
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure–activity relationships, product species distribution and the mechanism of effect of multi-component flue gas on Hg0 adsorption and oxidation over CuO/ACs†
Li Tong‡
a,
Penglai Zuo‡a,
Xiaoxi Zhanga,
Quanming Lianga,
Kun Wanga,
Yawen Yanga,
Jieyu Liua,
Haixin Guo *b and
Peng Zhang*c
*b and
Peng Zhang*c
aInstitute of Urban Safety and Environmental Science, Beijing Academy of Science and Technology, Beijing 100054, China
bAgro-Environmental Protection Institute, Ministry of Agriculture and Rural Affairs, Tianjin 300191, China. E-mail: haixin_g@126.com
cNational Center for Occupational Safety and Health, National Health Commission of the People's Republic of China, Beijing 102308, China. E-mail: zhangpengbd@163.com
First published on 19th July 2024
Abstract
A series of Cu-doped activated cokes (CuO/ACs) were synthesized via an impregnation method and applied for the removal of elemental mercury (Hg0). Structure–activity relationships between Hg0 removal and CuO/AC surface characteristics were identified. Hg0 removal over CuO/AC occurs through a combination of physisorption and chemisorption and is mainly dominated by chemisorption. It was found that 1 nm micropores facilitate Hg0 physisorption. Hg0 could weakly adsorb onto an O-terminated crystal layer, whereas strongly adsorb onto Cu-terminated single highly dispersed, clustered and bulk CuO (110) crystal planes via the Mars–Maessen mechanism. Product distributions and mechanisms of Hg0 adsorption and oxidation over the CuO/AC catalyst under multi-component flue gases are also discussed. O2 enhances both physisorption and chemisorption toward Hg0 by 38%. Inhibition of Hg0 removal by SO2 originates from the competitive adsorption and deactivation of CuO cation vacancies, whereas the impact is weakened by O2 through generating 20% of physically adsorbed mercury product species. NO and O2 promote Hg0 chemisorption efficiency by 93% to form Hg(NO3)2. HOCl and/or Cl2 produced by HCl can oxidize 100% of Hg0 to HgCl2, and the catalytic oxidation efficiency is approximately 29%, but O2 slightly lowers the Hg0 catalytic oxidation efficiency by 8%. The affinity ability between various flue gases and Hg0 follows the order O2 < NO < HCl.
1. Introduction
Mercury derived from fuel combustion has aroused wide concern owing to its unique chemical properties and has adverse effects on human health.1 Among the various mercury species, elemental mercury (Hg0) is the hardest to capture using currently available pollution control devices.2,3Numerous efforts have been undertaken for mercury removal by means of adsorption and oxidation, which proved to be helpful for controlling Hg0.4–7 To date, wet flue gas desulfurization (WFGD) has been regarded as an effective way for disposing SO2 and is adopted to remove Hg2+ after the oxidation of Hg0 in industry. However, the re-emission of mercury during the WFGD progress is a serious problem. Activated carbon injection (ACI) has been recognized as the mainly commercialized technology to remove mercury in the commercial application of coal-fired power plants, especially for activated carbons impregnated with halogen.8 However, several shortcomings, such as carbon consumption, larger occupied area and higher costs, hamper its practical applications. In recent years, the use of transition metal oxides as oxidants for heterogeneous Hg0 removal has been a growing field.9,10 Nevertheless, it is difficult to remold the existing flue gas flow control devices, and catalysts' mechanical strength should be high enough to avoid dust erosion. For this purpose, materials could be employed downstream from fabric filters or electrostatic precipitators where temperature is commonly less than 160 °C.8
Among the various low-cost transition metal catalysts, copper oxides have been most developed because of their high activity for multi-pollutants, such as SO2, NO and VOCs.11–13 However, pure CuO is almost inert to Hg0 removal. Therefore, Cu-based catalysts were employed to enhance Hg0 removal performance (see Table S1 in the ESI†).14–16 For example, Zhang et al.14 studied Hg0 adsorption over novel copper-based porous metal–organic frameworks (Cu-MOFs) with a chlorine-rich functional group and found that the resulting adsorbent had an Hg0 removal efficiency of 91.8% at 120 °C in an N2 atmosphere. Liu et al.15 indicated that CuO/g-C3N4 materials had the Hg0 removal efficiency exceeding 98.0% at 120 °C in an N2 atmosphere. As a result, the Hg0 removal performances over the Cu-based catalysts significantly vary in support. The active coke (AC), which is a common carbon-based material and has the characteristics of activated carbon, is typically considered a good candidate support for Hg0 removal at low temperatures due to the lower expense and excellent mechanical strength.17 Zhao et al.16 investigated the effects of various synthesis factors on HNO3 plus copper-modified ACs for Hg0 removal. Still, the crystal structure affecting the mercury removal performance has not been studied in detail. CuO supported on the AC as a material system for Hg0 removal had ever been demonstrated to be active for Hg0 removal by fewer researchers, but little attention had been paid to the occurrence state and shape of the copper element and the detailed bonding sites for Hg0 to reveal the structure–activity relationship between Hg0 removal performance and CuO/AC surface characteristics.
Actually, the pollutants' removal performance is a result of the interaction among pollutants, materials, and the co-existing flue gas. In particular, Hg0 is inferior in the gas volume and dipole moment, thus the removal performance is not mass transfer favored under the multi-component flue gas. Therefore, the Hg0 removal selectivity strongly depends on the reciprocal effect of the material structures and flue gas components. The flue gas composition is primarily determined by the coal matrix and significantly affects the Hg0 removal efficiency, particularly for O2, SO2, NO, and HCl. Laboratory experiments indicated that O2 or HCl showed enhancement in Hg0 oxidation.18,19 However, the impact of NO or SO2 on Hg0 removal has not reached a consensus,19–21 which mainly depends on the synthetic effect of competitive adsorption, poisoning and the newly generated active groups responsible for Hg0 removal. However, the information on the internal reaction mechanism of Hg0 and multi-component flue gases over CuO/AC was still insufficient for Cu-based AC catalysts.
Based on the above-mentioned statement, this work focuses on building the structure–activity relationship between Hg0 removal performance and CuO/AC surface characteristics, and investigates the effect of multi-component flue gas and product species distribution for Hg0 adsorption and oxidation over the CuO/AC. Furthermore, the affinity ability between different flue gases and Hg0 over the CuO/AC is discussed, and the probable reaction mechanisms are also proposed. In the present work, insights into the structure–activity relationships between the Hg0 removal performance and the CuO/AC provide guidance for the improvement of the current preparation method. Furthermore, exploring the reaction mechanism facilitates Hg0 removal through optimizing the proportion of flue gas components to enhance the Hg0 removal selectivity, and then probing into the product species distributions facilitates the regulation of the migration direction of product species, which oriently improve the adsorption efficiency or the catalytic oxidation efficiency instead of merely improving the material structure.
