column as can be seen with 175Yb and 161Tb elution curves, which is similar to the results containing only terbium. There were no trivalent lanthanides detected in the first elutions with water as mobile phase, whereas a brown coloured solution was collected in the first fractions, corresponding to Tb4+. When calculating the ratios of Tb3+ to Tb4+ in the feed solution, the integration of the peak areas of the elution curves were used, assuming that there was no back-reduction occurring during the separation process and the contribution of first elution peak was purely originating from Tb4+ complex whereas the second elution peak was only composed of Tb3+ complex. The amount of Tb4+ in the mixture was calculated to be ca. 8.6%, after 2 hours of electrolysis. This difference in the amount of Tb4+ can be explained by the difference in the electrolysis time. Therefore, Tb3+ was successfully oxidized to Tb4+ and stabilized in the aforementioned experimental conditions, as well as Tb4+ was separated from the other trivalent lanthanides.
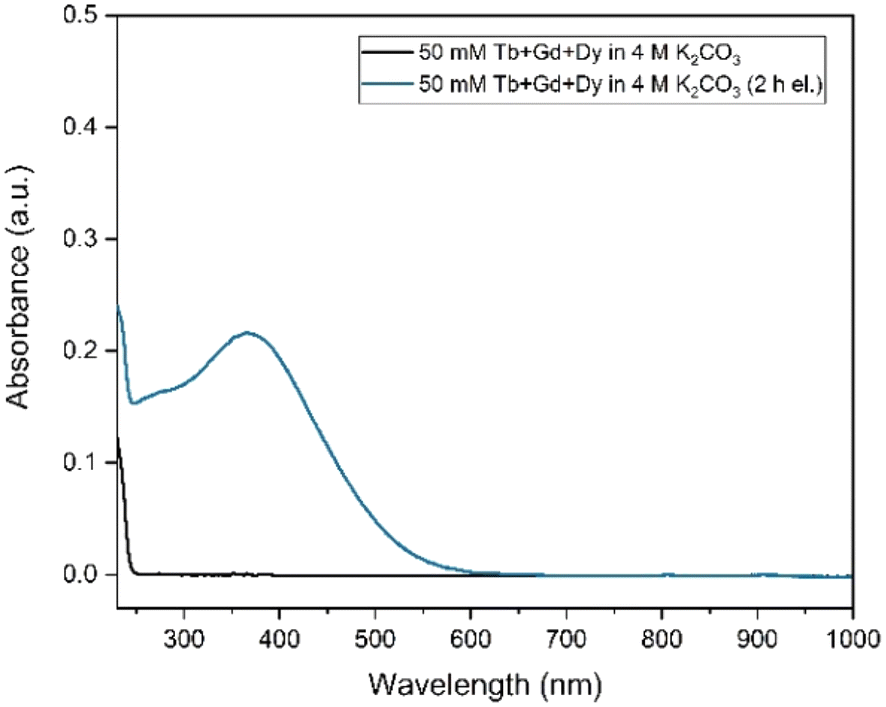 | ||
| Fig. 6 UV–Vis absorption spectra of 0.05 mol L−1 Tb3+, 0.05 mol L−1 Gd3+ and 0.05 mol L−1 Dy3+ in 4 mol L−1 K2CO3 at pH = 14, before and after the electrolysis. Electrolysis was performed at constant potential +1.3 V vs. Ag/AgCl for 2 h. The formation of Tb4+ was followed by the absorbance peak at 365 nm. Blank carbonate solution was used as the reference sample for baseline correction. The path length was 1 mm. | ||
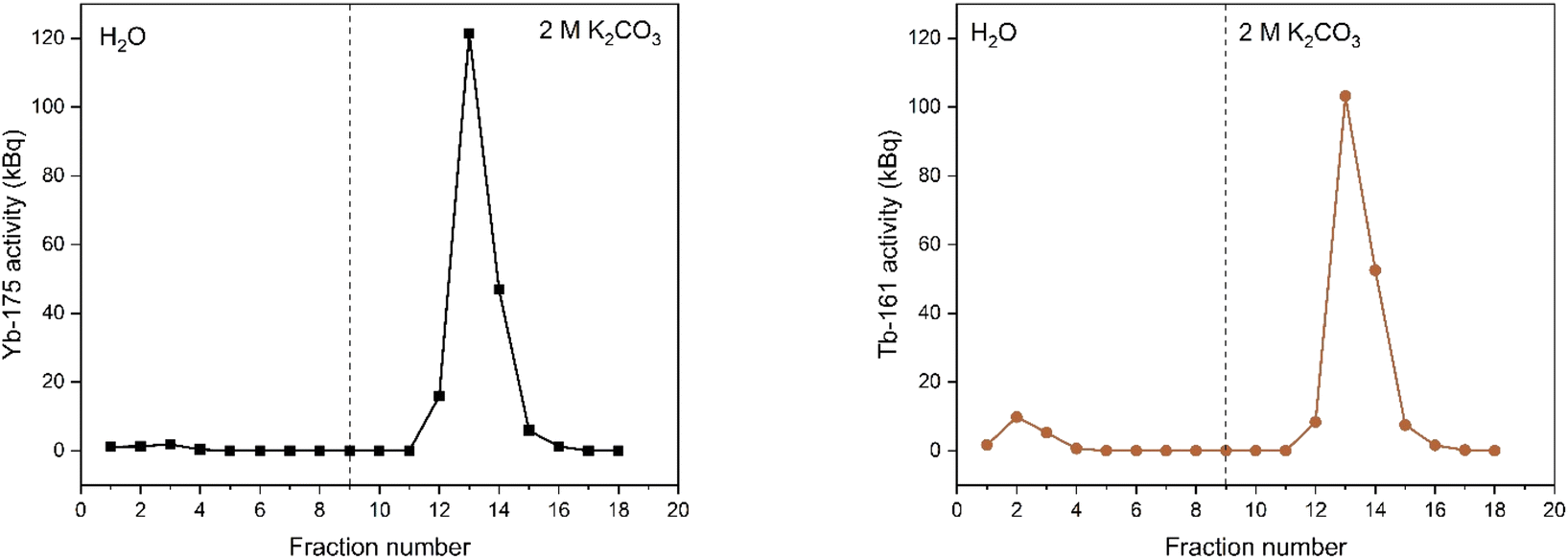 | ||
