
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInjectable, self-healing and degradable dynamic hydrogels with tunable mechanical properties and stability by thermal-induced micellization†
Chunqing Lina,
Leniu Chena,
Yuan Hea,
Wenlong Xianga,
Yujing Nie*a,
Baixue Cai*b and
Zanru Guo *a
*a
aCollege of Chemistry, Chemical Engineering and Environmental Science, Minnan Normal University, Zhangzhou 363000, PR China. E-mail: guozanru@mnnu.edu.cn; nieyujing@mnnu.edu.cn
bChongqing Academy of Metrology and Quality Inspection, Chongqing 401120, PR China. E-mail: baicai1901@163.com
First published on 20th May 2024
Abstract
Dynamic hydrogels possessing injectable, degradable and self-healing abilities have attracted considerable attention in the biomedical field in recent years, but it is difficult to tune the mechanical properties and stability of conventional dynamic hydrogels. In this work, we synthesized ABA-triblock copolymers via RAFT polymerization, where the A block consisted of thermo-sensitive poly(N-isopropylacrylamide-co-diacetone acrylamide) and the B block was hydrophilic poly(acrylamide). Subsequently, dynamic hydrogels were obtained based on the acylhydrazone bonds between the triblock copolymers and adipic acid dihydrazide (ADH). The obtained hydrogels exhibited injectable and self-healable abilities. In response to the thermal-induced micellization of their temperature-responsive blocks, the mechanical strength of the hydrogels not only increased, but also they exhibited high stability even at pH 2.0. Moreover, the hydrogel in the stable state could be degraded by the fracture of its trithiocarbonate groups. In addition, the hydrogels exhibited good cytocompatibility and controlled release behavior for doxorubicin (DOX). Considering these attractive tunable properties, these dynamic hydrogels show various potential applications in the biomedical field, such as drug carriers and cell or tissue engineering scaffolds.
1. Introduction
Hydrogels, which are typical soft materials filled with water, have attracted considerable attention given that they are similar to natural extracellular matrices,1 enabling their widespread application ranging from drug delivery vehicles,2 tissue engineering,3,4 and soft robotics5 to flexible electronics.6,7 In recent years, to expand the application of hydrogels to minimally invasive injection and autonomously repairing damage, hydrogels with injectable and self-healing abilities have been developed.8 Crosslinking via dynamic covalent bonds (DCBs) is one of the effective approaches to endow hydrogels with the abovementioned properties, given that they combine the reversibility of noncovalent bonds and the stability of covalent bonds.9–12 The controllable dynamic equilibrium of the formation and dissociation of DCBs usually endows hydrogels with sol–gel transition, shear-thinning and self-healing properties.11,13To date, several DCBs such as disulfde bonds, Schiff bases, phenylboronate esters and acylhydrazone,14 have been employed to construct dynamic hydrogels.15,16 Among them, acylhydrazone bonds, resulting from the condensation of hydrazides with carbonyl groups, are particularly promising considering their thermodynamic stability and wide designability of their component structures.17,18 Moreover, acylhydrazones show rapid kinetics in mild acidic environments or in the presence of nucleophilic catalysts and hydrolytic lability, enabling hydrogels to be in situ formed and biodegraded.19 These characteristics show that hydrogels based on acylhydrazone are suitable for application in biological systems.17–19 Deng's group20,21 prepared several dynamic hydrogels containing acylhydrazone and demonstrated their reversible sol–gel transitions and self-healable ability. Chen et al.17 developed self-healing and injectable polysaccharide-based hydrogels crosslinked by imine and acylhydrazone bonds. Zhang et al.18 constructed cellulose-based hydrogels through acylhydrazone linkages. The hydrogels displayed self-healing, injectable and sol–gel transition properties and were used as drug carriers and cell culture scaffolds owing to their biocompatibility.18 Besides the aldehyde groups used for the formation of acylhydrazone bonds, we and other researchers also employed polymers bearing ketone groups to form acylhydrazone-based hydrogels with self-healing ability and switchable sol–gel transition.13,22–26 It was found that the physicochemical properties of the established hydrogels were stable and unchangeable.22–26 Alternatively, their mechanical and stability properties were mainly controlled by varying the density of crosslinks. Generally, the mechanical and stability of the hydrogels were proportional to the number of crosslinks. However, the hydrogels with a low crosslinking density showed uncontrolled swelling and even disintegrated because DCBs are not as strong as covalent bonds.22,24 In contrast, phase separation was observed in the highly crosslinked networks.22 In addition, they could not survive under strong acidic conditions (such as pH 1–3 in the acidic stomach lumen for digestion) due to the breakage of DCBs.20–26 Thus, strategies capable of tuning the mechanical properties and stability of established DCB hydrogels by stimuli are highly attractive.
Thus, to regulate the mechanical properties and stability of dynamic hydrogels, temperature responsiveness was incorporated in acylhydrazone hydrogels.26–28 The temperature stimulus can be easily realized both in vitro and in vivo. Qin et al.25,26 obtained thermo-responsive self-healable hydrogels by crosslinking non-thermoresponsive polymers with acylhydrazone. Hoare's group24,27,28 prepared a series of temperature-responsive acylhydrazone-based hydrogels using poly(N-isopropylacrylamide) (PNIPAM) and poly(oligoethylene glycol methacrylate) (POEGMA). They found that the storage modulus of the hydrogels increased and their degradation slow down when the temperature was above the volume phase transition temperature (VPTT) of their components.27,28 This was be attributed to the fact that DCBs were wrapped by the temperature-responsive polymers.27 However, a phase transition happened throughout the network, resulting in the de-swelling and collapse of the hydrogel network,27 which may have a negative impact in bioapplications. In this case, if temperature-responsive components and DCBs are designed as domains, and then uniformly introduced in the hydrophilic hydrogel network, the crosslinking will be enhanced given that DCBs will be wrapped and “locked” by temperature-responsive blocks. Also, the water released from the temperature-responsive domains can remained in the hydrophilic hydrogel network. Thus, the mechanical properties and stability of dynamic hydrogels may be enhanced, while their volume shrinkage is negligible with an increase in temperature.
The formation of the structure containing domains is similar to ABA triblock copolymers, in which the thermal-responsive A blocks dehydrate and associate into crosslinking domains and the middle B blocks act as network bridges.29,30 Wang et al.29 reported the synthesis of a supramolecular hydrogel via the sol–gel transition of an ABA triblock copolymer containing a terminal poly(N-isopropylacrylamide) (PNIPAM) block and ureido pyrimidinone (UPy) moieties. Upon increasing the temperature, the PNIPAM segments assembled into micelles and provided a hydrophobic microenvironment for UPy dimerization to form cross-linking points. Similarly, Zeng et al.30 used thermal-responsive terminal blocks of ABA triblock copolymer to form hydrophobic interactions, resulting in a sol–gel transition and gelation. It is anticipated that DCB hydrogels are formed by the ABA triblock copolymer, in which the terminal A blocks contain thermal-responsive units and DCBs, and the DCB linkages can be wrapped by dehydrated temperature-responsive blocks. Thus, the mechanical properties and stability of the hydrogels would be enhanced because the DCBs are “locked” by hydrophobic domains. However, dynamic hydrogels based on DCBs with tunable mechanics and stability have not been investigated to date. Besides, degradability is another important property for stable hydrogels,26,31 where it is crucial for stable DCB hydrogels to be integrated with degradability.
