
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElucidating bis-pyrimidines as new and efficient mushroom tyrosinase inhibitors: synthesis, SAR, kinetics and computational studies†
Manazza Afzala,
Rabia Mehmooda,
Ehsan Ullah Mughal *b,
Nafeesa Naeemb,
Zaman Ashraf*c,
Yasir Nazird,
Fatma Mohsen Shalabye,
Amal El-Sayed Abd El Hadyf and
Amina Sadiq*a
*b,
Nafeesa Naeemb,
Zaman Ashraf*c,
Yasir Nazird,
Fatma Mohsen Shalabye,
Amal El-Sayed Abd El Hadyf and
Amina Sadiq*a
aDepartment of Chemistry, Govt College Women University, Sialkot-51300, Pakistan. E-mail: amina.sadiq@gcwus.edu.pk
bDepartment of Chemistry, University of Gujrat, Gujrat-50700, Pakistan. E-mail: ehsan.ullah@uog.edu.pk
cDepartment of Chemistry, Rawalpindi Women University, Rawalpindi-46300, Pakistan. E-mail: zaman.ashraf@f.rwu.edu.pk
dDepartment of Chemistry, University of Sialkot, Sialkot-51300, Pakistan
eKing Khalid University, Faculty of Sciences, Biology Department, Abha, Kingdom of Saudi Arabia
fDepartment of Biology, Faculty of Science, Majmaah University, Al Majma'ah 15341, Kingdom of Saudi Arabia
First published on 19th July 2024
Abstract
In this study, a series of novel bis-pyrimidine derivatives (1P–8P) were designed, synthesized, characterized, and investigated for their in vitro inhibitory activity against mushroom tyrosinase, an enzyme critical in melanin biosynthesis and implicated in various hyperpigmentation disorders. To the best of our knowledge, the bispyrimidine scaffold has been evaluated for the first time for its tyrosinase inhibitory activity. Their inhibitory activities were assessed, revealing inhibition with IC50 values in the micromolar range. Additionally, this series of compounds were found to inhibit tyrosinase activity in a mixed-type manner, with IC50 values ranging from 12.36 ± 1.24 to 86.67 ± 3.08 μM. To further elucidate the binding interactions, molecular docking simulations were performed, identifying key residues in the active site responsible for binding affinity. Furthermore, molecular dynamics (MD) simulations were conducted to assess the dynamic behavior, stability, and binding affinity of the most potent inhibitor, compound 6P. Quantitative Structure–Activity Relationship (QSAR) models were developed to correlate the structural features of the bis-pyrimidines with their inhibitory activity, providing insights into the structure–activity relationships (SAR) that govern their potency. The experimental and theoretical findings demonstrated excellent agreement. These findings pave the way for the development of novel bis-pyrimidine-based therapeutic agents for treating hyperpigmentation and related conditions.
1. Introduction
Tyrosinase (TYR; EC 1.14.18.1) is a copper-containing enzyme that is pivotal in the biosynthesis of melanin, the pigment responsible for the coloration of skin, hair, and eyes in humans.1 This enzyme catalyzes the oxidation of tyrosine to dihydroxyphenylalanine (DOPA) and subsequently to DOPA-quinone, which undergoes further reactions to produce melanin.2 TYR is widely distributed across various species, including plants, animals, and microorganisms, indicating its essential role in diverse biological processes.3 The activity of TYR is directly linked to several dermatological conditions and cosmetic concerns.4 Hyperactivity of TYR can lead to hyperpigmentation disorders such as melasma, lentigines, and post-inflammatory hyperpigmentation.5 Conversely, insufficient TYR activity is associated with hypopigmentation disorders like albinism.6 Due to its central role in pigmentation, TYR is a key target for developing therapeutic agents aimed at modulating melanin production.7 In the cosmetic industry, TYR inhibitors are extensively used in formulations designed to lighten skin and treat hyperpigmentation.8The quest for effective TYR inhibitors has led to the discovery of numerous natural and synthetic compounds.9 Natural inhibitors, such as kojic acid, arbutin, and licorice extract, are commonly used in cosmetic products.10,11 Numerous synthetic TYR inhibitors have been developed, including bis-salicylaldehyde, acetamides, chalcones, 3-hydroxyflavones, thiazolidinones, coumarin-resveratrol hybrids, benzofuran-thiosemicarbazone hybrids, flavone hydrazones, thioflavonols, thioflavones, benzothiazepines, benzopyrimidinone, oxadiazoles and oxazolones12–19 (Fig. 1). Despite the fact that many synthetic inhibitors have demonstrated inhibitory activity against mushroom TYR, only a few have shown effectiveness in inhibiting melanogenesis in cellular or skin models.20,21 Consequently, the search for novel TYR inhibitors and the development of more effective cytotoxic agents remain crucial steps in the drug discovery and development process.