| Fig. 7 Elution curves of tracer spiked electrolyzed lanthanide mixture solution (0.05 mol L−1 Tb3+, 0.05 mol L−1 Gd3+ and 0.05 mol L−1 Dy3+ in 4 mol L−1 K2CO3) solution before (left) and after (right) electrolysis at +1.3 V vs. Ag/AgCl, using a Dowex 1-X8 resin filled cartridge (ϕ: 1 cm, h: 2.5 cm, BV: 2 mL). Feed: 0.2 mL of a solution containing 0.018 mol L−1 Ln3+ in 1 mol L−1 K2CO3 solution (pH ≈ 12). The vertical dashed line denotes the change of mobile phase from water to 2 mol L−1 K2CO3. Flow rate: 0.3 mL min−1. | ||
In order for this method to be a viable route for therapeutic radiolanthanide preparation, we should briefly evaluate its compatibility with exisiting radiolabelling strategies. Thanks to the similar properties with clinically-used 177Lu, there has been many studies3,5,7,11 on the therapeutic potential of 161Tb. As a result, similar approaches of radiolabelling has been applied and compared to 177Lu. As the advantage of radiolanthanides, since the most stable oxidation state is trivalent, similar chemistry for radiolabelling could be applied. The most common chelators that are used for terbium isotopes are DOTA derivatives, whereas DTPA is also considered as alternative. The radiolabelling requires high temperature and a pH range of 4–5.5, where trivalent terbium is complexed with these chelators.16 Adjusting the pH of the eluted Tb4+ fractions to a value within the appropriate labelling window would thermodynamically favor reduction to Tb3+, thus making it even more analogues to the common labelling strategies used for 177Lu.