Herein, we report the preparation of injectable, self-healing, degradable DCB hydrogels possessing temperature-tunable mechanical properties and stability based on ABA triblock copolymers comprised of a ketone group-functionalized thermosensitive A block and hydrophilic B block, as illustrated in Scheme 1. This triblock copolymer poly[(N-isopropylacrylamide)-co-(diacetone acrylamide)]-b-poly[(acrylamide)-b-poly(diacetone acrylamide)-co-(N-isopropylacrylamide)], denoted as P(NIPAM-co-DAAM)-b-PAM-b-P(DAAM-co-NIPAM), was synthesized via reversible addition–fragmentation chain-transfer (RAFT) polymerization. The hydrogels were generated by mixing the triblock copolymers and adipic dihydrazide (ADH) via the condensation reaction between their ketone groups and hydrazide. Based on the dynamical acylhydrazone crosslinker, the hydrogels displayed a sol–gel transition, injectability and self-healing behaviors. Upon exposure to thermal stimulus, their mechanical properties and stability were enhanced because the DCBs were locked by temperature-induced micelles. Moreover, the hydrogel with enhanced stability could be degraded by the fracture of its trithiocarbonate groups (Scheme 1). In addition, the hydrogels exhibited good cytocompatibility and controlled release behavior for DOX. Considering these attractive characteristics, these dynamic hydrogels may find potential applications in the biomedical field, such as drug carriers and cell or tissue engineering scaffolds.
 | ||
| Scheme 1 Schematic of the fabrication of the sol–gel transition, injectable, self-healable and degradable hydrogels with tunable mechanical properties and stability by ABA triblock copolymers. | ||
2. Experimental
2.1 Materials
Acrylamide (AM, 99.5%), diacetone acrylamide (DAAM, 99%), N-isopropylacrylamide (NIPAM, 99%), and azobisisobutyronitrile (AIBN, 99%) were purchased from Aladdin Co., Ltd and purified by recrystallization. 2-(1-Carboxy-1-methyl-ethylsulfanylthio-carbonylsulfanyl)-2-methylpropionic acid (CMP) used as the chain transfer agent (CTA) was synthesized following a previous report.32 Adipic dihydrazide (ADH, 99%, Aladdin Co., Ltd) was used as received. The pH of the solutions was adjusted using 4 M NaOH and HCl. All other reagents were analytical grade and used as received.2.2 Characterization
1H NMR spectra were recorded on a Bruker 300 MHz spectrometer at room temperature using CDCl3 or DMSO-d6 as the solvent.Fourier transform infrared (FT-IR) spectra were recorded on a Nicolet MX-1E FTIR (USA) spectrophotometer. The dried samples were compressed into films with KBr.
A gel permeation chromatography (GPC) system (Waters 1515-717-2414) equipped with a refractive index detector was used to determine the molecular weight and molecular weight distribution of the polymers. CHCl3 and DMSO were used as the eluent (flow rate of 1.0 mL min−1) for macro-CTA and triblock copolymers at 30 °C, respectively. Mono-dispersed polystyrene was used as the standard to obtain the calibration curve.
Dynamic light scattering (DLS) measurements were performed on a Nano ZS90 Zetasizer (Malvern Instruments Ltd, Malvern, U.K.) equipped with an He–Ne laser (633 nm, 4 mW) at a detection angle of 90°.
SEM observation was performed on a JSM-6510 (JEOL, Japan). The hydrogel samples were frozen by immersing them into liquid nitrogen, and then lyophilized.
Rheological characterization was characterized on a Physica MCR 302 (Anton Paar, Austria) rheometer equipped with a parallel plate with a diameter of 50 mm. The experiments were measured in stress or strain-controlled mode. The temperature was increased at a rate of 1.0 °C min−1 during the modulus change upon increasing temperature.
Compression strain–stress tests were performed using a CMT4104 universal testing machine fitted with a 100 N load cell. The cylindrical hydrogels (18 mm in diameter and 14 mm in height) were tested by compression at a rate of 5 mm min−1.
2.3 Synthesis of macro-CTAs
Three macro-CTAs were synthesized though RAFT polymerization by varying the feeding ratio of NIPAM and DAAM (Scheme S1 and Table S1†), and their targeted degree of polymerization was 200. The procedure for the preparation of macro-CTA1 is as follows: NIPAM (5.0 g, 44.2 mmol), DAAM (0.831 g, 4.91 mmol), CMP (0.069 g, 0.246 mmol), AIBN (0.008 g, 0.0492 mmol) and 1,4-dioxane (16 mL) were added to a flask. The mixture solution was bubbled with nitrogen for 30 min to remove oxygen. Subsequently, the flask was submerged in water bath at 70 °C for 24 h under magnetic stirring. After that, the reaction mixture was precipitated into excess ice-cold n-hexane. The product was reprecipitated in excess ice-cold n-hexane. The resultant solid was dried by freeze-drying. Yield: 5.782 g (98%). 1H NMR (300 MHz, CDCl3), ppm: 0.9–1.2 ppm (CH3 in NIPAM units), 1.2–1.8 ppm (CH2 in main chains, (CH3)2C in DAAM units), 1.8–2.4 ppm (CH in main chains and CH3CO in DAAM units), 2.6–3.2 ppm (CH2 in DAAM units), 3.7–4.0 ppm (CH in NIPAM units).2.4 Synthesis of triblock copolymers P(NIPAM-co-DAAM)-b-PAM-b-P(NIPAM-co-DAAM)
ABA triblock copolymers were prepared via the polymerization of AM based on RAFT, and the targeted degree of polymerization of the AM block was 800. In a typical experiment, triblock copolymer P2 (Table 1, entry P2) was synthesized as follows: AM (7.1 g, 0.1 mol), macro-CTA1 (2.932 g, 0.125 mmol), AIBN (4.1 mg, 0.025 mmol), and DMSO (27.3 mL) were mixed in a 100 mL bottomed flask. The reaction mixture was purged with nitrogen for 30 min. Subsequently, the flask was immersed in water bath at 70 °C for 24 h under magnetic stirring. The product was precipitated from excess ether three times and dried under vacuum. P4 and P6 were prepared via a similar procedure under the initiation of macro-CTA2 and macro-CTA3, respectively. 1H NMR (DMSO-d6, ppm): 0.9–1.2 ppm (CH3 in NIPAM units), 1.2–1.8 ppm (CH2 in main chains, (CH3)2C in DAAM units), 1.8–2.4 ppm (CH in main chains and CH3CO in DAAM units), 2.6–3.2 ppm (CH2 in DAAM units), 3.7–4.0 ppm (CH in NIPAM units).| Copolymer | Preset composition (mol%) | Final compositiona (mol%) | Yield (%) | Mn, theob (g mol−1) | Mn, calc (g mol−1) | Mn,GPCd (g mol−1) | PDId | Cloudy point (°C) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NIPAM | AM | DAAM | NIPAM | AM | DAAM | |||||||
| a Calculated from 1H NMR spectra.b Calculated from polymerization stoichiometry.c Evaluated by monomer conversion.d Determined by GPC. | ||||||||||||
| P2 | 18.0 | 80.0 | 2.0 | 17.9 | 78.7 | 3.4 | 98% | 79![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 810 810 |
78674 |
63886 |
1.19 | 33.2 |
| P4 | 16.0 | 80.0 | 4.0 | 14.7 | 80.7 | 4.6 | 96% | 81772 |
79500 |
68820 |
1.20 | 32.0 |
| P6 | 14.0 | 80.0 | 6.0 | 13.6 | 79.1 | 7.3 | 96% | 81820 |
79548 |
76029 |
1.33 | 31.3 |
2.5 Fabrication of the hydrogels
The hydrogels were prepared by simply mixing the polymer solutions and adipic dihydrazide (0.5 equiv. to ketone groups of polymers) with 20 wt% solid content. The pH of the mixture solution was adjusted to 6.0 using 4 M HCl and 4 M NaOH. The viscous solution became a hydrogel after ca. 1 h at room temperature. The hydrogel was kept at room temperature for 24 h before testing. The obtained hydrogel was labeled as Hx (m represents the mole fraction of PDAAM in the polymer), i.e., H2, H4 and H6.2.6 Self-healing and injectable properties of the hydrogel
To thoroughly investigate the self-healing property of hydrogel H4, two disk-shaped hydrogels, one stained with rose red and the other stained with methyl blue for clarity, were cut into two halves. Thereafter, the two pieces of hydrogels were combined for 12 h at room temperature. The healing efficiency was estimated by measuring the compression strength of the original hydrogel and the self-healed hydrogel. The healing efficiency (HE) is defined as follows:
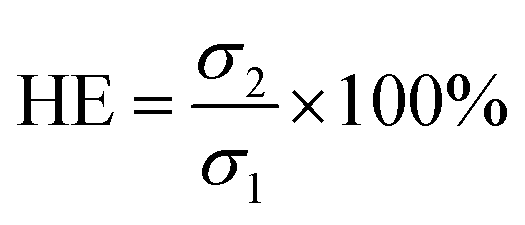 | (1) |
The injectability experiment of the hydrogel was implemented by extruding H4 from a syringe with a 20-G needle.
2.7 The stability of hydrogels after thermal responsiveness
The hydrogels (about 6 g) were prepared in a reagent bottle and stored at 37 °C. Their mass changes were recorded at predetermined times. The water loss rate was calculated using the following formula:
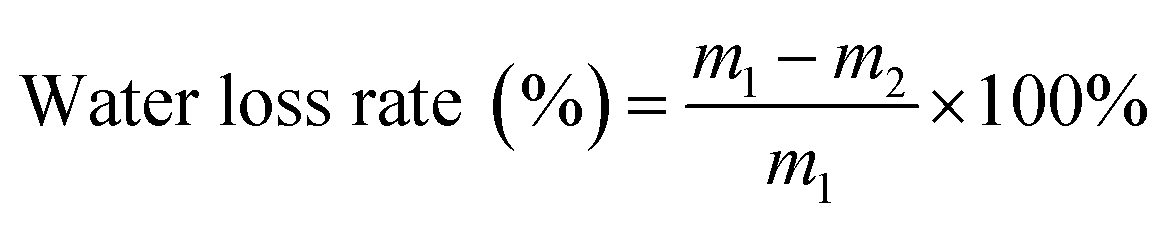 | (2) |
For the evaluation of the stability of the hydrogels at different pH and temperature, the hydrogels (about 1.2 g) were firstly placed at 5 °C or 37 °C for 5 min, and then were placed in 20 mL PBS buffer solution with different pH (2.0, 5.0, 7.4), where the PBS buffer solution was kept at the corresponding temperature (5 °C or 37 °C). At predetermined intervals, the hydrogels were removed from the PBS buffer solution and their weights measured. Weight changing (%) was calculated using the following formula:
 | (3) |
2.8 Cytocompatibility of hydrogels and controlled drug release from them
The procedure for the cytocompatibility evaluation was similar to our previous report.13 Maceration extracts were prepared by incubating the sterilized hydrogels with cell culture medium, and the ratio of hydrogel to medium concentration was 5.0 mg mL−1 at 37 °C for 24 h. Also, two other ratios (5.0 mg mL−1 and 10.0 mg mL−1) were chosen to test the cytotoxicity of the different concentrations.DOX was used as the model drug for controlled release from the hydrogels. To prepare the DOX-loaded hydrogels, 5.0 mg of DOX·HCl was added to 10 mL mixture solution of hydrogel. When DOX·HCl dissolved completely, the pH of the solution was adjusted to 6.0 and kept for 24 h at room temperature to obtain the DOX-loaded hydrogel. For the release of DOX, a cubic hydrogel (1.0 g) was picked up from the hydrogel and immersed in 50 mL PBS medium at different temperature (5.0 °C and 37 °C) and pH (2.0 and 7.4). The amount of released DOX at different times was determined by the UV absorbance of DOX at 485 nm.
3. Results and discussion
3.1 Synthesis of P(NIPAM-co-DAAM)-b-PAM-b-P(DAAM-co-NIPAM) through RAFT polymerization
RAFT polymerization was employed to prepare the ABA triblock copolymers given that it is one of the most powerful techniques for the preparation of block copolymer architectures.33,34 To obtain the ABA triblock copolymers and endow the hydrogel networks with degradability after enhancing their stability, CMP was used as the CTA because the polymer grows along both sides of the central degradable trithiocarbonate group. As shown in Scheme 1, NIPAM and DAAM were firstly copolymerized to form difunctional macro-CTAs, which are also the thermal-sensitive A block-containing ketone groups in the final triblock copolymer. To examine the effect of crosslinking degree on the properties of the hydrogels, macro-CTAs with different feed ratios of DAAM were synthesized. The macro-CTAs were characterized by 1H NMR spectroscopy (Fig. S1†) and their final composition is listed in Table S1.† To insert the hydrophilic B block into the copolymers, AM was polymerized based on macro-CTAs, which resulted in the formation of the P(NIPAM-co-DAAM)-b-PAM-b-P(DAAM-co-NIPAM) triblock copolymers. The composition of the copolymer was characterized by 1H NMR spectroscopy. The protons assigned to the three units can be seen in the 1H NMR spectra (Fig. 1a). Based on the integration of the intensity ratio of the protons in the corresponding unit, the molar ratio of AM, DAAM and NIPAM units was calculated using formula (4), as follows:|
CAM:CDAAM:CNIPAM = 2(A2, 7 − 2A6 − A3):A6:2A3
| (4) |
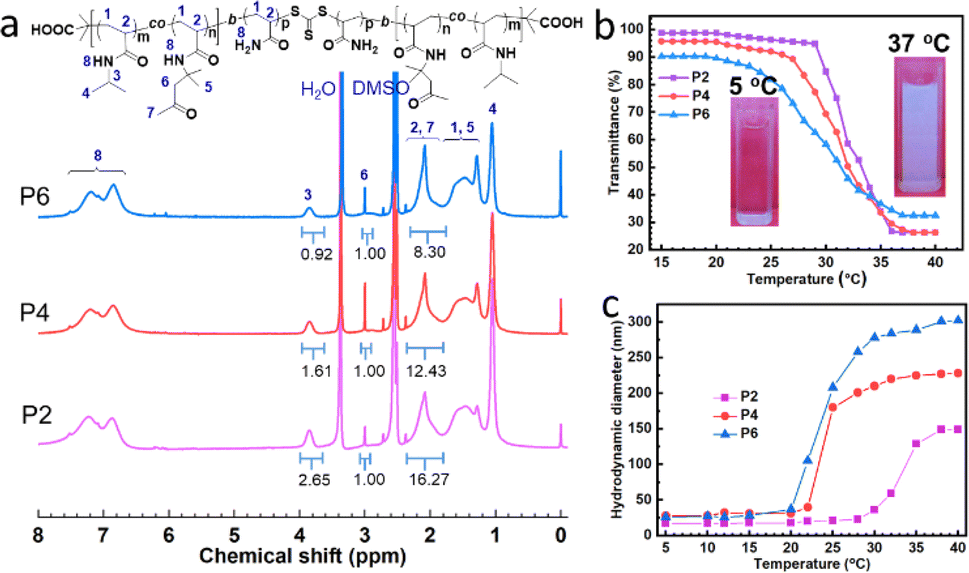 | ||
| Fig. 1 (a) 1H NMR spectra of copolymers. (b) Dependence of transmittance at 500 nm on temperature for aqueous solutions of copolymers. Inset is a photograph of P4 at 5 °C and 37 °C, respectively. (c) Variation in copolymer hydrodynamic diameter from DLS with increased temperature. | ||
3.2 The temperature responsiveness and micellization of the P(NIPAM-co-DAAM)-b-PAM-b-P(DAAM-co-NIPAM) triblock copolymers
(1) To endow the triblock copolymers with temperature responsiveness on, NIPAM units were incorporated into the A block P(DAAM-co-NIPAM), given that PNIPAM is a popular thermoresponsive polymer.27,29,35 To confirm whether the triblock copolymers retained their LCST behavior their solutions were observed at different temperatures, where the aqueous solution of P4 was transparent at 5 °C (Fig. 1b, inset images), while it transformed to milky white at 37 °C, indicating that the polymers showed obvious thermal responsiveness. The cloudy point of the triblock copolymer was determined by the variation in transmittance with temperature. Fig. 1b shows that the transmittance of the three copolymer solutions exhibited a sharp transition when the temperature was increased to ca. 30 °C, reflecting the dehydration of the polymers.35 The cloudy points (defined as the temperature at the midpoint of the decreasing transmittance curve) of P2, P4, and P6 were 33.2 °C, 32.0 °C and 31.3 °C, respectively. Thus, it can be concluded that the cloudy point of the triblock copolymers decreased with an increase in the number of DAAM units. This should be attribute to the hydrophobic DAAM units, which increased the hydrophobic attractive of the copolymers.27,29,35 When the A block of the copolymer exhibited thermal responsiveness and dehydration, the triblock copolymers transformed from hydrophilic to amphiphilic in aqueous solution, and self-assembled into micelles.29,30 Thus, to monitor the variation in the hydrodynamic diameter of the copolymers, dynamic light-scattering (DLS) measurements were carried out. As shown in Fig. 1c, the average diameters of the micelles of P2, P4 and P6 were 7 nm, 28 nm and 26 nm at 5 °C, respectively, which are consistent with the observation in a previous report.29 When the temperature increased to over 20 °C, their hydrodynamic diameters increased. Also, with an increase in the content of DAAM units, the micelle size increased, while the temperature response tended to decrease. The average diameters of the micelles formed by P2, P4 and P6 were 149 nm, 228 nm and 302 nm at 40 °C, respectively. These results suggest that the triblock copolymers could self-assemble into micelles by their dehydrated A blocks, and the micellization could be tuned by the number of DAAM units.3.3 Preparation of hydrogels from triblock copolymers
The dynamic acylhydrazone can be generated between ketone groups and hydrazide, which provides a crosslinker for dynamic hydrogels.20–26 Thus, the P(NIPAM-co-DAAM)-b-PAM-b-P(DAAM-co-NIPAM) triblock copolymers containing ketone groups were mixed with ADH, resulting in the formation of dynamic DCB hydrogels. As illustrated in Fig. 2a, the mixtures of copolymers and ADH changed from flowing solutions to gels and supported their weight when the pH of the solutions was adjusted to ca. 6 and maintained at the temperature of 5 °C for ca. 6 h. Furthermore, with an increase in the number of DAAM units, the hydrogel became transparent to slightly opaque, such as H6. This phenomenon is in good agreement with our previous observation.22 It should be ascribed to the higher crosslinking density, which caused the formation of a thick network skeleton. Thus, to confirm this, SEM was used to observe the microstructure of the hydrogels. As shown in Fig. 2c, freeze-dried H2 possessed uniform interconnected porous structures. However, with an increase in the number of DAAM units, its pore size increased and its skeleton became thicker (Fig. 2d and e). This can be explained by the fact that the increase in the number of DAAM units in the copolymers led to the formation of a larger crosslinking domain, resulting in a thicker skeleton and looser structure. This variation in the microstructure of the hydrogels is similar to that observed in our previous reports.22 However, macroscopic phase separation was not observed in H6. | ||
| Fig. 2 (a) Images of hydrogels from triblock copolymer and ADH with 20% gels. (b) FT-IR spectra of DAAM, triblock copolymers and corresponding hydrogels. SEM images of H2 (c), H4 (d) and H6 (e) hydrogels. | ||
To demonstrate the formation of the hydrogels by dynamic acylhydrazone bonds, the precursors and their hydrogels were analyzed by infrared spectroscopy (Fig. 2b). The triblock copolymers exhibited an obvious stretching band for ketone (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) at 1710 cm−1, which is consistent with the characteristic absorption band of DAAM. Moreover, the strength of the band for ketone increased from P2 to P6 due to the increase in the number of DAAM units. In contrast, the absorption from the ketone vibration (CO) of the copolymers at 1710 cm−1 fully disappeared in the spectrum of the dried hydrogels, while a new peak at 1662 cm−1 ascribed to CN from acylhydrazone bonds appeared in their curves, manifesting the reaction between the copolymers and ADH cross-linker.17,22
O) at 1710 cm−1, which is consistent with the characteristic absorption band of DAAM. Moreover, the strength of the band for ketone increased from P2 to P6 due to the increase in the number of DAAM units. In contrast, the absorption from the ketone vibration (CO) of the copolymers at 1710 cm−1 fully disappeared in the spectrum of the dried hydrogels, while a new peak at 1662 cm−1 ascribed to CN from acylhydrazone bonds appeared in their curves, manifesting the reaction between the copolymers and ADH cross-linker.17,22
Dynamic rheology measurements were performed to further confirm the formation of the hydrogels at 5 °C. As shown in Fig. 3a, oscillatory frequency sweep experiments revealed that the storage modulus (G′) was consistently higher than the loss modulus (G′′) in the tested frequency range, indicating that the hydrogels exhibited elastic solid characteristic.17,18 The G′ of the hydrogels increased with an increase in the number of DAAM units, which implies that the mechanical properties of the hydrogels increased with the increase in DAAM units.18 This should be attributed the denser crosslinking density because the greater the number of ketone carbonyl groups in the triblock copolymer, the greater the amount of crosslinking formed in the hydrogels. In the case of H2, G′′ was close to G′, suggesting the easy deformation of H2 due to its low crosslinking. In addition, the obtained hydrogels could withstand certain strain, which was measured by strain amplitude sweeps at a fixed frequency of 1 Hz at 5 °C. As illustrated in Fig. 3b, the G′ of the hydrogels was greater than G′′ at low strain. When the strain was greater than a critical value, G′′ surpassed G′, indicating that the gel network was disrupted and transferred to a sol state.17,18 The critical strain value for H2, H4 and H6 was 1900%, 956% and 304%, respectively. Although the highly crosslinked hydrogels (such as H6) crosslinked by acylhydrazone bonds have higher mechanical strength, they tended to brittle and could not withstand large strain; in contrast, the hydrogels with less cross-linkers (such as H2) were soft and could tolerate deformation. Hence, the results certified that the mechanical properties of the hydrogels could be tuned by varying the DAAM content.
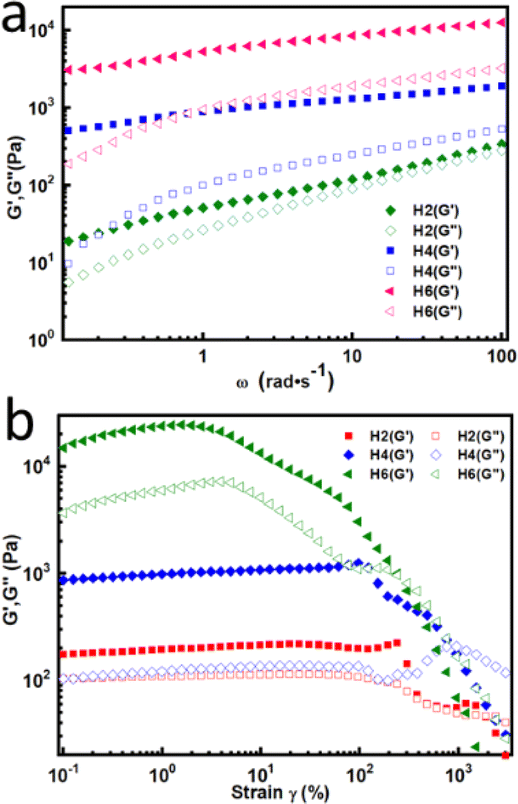 | ||
| Fig. 3 (a) G′ and G′′ of the hydrogels from oscillatory frequency sweep. (b) Strain sweep measurements of the hydrogels at 5 °C. | ||
3.4 Injectable and self-healing properties of the hydrogel
Hydrogels with injectable ability may have many advantages for biomedical applications, which can provide surgical operation with minimally invasive methods.27,36 Thus, to realize injectability, the gel viscosity should be thinned at high shear rates. Accordingly, viscosity measurements were performed for the hydrogels. As shown in Fig. 4a, the viscosity of H4 at 5 °C decreased with an increase in shear rate, indicating its apparent shear-thinning behavior.17,36 This was caused by the disruption of the acylhydrazone crosslinkers in the hydrogel network by shear. It was visualized that the gel could be extruded through a 20 G needle (Fig. 4a, inset). Also, the gel could be injected to form desired shapes (Fig. 4b and c). The direct use of injectable hydrogels replaces prepolymer-formed hydrogels via in situ gelation after injection, which may avoid the diffusion of precursors due to the flow of body fluids.27,28 | ||
| Fig. 4 (a) Viscosity and shear-thinning behavior of H4. The inset image is the gel injected with a conventional syringe using a 20 G needle. (b) and (c) Two different color H4 samples (one stained with rose red and the other stained with methyl blue) injected into a beaker from needles. | ||
Injectable hydrogels with self-healing property are crucial for practical application, given that the hydrogels should undergo structural recovery after removing the applied stress.36 Firstly, the self-healing ability of the hydrogels was visually examined. As illustrated in Fig. 5a, two hydrogel disks (one stained with rose red and the other stained with methyl blue) were cut into half, respectively. Then, the two different colored semicircle gels were placed in intimate contact along the cut line for 12 h without any external intervention. It was found that they completely healed into an integral hydrogel disk, and the resultant hydrogel maintained its integrity even upon exposure to tensile force. Furthermore, optical microscopy was employed to observe the self-healing process of the contacting surfaces (Fig. 5b). The gap between the two semicircles became indistinct at 6 h, and completely disappeared after 12 h. This should be attributed to the dynamic fracture and regeneration of the acylhydrazone bonds.22,26
 | ||
| Fig. 5 (a) Photographs of the self-healing process of hydrogel H4 at 5 °C for 12 h. One hydrogel stained with rose red and the other stained with methyl blue. (b) Optical microscopy images of the contacting surfaces during the self-healing process. (c) Step-strain measurements of H4 at 5 °C. (d) Compressive stress–strain curves of initial and healed hydrogels. The inset imagines show the process for the preparation of the healed hydrogel. | ||
Subsequently, step-strain measurements were performed to assess the self-healing behaviors of H4 (Fig. 5c). As mentioned above, the critical strain value (the G′ curve intersects with that of G′′) of H4 was 956%. When the applied shear strain was stepped from 1% to 900% and held for 100 s, G′′ approached G′, while they immediately recovered their initial values after the strain returned to 1%. With an increase in the strain to 1300%, the G′ dropped from about 900 to 100 Pa immediately and became lower than G′′, indicating that the gel network was disrupted, and subsequently shifted into a liquidlike state. However, G′ and G′′ were almost fully restored when the strain returned to 1%. Similarly, when the hydrogel alternately suffered from a large strain (1800%) and small strain (1%), G′ also quickly recovered to its initial value. This reversible recovery behaviour for the modulus implies the reconstruction of the hydrogel network, indicating the excellent self-healing performance of the hydrogel.17,36 In addition, compression tests were conducted to evaluate their self-healing efficiency (Fig. 5d). The compression strength of the original hydrogel was ca. 40.05 kPa. In contrast, the compression strength of the self-healed hydrogel from the gel fragments was 32.87 kPa. The healing efficiency was calculated to be 82%, indicating that the hydrogel could recover without considerable loss of its mechanical properties.
3.5 Thermal-induced reinforcement for hydrogels
Given that the above-illustrated A block in the copolymers showed thermal responsiveness, we investigated whether the properties of their hydrogels could be tuned by temperature. As shown in Fig. 6a, the hydrogels gradually turned white when the temperature increased from 5 °C to 37 °C. This phenomenon can be explained by the fact that the A block in the network dehydrated and collapsed to form hydrophobic microdomains,29,30 causing a decrease in the light transmission. This agrees well with the DLS results, where the triblock copolymers could form micelles upon heating. After the hydrogels were cooled to 5 °C, they returned to their original state, given that the PNIPAM chains stretched in the hydrogel network. | ||
| Fig. 6 (a) Images of temperature responsiveness of hydrogels. (b) Variation in modulus upon with an increase in temperature. (c) Compression stress–strain curves and (d) corresponding compressive stress of hydrogels before and after temperature responsiveness at 70% strain. (e) Water loss rate curves of the three hydrogel samples at 40 °C. | ||
To reveal the effect of temperature responsiveness on the properties of the hydrogels, dynamic rheological measurements were performed with an increase in temperature from 5 °C to 40 °C. As depicted in Fig. 6b, the value of G′ was invariably greater than that of G′′, and the G′ and G′′ of the three hydrogels presented a plateau at low temperature. As the temperature increased, their modulus increased dramatically at around 20 °C to 35 °C, suggesting that the mechanical properties of the hydrogels were enhanced. The responsive temperature range was consistent with that of the variation in the DLS hydrodynamic diameters. Meanwhile, the compression strength was compared evaluated both before and after heating. As illustrated in Fig. 6c and d, the compression strength of the H2, H4 and H6 hydrogels was 19.3 kPa, 32.99 kPa and 52.83 kPa at 5 °C, respectively, i.e., their compression strength increased with an increase in DAAM content, which is consistent with the above-mentioned rheological results, due to the increase in the crosslinking density. However, the strength of H2, H4 and H6 increased to 19.3 kPa, 32.99 kPa, and 52.83 kPa at 37 °C, respectively, indicating that their strength was enhanced more than once by thermal responsiveness. This may be explained by two reasons, i.e., on the one hand, the dehydration of the thermo-responsive A blocks in the network of the triblock copolymers caused micellization, resulting in the formation of hydrophobic microdomains,29,30 which then locked the dissociation of the acylhydrazone bonds due to the lower water accessibility for hydrolysis.27 On the other hand, the micelles in the network enhanced their mechanical properties via the energy dissipation mechanism.37,38
Generally, hydrogels formed by thermal-responsive random copolymers show obvious de-swelling upon temperature stimulus due to the collapse of their polymer chain.24,27 Previous reports showed that the water loss rate was up to 80% for hydrogels based on random copolymers of AM and DAAM and NIPAM units.39 In contrast, our hydrogels kept their shapes and did not exhibit de-swelling and macroscopic phase separation after heating. As shown in Fig. 6e, the hydrogels (H2, H4 and H6) were kept at 37 °C for 24 h, and their water loss rate was less than 1%. Although the block with PNIPAM units shrank and dehydrated, the hydrophilic PAM blocks could trap the water and maintain the 3D network structure. The microstructure of the hydrogels at 37 °C was observed by SEM (Fig. S4†). It was found that the network structure of the hydrogels was similar to that of at 5 °C, indicating their resistance to temperature changes.
3.6 The stability for hydrogels upon thermal responsiveness
The stability of hydrogels in different environments is an important consideration for their bioapplication,40 given that the microenvironments of organs possess a characteristic pH level.41 The swelling or degradation kinetics were compared at different pH (2.0, 5.0 and 7.4) both before and after thermal responsiveness (Fig. 7). The chosen pH values were used to simulate the conditions in the acidic stomach lumen, duodenum, and ileum,40 respectively. As shown in Fig. 7a–c, at 5 °C, the hydrogels gradually swelled and still remained stable at pH 7.4, and their weight change increased with a decrease in crosslinking degree. Generally, dynamic hydrogels with a low crosslinker degree would degrade by swelling,17,22,27,28 although the acylhydrazone bonds are kinetically “locked” under neutral conditions.17,22,27,28 This indicates that the stability of the hydrogels was improved by the triblock copolymers, which may be related to the enhancement in their linkages due to the physical wrapping of the acylhydrazone bonds within the NIPAM units (Scheme 1)29,30 and the formation of hydrogen bonds between the acylhydrazone bonds and NIPAM units.42 When the hydrogels were placed in pH 5.0 PBS (Fig. 7b), they dramatically swelled first, and then degraded due to the rapid breaking and reforming of their acylhydrazone linkages under the mild acidic conditions.17,18 However, the degradation became slower than that with a higher crosslinking degree.22 When the hydrogels were incubated in pH 2.0 solution (Fig. 7c), their degradation occurred faster, which is consistent with the degradation of hydrazones under strong acidic conditions.22,24,27 | ||
| Fig. 7 Stability of the hydrogels at different pH and temperatures. | ||
In contrast, the stability of these hydrogels was evaluated at 37 °C (physiological temperature), in which they suffered from thermal responsiveness. At pH 7.4, the variation in weight change was similar with that of at 5 °C, but the swelling decreased (Fig. 7d). This should be attributed to the hydrophobic domains from the dehydrated A blocks, which tightened the gel networks and their reduced swelling.29 Compared with the degradation of the hydrogels in pH 5.0 PBS at 5 °C, H4 and H6 just swelled and did not degrade at pH 5 (Fig. 7e). In contrast, H2 was degraded, but its degradation time was 12 times longer than that at 5 °C. These results suggest that the stability of the hydrogels was enhanced upon introducing thermal responsiveness, which results from the hydrophobic microdomains locking the dissociation reaction of the acylhydrazone bonds due to the lower water accessibility for hydrolysis.27 In the case of H2, its crosslinking degree was too low to maintain its network structure. When the pH decreased to 2.0, the three hydrogels also swelled firstly, and degradation occurred on H2 and H6 after a longer survival time than that at 5 °C, while H4 remained stable and only had a slight degradation after swelling for 30 days (Fig. 7f). Despite the instability of the acylhydrazone bonds under strong acidic conditions,17,18,22 the linkages were “locked” by the collapse of the PNIPAM chains when the temperature was higher than the cloud point (Scheme 1), which improved their stability. Hydrogel H2 could not resist swelling, which may be ascribed to the fact that its hydrophobic domains were not strong enough due to its low number of acylhydrazone cross-link points. By comparison, the acylhydrazone bonds hydrogel H6 tended to dissociate, which can be attributed to the fact that the acylhydrazone linkages could not be protected by the domains owing to the decrease in the number of NIPAM units. These results suggest that H4 has the optimal stability even in strong acidic conditions, and thus can be applied in the stomach environment.
3.7 Degradation of the hydrogel after responsiveness
As demonstrated above, the hydrogels (such as H4) exhibited injectability, self-healing ability, and became very stable at 37 °C. In this case, a stable hydrogel with another degradation mechanism is an important factor for its application. The triblock copolymers were prepared by RAFT polymerization, and the trithiocarbonate group was located in the middle of the polymer chains. The trithiocarbonate group could be cleaved by a nucleophilic reagent, such as ethanolamine,43 which may induce the degradation of the hydrogels. Thus, to examine their degradation, the H4 samples were immersed in a solution of pH 7.4 (Fig. S5†), and ethanolamine (10 equiv. of trithiocarbonate groups) was added to one of them. It was observed that the hydrogel degraded in the presence of ethanolamine after 2 h, while the other was just slight swollen in the absence of ethanolamine. These results confirmed that the cleavage of the trithiocarbonate groups enabled the degradation of the hydrogel, as shown in Scheme 1.3.8 Cytocompatibility and controlled drug release from hydrogels
Biocompatibility is an essential factor for hydrogels used in biomedical application.17,18 Here, the biocompatibility of the hydrogels was first evaluated in L929 fibroblast cells using the MTT assay. The cells were incubated in macerating solutions of hydrogels for 24, 48 and 72 h, and their relative cell viability was measured, which was higher than 95% (Fig. 8a). In the case of the MTT assay on the maceration extract of different mass ratios of H4, the relative viability was still greater than 95% at 24, 48 and 72 h (Fig. 8b). To further test the cell compatibility, L929 cells were cultured on H4. The results of the live/dead staining of the cells indicated that no significant red fluorescence (dead cells) was observed and the rounded cells could adhere to the hydrogel and proliferate with an increase in the culture time (Fig. 8c–e). In addition, predominantly viable cells (greater than 95%) were apparently observed after incubation for 12, 24, and 48 h (Fig. 8c–e). These results indicate that the hydrogels have non-cytotoxicity against L929 cells and can serve as a platform for cell carriers, implying their potential application in the biomedical field. | ||
| Fig. 8 MTT assays for cell viability of L929 cells incubated with maceration extracts from different hydrogels (a) and different mass ratios (b) of H4 to medium at 37 °C. (c)–(e) Live/dead staining of L929 cells seeded on H4 for 24 h, 48 h and 72 h, respectively (viable cells: green and dead cells: red). (f) Release curves of DOX from H4 in different buffer solutions and temperatures. | ||
In view of the above-mentioned multiple properties, the hydrogel was applied for controllable drug release. Fig. 8f shows the release profiles of the model drug DOX from hydrogel H4 under various conditions. At the physiological temperature of 37 °C, the hydrogel exhibited controllable release behaviour for DOX, even at a pH close to that of the stomach environment, which is attributed to the stability of the hydrogel and the diffusion of DOX from its gel network. The release rate of DOX was slightly faster at pH 2.0 than that at pH 7.4. This may be explained by the fact that DOX is more soluble in an acidic environment44 and H4 had a slight weight loss at pH 2.0. However, it should be noted that DOX was not completely released from the hydrogel, given that the drug was possibly wrapped in its hydrophobic domains.45 In comparison, DOX in the hydrogel was rapidly released in buffer solution of pH 2.0 at 5 °C, and DOX was almost completely released within 3 h. This can be explained by the fact that H4 swelled rapidly, and then dissociated due to the breakage of its acylhydrazone linkages at pH 2.0 and 5 °C, which is consistent with the above-mentioned results.
In addition, the data for DOX release from the hydrogel under different conditions were fitted with zero-order, first-order, Higuchi and Ritger–Pappas models (formulas are listed in the ESI†) to elucidate its release mechanism,46 respectively. As presented in Table S2,† the Ritger–Pappas equation showed the best fit among the models based on the correlation coefficients (R2). In the Ritger–Pappas model, the n value indicates the release mechanism.47 At 37 °C, the n value for DOX release at pH 2.0 and pH 7.4 (0.303 and 0.204, respectively) was less than 0.45, indicating that the release of DOX from H4 was controlled by the Fickian diffusion mechanism.48 However, the n value (0.454) for DOX release at pH 2.0 at 5 °C decreased in the range of 0.45 < n < 0.89, which can be classified as non-Fickian or anomalous transport47 owing to the combination diffusion and swelling.47
4. Conclusions
We prepared well-defined ABA triblock copolymers via RAFT polymerization. The triblock copolymers were comprised of a middle PAM block and terminal PNIPAM block with randomly incorporated DAAM units, which could assemble into micelles with an increase in temperature. Based on the condensation reaction between the ketone groups and hydrazide, dynamic hydrogels were generated by mixing the triblock copolymers and adipic dihydrazide (ADH). The hydrogels displayed a sol–gel transition, self-healing, and injectability. Upon exposure to thermal stimulus, their mechanical properties and stability were enhanced because the DCB acylhydrazone were wrapped and “locked” by the temperature-induced micelles, and the hydrogel could even survive at pH 2.0. Moreover, despite their enhanced stability, the hydrogels could be degraded by the fracture of the trithiocarbonate groups. In addition, the hydrogels exhibited good cytocompatibility and controlled release behavior for DOX. Considering these attractive characteristics, these dynamic hydrogels may find potential applications in the biomedical field, such as drug carriers and cell or tissue engineering scaffolds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project is supported by the Natural Science Foundation of Fujian Province, China (2022J01900, 2023J01916), National Natural Science Foundation of China (21802041), and Natural Science Foundation of Jiangxi Province, China (20202ACBL214001).References
- Z. Wang, H. Wei, Y. Huang, Y. Wei and J. Chen, Chem. Soc. Rev., 2023, 52, 2992–3034 RSC.
- P. Lavrador, M. R. Esteves, V. M. Gaspar and J. F. Mano, Adv. Funct. Mater., 2021, 31, 2005941 CrossRef CAS.
- S. Hu, C. Zeng, Y. Jiang, W. Kong and M. Zhu, Chem. Mater., 2024, 36, 1054–1087 CrossRef CAS.
- C. D. Spicer, Polym. Chem., 2020, 11, 184–219 RSC.
- Y. Chen, Y. Zhang, H. Li, J. Shen, F. Zhang, J. He, J. Lin, B. Wang, S. Niu, Z. Han and Z. Guo, Nano Today, 2023, 49, 101764 CrossRef CAS.
- Y. Zhang, Y. Tan, J. Lao, H. Gao and J. Yu, ACS Nano, 2023, 17, 9681–9693 CrossRef CAS PubMed.
- L. Hu, P. L. Chee, S. Sugiarto, Y. Yu, C. Shi, R. Yan, Z. Yao, X. Shi, J. Zhi, D. Kai, H.-D. Yu and W. Huang, Adv. Mater., 2023, 35, 2205326 CrossRef CAS PubMed.
- P. Bertsch, M. Diba, D. J. Mooney and S. C. G. Leeuwenburgh, Chem. Rev., 2023, 123, 834–873 CrossRef CAS PubMed.
- S. Amirthalingam, A. K. Rajendran, Y. G. Moon and N. S. Hwang, Mater. Horiz., 2023, 10, 3325–3350 RSC.
- S. Huang, X. Kong, Y. Xiong, X. Zhang, H. Chen, W. Jiang, Y. Niu, W. Xu and C. Ren, Eur. Polym. J., 2020, 141, 110094 CrossRef CAS.
- S. Tang, B. M. Richardson and K. S. Anseth, Prog. Mater. Sci., 2021, 120, 100738 CrossRef CAS.
- M. M. Perera and N. Ayres, Polym. Chem., 2020, 11, 1410–1423 RSC.
- Z. Guo, H. Gu, Y. He, Y. Zhang, W. Xu, J. Zhang, Y. Liu, L. Xiong, A. Chen and Y. Feng, Chem. Eng. J., 2020, 388, 124282 CrossRef CAS.
- A. W. Jackson and D. A. Fulton, Polym. Chem., 2013, 4, 31–45 RSC.
- M. Rizwan, A. E. G. Baker and M. S. Shoichet, Adv. Healthcare Mater., 2021, 10, 2100234 CrossRef CAS PubMed.
- J. Ye, S. Fu, S. Zhou, M. Li, K. Li, W. Sun and Y. Zhai, Eur. Polym. J., 2020, 139, 110024 CrossRef CAS.
- Z. Wei, J. H. Yang, Z. Q. Liu, F. Xu, J. X. Zhou, M. Zrínyi, Y. Osada and Y. M. Chen, Adv. Funct. Mater., 2015, 25, 1352–1359 CrossRef CAS.
- X. Yang, G. Liu, L. Peng, J. Guo, L. Tao, J. Yuan, C. Chang, Y. Wei and L. Zhang, Adv. Funct. Mater., 2017, 27, 1703174 CrossRef.
- E. Bakaic, N. M. B. Smeets and T. Hoare, RSC Adv., 2015, 5, 35469–35486 RSC.
- G. Deng, F. Li, H. Yu, F. Liu, C. Liu, W. Sun, H. Jiang and Y. Chen, ACS Macro Lett., 2012, 1, 275–279 CrossRef CAS PubMed.
- G. Deng, C. Tang, F. Li, H. Jiang and Y. Chen, Macromolecules, 2010, 43, 1191–1194 CrossRef CAS.
- Z. Guo, W. Ma, H. Gu, Y. Feng, Z. He, Q. Chen, X. Mao, J. Zhang and L. Zheng, Soft Matter, 2017, 13, 7371–7380 RSC.
- W. Ma, X. Yang, H. Liu, Z. Guo, J. Zhang, G. Kang, C. Yu and H. Wei, React. Funct. Polym., 2023, 184, 105513 CrossRef CAS.
- M. Patenaude, S. Campbell, D. Kinio and T. Hoare, Biomacromolecules, 2014, 15, 781–790 CrossRef CAS PubMed.
- J. Shen, L. Chang, D. Chen, Y. Wang, W. Li, Y. He and J. Qin, J. Polym. Res., 2021, 28, 132 CrossRef CAS.
- R. Chang, H. An, X. Li, R. Zhou, J. Qin, Y. Tian and K. Deng, Polym. Chem., 2017, 8, 1263–1271 RSC.
- N. M. B. Smeets, E. Bakaic, M. Patenaude and T. Hoare, Acta Biomater., 2014, 10, 4143–4155 CrossRef CAS PubMed.
- E. Bakaic, N. M. B. Smeets, H. Dorrington and T. Hoare, RSC Adv., 2015, 5, 33364–33376 RSC.
- G. Zhang, Y. Chen, Y. Deng, T. Ngai and C. Wang, ACS Macro Lett., 2017, 6, 641–646 CrossRef CAS PubMed.
- L. Li, B. Yan, J. Yang, W. Huang, L. Chen and H. Zeng, ACS Appl. Mater. Interfaces, 2017, 9, 9221–9225 CrossRef CAS PubMed.
- P. M. Kharkar, K. L. Kiick and A. M. Kloxin, Chem. Soc. Rev., 2013, 42, 7335–7372 RSC.
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754–6756 CrossRef CAS.
- G. K. K. Clothier, T. R. Guimarães, S. W. Thompson, J. Y. Rho, S. Perrier, G. Moad and P. B. Zetterlund, Chem. Soc. Rev., 2023, 52, 3438–3469 RSC.
- G. Moad, Polym. Chem., 2017, 8, 177–219 RSC.
- Z. Guo, H. Yin, Y. Feng and S. He, RSC Adv., 2016, 6, 37953–37964 RSC.
- V. Yesilyurt, M. J. Webber, E. A. Appel, C. Godwin, R. Langer and D. G. Anderson, Adv. Mater., 2016, 28, 86–91 CrossRef CAS PubMed.
- Y. N. Sun, G. R. Gao, G. L. Du, Y. J. Cheng and J. Fu, ACS Macro Lett., 2014, 3, 496–500 CrossRef CAS PubMed.
- X. Zhao, Soft Matter, 2014, 10, 672–687 RSC.
- Y. HE, Y. Luo, T. Liu, Y. Zhang, Z. Guo and J. Zhang, J. Funct. Polym., 2022, 35, 93–100 CAS.
- K. Zhang, Q. Feng, Z. Fang, L. Gu and L. Bian, Chem. Rev., 2021, 121, 11149–11193 CrossRef CAS PubMed.
- W. Gao, J. M. Chan and O. C. Farokhzad, Mol. Pharmaceutics, 2010, 7, 1913–1920 CrossRef CAS PubMed.
- Y. Katsumoto, T. Tanaka, K. Ihara, M. Koyama and Y. Ozaki, J. Phys. Chem. B, 2007, 111, 12730–12737 CrossRef CAS PubMed.
- P. T. Boeck, J. Tanaka, W. You, B. S. Sumerlin and A. S. Veige, Polym. Chem., 2023, 14, 2592–2598 RSC.
- M. Chen, F. Song, Y. Liu, J. Tian, C. Liu, R. Li and Q. Zhang, Nanoscale, 2019, 11, 3814–3826 RSC.
- F. Hou, B. Xi, X. Wang, Y. Yang, H. Zhao, W. Li, J. Qin and Y. He, Colloids Surf., B, 2019, 183, 110441 CrossRef CAS PubMed.
- Y. Liang, J. Song, H. Dong, Z. Huo, Y. Gao, Z. Zhou, Y. Tian, Y. Li and Y. Cao, Sci. Total Environ., 2021, 787, 147422 CrossRef CAS PubMed.
- A. Olejnik, I. Nowak and G. Schroeder, Int. J. Environ. Sci. Technol., 2019, 16, 5623–5634 CrossRef CAS.
- S. Mongkolkitikul, N. Paradee and A. Sirivat, Eur. J. Pharm. Sci., 2018, 112, 20–27 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR of Macro-CTAs, GPC traces of polymers, SEM of hydrogels at 37 °C, the images of hydrogel degradation, fitting models for release data and its fitting results. See DOI: https://doi.org/10.1039/d4ra02480j |
| This journal is © The Royal Society of Chemistry 2024 |