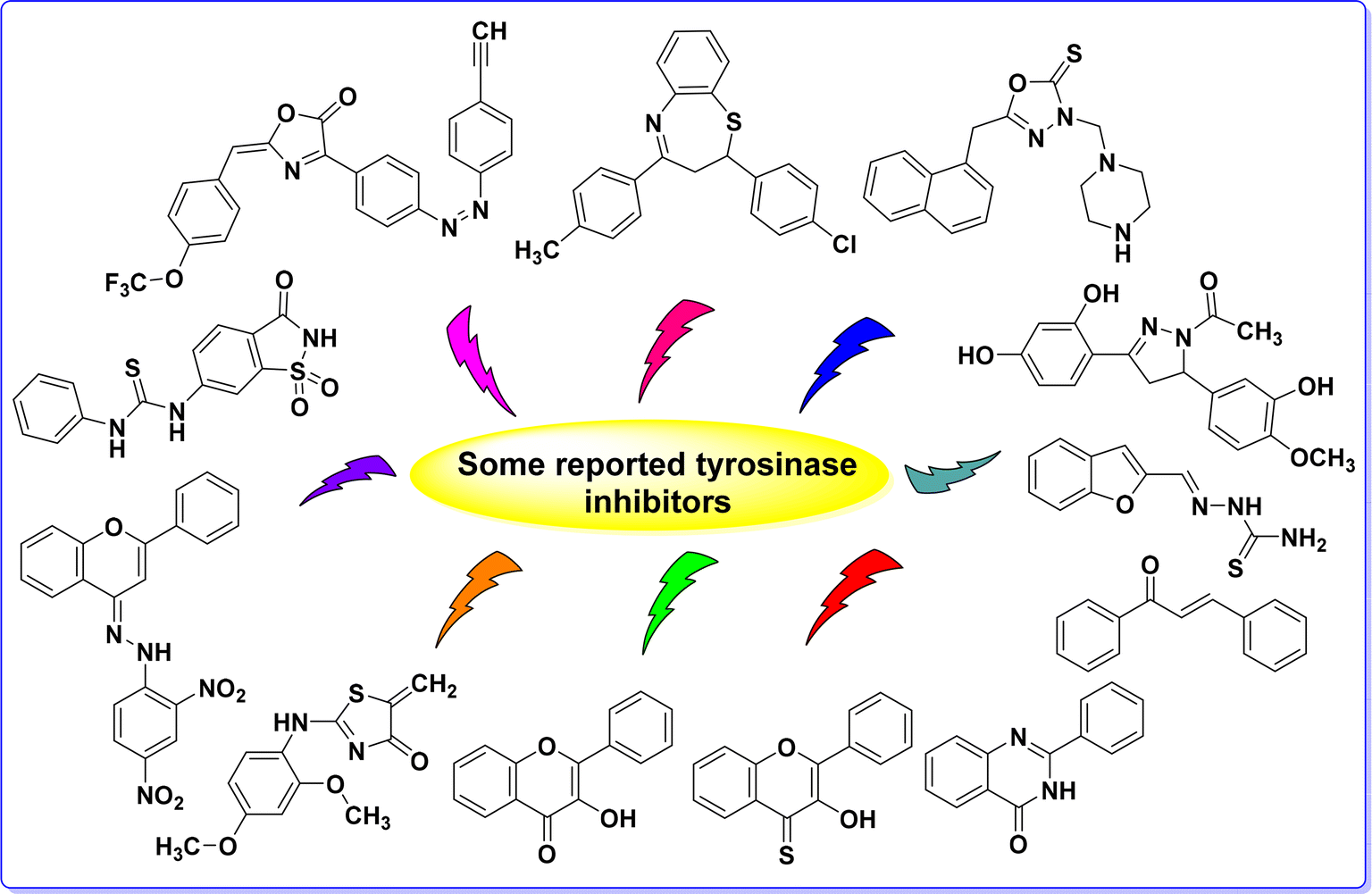 | ||
| Fig. 1 Some recently developed TYR inhibitors. | ||
Bis-chalcones (Fig. 2a), a subclass of chalcones, are abundant in plants and widely present in various organisms, serving as precursors to flavonoids and several other heterocyclic pharmacophores.22,23 These compounds have demonstrated a range of bioactivities, including anti-tubercular, anti-viral, anti-cancer, anti-inflammatory, anti-tumor, anti-fungal properties, and TYR inhibition.24–26 The structural flexibility of chalcones allows for extensive modification, which can lead to enhanced biological activity and specificity.27 The cyclization of bischalcones leads to the formation of diverse heterocyclic scaffolds, comprising six- and seven-membered rings with various heteroatoms, including compounds such as pyrimidines, dihydropyrimidines, benzothiazepines, benzodiazepines and pyrazoline, etc.28 Among these, nitrogen-containing heterocyclic compounds are especially notable for their significance in bioactivity studies.23 Nitrogen-containing heterocycles are prominent chemical structures that have gained substantial interest from researchers owing to their wide array of biological applications.29,30
 | ||
| Fig. 2 Chemical structures of the bis-chalcone (a) and bis-pyrimidine (b). | ||
Bis-pyrimidines are a class of compounds featuring two pyrimidine rings, which are six-membered heterocyclic structures containing two nitrogen atoms31 (Fig. 2b). These compounds have attracted considerable interest in medicinal chemistry on account of their diverse and potent biological activities and potential therapeutic applications.32–34 Among various chemical motifs explored for TYR inhibition, the pyrimidine scaffold has emerged as a highly potent and versatile framework.35,36 The presence of amino groups at both poles of the bis-pyrimidine scaffold is crucial for effective TYR inhibition, as they enhance binding affinity through hydrogen bonding and interaction with the enzyme's active site residues and also attributed to their ability to chelate the copper (Cu2+) ions at the enzyme's active site.37,38 Their ability to interact with various biological targets is attributed to the electronic and steric characteristics imparted by the pyrimidine rings.39–41 Furthermore, bis-pyrimidines have shown favorable drug-like properties, including good bioavailability and metabolic stability, making them attractive candidates for further development in medicinal chemistry.42
Encouraged by the biological significance of pyrimidine motif and the increasing demand for novel, harmless, and potent TYR inhibitors, we aimed to synthesize bis-pyrimidines, anticipating that these products would exhibit enhanced TYR inhibitory activity. To the best of our knowledge, the TYR inhibitory potential of bis-pyrimidine derivatives has not been explored previously. Therefore, we present a simple multi-component reaction strategy for the efficient synthesis of bis-pyrimidines, targeting their application as anti-TYR agents. To effectively address the challenges posed by skin disorders such as melasma, senile lentigines and ephelides it is crucial to discover novel and safe inhibitors for this enzyme. Additionally, all experimental findings have been corroborated by comprehensive computational studies.
2. Materials and methods
All chemicals were sourced from Merck (Germany) and utilized without further purification. Melting points were determined with an uncorrected electrothermal apparatus. Infrared (IR) spectra were recorded on a Bio-Rad spectrophotometer. Nuclear Magnetic Resonance (NMR) spectra were collected on a Bruker DRX 600 spectrometer, operating at 600 MHz for 1H and 151 MHz for 13C. UV-visible absorption spectra were measured using a Jasco V-670 spectrophotometer with a quartz cell.3. General procedure for the synthesis of bis-chalcones (1C–8C) and bis-pyrimidines (1P–8P)29
The synthesis of bis-chalcones (step-1) was carried out following the method previously described in the literature.29In step-2, to synthesize bis-pyrimidines, a bis-chalcone (1.0 mmol) was combined with guanidine hydrochloride (4.0 mmol) in an aqueous potassium hydroxide solution (5–6 mL, 50%) and the mixture was refluxed in ethanol (15 mL) for 4–5 hours. The completion of the reaction was monitored using thin-layer chromatography (TLC). After cooling the reaction mixture, the solvent was evaporated under reduced pressure to obtain the crude product. This crude material was then purified by recrystallization from methanol, resulting in the formation of bis-pyrimidines (1P–8P).