2. Experimental
2.1. Material preparation
The cylinder-shaped support with a diameter of 8 mm is a kind of commercial activated coke. The preparation procedure is as follows:(1) Prior to use, virgin ACs were ground and crushed to 40–80 mesh. Afterwards, they were washed and rotated with distilled water and quickly dried at 110 °C for 14 h.
(2) Then, the CuO/ACs were synthesized by an equivalent-volume impregnation method with cupric nitrate as the precursor. The ACs were added into a solution with a required amount of cupric nitrate with the assistance of an ultrasonic cleaner for 0.5 h.
(3) Finally, the materials were dried at 110 °C for 14 h, followed by calcination under an Ar atmosphere at 300 °C for 3 h.
The catalyst is named M% CuO/AC, where M represents the mass ratio of copper oxide to AC.
2.2. Characterization
The samples' porosity characteristics were obtained using the nitrogen (N2) adsorption–desorption method (Autosorb iQ, Quantachrome). Before measurement, all the CuO/ACs were outgassed at 120 °C for 12 h. X-ray diffraction (XRD) measurements were carried out using a powder diffractometer (Rigaku D/Max-RA). An ESCALAB 250 X-ray photoelectron spectrometer equipped with a monochromatic Al Kα source was employed to determine the elemental chemical states of the materials. Temperature Programmed Reduction (TPR) profiles of the samples were collected using an ASAP ChemiSorb 2920.The technique of temperature-programmed desorption coupled with a Hg0 analyzer (Hg0-TPD) was used to analyze the adsorbed mercury product species and its amount on the catalysts. After the materials were placed in a Hg0-containing gas flow for 2 h, the desorption process was recorded from 40 °C to 600 °C at a 10 °C min−1 heating rate in N2.
2.3. Activity test
The removal performances of the samples were evaluated using a quartz microreactor, and the experimental device is referenced to our previous research.20 Approximately 50 mg catalysts were used in each test. The reaction temperature is modulated through a heating furnace outside. A sealed Hg0 permeation device was employed to provide a constant Hg0 concentration. To shorten the experimental time, a higher initial Hg0 concentration was chosen as 270 ± 5 μg m−3. The Lumex RA-915M+ Hg0 analyzer conventionally monitors the outlet Hg0 concentration. The simulated flue gases at a total flow rate of 300 mL min−1, consisting of NO, HCl, SO2 or O2, balanced in N2 were accurately accommodated by the mass flowmeters. The weight hourly space velocity (WHSV) was calculated to be 3.6 × 105 mL g−1 h−1. The pipeline was wrapped with heating tape to prevent Hg0 deposition by heating the pipe temperature to 80 °C. To ensure equal experimental conditions, the CuO/AC catalyst was purged with N2 for 20 min before each test. Moreover, a needle valve was added into the gaseous channel for the determination of the initial Hg0 concentration to eliminate the pressure drop caused by adding the CuO/AC samples to the empty quartz microreactor. In this work, we defined three terms: total removal efficiency, adsorption efficiency and catalytic oxidation efficiency, which were calculated based on the 2 h experimental results; it therefore referred to the cumulative efficiency. The cumulative total removal efficiency (ηcap) was calculated based on the difference in the integral area of inlet and outlet Hg0, and the cumulative adsorption efficiency (ηads) was calculated on the basis of the integral area of the Hg0-TPD curve after pre-adsorption in a mercury-containing flue gas for 2 h. Therefore, the cumulative catalytic oxidation efficiency (ηoxi) is defined as (ηcap) − (ηads). The breakthrough ratio of the samples is defined as follows:
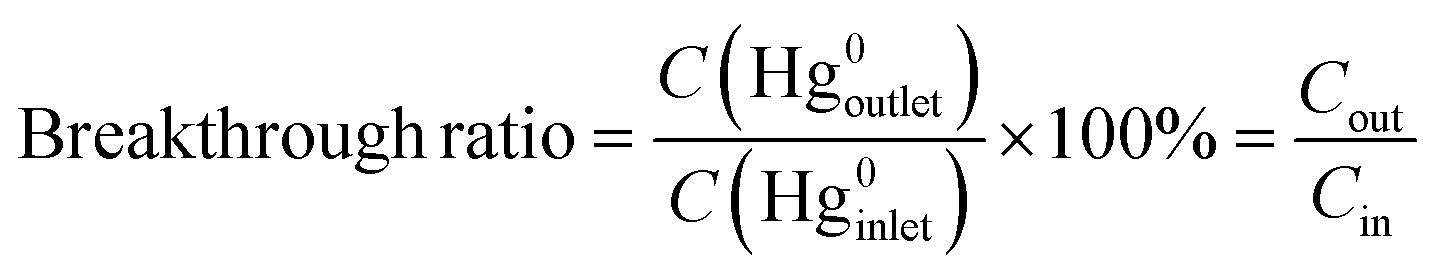 | (1) |
3. Results and discussion
3.1. Structure–activity relationship between the Hg0 removal performance and the surface properties of CuO/AC
 | ||
| Fig. 1 Breakthrough curves of Hg0 over virgin AC and CuO/AC with different CuO contents balanced in a pure N2 atmosphere at 120 °C. | ||
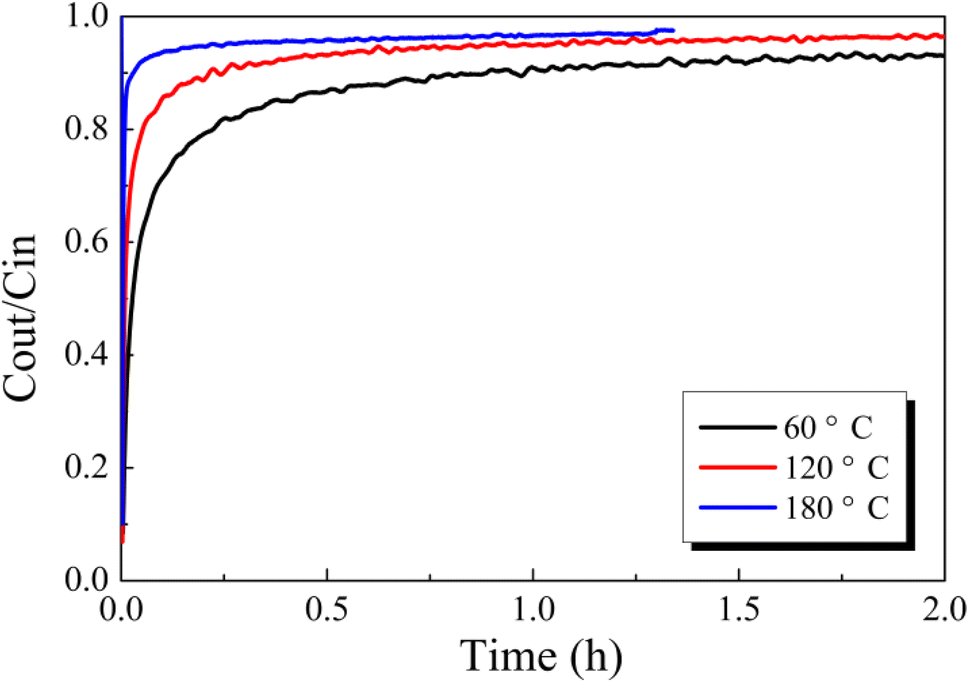 | ||
| Fig. 2 Effect of reaction temperature on Hg0 removal over 12% CuO/AC under a pure N2 atmosphere. | ||
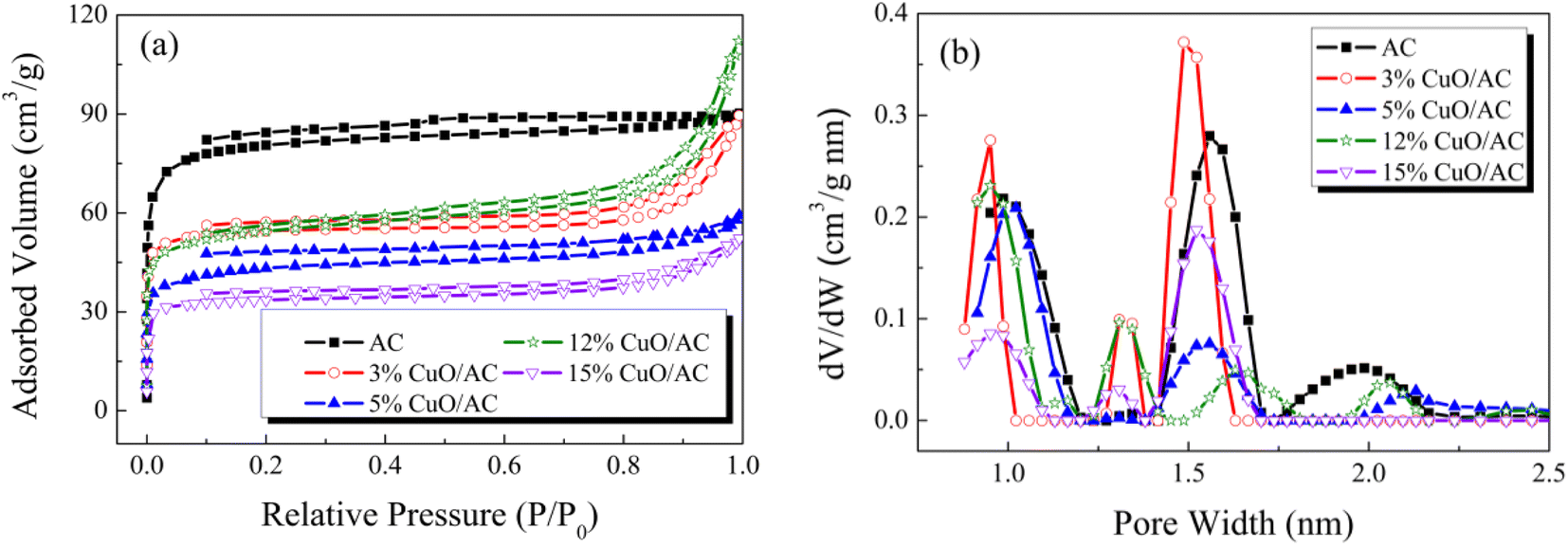 | ||
| Fig. 3 (a) N2 adsorption–desorption isotherms and (b) pore size distribution of the catalysts with different CuO contents. | ||
| Samples | BET surface area (m2 g−1) | Total pore volume (cm3 g−1) | Average pore diameter (nm) |
|---|---|---|---|
| a The BET surface area was calculated from the N2 adsorption isotherms using the BET equation. The total pore volume was accessed from the amount of N2 adsorbed at p/p0 = 0.99. The average pore diameter was calculated based on four times the total pore volume divided by the specific surface area. | |||
| AC | 251 | 0.138 | 2.20 |
| 1% CuO/AC | 246 | 0.123 | 2.20 |
| 3% CuO/AC | 193 | 0.131 | 2.72 |
| 5% CuO/AC | 191 | 0.109 | 2.29 |
| 7% CuO/AC | 160 | 0.102 | 2.71 |
| 12% CuO/AC | 138 | 0.090 | 2.59 |
| 15% CuO/AC | 121 | 0.077 | 2.56 |
| 20% CuO/AC | 93 | 0.070 | 2.99 |
Therefore, based on the comprehensive analysis above, it could be deduced that physisorption facilitates the Hg0 removal but is not the only factor.
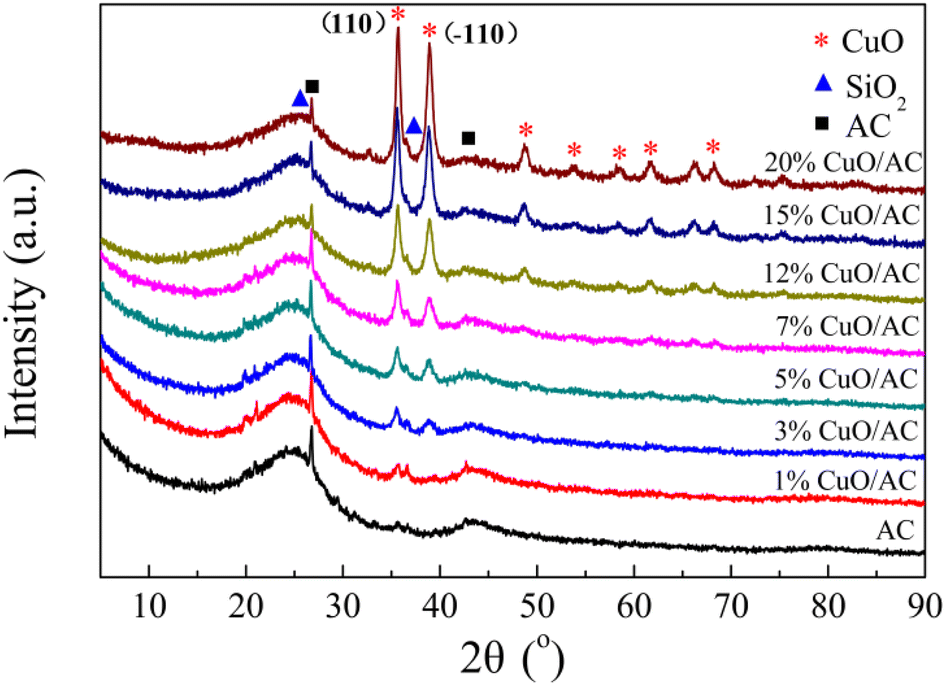 | ||
| Fig. 4 X-ray diffraction patterns of CuO/AC catalysts with different CuO contents. | ||
 | ||
| Fig. 5 H2-TPR profiles of virgin AC and the 12% CuO/AC catalyst. | ||
The environment of the copper ions and/or their structures is quite complicated and can be classified into two types of form: Cu2+, including the isolated form (highly-dispersed) and clustered form (bulk-like), as well as Cu+. However, the two peaks could not be explained through the step-wise reductions of the supported copper species, such as Cu2+ → Cu+ and Cu+ → Cu0, because the areas for each peak are apparently distinct, and the reduction of Cu+ usually appears at high temperatures.29 Moreover, many researchers29,30 confirmed that most reduction of CuO occurred in a representative two-stage pattern at lower temperatures, where all the crystalline-supported Cu2+ ions reduce. Therefore, it can be concluded that the peak in the overlapped TPR profiles at 195 °C is associated with the reduction of the small two- and/or three-dimensional bulk CuO, weak magnetic associates, and isolated Cu2+ ions, whereas the peak centered at 258 °C corresponds to the reduction of large three-dimensional clusters of CuO and/or the bulk CuO phase. The lowering of reduction temperature evidences the redox ability of the supported copper oxide, which is greatly improved after modification. This is because the couple of Cu/Cu+ facilitates the reduction of the energy required for electron transport and/or boosts the mobility of the active oxygen, thus promoting the reactant activation and/or generation of surface oxygen vacancies.31,32 The surface oxygen vacancies further result in structural distortion, which is in accordance with the XRD results.