XANES measurements
To explore the oxidation states of terbium ions during the course of electrolysis, XANES experiments have been carried out at the Tb L3-edge. With the starting solution of 0.07 Tb3+ mol L−1 Tb in 4 mol L−1 K2CO3 at pH = 14, electrolysis was performed and recorded every 30 minutes. The normalized spectra at various electrolysis times can be seen in Fig. 8. The white lines of Tb-L3 were observed with a maximum at 7519 eV, whereas a shoulder appeared at between 7525–7530 eV, which is indication of Tb4+ (ESI Fig. S3†). As the electrolysis time increases, the decrease in the white line could explain the conversion of Tb3+ to Tb4+, and the final spectrum containing a mixture of both valence states. In order to quantify the conversion, linear combination fitting (LCF) was performed, using initial Tb3+ solution without electrolysis performed (0.07 Tb3+ mol L−1 Tb in 4 mol L−1 K2CO3 at pH = 14) and Tb4O7 as the reference materials (ESI Fig. S4†). The final fitting using these references resembles the shape of the spectrum recorded after 24 hours of electrolysis. The conversion was calculated as 15% based on the fitting, which was in line with the results from the extraction chromatography experiments. On the other hand, even after 24 hours of electrolysis, the oxidation of Tb3+ to Tb4+ was only partial, meaning that after the oxidation the solution consists of a mixture of Tb3+ and Tb4+ complexes. Therefore, the uncertainty on the quantification based on LCF was accepted as high. The EXAFS spectrum of this mixture couldn't be analyzed due to the noise resulting from low terbium concentrations.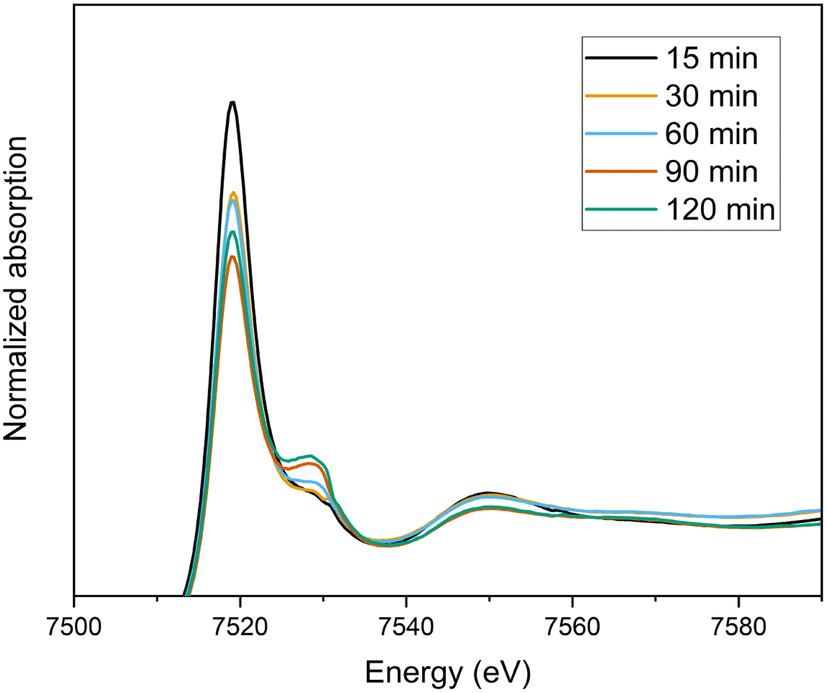 | ||
| Fig. 8 Normalized XANES spectra of the oxidation of 0.07 mol L−1 Tb3+ in a 4 mol L−1 K2CO3 solution as a function of time (15 min–120 min). | ||
Ab initio simulation of L3 XANES spectra was carried out by considering the structures reported by Michiba et al.52 (ICSD 422736) and Dixey and Saines53 (ICSD 7183) to gain more insight into the electronic and local structural features of terbium (Fig. 9). As can be seen in Fig. 10, there are five carbonato and two hydroxo ligands coordinated to Tb in the structure reported by Dixey and Saines,53 (ICSD 7183) where only one carbonato is a bidentate ligand and the others are monodentate. Similarly, in the structure that was reported by Michiba et al.,52 the lanthanides were coordinated with five carbonato ligands (one bidentate and four monodentate), whereas the only difference was a third hydroxo ligand present in the structure. Although both structures contain Tb3+ surrounded by carbonates and hydroxides, the different local environment around terbium determines differences in the simulated L-edge. For ICSD 7183, terbium has an overall coordination number of 8, with bond valence sum of 2.85 (considering a bond search factor of 0.87). At the same time, the ICSD 422736 structure features terbium in a nine-fold coordination environment with average bond valence sum of 3.06 (bond search factor 0.87). Thus, the electronic distribution for the two structures is different to a small extent, with terbium in ICSD 422736 having a slightly higher effective charge. This can also be verified in the small shift of the edge toward higher energy in the simulated ICSD 422736 spectrum when compared to the respective one for ICSD 7183 (Fig. 9).