3.1. Enzyme inhibition activity
3.2. Kinetic analysis
The methodology employed for analyzing the kinetic activity has been thoroughly described in our previous publication.43,44 Please refer to the procedure outlined in the ESI† file for detailed instructions.3.3. Molecular docking study
The methodology for ligand and protein preparations and docking simulations were performed following our already reported methods.45 For specific instructions, please refer to the procedure outlined in the ESI† file.3.4. MD simulation analysis
The methodology used for MD simulation analysis is detailed in our previous publications.46–52 For specific instructions, please refer to the procedure outlined in the ESI† file.3.5. QSAR analysis
The methodology used to analyze the kinetic activity is comprehensively detailed in our previous publications.42 For specific instructions, please refer to the procedure outlined in the ESI† file.4. Results and discussion
4.1. Chemistry
To synthesize bis-pyrimidine derivatives (1P–8P), the terephthalaldehyde was taken as starting material, and thus allowed to react with various substituted aromatic acetophenones in the presence of sodium hydroxide (NaOH) dissolved in methanol. Subsequently, the reaction mixture was stirred for 4–5 hours at 0–5 °C to produce the bis-chalcones (1C–8C) in moderate to good yield (Scheme 1). These intermediate compounds were purified through recrystallization by ethanol (EtOH) before proceeding for the next step. Thereafter, the bis-chalcones (1C–8C) were then cyclized with guanidine hydrochloride in the presence of aqueous solution of NaOH and ethanol after refluxing for 3–4 h in order to obtain the target bis-pyrimidines (1P–8P) in moderate to excellent yield (Scheme 1 and Table 1). The target compounds were purified through recrystallization from EtOH. The bis-pyrimidines were found to be prone to oxidation for longtime standing under the air. | ||
| Scheme 1 Synthetic route for the synthesis of bis-chalcones and bis-pyrimidines. | ||
| Sr. no. | Chemical structure | Yield (%) |
|---|---|---|
| 1P |  |
80% |
| 2P |  |
75% |
| 3P | 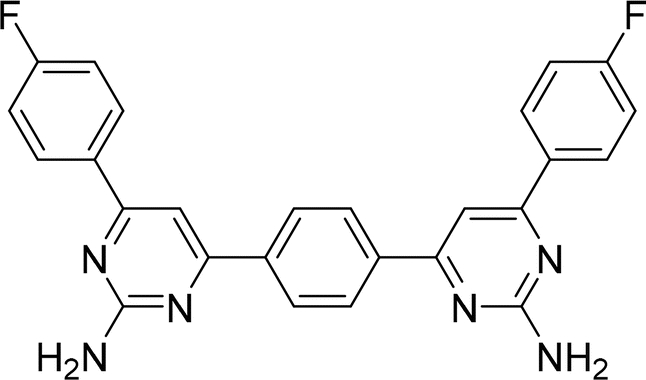 |
75% |
| 4P |  |
75% |
| 5P |  |
80% |
| 6P |  |
79% |
| 7P |  |
83% |
| 8P |  |
79% |
All the newly synthesized target compounds (1P–8P) were thoroughly characterized using common spectroscopic techniques, including IR, UV-vis, NMR and mass spectrometry. For example, all compounds (1P–8P) exhibited distinct bands in the range of 1550–1630 cm−1 attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching, confirming the ring closure formation. Additionally, absorption bands at 1220–1300 cm−1 indicated C–N stretching vibrations, further confirming the formation of the desired pyrimidine ring in the target compounds. There is another distinctive stretching band with two spikes in the range of 3190–3320 cm−1 on account of the amino (–NH2) group. Similarly, UV-vis spectra were recorded in methanol solution, revealing two absorption bands in the ranges of 240–260 nm and 325–350 nm, corresponding to the aromatic chromophores.
N stretching, confirming the ring closure formation. Additionally, absorption bands at 1220–1300 cm−1 indicated C–N stretching vibrations, further confirming the formation of the desired pyrimidine ring in the target compounds. There is another distinctive stretching band with two spikes in the range of 3190–3320 cm−1 on account of the amino (–NH2) group. Similarly, UV-vis spectra were recorded in methanol solution, revealing two absorption bands in the ranges of 240–260 nm and 325–350 nm, corresponding to the aromatic chromophores.
The structural confirmation of the compounds was primarily established using 1H and 13C-NMR spectroscopy. For instance, the 1H-NMR spectrum (Fig. 3) of compound 4P demonstrated distinctive signal patterns. A singlet at δ 8.36 ppm was observed for the four chemically and magnetically equivalent aromatic protons (Ha) belonging to the central aromatic ring (ring A). Adjacent to this, a doublet appeared at δ 8.16 ppm, attributed to another set of four equivalent aromatic protons (Hb), which are deshielded due to the nearby electronegative pyrimidine ring (ring B). A singlet at δ 7.77 ppm was characteristic of two chemically equivalent aromatic protons (Hc), followed by a doublet at δ 7.36 ppm for four aromatic protons (Hd), which experience upfield shifts due to ortho-methyl groups. The presence of two amino (–NH2) groups was confirmed by a singlet at δ 6.76 ppm, corresponding to four chemically equivalent amino protons (He). Additionally, a highly upfield singlet at δ 2.39 ppm indicated the presence of two methyl groups attached to aromatic ring C. Further evidence from the 13C-NMR spectra supported the formation of pyrimidine analogues, where signals at δ 164.4–163.5 ppm and δ 162.8–160.8 ppm were assigned to CN and CC, respectively, across all synthesized compounds. The characteristic signals for C–H of the pyrimidine ring were observed in the range of δ 102.1–105.7 ppm, consistent with the formation of the pyrimidine nucleus in all synthesized compounds (See ESI† file). The spectral data obtained are in agreement with the structures of the newly synthesized target compounds (1P–8P).
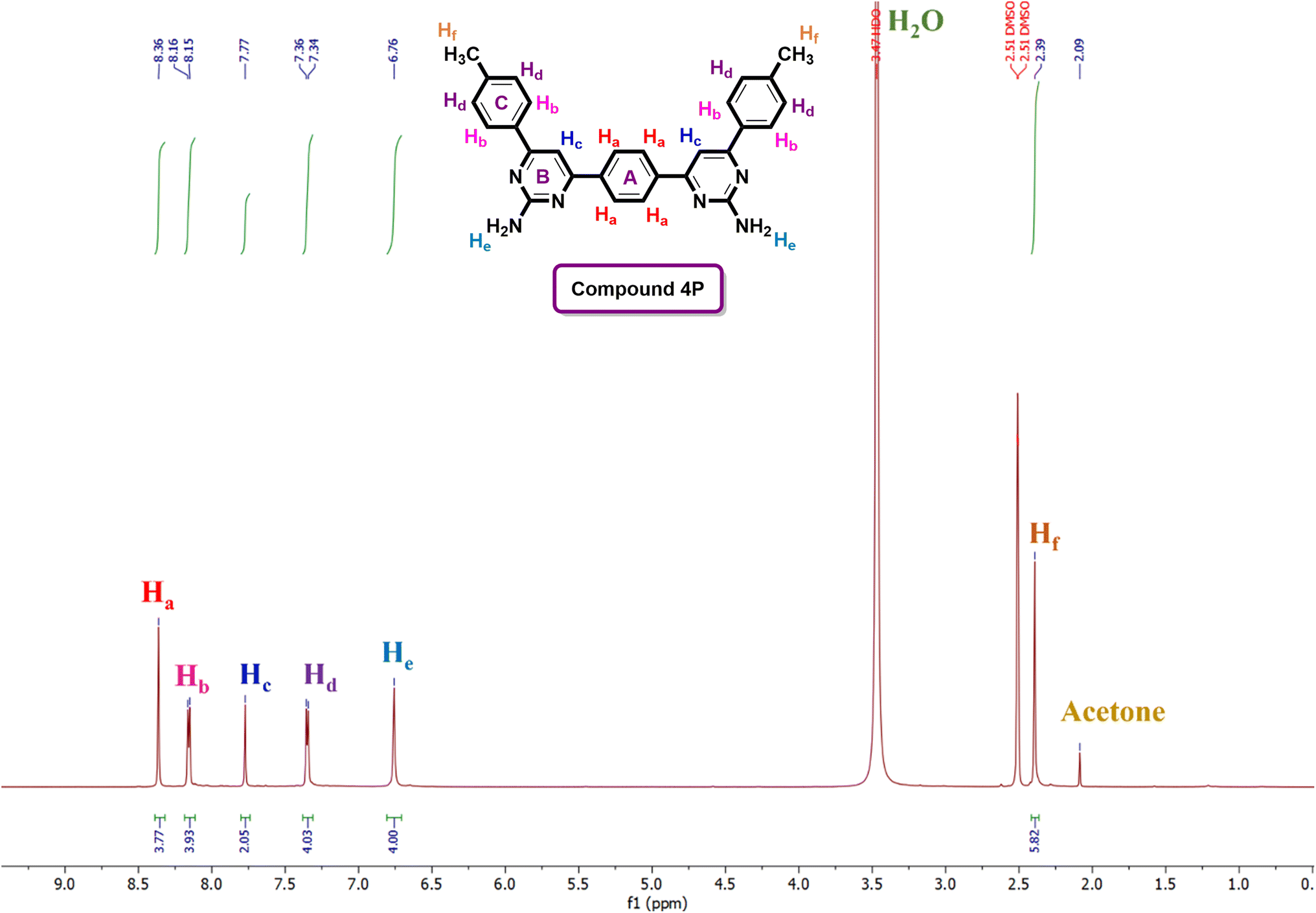 | ||
| Fig. 3 1H-NMR spectrum of the compound 4P. | ||
The spectroscopic data of bis-chalcones (1C–6C & 8C) are already reported in the literature.53–59 Nevertheless, the spectral data of all the newly synthesized target compounds is given in ESI† file.