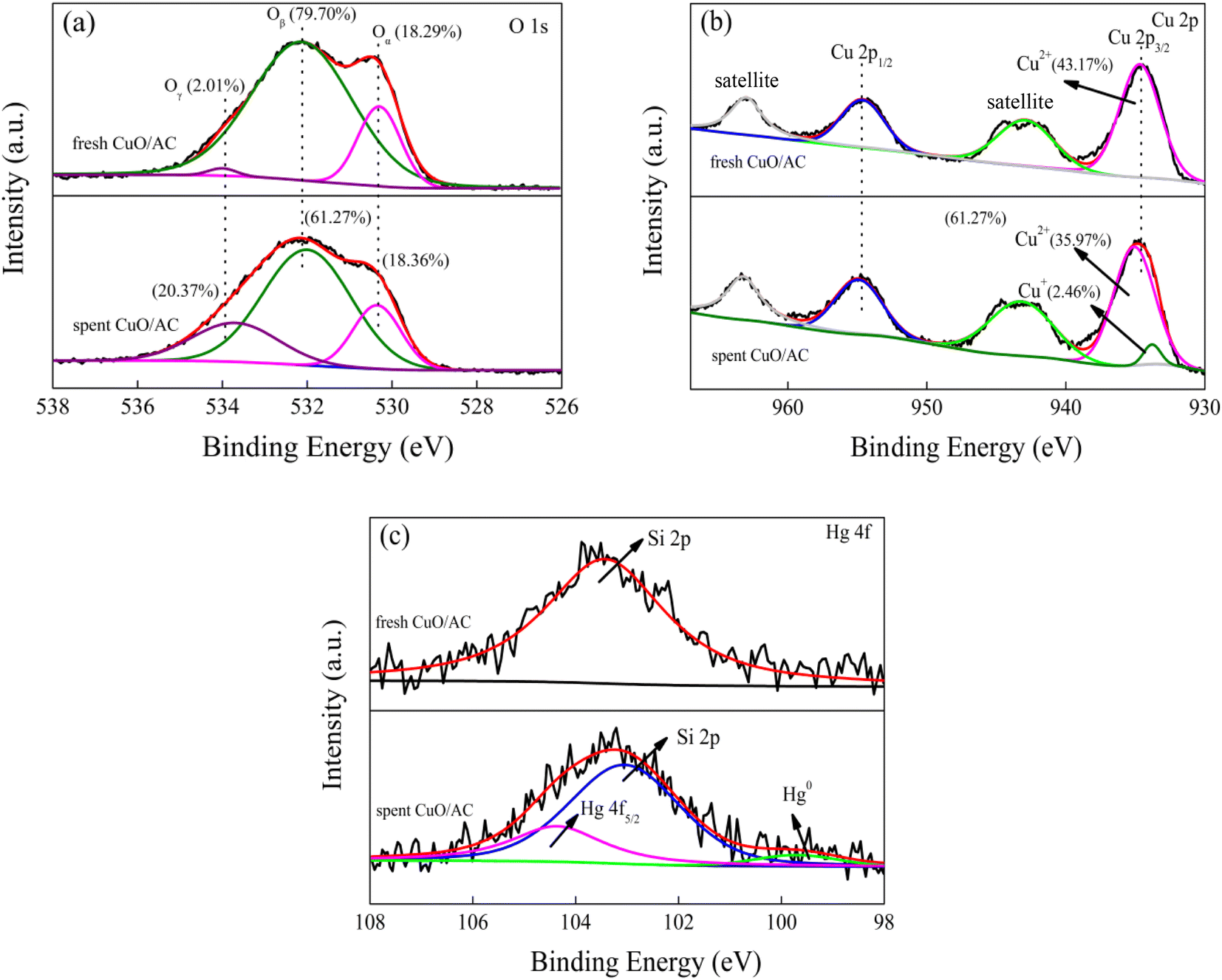 | ||
| Fig. 6 (a) O 1s, (b) Cu 2p and (c) Hg 4f profiles of fresh AC and spent 12% CuO/AC catalysts. | ||
Fig. 6(b) presents the Cu 2p XPS spectra and their Gaussian fitting patterns of fresh and spent 12% CuO/AC samples. The Cu 2p profile was typically deconvoluted into four peaks, which could be assigned to Cu 2p3/2, Cu 2p1/2 and two satellite peaks. For the fresh 12% CuO/AC, the Cu 2p3/2, Cu 2p1/2 and two satellite peaks are the three primary characteristic peaks of Cu2+,33 which echos the XRD and H2-TPR results. However, after the adsorption of Hg0 onto the surface of CuO/AC, the Cu 2p3/2 peak divided into two valence states. The binding energy in the range of 934.6–935 eV indicates the presence of CuO, while that centered at 933.8 eV is close to the Cu2O peak,25 demonstrating that CuO participates in removal progress and sectional species transform to Cu2O after reaction. However, this result cannot exclude the simultaneous possibility of an adsorption behavior and catalytic oxidation during the Hg0 removal.
The Hg 4f XPS profiles of the spent 12% CuO/AC are shown in Fig. 6(c). A single peak centered at 103.3 eV in fresh 12% CuO/AC was ascribed to Si 2p and in line with the XRD results. After the reaction, an intense peak at 104.4 eV corresponding to Hg 4f5/2 was attributed to HgO, and another small peak at 99.6 eV was assigned to Hg0.33 This result demonstrates that the mercury species over the spent CuO/ACs consists of Hg0 and HgO. Therefore, the results manifest that the Hg removal activities are restrained not only by the carbon texture, but also by the chemical properties, especially the lattice oxygen and chemisorbed oxygen groups.