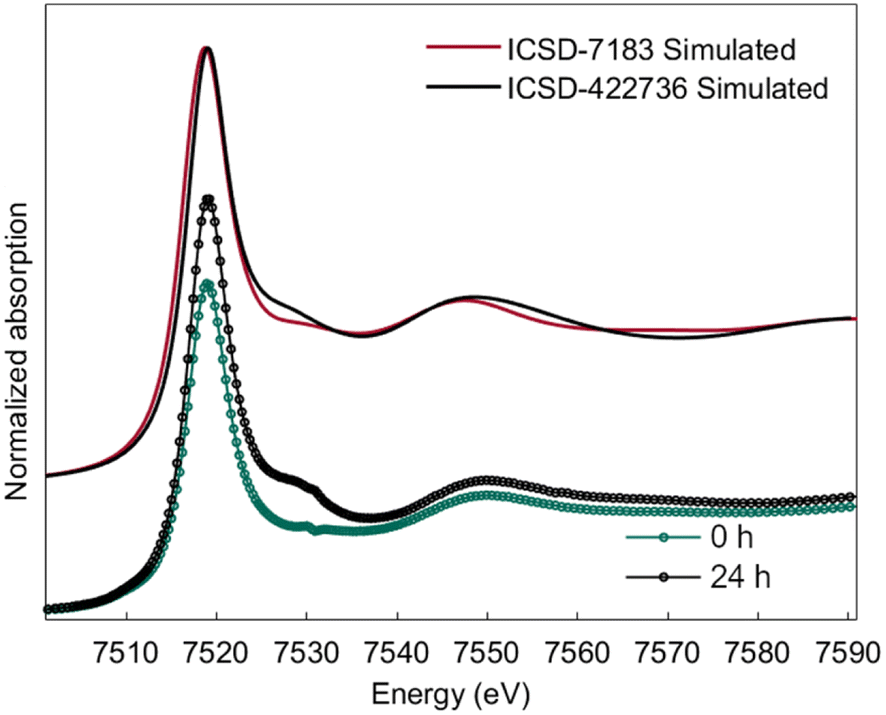 | ||
| Fig. 9 Simulated L3-edge spectra for reference structures ICSD-7183 and ICSD-422736 and comparison with experimental XANES traces of 0 h and 24 h electrolysis. | ||
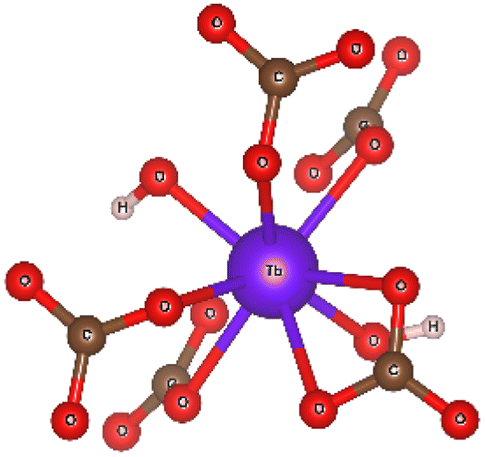 | ||
| Fig. 10 The coordination environment of terbium in the Tb(OH)CO3 crystal (ICSD 7183), as published by Dixey and Saines et al.,48 visualized as a ball and stick model. | ||
Interestingly, the experimental XANES trace recorded after 24 h of electrolysis displays more similarities with ICSD 422736, while the experimental spectrum at time zero (0 h) is closer to ICSD 7183. This might be related to a partially higher oxidation state of Tb at 24 h, as discussed above, as well as a different coordination environment, possibly a nine-fold coordination for the Tb4+/Tb3+ complex after 24 hours of electrolysis, vs. an eight-fold coordination for the Tb3+ complex.