4.2. Mushroom TYR inhibition study
The TYR inhibitory activity of the synthesized compounds was assessed and compared to the standard inhibitor, kojic acid. The half-maximal inhibitory concentration (IC50) values, expressed in micromolar (μM) along with the standard error of the mean (SEM), are given in Table 2.| Compound no. | Tyrosinase inhibitory activity IC50 (μM ± SEM) |
|---|---|
| 1P | 86.67 ± 3.08 |
| 2P | 30.56 ± 1.62 |
| 3P | 45.98 ± 2.05 |
| 4P | 37.21 ± 1.92 |
| 5P | 40.87 ± 2.08 |
| 6P | 12.36 ± 1.24 |
| 7P | 17.19 ± 1.69 |
| 8P | 36.56 ± 1.89 |
| Kojic acid (standard) | 16.69 ± 2.81 |
Among the tested compounds, 6P exhibited the most potent TYR inhibitory activity with an IC50 value of 12.36 ± 1.24 μM, surpassing the standard kojic acid (IC50 = 16.69 ± 2.81 μM). Other compounds, such as 7P (IC50 = 17.19 ± 1.69 μM) and 8P (IC50 = 36.56 ± 1.89 μM), also demonstrated significant inhibitory activities. Compound 1P showed the least inhibition with an IC50 value of 86.67 ± 3.08 μM. These results suggest that compound 6P is a promising candidate for further development as a TYR inhibitor.
4.3. Kinetic study
Compound 6P exhibited the highest TYR inhibitory potential, prompting an investigation into its inhibition pattern through in vitro kinetic study using Lineweaver–Burk and Dixon plots. The analysis revealed that 6P acts as a mixed-type inhibitor of TYR, with a Ki value of 16 μM (Fig. 4 and 5). In mixed-type inhibition, both Km and Vmax are altered, with Vmax consistently decreasing, while Km can either increase or decrease.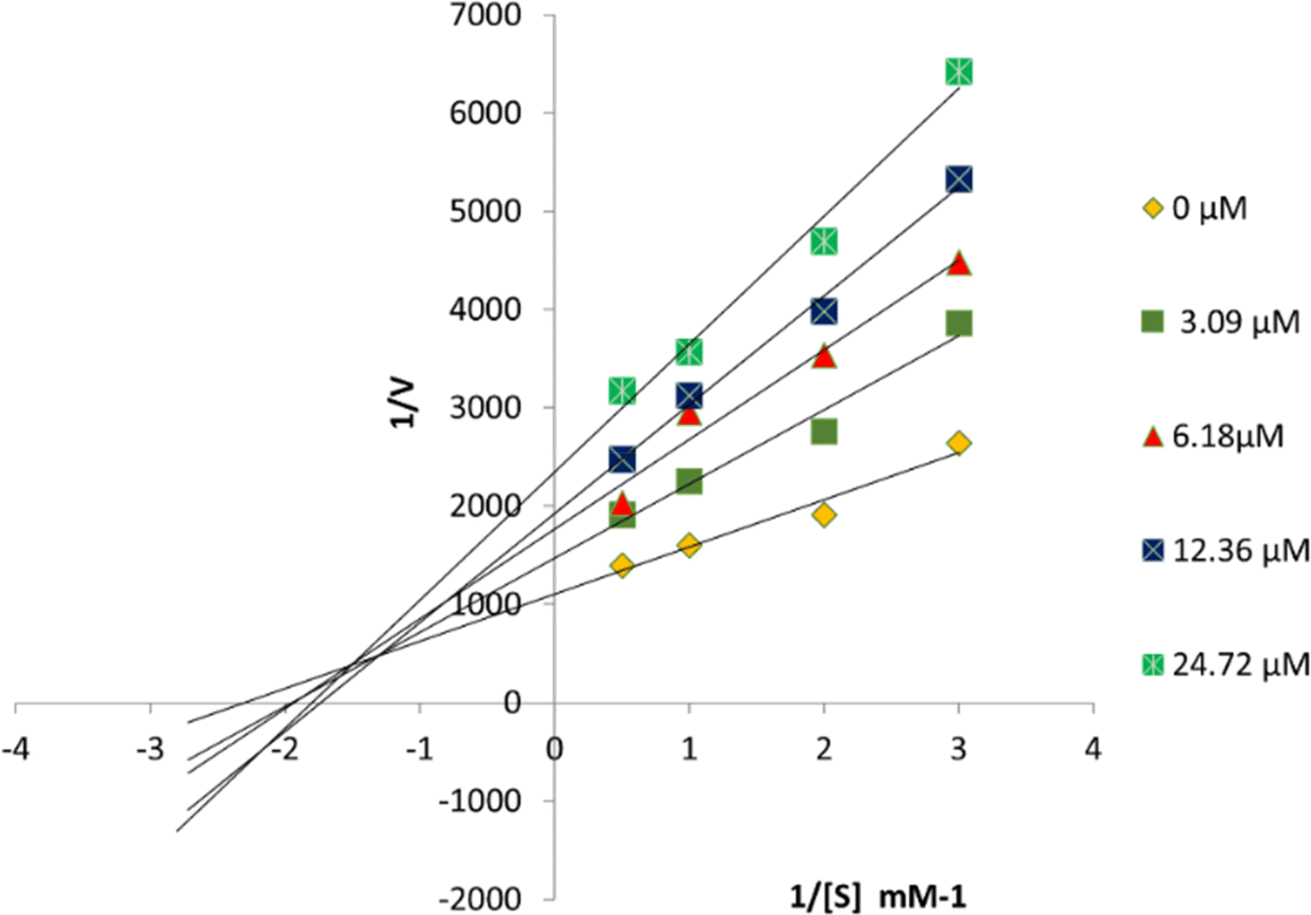 | ||
| Fig. 4 Lineweaver–Burk plots for TYR inhibition were generated in the presence of 6P at concentrations of 0, 3.09, 6.18, 12.36, and 24.72 μM, with substrate L-DOPA concentrations ranging from 0.5 to 3 mM. | ||
 | ||
| Fig. 5 Dixon plots for TYR inhibition were created using 6P at concentrations of 0, 3.09, 6.18, 12.36, and 24.72 μM, with substrate L-DOPA concentrations ranging from 0.5 to 3 mM. | ||
4.4. Molecular docking studies
Molecular docking studies revealed Pro, Asn, Leu, His, Arg, Ala, Glu, Val, Met, Gly and Phe and conserved copper (Cu2+) ions as main binding receptors. The docking energies were little fluctuated within −5.95 to −7.95 (kcal mol−1) (Table 3). The interaction of synthesized compounds (1P–8P) with crystal structure of mushroom tyrosinase (PDB ID 2Y9X) was simulated using molecular docking.| Compound no. | Docking score (kcal mol−1) PDB ID 2Y9X |
|---|---|
| 1P | −5.95 |
| 2P | −6.51 |
| 3P | −6.07 |
| 4P | −7.07 |
| 5P | −6.53 |
| 6P | −7.95 |
| 7P | −7.51 |
| 8P | −6.86 |
| Kojic acid (standard) | −4.99 |
The docked complexes predicted 6P (6,6′-(1,4-phenylene)bis[4-(4-aminophenyl)pyrimidin-2-amine]) binding inside enzymatic pocket of tyrosinase by picking up strong hydrogen bonds and other molecular interactions (Fig. 6). The aniline proton on pyrimidine ring is interacting through H-bond with active site residue Glu322 (2.54 Å) and this phenyl ring is further stabilized by π–π stacking with His244. The –NH2 on pyrimidine is forming another H-bond with Asn260 (2.26 Å). The docked complex predicted other part of 6P forming hydrophobic interactions with side chain residue His285 making it a stable ligand–protein complex. The other part of this ligand is further stabilized by π-interaction with active site residue His263 and is lying closer to Phe192, Glu185, Pro186 and Thr187.
 | ||
| Fig. 6 The docked complex of 6P (6,6′-(1,4-phenylene)bis[4-(4-aminophenyl)pyrimidin-2-amine]) with tyrosinase PDB ID 2Y9X. | ||