3.2. Effect of multi-component gases and product species analysis
 | ||
| Fig. 7 Effect of O2 concentration balanced in N2 on Hg0 removal over the 12% CuO/AC catalyst at 120 °C. | ||
To reveal the reaction path toward Hg0 removal under the O2 atmosphere, the Hg0-TPD profiles were collected. As shown in Fig. 8, irrespective of the presence of O2, the Hg0 decomposition peaks were in a similar position, beyond that the peak intensities were different. Therefore, it is inferred that the presence of O2 will not alter the reaction path toward Hg0 removal but will affect its removal efficiency. Two decomposition peaks appeared on the TPD spectra at around 144 °C and 178 °C. Nevertheless, after the samples were purged with N2 for 1 h before the Hg0-TPD experiments, the desorption peak vanished at a lower temperature (shown in Fig. 8(b)). Therefore, the peak at 144 °C was assigned to the decomposition of the physically adsorbed mercury species when combined with XPS characterization results, while that peak centered at 178 °C was assigned to the HgO desorption. The Hg0-TPD profiles shown in Fig. 8(a) were further deconvoluted into two peaks based on the Gaussian method to distinguish the primary interaction mechanism, as shown in Fig. S2.† Interestingly, the area of HgO decomposition peak prevails regardless of the presence of O2 in the pre-adsorption atmosphere, which suggests that chemisorption is the primary adsorption mechanism rather than physisorption.
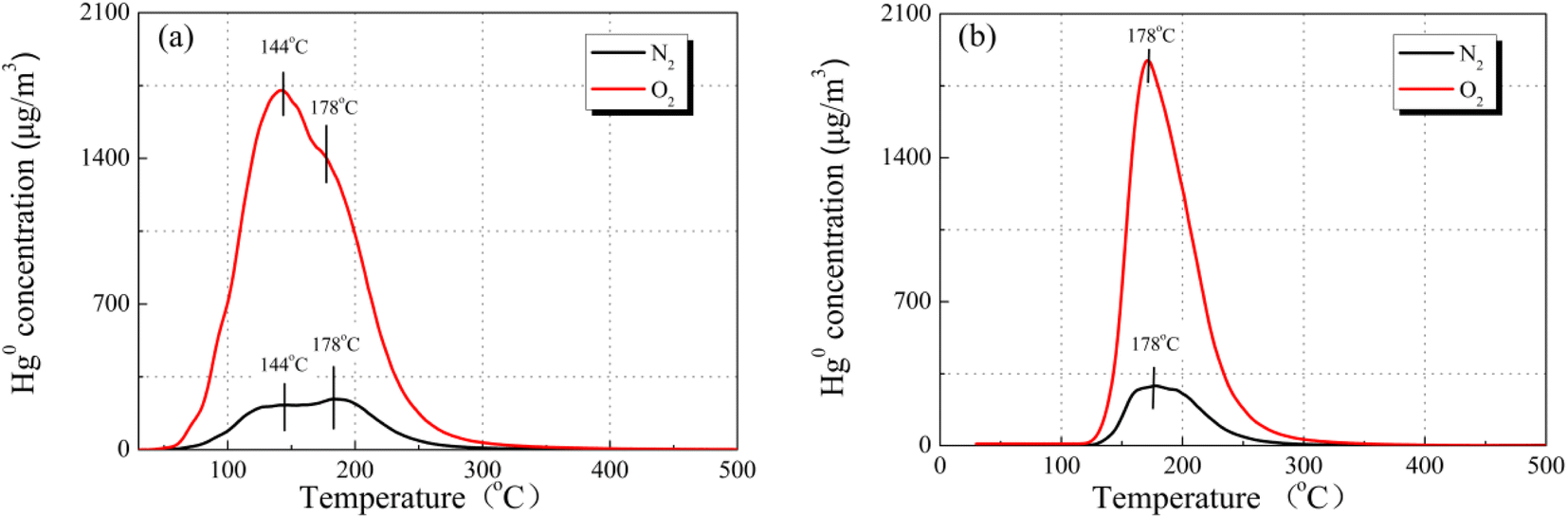 | ||
| Fig. 8 Hg0-TPD patterns of the 12% CuO/AC catalyst after pre-adsorption in an N2 or O2 atmosphere (a) without N2 purge and (b) with N2 purge at 120 °C (pre-adsorption reaction conditions: 7% O2 and N2 as the balance). | ||
Previously, Xiang et al.35 have also carried out similar research for the Hg0 adsorption mechanism on the CuO (110) crystal planes to evaluate the applicability of the SCR system for Hg0 removal. In their research, they found that Hg0 could strongly adsorb onto the Cu-terminated CuO (110) crystal planes, whereas weakly adsorb onto the O-terminated crystal layer, and the former bonding way was predicted to be the main adsorption mode. Accordingly, their conclusions are well consistent with our results obtained in this work.
 | ||
| Fig. 9 Effect of SO2 concentration balanced in an (a) N2 or (b) O2 atmosphere on Hg0 removal over the 12% CuO/AC catalyst at 120 °C. | ||
Desorption experiments were conducted to understand the Hg0 removal mechanism after pre-adsorption in SO2. As exhibited in Fig. 10, the Hg0 decomposition peak appeared at approximately 142 °C. As previously reported,37 the HgSO4 desorption temperature was much higher than that of HgO. In comparison with the case of the O2 atmosphere, we assume that the peak at 142 °C originates from the decomposition of physically adsorbed mercury species.
 | ||
| Fig. 10 Hg0-TPD patterns of the 12% CuO/AC catalyst after pre-adsorption in SO2 at 120 °C (pre-adsorption reaction conditions: 200 ppm of SO2, 7% O2, and N2 as the balance). | ||
Therefore, it is demonstrated that the presence of O2 weakens the suppression in physisorption toward Hg0, but the chemisorption is completely constrained by the presence of SO2. This result supports the above-mentioned viewpoint that the competitive adsorption between SO2 and Hg0 and the damage in the surface cation vacancies of the metal oxides take charge of the decrease in Hg0 removal.
 | ||
| Fig. 11 Effect of NO concentration balanced in an (a) N2 or (b) O2 atmosphere on Hg0 removal over the 12% CuO/AC catalyst at 120 °C. | ||
Breakthrough experiments of Hg0 over 12% CuO/AC were contrasted to identify the stimulative mechanisms for Hg0 activity in the presence of NO. As demonstrated in Fig. 11, introducing O2 into a NO atmosphere led to a prominent drop in the outlet Hg0 concentration compared with that of an individual NO or O2 atmosphere, indicating that the coexistence of O2 and NO plays a synergistic role in the removal of Hg0. The formed NO2 species gradually increases after the O2-containing gas was switched in, hence enhancing the Hg0 removal over the CuO/ACs. Furthermore, the Hg0 removal performance in NO obviously excels in that of O2, which implies that the reaction paths for Hg0 removal under O2 and NO are different.