Conclusions
161Tb tracer spiked solutions were successfully electrolyzed, and further diluted without affecting the stability of Tb4+. The separation of trivalent and tetravalent terbium was achieved by using Dowex 1-X8 anion exchange resin for the first time to the best of our knowledge. Tb4+ had no interaction with the column and eluted in the initial fractions, whereas the anionic carbonate complex of Tb3+ was retained on the column until the eluent is switched from water to concentrated carbonate solution. Full separation was achieved without any back-reduction of Tb4+ to Tb3+, proven by UV-Vis absorption spectroscopy results. Based on these results, an estimation of the electrolysis yield was done, which was calculated as ≈11.2% after 3 h of electrolysis at a constant potential at +1.3 V vs. Ag/AgCl. The separation method was proven also successfully in the presence of other lanthanides in the feed solution. Similar results were obtained when the same protocol was applied with an equimolar mixture of Gd3+, Tb3+ and Dy3+. Therefore, this separation method was found to be applicable for intergroup lanthanide separation. Finally, X-ray absorption spectroscopy results showed some minor differences in the structure of the Tb3+ and Tb4+ carbonate complexes, where different coordination numbers were obtained based on the simulations. As a result, the separation method looks promising towards the purification of 161Tb for medical use.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
M. Ö. A.: conceptualization, methodology, analysis, investigation, visualization, writing – original draft, writing – review & editing. A. M.: XAS analysis, writing – original draft, writing – review & editing. B. G.: conceptualization, methodology, analysis, investigation, writing – review & editing. K. V. H.: conceptualization, methodology, analysis, investigation, writing – review & editing. G. D.: XAS experiments, writing – original draft, writing – review & editing. G. A.: XAS experiments, writing – original draft, writing – review & editing. K. B.: conceptualization, methodology, analysis, investigation, writing – review & editing. T. C.: conceptualization, methodology, analysis, investigation, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research is funded by Research Foundation – Flanders (FWO) – SBO project Terbium Isotopes for Medical Applications in Flanders (Tb – IRMA – V, grant number S005019N). A. M. acknowledges Research Foundation – Flanders (FWO) for funding (fellowship no. 1228622N). The beamline team at Elettra Sincrotrone Trieste are acknowledged for granting the possibility to conduct XAS studies (with proposal number 20210288). M. Ö. A. is grateful to Robin Aerts, Wout Van Craenendonck and Dr Peter Zsabka for the support in XAS and gamma spectrometry measurements. Stijn Rameakers and Dr Michiel Van de Voorde are acknowledged for the production of radiotracers used in this work.Notes and references
- CERN Courier, Terbium: a new ‘Swiss army knife’ for nuclear medicine, https://cerncourier.com/a/terbium-a-new-swiss-army-knife-for-nuclear-medicine/, accessed 16 March 2020.
- C. Müller, K. Zhernosekov, U. Köster, K. Johnston, H. Dorrer, A. Hohn, N. T. van der Walt, A. Türler and R. Schibli, J. Nucl. Med., 2012, 53, 1951–1959 CrossRef PubMed.
- S. Lehenberger, C. Barkhausen, S. Cohrs, E. Fischer, J. Grünberg, A. Hohn, U. Köster, R. Schibli, A. Türler and K. Zhernosekov, Nucl. Med. Biol., 2011, 38, 917–924 CrossRef CAS PubMed.
- A. Al-Ibraheem and A. M. Scott, Nucl. Med. Mol. Imaging, 2023, 57, 168–171 CrossRef PubMed.
- J. Grünberg, D. Lindenblatt, H. Dorrer, S. Cohrs, K. Zhernosekov, U. Köster, A. Türler, E. Fischer and R. Schibli, Eur. J. Nucl. Med. Mol. Imaging, 2014, 41, 1907–1915 CrossRef PubMed.
- C. Müller, J. Reber, S. Haller, H. Dorrer, P. Bernhardt, K. Zhernosekov, A. Türler and R. Schibli, Eur. J. Nucl. Med. Mol. Imaging, 2014, 41, 476–485 CrossRef PubMed.
- C. Müller, C. A. Umbricht, N. Gracheva, V. J. Tschan, G. Pellegrini, P. Bernhardt, J. R. Zeevaart, U. Köster, R. Schibli and N. P. Van Der Meulen, Eur. J. Nucl. Med. Mol. Imaging, 2019, 46, 1919–1930 CrossRef PubMed.
- R. P. Baum, A. Singh, H. R. Kulkarni, P. Bernhardt, T. Rydén, C. Schuchardt, N. Gracheva, P. V Grundler, U. Köster, D. Müller, M. Pröhl, J. R. Zeevaart, R. Schibli, N. P. Van Der Meulen and C. Müller, J. Nucl. Med., 2021, 62, 1391–1397 CrossRef PubMed.
- M. Van de Voorde, K. Van Hecke, T. Cardinaels and K. Binnemans, Coord. Chem. Rev., 2019, 382, 103–125 CrossRef CAS.
- E. J. Monroy-Guzman and F. Salinas, J. Mex. Chem. Soc., 2015, 59, 143–150 Search PubMed.
- N. Gracheva, C. Müller, Z. Talip, S. Heinitz, U. Köster, J. R. Zeevaart, A. Vögele, R. Schibli and N. P. Van Der Meulen, EJNMMI Radiopharm. Chem., 2019, 4, 12 CrossRef PubMed.
- F. Rösch, Radiochim. Acta, 2007, 95, 303–311 CrossRef.
- R. D. Teo, J. Termini and H. B. Gray, J. Med. Chem., 2016, 59, 6012–6024 CrossRef CAS PubMed.
- Z. Talip, C. Favaretto, S. Geistlich and N. P. Van Der Meulen, Molecules, 2020, 25, 966 CrossRef CAS PubMed.
- K. Mishiro, H. Hanaoka, A. Yamaguchi and K. Ogawa, Coord. Chem. Rev., 2019, 383, 104–131 CrossRef CAS.
- T. I. Kostelnik and C. Orvig, Chem. Rev., 2019, 119, 902–956 CrossRef CAS PubMed.
- J. M. Schwantes, R. Sudowe, H. Nitsche and D. C. Hoffman, J. Radioanal. Nucl. Chem., 2008, 276, 543–548 CrossRef CAS.
- S. A. Sayed, K. A. Rabie and I. E. Salama, Sep. Purif. Technol., 2005, 46, 145–154 CrossRef CAS.
- M. Van de Voorde, K. Van Hecke, K. Binnemans and T. Cardinaels, Sep. Purif. Technol., 2020, 232, 115939 CrossRef CAS.
- D. E. Hobart, K. Samhoun, J. P. Young, V. E. Norvell, G. Mamantov and J. R. Peterson, Inorg. Nucl. Chem. Lett., 1980, 16, 321–328 CrossRef CAS.
- P. G. Varlashkin, G. M. Begun and J. R. Peterson, J. Less-Common Met., 1985, 109, 123–134 CrossRef CAS.
- S. Qiang, W. Zhijian and G. Chongli, Solvent Extr. Ion Exch., 1995, 13, 275–288 CrossRef CAS.
- M. O. Arman, B. Geboes, K. Van Hecke, K. Binnemans and T. Cardinaels, J. Appl. Electrochem., 2022, 52, 583–593 CrossRef CAS.
- M. Gras, N. Papaiconomou, E. Chainet, F. Tedjar and I. Billard, Sep. Purif. Technol., 2017, 178, 169–177 CrossRef CAS.
- M. Atanassova, Solvent Extr. Ion Exch., 2009, 27, 159–171 CrossRef CAS.
- M. Krättli, T. Müller-Späth, N. Ulmer, G. Ströhlein and M. Morbidelli, Ind. Eng. Chem. Res., 2013, 52, 8880–8886 CrossRef.
- K. L. Nash, Handb. Phys. Chem. Rare Earths, 1994, 18, 197–238 Search PubMed.
- K. Shimojo, N. Aoyagi, T. Saito, H. Okamura, F. Kubota, M. Goto and H. Naganawa, Anal. Sci., 2014, 30, 263–269 CrossRef CAS PubMed.
- N. Sivaraman, R. Kumar, S. Subramaniam and P. R. Vasudeva Rao, J. Radioanal. Nucl. Chem., 2002, 252, 491–495 CrossRef CAS.
- B. Gupta, P. Malik and A. Deep, Solvent Extr. Ion Exch., 2007, 21, 239–258 CrossRef.
- V. V Smirnov, M. D. Karavan, M. V Logunov, I. G. Tananaev and B. F. Myasoedov, Radiochemistry, 2018, 60, 404–419 Search PubMed.
- E. V. Kuzovkina, E. A. Lavrinovich, A. P. Novikov, E. S. Stepanova, M. D. Karavan and I. V. Smirnov, J. Radioanal. Nucl. Chem., 2017, 311, 1983–1989 CrossRef CAS.
- I. V. Smirnov, E. S. Stepanova, N. M. Ivenskaya, M. D. Karavan, S. R. Zaripov, S. R. Kleshnina, S. E. Solovieva and I. S. Antipin, J. Radioanal. Nucl. Chem., 2017, 317, 1257–1265 CrossRef.
- Z. K. Karalova, T. V Bukina, E. A. Devirts, Z. Z. Agaev and B. F. Myasoedov, Radiokhimiya, 1987, 29, 767–772 CAS.
- Z. K. Karalova, E. A. Lavrinovich, B. F. Myasoedov, L. A. Fedorov and S. A. Sokolovskii, Radiokhimiya, 1988, 31, 38–45 Search PubMed.
- E. V Kuzovkina, E. A. Lavrinovich, A. P. Novikov, E. S. Stepanova, M. D. Karavan and I. V Smirnov, J. Radioanal. Nucl. Chem., 2018, 315, 639–642 CrossRef.
- A. V. Boyarintsev, G. V. Kostikova, S. I. Stepanov, A. A. Shoustikov, A. M. Chekmarev and A. Y. Tsivadze, Solvent Extr. Ion Exch., 2021, 39, 745–763 CrossRef.
- W. E. Clifford, E. P. Bullwinkel, L. A. McClaine and P. Noble, J. Am. Chem. Soc., 1958, 80, 2959–2961 CrossRef CAS.
- A. V. Boyarintsev, S. A. Perevalov, S. I. Stepanov and A. M. Chekmarev, J. Radioanal. Nucl. Chem., 2020, 327, 385–393 CrossRef.
- Z. K. Karalova, B. F. Myasoedov, T. V Bukina and L. M. Rodionova, Radiokhimiya, 1985, 27, 47–49 CAS.
- Z. K. Karalova, B. F. Myasoedov, L. M. Rodionova and S. A. Kuznetsova, Radiokhimiya, 1981, 25, 187–191 Search PubMed.
- H. S. Sherry and J. A. Marinsky, Inorg. Chem., 1964, 3, 330–335 CrossRef CAS.
- H. S. Sherry and J. A. Marinsky, Inorg. Chem., 1963, 2, 957–959 CrossRef CAS.
- A. Di Cicco, G. Aquilanti, M. Minicucci, E. Principi, N. Novello, A. Cognigni and L. Olivi, J. Phys.: Conf. Ser., 2009, 190, 012043 CrossRef.
- Y. Joly, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 125120 CrossRef.
- L. Hedin, B. I. Lundqvist and S. Lundqvist, Solid State Commun., 1971, 9, 537–541 CrossRef CAS.
- N. Saito and T. Sekine, Nature, 1957, 180, 753 CrossRef CAS.
- S. Misumi and T. Taketatsu, Bull. Chem. Soc. Jpn., 1958, 32, 876–879 CrossRef.
- T. Taketatsu, Bull. Chem. Soc. Jpn., 1963, 36, 549–553 CrossRef CAS.
- T. Taketatsu, Talanta, 1963, 10, 1077–1082 CrossRef CAS.
- R. S. Gärtner and G. J. Witkamp, Sep. Sci. Technol., 2005, 40, 2391–2410 CrossRef.
- K. Michiba, T. Tahara, I. Nakai, R. Miyawaki and S. Matsubara, Z. Kristallogr., 2011, 226, 518–530 CAS.
- R. J. C. Dixey and P. J. Saines, Inorg. Chem., 2018, 57, 12543–12551 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra02694b |
| This journal is © The Royal Society of Chemistry 2024 |