The second highest docked score ligand 7P [6,6′-(6,6′-(1,4-phenylene)bis(2-aminopyrimidine-6,4-diyl))bis(2-bromo-4-chlorophenol)] in the current synthesized series is predicted to interact through 2 strong hydrogen bond interactions (2.22 Å, 2.01 Å) with active site residues (Fig. 7). Asn260 and Arg268 are crucial amino acids in the tyrosinase signature and any interaction with these residues leads to stable ligand–protein complex. Moreover, 7P is picking other hydrophobic interactions inside catalytic site. Pyrimidines rings are forming π–π interactions with His244 and Phe264 and further stabilizing the complex through cationic bond. The chlorine of 7P forms a halogen bond with Ala246. All these interactions may provide a basis for development of effective inhibitors/activators of tyrosinase.
 | ||
| Fig. 7 The docked complex of 7P [6,6′-(6,6′-(1,4-phenylene)bis(2-aminopyrimidine-6,4-diyl))bis(2-bromo-4-chlorophenol)] with tyrosinase PDB ID 2Y9X. | ||
4.5. MD simulation
The Root Mean Square Deviation (RMSD) of the carbon alpha atoms of the protein was assessed to investigate the stability of the complex (compound 6P) (Fig. 8a). The RMSD of protein gradually increased to 0.2 nm at nm and then showed a minor deviation of 0.5 nm at 30 ns where it decreased to 0.15 nm. The protein showed deviations in the range of 0.15 to 0.2 nm till 50 ns and then showed stability in the trend as after 50 ns, the average RMSD remained in the range of 0.18 to 0.2 nm. To assess the flexibility of protein residues, root mean square fluctuation (RMSF) was calculated and depicted in Fig. 8b. Higher RMSF values indicate flexible residues, whereas lower values denote rigid residues. In the RMSF plot, residues spanning from 70 to 90, 240 to 250, and 320 to 340 exhibited significant fluctuations due to loop regions, while other residues maintained minimal fluctuation throughout the simulation, indicating rigidity. Similarly, the radius of gyration (Rg) was computed over the simulation period to evaluate the compactness of the protein structure. Elevated Rg values signify unfolding events within the protein during simulation. Fig. 8c illustrates the stability of Rg throughout the simulation, maintaining a consistent range from approximately 2.05 to 2.06 nm, suggesting stability in the protein structure. Furthermore, hydrogen bonding between the protein and ligand 6P was analyzed to assess complex stability. The dynamics of hydrogen bond formation and distortion were observed during the simulation. Fig. 8d displays the fluctuation in the number of hydrogen bonds formed between the protein and ligand over time. Initially, up to 4 hydrogen bonds were formed, gradually stabilizing to an average of 2.5 bonds throughout the simulation period, indicating strong binding and complex stability. | ||
| Fig. 8 The MD trajectory analysis measures the RMSD, RMSF, Rg and hydrogen bonds. (a) The root means square deviation of protein backbone atoms. (b) The flexibility protein residues with bound ligand 6P. (c) Rg analysis to measure the compactness of protein. (d) The number of hydrogen bonds formed between protein and ligand 6P during simulation. | ||
4.6. QSAR studies
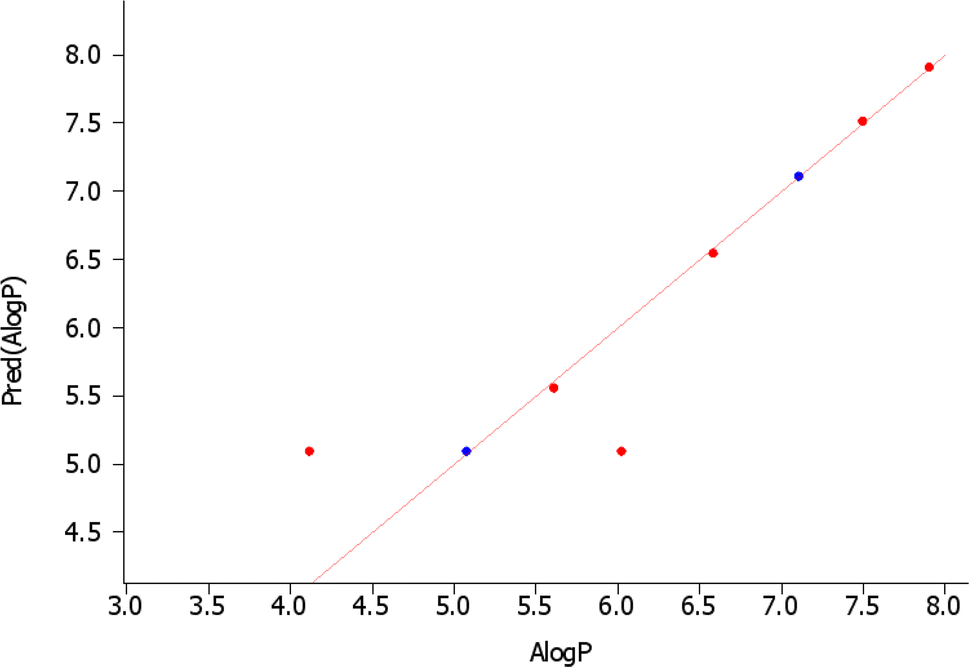
This study explored the structure–activity relationships (SAR) of bispyrimidines for inhibitory effects against TYR using chemical descriptors. A high correlation coefficient (r = 0.9553) was found, indicating strong alignment between observed parameters (Fig. 9). The QSAR model employed log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P (o/w) as the activity field with SlogP as the descriptor. QSAR models, considering hydrogen bonding, hydrophobic interactions, and induced fit patterns, were developed and split into training and test sets, showing a positive correlation between logP values and activities. Partial Least Squares (PLS) regression yielded a Root Mean Square Error (RMSE) of 0.0153426 and an R-squared (R2) value of 0.806308, while Multiple Linear Regression (MLR) confirmed model quality with a satisfactory cross-validated coefficient (q2) and R2.