Accordingly, the Hg0-TPD experiments were performed to clarify the mechanism after pre-adsorption in the presence of NO and O2. As can be seen in Fig. 12, after pre-adsorption in the presence of NO, the positions of the primary Hg0 desorption peaks were basically identical irrespective of the presence of O2, suggesting the same mercury product species, whose temperature is lower than that of HgO decomposition. Taking account of the flue gas components, the desorption peak center temperature merely possibly corresponds to Hg(NO3)2 or HgO. Moreover, the Hg(NO3)2 decomposition temperature was lower than that of HgO.41 Hence, the stable product species toward Hg0 after pre-adsorption in NO is identified as Hg(NO3)2.
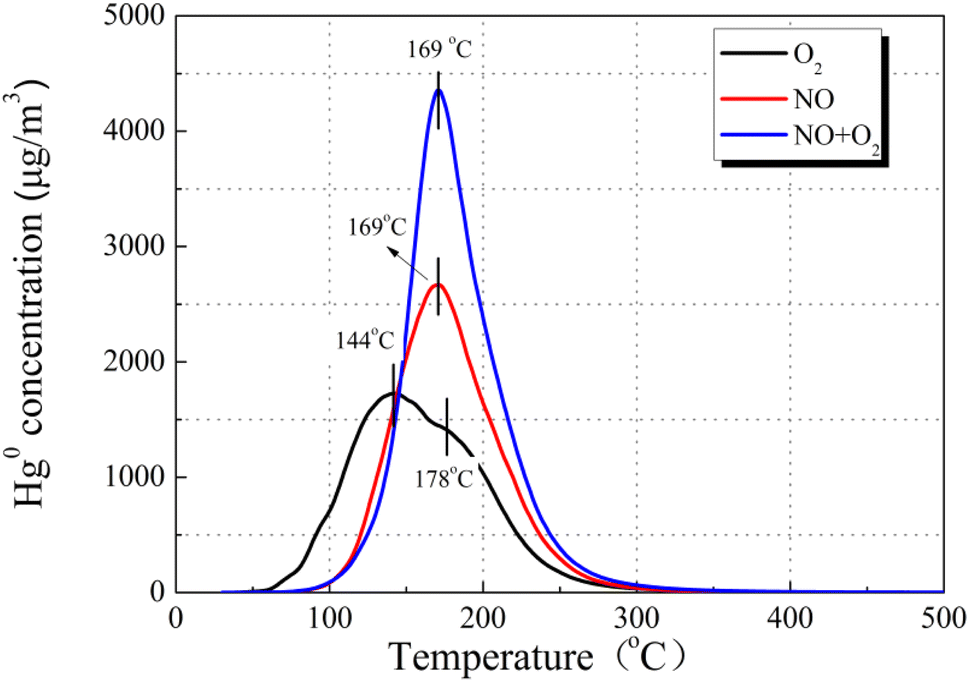 | ||
| Fig. 12 Hg0-TPD patterns of the 12% CuO/AC catalyst after pre-adsorption in NO at 120 °C (pre-adsorption reaction conditions: 500 ppm of NO, 7% O2, and N2 as the balance). | ||
 | ||
| Fig. 13 Effect of HCl concentration balanced in N2 on Hg0 removal over the 12% CuO/AC catalyst at 120 °C. | ||
To better comprehend the nature of the interaction between HCl and Hg0, we examined Hg0-TPD patterns for CuO/AC after pre-adsorption in HCl at 120 °C. Fig. 14 displays the Hg0 removal species after pre-adsorption in 15 ppm HCl regardless of the presence of O2. The peaks after pre-adsorption in HCl and O2 were observed at approximately 10 °C higher than the HCl-containing atmosphere, but the peak shape was extremely similar. As for the desorption characteristics of HgClx, it had been reported that Hg2Cl2 decomposition occurred in two steps, while HgCl2 decomposition occurred at a single peak.44 Therefore, HgCl2 is the most plausible product formed after pre-adsorption in HCl, regardless of the presence of O2. The peak shift should be attributed to the difference in bonding sites or molecular configuration between the Hg–Cu–Cl–O system on account of the O2 atmosphere.
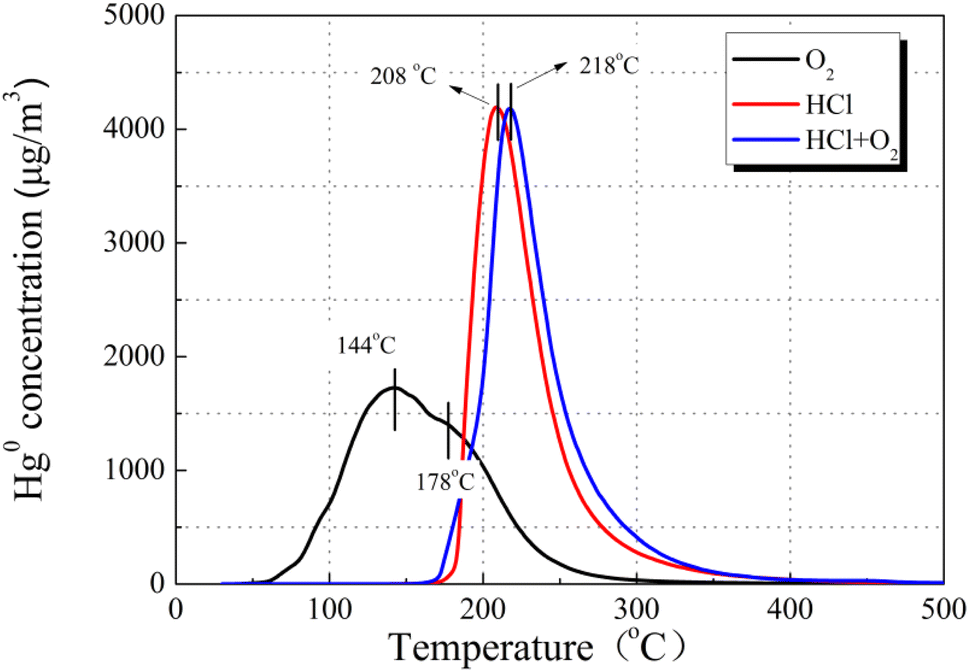 | ||
| Fig. 14 Hg0-TPD patterns of the 12% CuO/AC catalyst after pre-adsorption in HCl at 120 °C (pre-adsorption reaction conditions: 15 ppm of HCl, 7% O2, and N2 as the balance). | ||
3.3. Distribution laws toward mercury removal species and proposed reaction mechanism
To comprehensively gain insights into the different flue gas components on distribution laws toward Hg0 product species, the cumulative Hg0 adsorption and oxidation efficiency was qualitatively calculated, and the results are shown in Fig. 15. As depicted, HCl was confirmed as the most effectual flue gas component contributing for Hg0 removal, and the removal efficiency almost reached 100%. O2 enhances Hg0 adsorption by 38% compared to the N2 atmosphere, while SO2 and O2 decrease Hg0 adsorption by 24% compared to pure O2. Under the coexistence of NO and O2 atmospheres, 93% of Hg0 was captured, showing the most outstanding enhancement in terms of Hg0 adsorption, whereas the HCl atmosphere affords the most excellent enhancement by 29% for Hg0 catalytic oxidation activity. The enhancement in cumulative Hg0 removal efficiency under various flue gas components is sequenced: O2 < NO < HCl, but this sequence is inversely proportional to their gas concentration. As a result, we assume that the affinity ability rather than the gas concentration determines the removal activities toward Hg0, which follows the same trend as that of the cumulative Hg0 removal efficiency. This conclusion can be employed as a reference for adjusting the proportion of flue gas components to increase the removal selectivity toward Hg0, further to improve the adsorption efficiency or the catalytic oxidation efficiency oriently. | ||
| Fig. 15 Effect of individual flue gas components on Hg0 adsorption and oxidation over the 12% CuO/AC catalyst at 120 °C. | ||
Generally, the Hg0 removal over the CuO/AC catalyst results from a combination of physisorption, chemisorption, and catalytic oxidation. The detailed Hg0 removal mechanisms under various flue gases over the CuO/ACs catalyst are presented in Fig. 16.
 | ||
| Fig. 16 Proposed removal mechanism toward Hg0 under various simulated flue gases over CuO/AC. | ||
First, the gaseous Hg0 physically adsorbs onto the O-terminated CuO (110) crystal planes and chemically bonds onto the Cu-terminated highly dispersed, clustered and bulk CuO surface. Then, it reacts with chemisorbed oxygen to from HgO following the Mars–Maessen mechanism.
The presence of O2 enhances both the chemisorption at the Oβ sites and physisorption toward Hg0 and the reaction is principally controlled by chemisorption through the generation of HgO. The competitive interaction between Hg0 and SO2, as well as the deactivation of cation vacancies of CuO leads to an inhibition in Hg0 removal, whereas the presence of O2 could weaken the damage by compensating the physically adsorbed mercury product species. In the presence of NO, the oxidizing NO2 species and/or bidentate nitrate species formed on the metal oxide and carbon matrix are favorable for Hg0 adsorption to produce Hg(NO3)2. In the presence of HCl, the generation of HgCl2 over the CuO/ACs catalyst involves HOCl or Cl2 oxidizing Hg0, and a small brunch of that of HgCl2 further escapes into the gas phase. The addition of the O2 atmosphere to the HCl-containing gas causes a decrease in the Hg0 oxidation efficiency.
4. Conclusions
CuO/ACs were prepared to remove Hg0 at lower temperatures. It was found that 12% CuO/AC exhibits higher activity toward Hg0 at 120 °C. Characterization techniques and bench-scale experiments were performed to gain insights into the structure–activity relationships, product species distribution law and the mechanism of impact of multi-component flue gases on Hg0 adsorption and oxidation over the CuO/ACs. The main conclusions from this experimental study are summarised as follows:• Hg0 removal under a N2 atmosphere over CuO/ACs is dominated by both physisorption and chemisorption, and it occurs primarily through chemisorption.
• Micropores centered at 1 nm facilitate the Hg0 physisorption. The active component of the O-terminated CuO (110) crystal planes can physically adsorb Hg0 and can chemically adsorb Hg0 through the Mars–Maessen mechanism.
• The presence of O2 enhances both physisorption and chemisorption of mercury by 38%, and the chemisorption occurring at the Oβ sites preponderates with the product of HgO.
• Competitive adsorption and the deactivation of cation vacancies of CuO contribute to the decrease in Hg0 removal by SO2, whereas O2 can weaken the deactivation by augmenting 20% of physically adsorbed mercury.
• NO and O2 make for the Hg0 chemisorption due to the generation of the NO2 and/or bidentate nitrate species on the metal oxide or carbon matrix, and the removal efficiency reached 93%.
• The Hg0 catalytic oxidation appears when HCl is converted into HOCl and/or Cl2, but the catalytic oxidation efficiency of Hg0 decreases in the presence of O2 by 8%.
• The affinity abilities between various flue gases and Hg0 are in the following order: O2 < NO < HCl.
In future studies, it is recommended that quantum chemistry methods based on density functional theory (DFT) calculations be used to simulate the reaction progress further to identify the adsorption products under multi-component flue gases and clarify the interaction mechanism in heterogeneous systems. This suggestion will be addressed in the next work.
Data availability
The data that support the findings of this study are available from the corresponding author, Haixin Guo, upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 21607009), the Science and Technology Research and Development Program for Handan City (2342071019) the Financial Program of BJAST (24CB001-13) and the IUSE Budding Talent Program (DQ-RCB-230432).References
- L. Tong, W. Q. Xu, Y. Yang, R. H. Liu, Y. H. Wu and T. Y. Zhu, RSC Adv., 2016, 6, 78743–78749 RSC.
- R. L. Hao, X. H. Dong, Z. Wang, L. Fu, Y. Han, B. Yuan, Y. P. Gong and Y. Zhao, Environ. Sci. Technol., 2019, 53, 8324–8332 CrossRef CAS PubMed.
- H. T. Zhao, X. Luo, J. He, C. Peng and T. Wu, Fuel, 2015, 147, 67–75 CrossRef CAS.
- B. A. Dranga, L. Lazar and H. Koeser, Catalysts, 2012, 2, 139–170 CrossRef CAS.
- R. L. Hao, Z. Wang, Y. P. Gong, Z. Ma, Z. Qian, Y. C. Luo, B. Yuan and Y. Zhao, J. Hazard. Mater., 2020, 383, 121135 CrossRef CAS.
- H. Zhao, C. I. Ezeh, S. Yin, Z. Xie, C. H. Pang, C. Zheng, X. Gao and T. Wu, Appl. Catal., B, 2020, 263, 117829 CrossRef CAS.
- W. Xu, J. Pan, B. Fan and Y. Liu, J. Cleaner Prod., 2019, 216, 277–287 CrossRef CAS.
- Z. Q. Tan, J. Xiang, S. Su, H. C. Zeng, C. S. Zhou, L. S. Sun, S. Hu and J. R. Qiu, J. Hazard. Mater., 2012, 239, 160–166 CrossRef PubMed.
- Y. Ma, B. Mu, X. Zhang, D. Yuan, C. Ma, H. Xu, Z. Qu and S. Fang, Chem. Eng. J., 2019, 358, 1499–1506 CrossRef CAS.