P (o/w) as the activity field with SlogP as the descriptor. QSAR models, considering hydrogen bonding, hydrophobic interactions, and induced fit patterns, were developed and split into training and test sets, showing a positive correlation between logP values and activities. Partial Least Squares (PLS) regression yielded a Root Mean Square Error (RMSE) of 0.0153426 and an R-squared (R2) value of 0.806308, while Multiple Linear Regression (MLR) confirmed model quality with a satisfactory cross-validated coefficient (q2) and R2.
 | ||
| Fig. 9 QSAR analysis of synthesized bispyrimidines (1P–8P). | ||
5. Structure–activity relationship (SAR) based on IC50 and docking studies of tyrosinase
All synthesized analogues (1P–8P) were evaluated for their in vitro tyrosinase inhibitory activity. The inhibitory activity data, as shown in Table 1, facilitated the creation of an initial SAR model (Fig. 10). This model was developed to examine the impact of substituents on the aryl ring and the influence of various substituent types on the activity profile, providing insights that could be further explored. | ||
| Fig. 10 SAR based TYR inhibition and molecular docking. | ||
Interestingly, among the synthesized compounds, compound 6P demonstrated the most potent TYR inhibitory activity with an IC50 value of 12.36 ± 1.24 μM, which is more effective than the standard inhibitor kojic acid (IC50 = 16.69 ± 2.81 μM). This high activity could be attributed to the presence of amino groups at the para position of the phenyl ring, which may enhance the interaction with the enzyme's active site through hydrogen bonding or other electronic effects.
Other compounds, such as 7P and 8P, also exhibited significant TYR inhibitory activities with IC50 values of 17.19 ± 1.69 μM and 36.56 ± 1.89 μM, respectively. The presence of bulky substituents and halogen atoms in 7P may contribute to its activity by increasing the compound's affinity for the enzyme through hydrophobic interactions or halogen bonding. Compound 7P, which has –OH, –Br and –Cl groups at positions 2, 3, and 5 on both rings of the scaffold, demonstrates a structure that fits well and interacts strongly with the active site of the target enzyme. In case of compound 8P, the results clearly indicate that the hydroxy groups at the 4th position on both rings of the bispyrimidine motif are responsible for the enhanced inhibitory activity.
Conversely, 1P showed the least inhibitory activity with an IC50 value of 86.67 ± 3.08 μM. The lower activity of compound 1P, which lacks substitution, suggests that the unsubstituted bispyrimidine results in reduced inhibitory activity. This indicates that the various substituents enhance the IC50 values more effectively than the NH2 group on the bispyrimidine scaffold. Therefore, the NH2 group on the pyrimidine scaffold does not play a significant role in tyrosinase inhibition.
Furthermore, the other compounds, 2P, 3P, 4P and 5P, displayed moderate TYR inhibitory activities with IC50 values ranging from 31.56 ± 1.62 μM to 45.98 ± 2.05 μM. The variations in their inhibitory activities could be attributed to the different electronic and steric properties of the substituents on the phenyl rings, which affect their binding affinities to the enzyme. The compounds 2P, 3P, 4P and 5P in bispyrimidine series exhibited lower activity against the target enzyme. This indicates that the presence of highly hydrophilic groups (such as –I, –Br, –F, –CH3) on both rings of the motif is responsible for their reduced activity due to diminished interactions with the enzyme.
Henceforth, the SAR analysis suggests that the presence and position of amino groups, halogen atoms, and other substituents play crucial roles in determining the TYR inhibitory activity of these compounds. Compound 6P, with its p-amino phenyl substituents, emerged as the most promising candidate for further development as a TYR inhibitor.
The SAR based on the docking scores of synthesized bispyrimidines with PDB ID 2Y9X is as follows:
Compound 1P, with no substitution, exhibited the lowest docking score of −5.95 kcal mol−1, indicating the least binding affinity. Compounds with substitutions generally showed improved docking scores compared to the unsubstituted compound. For instance, 2P and 3P, with scores of −6.51 and −6.07 kcal mol−1 respectively, displayed better binding affinity than 1P. Compound 4P had a docking score of −7.07 kcal mol−1, suggesting a significant increase in binding affinity due to its specific substitution pattern. Compound 5P’s score of −6.53 kcal mol−1 also indicates improved binding compared to the unsubstituted compound. Moreover, the most notable improvement was seen with compounds 6P and 7P, having scores of −7.95 and −7.51 kcal mol−1 respectively, demonstrating the highest binding affinities. Compound 8P had a docking score of −6.86 kcal mol−1, further confirming that substitutions enhance binding affinity. Comparatively, kojic acid, the standard, had a docking score of −4.99 kcal mol−1, indicating that all substituted bispyrimidines exhibited better binding affinities than kojic acid. It is concluded that the introduction of various substituents on the bispyrimidine scaffold significantly enhances the binding affinity, as reflected by the improved docking scores, with compounds 6P and 7P showing the most potent interactions.
Furthermore, the moderate to good IC50 values and docking scores of halogen-substituted compounds compared to other substituents can be attributed to the combined effects of strong electron withdrawal (especially with fluorine), potential steric hindrance (especially with bromine and iodine), increased hydrophobicity, and less favorable polarizability effects. These factors can lead to suboptimal interactions with the protein binding site, reducing the overall binding affinity and biological activity (Fig. 10).
6. Conclusions
In this study, we have developed, synthesized, and evaluated a series of bis-pyrimidine derivatives (1P–8P) targeting the tyrosinase enzyme. Our findings indicate that these compounds exhibit moderate to good inhibitory activity against tyrosinase, with IC50 values ranging from 12.36 ± 1.24 to 86.67 ± 3.08 μM. All tested compounds (1P–8P) demonstrated significant inhibitory potential against tyrosinase compared to the standard, kojic acid (IC50 = 16.69 ± 2.81 μM). Remarkably, compound 6P emerged as a potent inhibitor with an IC50 value of 12.36 ± 1.24 μM. Furthermore, compounds 2P, 4P, 7P and 8P also showed moderate to good tyrosinase inhibitory activity. Kinetic analysis revealed that compound 6P exhibits mixed-type inhibition against tyrosinase. SAR analysis revealed that the presence and placement of electron-withdrawing and electron-donating substituents exerted a notable influence on inhibitory activity. Findings from molecular docking studies were consistent with those observed in vitro biological assays. Additionally, MD simulations corroborated the stability of the docked structures within the binding sites. QSAR analysis showed a strong correlation between the structures of compounds 1P–8P and their inhibitory activities (IC50), with a correlation coefficient (R2) of 0.806308.Data availability
All the spectroscopic data is provided in ESI† file.Author contributions
Manazza Afzal: experimental work performance; Rabia Mehmood: experimental work performance; Ehsan Ullah Mughal: main idea, final writing the manuscript; Nafeesa Naeem: data analysis and collection, first-draft preparation; Zaman Ashraf: performed enzyme inhibition activity and kinetic analysis; Yasir Nazir: performed molecular docking and QSAR studies; Fatma Mohsen Shalaby: formal analysis; Amal El-Sayed Abd El Hady: formal analysis; Amina Sadiq: supervision.Conflicts of interest
The authors declare that they have no conflicts of interest.Acknowledgements
The authors extend their appreciation to the Deanship of Research and Graduate Studies at King Khalid University for funding this work through Large Research Project under grant number RGP2/401/45. The authors also thank to the Higher Education Commission (HEC), Islamabad, Pakistan for the financial support under the NRPU project (Ref No. 20–15800/NRPU/R&D/HEC/2021).References
- Z. Peng, G. Wang, Q.-H. Zeng, Y. Li, H. Liu, J. J. Wang and Y. Zhao, Crit. Rev. Food Sci. Nutr., 2022, 62, 4053–4094 CrossRef CAS PubMed
.
- R. Pal, G. Teli, G. S. P. Matada and P. S. Dhiwar, J. Mol. Struct., 2023, 136021 CrossRef CAS
- S. Y. Lee, N. Baek and T.-g. Nam, J. Enzyme Inhib. Med. Chem., 2016, 31, 1–13 CrossRef CAS
- R. J. Obaid, E. U. Mughal, N. Naeem, A. Sadiq, R. I. Alsantali, R. S. Jassas, Z. Moussa and S. A. Ahmed, RSC Adv., 2021, 11, 22159–22198 RSC
- J. Li, L. Feng, L. Liu, F. Wang, L. Ouyang, L. Zhang, X. Hu and G. Wang, Eur. J. Med. Chem., 2021, 224, 113744 CrossRef CAS PubMed
- H. Hosseinpoor, S. M. Farid, A. Iraji, M. S. Asgari, N. Edraki, S. Hosseini, A. Jamshidzadeh, B. Larijani, M. Attarroshan and S. Pirhadi, Bioorg. Chem., 2021, 114, 104979 CrossRef CAS PubMed
- T. Pillaiyar, M. Manickam and V. Namasivayam, J. Enzyme Inhib. Med. Chem., 2017, 32, 403–425 CrossRef CAS PubMed
- A. Mermer and S. Demirci, Eur. J. Med. Chem., 2023, 115655 CrossRef CAS
- E. Beltran, M. R. Serafini, I. A. Alves and D. M. Aragón Novoa, Curr. Med. Chem., 2024, 31, 308–335 CrossRef CAS PubMed
- S. Zolghadri, A. Bahrami, M. T. Hassan Khan, J. Munoz-Munoz, F. Garcia-Molina, F. Garcia-Canovas and A. A. Saboury, J. Enzyme Inhib. Med. Chem., 2019, 34, 279–309 CrossRef CAS PubMed
- M. N. Masum, K. Yamauchi and T. Mitsunaga, Rev. Agric. Sci., 2019, 7, 41–58 CrossRef
- S. Vittorio, C. Dank and L. Ielo, Int. J. Mol. Sci., 2023, 24, 9097 CrossRef CAS
- E. U. Mughal, J. Ashraf, E. M. Hussein, Y. Nazir, A. S. Alwuthaynani, N. Naeem, A. Sadiq, R. I. Alsantali and S. A. Ahmed, ACS Omega, 2022, 7, 17444–17461 CrossRef CAS PubMed
- J. Ashraf, E. U. Mughal, R. I. Alsantali, R. J. Obaid, A. Sadiq, N. Naeem, A. Ali, A. Massadaq, Q. Javed and A. Javid, Bioorg. Med. Chem., 2021, 35, 116057 CrossRef CAS
- J. Ashraf, E. U. Mughal, A. Sadiq, M. Bibi, N. Naeem, A. Ali, A. Massadaq, N. Fatima, A. Javid and M. N. Zafar, J. Biomol. Struct. Dyn., 2021, 39, 7107–7122 CrossRef CAS
- N. A. Alshaye, E. U. Mughal, E. B. Elkaeed, Z. Ashraf, S. Kehili, Y. Nazir, N. Naeem, N. Abdul Majeed and A. Sadiq, J. Biomol. Struct. Dyn., 2023, 41, 8307–8322 CrossRef CAS
- R. I. Alsantali, E. U. Mughal, N. Naeem, M. A. Alsharif, A. Sadiq, A. Ali, R. S. Jassas, Q. Javed, A. Javid and S. H. Sumrra, J. Mol. Struct., 2022, 1251, 131933 CrossRef CAS
- M. M. Al-Rooqi, A. Sadiq, R. J. Obaid, Z. Ashraf, Y. Nazir, R. S. Jassas, N. Naeem, M. A. Alsharif, S. W. A. Shah and Z. Moussa, ACS Omega, 2023, 8, 17195–17208 CrossRef CAS PubMed
- M. R. Bhosle, L. D. Khillare, J. R. Mali, A. P. Sarkate, D. K. Lokwani and S. V. Tiwari, New J. Chem., 2018, 42, 18621–18632 RSC
- S. Carradori, F. Melfi, J. Rešetar and R. Şimşek, Metalloenzymes, 2024, 533–546 Search PubMed
- B. Schuster and A. Sammain, JEADV Clinical Practice, 2024 Search PubMed
- R. Pereira, A. M. Silva, D. Ribeiro, V. L. Silva and E. Fernandes, Eur. J. Med. Chem., 2023, 252, 115280 CrossRef CAS
- K. Mezgebe, Y. Melaku and E. Mulugeta, ACS Omega, 2023, 8, 19194–19211 CrossRef CAS
- J.-C. Jung, Y. Lee, D. Min, M. Jung and S. Oh, Molecules, 2017, 22, 1872 CrossRef
- A. Modzelewska, C. Pettit, G. Achanta, N. E. Davidson, P. Huang and S. R. Khan, Bioorg. Med. Chem., 2006, 14, 3491–3495 CrossRef CAS PubMed
- A. M. Kuttithodi, D. Nikhitha, J. Jacob, A. Narayanankutty, M. Mathews, O. J. Olatunji, R. Rajagopal, A. Alfarhan and D. Barcelo, Molecules, 2022, 27, 8209 CrossRef CAS PubMed
- N. A. Elkanzi, H. Hrichi, R. A. Alolayan, W. Derafa, F. M. Zahou and R. B. Bakr, ACS Omega, 2022, 7, 27769–27786 CrossRef CAS PubMed
- B. Ardiansah, J. Appl. Pharm. Sci., 2019, 9, 117–129 CAS
- H. Parveen, F. Hayat, S. Mukhtar, A. Salahuddin, A. Khan, F. Islam and A. Azam, Eur. J. Med. Chem., 2011, 46, 4669–4675 CrossRef CAS
- S. Farooq and Z. Ngaini, J. Heterocycl. Chem., 2021, 58, 1209–1224 CrossRef CAS
- K. Kumar, J. Heterocycl. Chem., 2022, 59, 205–238 CrossRef CAS
- J. Basha and N. M. Goudgaon, J. Mol. Struct., 2021, 1246, 131168 CrossRef
- F. Jubeen, S. Z. Iqbal, N. Shafiq, M. Khan, S. Parveen, M. Iqbal and A. Nazir, Synth. Commun., 2018, 48, 601–625 CrossRef CAS
- M. Devi, S. Jaiswal, S. Jain, N. Kaur and J. Dwivedi, Curr. Org. Synth., 2021, 18, 790–825 CrossRef CAS
- S. S. Mirmortazavi, M. Farvandi, H. Ghafouri, A. Mohammadi and M. Shourian, Drug Des., Dev. Ther., 2019, 2169–2178 CrossRef CAS PubMed
- A. Chiriapkin, I. Kodonidi and D. Pozdnyakov, Iran. J. Pharm. Res., 2022, 21, 124–132 Search PubMed
- M. Debbabi, V. D. Nimbarte, S. Chekir, S. Chortani and A. Romdhane, Bioorg. Chem., 2019, 82, 129–138 CrossRef CAS
- D. Bayramoğlu, G. Kurtay and M. Güllü, Synth. Commun., 2020, 50, 649–658 CrossRef
- S. M. Sondhi, S. Jain, M. Dinodia, R. Shukla and R. Raghubir, Bioorg. Med. Chem., 2007, 15, 3334–3344 CrossRef CAS PubMed
- E. U. Mughal, Q. A. Raja, A. Y. A. Alzahrani, N. Naeem, A. Sadiq and E. Bozkurt, Dyes Pigm., 2023, 220, 111762 CrossRef CAS
- A. R. Guerroudj, E. U. Mughal, N. Naeem, A. Sadiq, J. H. Al-Fahemi, B. H. Asghar, N. Boukabcha, A. Chouaih and S. A. Ahmed, Spectrochim. Acta, Part A, 2024, 124093 CrossRef
- E. U. Mughal, S. Amjid, A. Sadiq, N. Naeem, Y. Nazir, H. Alrafai, A. A. Hassan, S. Y. Al-Nami, A. A. Abdel Hafez and S. W. Ali Shah, J. Biomol. Struct. Dyn., 2024, 42, 244–260 CrossRef CAS
- Z. Ashraf, M. Rafiq, S.-Y. Seo, K. S. Kwon and M. M. Babar, Eur. J. Med. Chem., 2015, 98, 203–211 CrossRef CAS PubMed
- Y. Nazir, H. Rafique, S. Roshan, S. Shamas, Z. Ashraf, M. Rafiq, T. Tahir, Z.-U.-R. Qureshi, A. Aslam and M. H. H. B. Asad, BioMed Res. Int., 2022, 2022, 1040693 Search PubMed
- W. T. Ismaya, H. J. Rozeboom, A. Weijn, J. J. Mes, F. Fusetti, H. J. Wichers and B. W. Dijkstra, Biochemistry, 2011, 50, 5477–5486 CrossRef CAS PubMed
- W. Jorgensen, J. Chandrasekhar, J. Madura, R. Impey and M. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS
- I. Qureshi, S. L. Pan and Y. Zheng, Info. Sys. J., 2021, 31 Search PubMed
- B. Hess, H. Bekker, H. J. Berendsen and J. G. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS
- H. Grubmüller, H. Heller, A. Windemuth and K. Schulten, Mol. Simul., 1991, 6, 121–142 CrossRef
- U. Essmann and L. Perera, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS
- B. Grant and L. Skjærven, The Bio3D packages for structural bioinformatics, Protein Science, 2021, 30, 20–30 CrossRef CAS PubMed
- N. Agrawal and A. A. Skelton, Mol. Pharmaceutics, 2018, 15, 289–299 CrossRef CAS
- A. F. Abbas, Synthesis, 2015, 7, 70–75 Search PubMed
- H. C. Kwong, A. J. Sim, C. Kumar, C. K. Quah, S. Chantrapromma, S. Naveen and I. Warad, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2018, 74, 835–839 CrossRef CAS PubMed
- B. Jiang, F. Han, M.-H. Lü, Z.-P. Wang, W. Liu, Y.-X. Zhang, J. Xu and R.-J. Li, Eur. J. Med. Chem., 2022, 239, 114529 CrossRef CAS
- C. Kumar, C. K. Quah, S. Chandraju, N. Lokanath, S. Naveen and M. Abdoh, IUCrData, 2017, 2, x170238 CrossRef
- S. Kumar, B. Narasimhan, S. M. Lim, K. Ramasamy, V. Mani and S. A. Shah, Mini-Rev. Med. Chem., 2019, 19, 609–621 CAS
- A. A. Al-Khalaf, A. F. Abbas and H. S. Al-Lami, Bas. J. Sci., 2022, 40, 437–464 Search PubMed
- Y. Wang, L. Li, T. Ma, X. Cheng and D. Liu, Anti-Cancer Agents Med. Chem., 2022, 22, 2116–2124 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04652h |
| This journal is © The Royal Society of Chemistry 2024 |