- B. Shen, S. Zhu, X. Zhang, G. Chi, D. Patel, M. Si and C. Wu, Fuel, 2018, 224, 241–249 CrossRef CAS.
- W. Q. Xu, H. R. Wang, X. Zhou and T. Y. Zhu, Chem. Eng. J., 2014, 243, 380–385 CrossRef CAS.
- M. A. Fayad, A. M. Abed, S. H. Omran, A. A. Jaber, A. A. Radhi, H. A. Dhahad, M. T. Chaichan and T. Yusaf, Int. J. Renewable Energy Dev., 2022, 11, 1068–1077 CrossRef.
- M. A. Fayad, S. I. Ibrahim, S. H. Omran, F. J. Martos, T. Badawy, A. M. Al Jubori d, H. A. Dhahad and M. T. Chaichan, Fuel, 2023, 331, 125549 CrossRef CAS.
- Z. Zhang, J. Liu, Z. Wang and Y. J. Yang, Fuel, 2021, 289, 119791 CrossRef CAS.
- D. J. Liu, C. Lu and J. Wu, J. Nanopart. Res., 2018, 20, 277 CrossRef.
- B. Zhao, H. Yi, X. Tang, Q. Li, D. Liu and F. Gao, Chem. Eng. J., 2016, 286, 585–593 CrossRef CAS.
- Y. Xiao, D. Pudasainee, R. Gupta, Z. H. Xu and Y. F. Diao, J. Hazard. Mater., 2017, 336, 232–239 CrossRef CAS PubMed.
- H. Y. Wu, C. T. Li, L. K. Zhao, J. Zhang, G. M. Zeng, Y. e. Xie, X. N. Zhang and Y. Wang, Energy Fuels, 2015, 29, 6747–6757 CrossRef CAS.
- H. L. Li, C. Y. Wu, Y. Li, L. Q. Li, Y. C. Zhao and J. Y. Zhang, J. Hazard. Mater., 2012, 243, 117–123 CrossRef CAS PubMed.
- K. L. Zhao, C. T. Li, S. H. Li, Y. Wang, J. Y. Zhang, T. Wang and G. M. Zeng, Appl. Catal., B, 2016, 198, 420–430 CrossRef.
- L. Tong, T. Yue, P. Zuo, X. Zhang, C. Wang, J. Gao and K. Wang, Fuel, 2017, 197, 1–7 CrossRef CAS.
- Y. Wu, W. Xu, Y. Yang, J. Wang and T. Zhu, Catal. Sci. Technol., 2018, 8, 297–306 RSC.
- D. Li, X. Tang, H. Yi, D. Ma and F. Gao, Ind. Eng. Chem. Res., 2015, 54, 9097–9103 CrossRef CAS.
- B. Zhao, H. Yi, X. Tang, Q. Li, D. Liu and F. Gao, J. Hazard. Mater., 2019, 364, 700–709 CrossRef CAS PubMed.
- W. Du, L. Yin, Y. Zhuo, Q. Xu, L. Zhang and C. Chen, Fuel Process. Technol., 2015, 131, 403–408 CrossRef CAS.
- M. Trépanier, A. Tavasoli, A. K. Dalai and N. Abatzoglou, Appl. Catal., A, 2009, 353, 193–202 CrossRef.
- L. K. Herrera, A. Justo and J. L. Perez-Rodriguez, J. Nano Res., 2009, 8, 99–107 CAS.
- G. Q. Zhang, Z. Li, H. Y. Zheng, T. J. Fu, Y. B. Ju and Y. C. Wang, Appl. Catal., B, 2015, 179, 95–105 CrossRef CAS.
- C. Liu, S. L. Nauert, M. A. Alsina, D. Wang, A. Grant, K. He, E. Weitz, M. Nolan, K. A. Gray and J. M. Notestein, Appl. Catal., B, 2019, 255, 117754 CrossRef CAS.
- A. Kubacka, M. J. Munoz-Batista, M. Fernandez-Garcia, S. Obregon and G. Colon, Appl. Catal., B, 2015, 163, 214–222 CrossRef CAS.
- L. Gao, C. Li, S. Li, W. Zhang, X. Du, L. Huang, Y. Zhu, Y. Zhai and G. Zeng, Chem. Eng. J., 2019, 371, 781–795 CrossRef CAS.
- T. Boningari, P. R. Ettireddy, A. Somogyvari, Y. Liu, A. Vorontsov, C. A. McDonald and P. G. Smirniotis, J. Catal., 2015, 325, 145–155 CrossRef CAS.
- L. Tang, C. Li, L. Zhao, L. Gao, X. Du, J. Zeng, J. Zhang and G. Zeng, Fuel, 2018, 218, 366–374 CrossRef CAS.
- S. H. Qiao, J. Chen, J. F. Li, Z. Qu, P. Liu, N. Q. Yan and J. Q. Jia, Ind. Eng. Chem. Res., 2009, 48, 3317–3322 CrossRef CAS.
- W. Xiang, J. Liu, M. Chang and C. Zheng, Chem. Eng. J., 2012, 200–202, 91–96 CrossRef CAS.
- H. B. Fu, X. Wang, H. B. Wu, Y. Yin and J. M. Chen, J. Phys. Chem. C, 2007, 111, 6077–6085 CrossRef CAS.
- M. A. Lopez-Anton, R. Perry, P. Abad-Valle, M. Díaz-Somoano, M. R. Martínez-Tarazona and M. M. Maroto-Valer, Fuel Process. Technol., 2011, 92, 707–711 CrossRef CAS.
- S. J. Yang, Y. W. Fu, Y. Liao, S. C. Xiong, Z. Qu, N. Q. Yan and J. H. Li, Catal. Sci. Technol., 2014, 4, 224–232 RSC.
- L. Tong, W. Q. Xu, X. Zhou, R. H. Liu and T. Y. Zhu, Energy Fuels, 2015, 29, 5231–5236 CrossRef CAS.
- Y. Yang, W. Xu, J. Wang and T. Zhu, Fuel, 2019, 249, 178–187 CrossRef CAS.
- M. Rumayor, M. Diaz-Somoano, M. A. Lopez-Anton and M. R. Martinez-Tarazona, Talanta, 2013, 114, 318–322 CrossRef CAS.
- A. Murakami, M. A. Uddin, R. Ochiai, E. Sasaoka and S. Wu, Energy Fuels, 2010, 24, 4241–4249 CrossRef CAS.
- W. M. Chen, Y. Pei, W. J. Huang, Z. Qu, X. F. Hu and N. Q. Yan, Environ. Sci. Technol., 2016, 50, 2564–2572 CrossRef CAS.
- M. A. Lopez-Anton, Y. Yuan, R. Perry and M. M. Maroto-Valer, Fuel, 2010, 89, 629–634 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra02483d |
| ‡ Co-first author: Li Tong and Penglai Zuo contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |