
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDFT study of Ti3C2 MXene nanosheets as a drug delivery system for 5-fluorouracil†
Maryam Sadeghi and
Bahram Khoshnevisan*
and
Bahram Khoshnevisan*
Faculty of Physics, University of Kashan, Kashan, Iran. E-mail: b.khosh@kashanu.ac.ir
First published on 25th June 2024
Abstract
In this study, we modeled a drug delivery system consisting of Ti3C2 MXene nanosheets as a carrier and 5-fluorouracil (FU) as a selected drug molecule using density functional theory (DFT) computations. During the adsorption procedure, electronic, magnetic and structural properties were calculated. Our results showed that the adsorption of FU drugs on the Ti3C2 surface is thermodynamically favorable. Our spin-polarized calculations also determined that the magnetization of Ti3C2 after FU adsorption does not change significantly, which is an important factor for magnetic hyperthermia and drug delivery. In addition, our calculations indicate that in the slightly acidic environment of tumor tissue, FU could start to be released (by increasing distance from the MXene surface and then instability of the complex) from the Ti3C2 surface without any substantial change in the structural properties. This study could provide a deep understanding of the interaction mechanism of 2-dimensional (2D) MXene materials with drugs at the atomistic scale and have an important contribution to the discovery and application of novel 2D materials as drug delivery systems.
1. Introduction
The use of nanotechnology can be a promising innovation and transformation in medicine and especially in the design of new drugs for cancer treatment. Specifically, 2D nanomaterials are interesting in the field of drug delivery due to their physical, chemical and biological properties that often differ from their bulk structure.1–4 Graphene and its derivatives and some other layered materials for instance hexagonal boron nitride (hBN), transition metal dichalcogenides (TMDs), transition metal oxides (TMOs), and black phosphorus (BP) are used in nanomedicine.5–10Another group of 2D layered materials discovered by two groups of researchers from Drexel University are MXenes.11 The new family with the general formula of Mn+1AXn is called the MAX phase and includes carbide, nitride and carbonitride of transition metals. In this formula, M, A and X are: transition metal atom, a main group element (mostly group 13 or 14 element from the periodic table), and carbon or nitrogen atoms, respectively, and n can be 1 to 3. MXenes are obtained by the selective etching of the A-layers of the MAX phase either in pure form (Mn+1Xn), or with surface functional groups, Mn+1XnTx, where Tx can be fluorine (F), hydroxyl (OH) and oxygen(O).12
MXenes have excellent properties in terms of electrical, optical and thermal stability, so most conducted studies on them have focused on energy storage,13,14 catalysis,15 and sensors.16,17 MXenes also have (i) high specific surface area, which is an efficient factor for high drug loading, (ii) tunable layered structure, and (iii) hydrophilic nature, which make them to be considered as new inorganic nanostructures for biological and biomedical applications.18 MXenes also have high photothermal conversion efficiency, which makes these materials suitable for photothermal therapy and hyperthermia.19,20
Ti3C2 is the first MXene that was discovered in 2011 by Michael Naguib et al.21 So far, many theoretical and experimental studies have been performed on it in various areas. In 2017 and 2020, A. M. Jastrzębska et al. investigated the cytotoxic effects of Ti3C2. The results revealed that these nanosheets exhibited the highest cytotoxic effect on the cancerous cell line of A549 while the normal cell line HaCaT showed no changes across all concentrations and remained unharmed.22 Furthermore, they deposited a layer of Ti2O3 on the Ti3C2Tx MXene using ultrasound and mild thermal oxidation after the synthesis process and indicated that it could probably regulate the cytotoxicity of samples to cancer cells. These samples were toxic to all cancer cell lines up to 375 mg l−1.23 In 2018, C. Xing et al. developed a MXene/DOX@cellulose hydrogel nano-platform, where MXene comprised Ti3C2 nanosheets, to study the release of the DOX model drug and its photothermal performance. This platform showed desirable biocompatibility, good photothermal efficiency and high capacity for Dox drug loading, and it was useful for immediate tumor destruction and preventing its recurrence.24 For the first time, the performance of Ti3C2 MXene for chemotherapy drug delivery was investigated by X. Han in 2018. The drug was released by pH-responsive method and NIR laser technique, and a high synergistic therapeutic outcome was obtained in the treatment of cancer.25 G. Liu et al. used a multifunctional nanoplatform (Ti3C2–DOX), in which the nanosheets have a small lateral size (∼100 nm) and included the stable surface functional group Al(OH)4. The work offered a new effective strategy for cancer therapy based on surface-modified Ti3C2 nanosheets.26 B. Zhu et al. combined gold nanorods (GNRs) with Ti3C2 nanosheets and prepared intelligent sandwich-like Ti3C2@GNRs/PDA/Ti3C2 nanohybrids, which were used for drug delivery with synergistically enhanced NIR drug release behavior. The results showed that this platform had pH/NIR-responsive drug release. Overall, it was promising for use in photothermal therapy and in remote controllable drug delivery.27 In 2020, Y. Liu et al. developed a heterostructured titanium carbide–cobalt nanowires (Ti3C2–CoNWs) nanocarrier. When DOX was used as a model drug, this nanocarrier had a high drug loading ability and showed drug release behavior induced by pH/NIR stimulations.28 In 2023, Z. Bai et al. showed that under the irradiation of a NIR-I laser, the Ti3C2 core in the DOX/Ti3C2/Apt-M therapeutic platform enhanced the effect of PTT and promoted the release of the DOX to enhance the chemotherapy effect.29
To the best of our knowledge, theoretical calculations have not been reported so far on using Ti3C2 as a drug delivery system for fluorouracil. Fluorouracil (abbreviated: 5-FU or FU) is one of the common cancer treatment drugs for some cancers, such as breast, stomach and colon.30
In this work, based on Density Functional Theory (DFT), we have modeled Ti3C2 MXene nanosheets as a carrier for FU. First, we investigated the adsorption behavior of 5-FU on Ti3C2 nanosheets. Then, we calculated the electronic structure and magnetic properties of Ti3C2@FU systems. Finally, drug release from the carrier surface, which is an important part of the drug delivery process, has been investigated. We hope this study provides a deep understanding of the interaction mechanism at the atomistic scale, and provides some insight for biomedical applications of Ti3C2 MXene nanosheets.
2. Computational methods
The calculations were done based on density functional theory (DFT) using the Quantum-ESPRESSO package. The exchange correlation interaction was considered within the generalized gradient approximation (GGA), Perdew–Burke–Ernzerhof (PBE).31,32 The vacuum space was considered to be 25 Å along the z-direction to prevent interaction with the periodic image of nanosheets. The energy cutoff for plane waves was set to 480 eV, and the convergence in energy and force were 10−4 eV and 10−4 eV Å−1, respectively. The van der Waals interactions, as a semiempirical correction, were also added through the “DFT-D2” treatment suggested by Grimme.33,343. Result and discussion
3.1 Structural properties of Ti3C2 and FU
In our study, Ti3C2 MXene nanosheets serve as the drug carrier. To simulate the primary structure of this MXene, its bulk structure was used as a model and a relaxation calculation was then performed for the Ti3C2 monolayer. Fig. 1 shows the Ti3C2 monolayer structure. The single layer of Ti3C2 includes two of the same groups of Ti atoms in the upper and lower regions of the layer, and one row of Ti atoms in the center that we will call Ti1 and Ti2, respectively (Fig. 1). It should be noted that Ti1 and Ti2 are different in magnetic and electronic properties.35 There are two rows of C atoms in the middle of the titanium atoms that bonded with Ti1 and Ti2.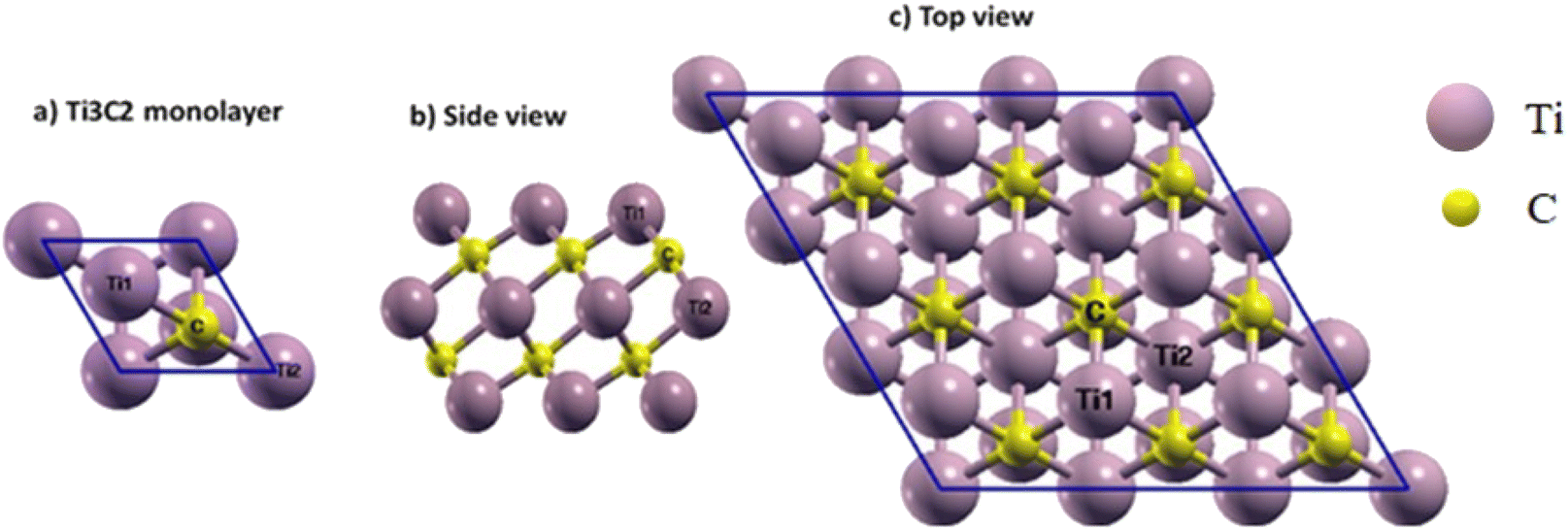 | ||
| Fig. 1 (a) Ti3C2 unit cell, (b) side and (c) top views of the 3 × 3 × 1 supercell of Ti3C2 MXene. Three special sites for the absorption process are marked on the (c). | ||
In the relaxed structure of the Ti3C2 monolayer, the a-lattice parameter of its hexagonal structure was determined to be 3.07 Å, and the bond lengths of Ti1–C and Ti2–C were found to be 2.05 and 2.20 Å, respectively. The thickness of the sheet (distance between Ti1 atoms at the upper and lower level of the surface) was 4.72 Å. For comparison, other theoretical and experimental results are summarized in Table 1, and it can be seen that our results are in good agreement with them.36–48
| a (Å) | Ti1–C (Å) | Ti2–C (Å) | Thickness (Å) | Magnetization (μB) | |
|---|---|---|---|---|---|
| a * means experimental reports. | |||||
| This work | 3.07 | 2.05 | 2.20 | 4.72 | 1.85 |
| Other reports | 3.07 (ref. 36 and 48) | 2.064 (ref. 47) | 2.21 (ref. 36) | 4.66 (ref. 36) | 1.9 (ref. 36), 1.93 (ref. 47) |
| 3.10 (ref. 46) | 2.062 (ref. 37) | 2.22 (ref. 37) | 4.64 (ref. 37) | 1.87 (ref. 38) | |
| 3.08 (ref. 39) | 2.083 (ref. 39) | 2.21 (ref. 40) | 4.60 (ref. 41) | 2.2 (ref. 42) | |
| *3.08 (ref. 43), *3.09 (ref. 44) | *1.87 (ref. 39 and 45) | ||||
FU is one of the common drugs used to treat cancer and has five different structures: the Diketo (contains two carbonyl groups), Dienol (contains two hydroxyl groups), and three Ketoenol tautomers (with one carbonyl and one hydroxyl groups). We have investigated all five structures in our simulations. It also must be noted that Ketoenol has three different configurations, and we took the most stable of them in our study (Fig. 2).49,50
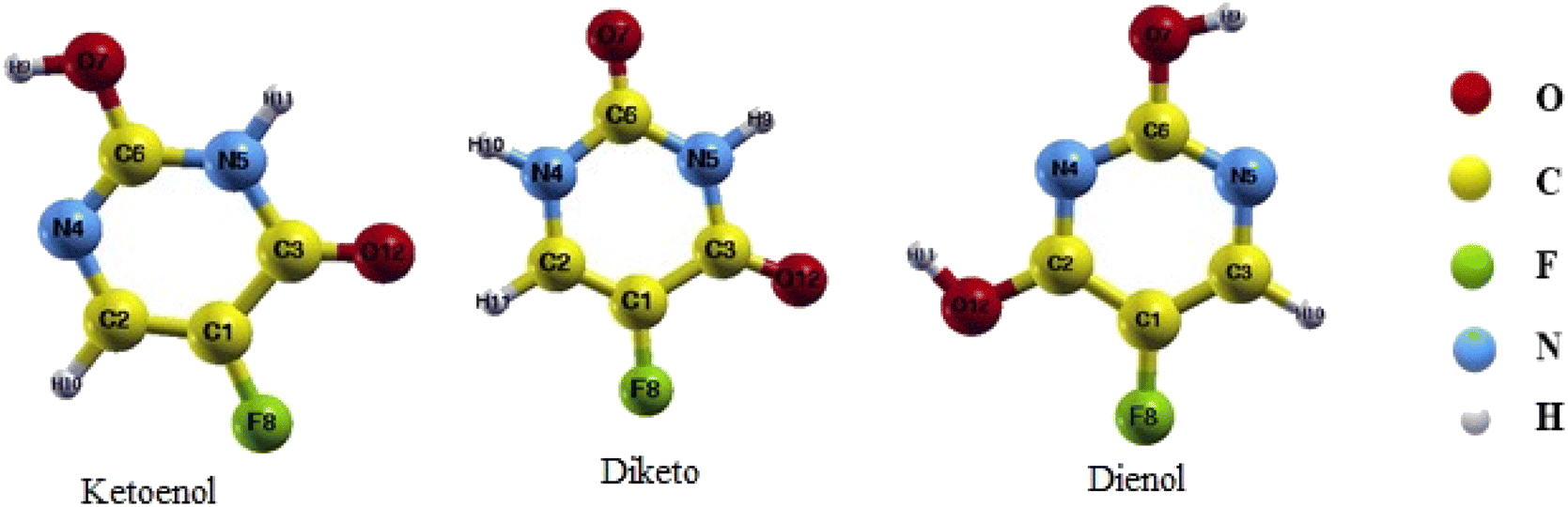 | ||
| Fig. 2 Different structures of the 5-FU drug. | ||
At first, each structure was simulated and optimized, and the bond lengths after relaxation calculations for each of the three drug forms are given in the first row of Tables 2–4. In comparison with the literature, there is a good agreement between our results and the previous theoretical and experimental reports.51–53
| N5–H9 | C3–O12 | C1–F8 | C2–H11 | N4–H10 | C6–O7 | C6–N5 | N5–C3 | C3–C1 | C1–C2 | C2–N4 | N4–C6 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diketo | 1.01 | 1.23 | 1.35 | 1.09 | 1.01 | 1.22 | 1.39 | 1.40 | 1.46 | 1.35 | 1.37 | 1.39 |
| Ref. 51 | — | 1.22 | 1.34 | — | — | 1.22 | 1.40 | 1.40 | 1.47 | 1.35 | 1.38 | 1.39 |
| ⊥Ti1 | 1.02 | 1.23 | 1.37 | 1.09 | 1.01 | 1.22 | 1.39 | 1.40 | 1.45 | 1.35 | 1.37 | 1.39 |
| ⊥Ti2 | Not converge | — | — | — | — | — | — | — | — | — | — | — |
| ⊥C | 1.02 | 1.31 | 1.37 | 1.08 | 1.01 | 1.22 | 1.39 | 1.39 | 1.40 | 1.36 | 1.39 | 1.39 |
| After release ⊥C | 1.02 | 1.31 | 1.36 | 1.08 | 1.01 | 1.22 | 1.39 | 1.38 | 1.41 | 1.36 | 1.38 | 1.39 |
| H9–O7 | O7–C6 | C6–N5 | N5–C3 | C3–C1 | C1–C2 | C2–N4 | N4–C6 | C2–H10 | C1–F8 | C3–O12 | N5–H11 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ketoenol | 0.98 | 1.35 | 1.36 | 1.42 | 1.45 | 1.36 | 1.37 | 1.30 | 1.09 | 1.35 | 1.23 | 1.02 |
| Ref. 52 and 53 | 0.98 | 1.34 | 1.36 | 1.42 | — | 1.37 | — | 1.30 | — | 1.35 | 1.22 | 1.02 |
| ⊥Ti1 | 0.98 | 1.34 | 1.36 | 1.42 | 1.44 | 1.36 | 1.37 | 1.30 | 1.09 | 1.38 | 1.23 | 1.02 |
| ⊥Ti2 | 0.98 | 1.35 | 1.36 | 1.42 | 1.44 | 1.36 | 1.37 | 1.30 | 1.09 | 1.37 | 1.23 | 1.02 |
| ⊥C | 0.98 | 1.34 | 1.36 | 1.42 | 1.45 | 1.36 | 1.37 | 1.30 | 1.09 | 1.36 | 1.23 | 1.02 |
| After release ⊥C | 0.98 | 1.34 | 1.36 | 1.4 | 1.45 | 1.36 | 1.37 | 1.3 | 1.1 | 1.35 | 1.23 | 1.02 |
| H9–O7 | O7–C6 | C6–N4 | N4–C2 | C2–O12 | O12–H11 | C2–C1 | C1–C3 | C1–F8 | C3–H9 | C3–N5 | N5–C6 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dienol | 0.98 | 1.35 | 1.33 | 1.33 | 1.34 | 0.98 | 1.40 | 1.38 | 1.35 | 1.09 | 1.34 | 1.34 |
| Ref. 52 and 53 | 0.98 | 1.35 | 1.34 | 1.33 | — | — | — | 1.38 | 1.35 | — | — | 1.34 |
| ⊥Ti1 | 0.98 | 1.34 | 1.34 | 1.32 | 1.34 | 0.98 | 1.40 | 1.38 | 1.38 | 1.09 | 1.34 | 1.34 |
| ⊥Ti2 | 0.98 | 1.34 | 1.33 | 1.32 | 1.36 | 0.10 | 1.39 | 1.38 | 1.38 | 1.09 | 1.34 | 1.34 |
| ⊥C | 0.98 | 1.34 | 1.34 | 1.32 | 1.34 | 0.98 | 1.40 | 1.38 | 1.38 | 1.09 | 1.34 | 1.34 |
| After release ⊥Ti2 | 0.98 | 1.35 | 1.34 | 1.33 | 1.35 | 0.98 | 1.40 | 1.38 | 1.36 | 1.09 | 1.34 | 1.34 |
3.2 FU adsorption on the Ti2C3 surface (Ti3C2@ FU)
To consider the FU adsorption on the Ti2C3 surface, we made a 3 × 3 × 1 supercell of Ti3C2. It is well known that according to the crystal structural symmetry of the Ti3C2 sheet, there are three adsorption sites for adding drugs and they are marked in Fig. 1.47,54 The first site is on the top of the Ti atom on the surface (⊥Ti1), the second is above the C atoms (⊥C) and the last one is on the top of the middle row of Ti atoms (⊥Ti2).The selected drug molecules were approached to the Ti3C2 surface at the ⊥Ti1, ⊥Ti2 and ⊥C sites. For example, Fig. 3 displays the most stable Ti3C2@ FU systems before and after the relaxation calculations, and other configurations before and after the relaxation can be seen in Fig. 1S–3S in the ESI† Section (Appendix). In the cases of Ketoenol and Diketo, ⊥C is the most stable configuration. Meanwhile, in the Dienol case, ⊥Ti2 is the most stable site for the drug. The bond lengths of the drug molecules after adsorption were calculated and are summarized in Tables 2–4. It can be seen that the bond lengths of the drugs have not changed after adsorption, which confirmed that the structures of the drugs were preserved.
 | ||
| Fig. 3 (a) Ti3C2@Dienol, (b) Ti3C2@Ketoenol, and (c) Ti3C2@Diketo before (left) and after (right) the adsorption process in the most stable configurations. | ||
In order to obtain the most stable structure, the adsorption energy was calculated for each of the three configurations. The formula for calculating adsorption energy is:
| Ead = (ETi3C2@FU) − (ETi3C2 − EFU) | (1) |
The structure with the highest negative adsorption energy was known as the most stable Ti3C2@drug system. Table 5 shows the calculated adsorption energies of each structure in the considered adsorption sites. Ead of the Diketo on the ⊥C site was calculated to be −3.39 eV, which is the most stable system. Moreover, ⊥the Ti2 site was the most stable configuration for Dienol with Ead equal to −2.36 eV. For Ketoenol, the ⊥C site with Ead: −2.03 eV was the most stable structure.
| Ti3C2@Diketo | Ti3C2@Ketoenol | Ti3C2@Dienol | |
|---|---|---|---|
| ⊥C | −3.39 | −2.03 | −2.01 |
| ⊥Ti2 | Not converged | −1.94 | −2.36 |
| ⊥Ti1 | −2.01 | −1.99 | −2.01 |
We compared our results with literature (Table 6), and our calculated Ead were higher than the adsorption of the FU drug on some nanosheets, such as SiG, BN, InN, AlN, ZnO, SWCNTs and also BNNT.50,55–60 In these nanosheets and nanotubes, it is necessary to increase the adsorption energy using some process such as doping because the Ead is low. On the other hand, our results were lower than those from the Diketo adsorption on GaN.62 In this case, there are problems with the drug releasing procedure. Our Ead were comparable with FU adsorbed on GNS and in some sites on graphene oxide, respectively.61,63
| Ead (eV) | Carrier surface | Form of FU drug | Ref. |
|---|---|---|---|
| −0.083, −0.172, −0.082 | (SiG) silicon graphene nanosheet | Diketo/Ketoenol/Dienol | 50 |
| −0.56 | BNNS (boron nitride nanosheet) | Diketo | 55, and 56 |
| −1.3, 0.81, −0.84 | InN, AlN, GaN (nanosheet) | Diketo | 55 |
| −0.423 | ZnONS (zinc oxide nanosheet) | Diketo | 57 |
| −0.358 | (4,0) SWCNT | Diketo | 58 |
| −0.13 | (8,0) BNNT (boron nitride nanotube) | Diketo | 59, and 60 |
| −1.74 | (GONS) graphene oxide nanosheets | Diketo | 61 |
| −5.41, −4.29, −1.37 | (GaN) gallium nitride | Diketo/Ketoenol/Dienol | 62 |
| −3.116 | GNS (graphene nanosheet) | Diketo | 63 |
3.3 Electronic and structural stability properties
Furthermore, the electronic density of state (DOS) is the main criterion for better understanding what happens during the drug interaction with the carrier surface. For this purpose, the DOS was calculated for the Ti3C2@FU systems. Furthermore, the DOS diagrams of bare Ti3C2 and FU were plotted for comparison.Fig. 4 shows the results of the partial DOS and total DOS diagrams of the Ti3C2@FU complexes. A careful comparison between the DOS of Ti3C2 and Ti3C2@FU systems reveals that the insertion of a drug into Ti3C2 altered the amount of the DOS at the Fermi surface. Actually, the amount of DOS at the Fermi surface increased slightly with respect to the Ti3C2 surface; these new states belonged to the drugs. Further investigation showed that the partial DOS of the drugs did not change after adsorption. This means that the electronic properties of the drug were slightly affected during the interaction, and it can be said that the structure of the drug was preserved during the adsorption.
 | ||
| Fig. 4 (a) The partial density of state (PDOS) of the optimized Ti3C2 MXene. (b–d) The density of state (DOS) of the Ti3C2@FU surface complex. | ||
The DOS diagram of the bare Ti3C2 shows the metallic behavior, and the Ti1 atoms have the main contribution on the Fermi level (Fig. 4a).
The molecular structure stability and reactivity could be predicted by chemical potential and chemical hardness. We have calculated the chemical potential and hardness using the following equations:
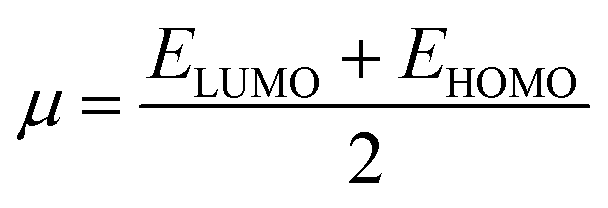 | (2) |
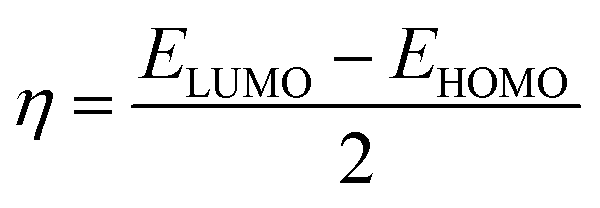 | (3) |
The results are shown in Table 7. It is found that after the adsorption, the values of the HOMO–LUMO energy gap (H/L gap) and chemical hardness for all of the systems were reduced. This indicated that the chemical stability of the drugs would be diminished; therefore, its chemical activity would be increased.
| HOMO (eV) | LUMO (eV) | H/L gap | η (eV) | η (eV) | ΔN | |
|---|---|---|---|---|---|---|
| Ti3C2 surface | 1.31 | 1.32 | 0.01 | 0.005 | 1.32 | |
| Diketo (⊥C) | 2.09 | 2.09 | 0.00 | 0.002 | 2.09 | 3.05 |
| Ketoenol (⊥C) | 2.006 | 2.009 | 0.003 | 0.002 | 2.08 | 2.80 |
| Dienol (⊥Ti2) | 2.10 | 2.11 | 0.01 | 0.005 | 2.11 | 2.68 |
To consider the charge transfer between the Ti3C2 surface and adsorbed drug molecule, we also calculated the ΔN parameter, which shows the fractional electron's charge that transfers from system A to system B:
| ΔN = (μB − μA)/(ηB + ηA) | (4) |
Iso surfaces of the most stable structures are also shown in Fig. 5. We can see the charge transfer between the drug and the surface during the adsorption process, which implied the interaction between the adsorbent and the surface in each of the three states.
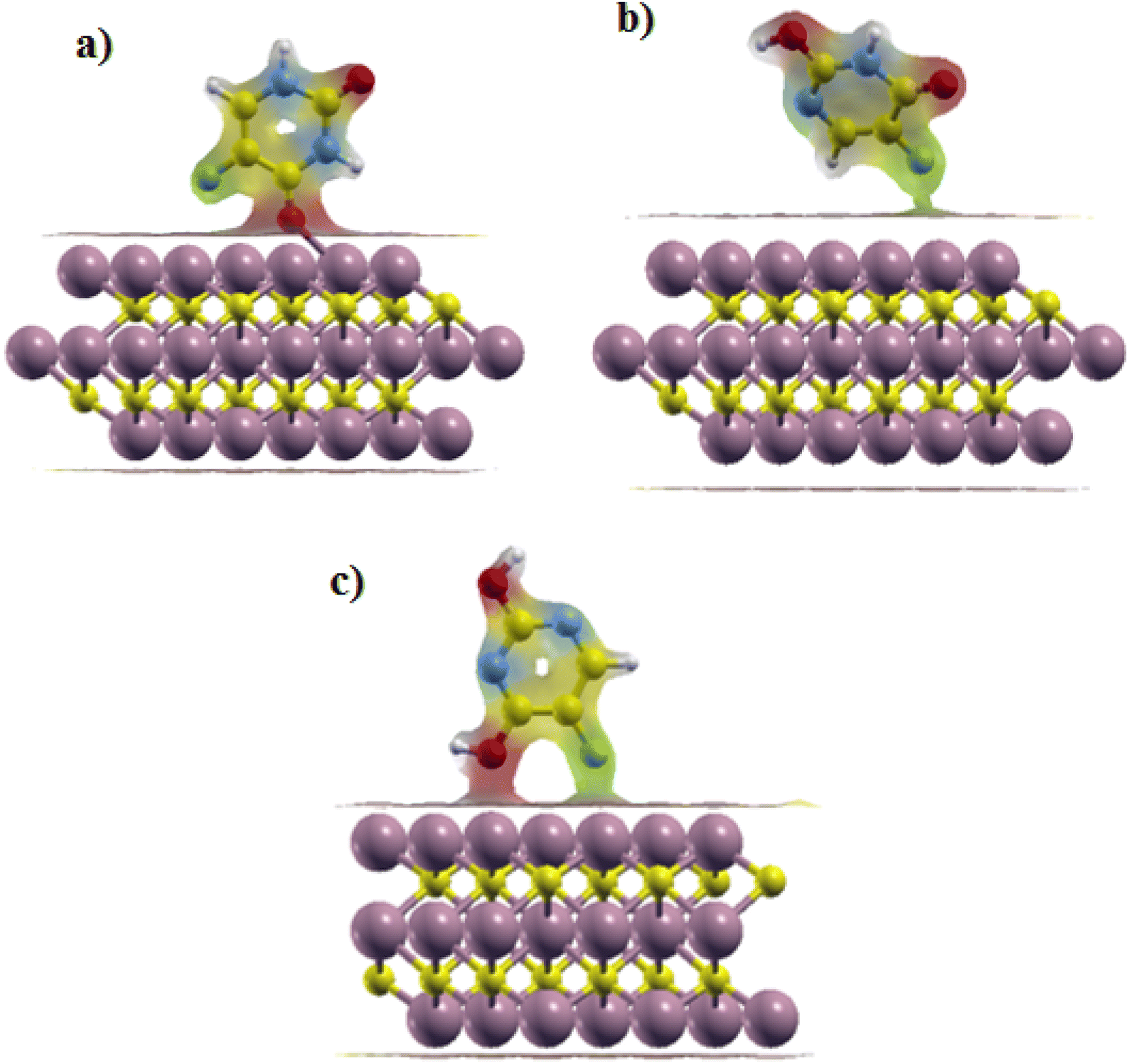 | ||
| Fig. 5 Iso surfaces of the most stable structures: (a) Ti3C2@Diketo, (b) Ti3C2@Ketonel and (c) Ti3C2@Dienol. | ||
3.4 Magnetic properties
The magnetic property is one of the most important features for drug delivery in order to achieve magnetic fluid hyperthermia. In this regard, the ideal nanoparticles for magnetic hyperthermia should show a satisfactory heating value at low magnetic fields, where the heating power is proportional to the magnetization squared.To explore the magnetic behavior of Ti3C2 during the adsorption process, we performed spin-polarized density functional theory calculations. The total magnetic moment of the 3 × 3 × 1 supercell of the Ti3C2 surface was calculated to be 20.28 μB per cell. Conversely, the average magnetic moment of each Ti1 atom on the Ti3C2 surface was about 0.49 μB. For Ti2 atoms in the middle of the surface, the calculated value was ∼0.024 μB, which confirms that the magnetic properties of Ti3C2 are primarily due to the Ti1 atoms.35,47,65,66
Fig. 6 shows the total magnetic moment of the most stable Ti3C2@FU systems. As it can be seen, the total magnetic moments varied in the range of 17.87 to 20.32 μB per cell. For the Ti3C2@Ketonel and Ti3C2@Dienol systems, the total magnetic moment did not change significantly before and after drug adsorption. In other words, drug adsorption did not destroy the magnetic properties of these systems. For the Ti3C2@Diketo case on the ⊥C site of the surface, the total magnetic moment value decreased to 17.82 μB per cell. Ti3C2@Diketo has the highest Ead. The oxygen atom of the Diketo bonded with one of the surface titanium atoms during the adsorption process, and caused the decrease of the magnetization. In contrast, for the two former cases, there was not any oxygen atom interactions. So, the bonding state plays a key role in the magnetic moment values. In all cases, after the drug adsorption on the carrier surface, in spite of the Ti1 atoms being around the drug adsorption site, the value of the magnetic moment of the other Ti1 atoms did not change significantly. However, Ti1 atoms that were located around the drug adsorption site on the surface during the adsorption process showed a change in the range of 0.33 to 0.55 μB.
 | ||
| Fig. 6 Total magnetic moment of the Ti1 atom and Ti3C2@FU systems at the most stable adsorption sites. | ||
3.5 Drug release
The release of the drug from the carrier inside the target cells is the most vital step during the drug delivery process. In order to consider the Ti3C2 surface as a candidate for a drug delivery system, the drug release process had to be investigated. In the literature, the NIR laser was introduced as one method in this regard. However, in this work, we used the feature of the microenvironment inside the tumor, which has a lower pH than the surrounding healthy tissues.25,28,67 For this reason, we calculated the release energy of the drug by adding protons around the binding site in the complex of the drug and the surface to weaken them. This could happen by adsorbing the free protons from the tumor matrix.The added protons can attack different places of the complex, including nucleophilic and the other sites as well.56,68 Therefore, we investigated the drug release behavior by considering the protons' attack on the drug, as well as the carrier surface around the adsorption site in the most stable structures.
In our modeling, the numbers of hydrogen atoms were added first to the F atom of the drug, but there was no sign of drug release. Therefore, we concluded that to weaken the surface Ti bindings with the drug, they should be attacked with protons gradually until the release features have been started.
The proton number was selected based on the sites expected to impact the release process, and the release energy was calculated per proton. According to the calculations in Section 3.3, it can be expected that the added protons and the carrier surface interact with each other. The charge is transferred from the surface to the protons, weakening (or breaking) the bond between the drug and the surface. After relaxation calculation of the Ti3C2@FU systems (Fig. 7), the calculated drug release energy was as follows: for the Ketoenol form in the ⊥C state, the release energy was obtained at +1.75 eV and the distance of the drug from the surface during the structural optimization was increased by 0.87 Å. The F bond with the Ti3C2 surface weakened and finally broke, which gave rise to the release of Ketoenol from the Ti3C2 surface.
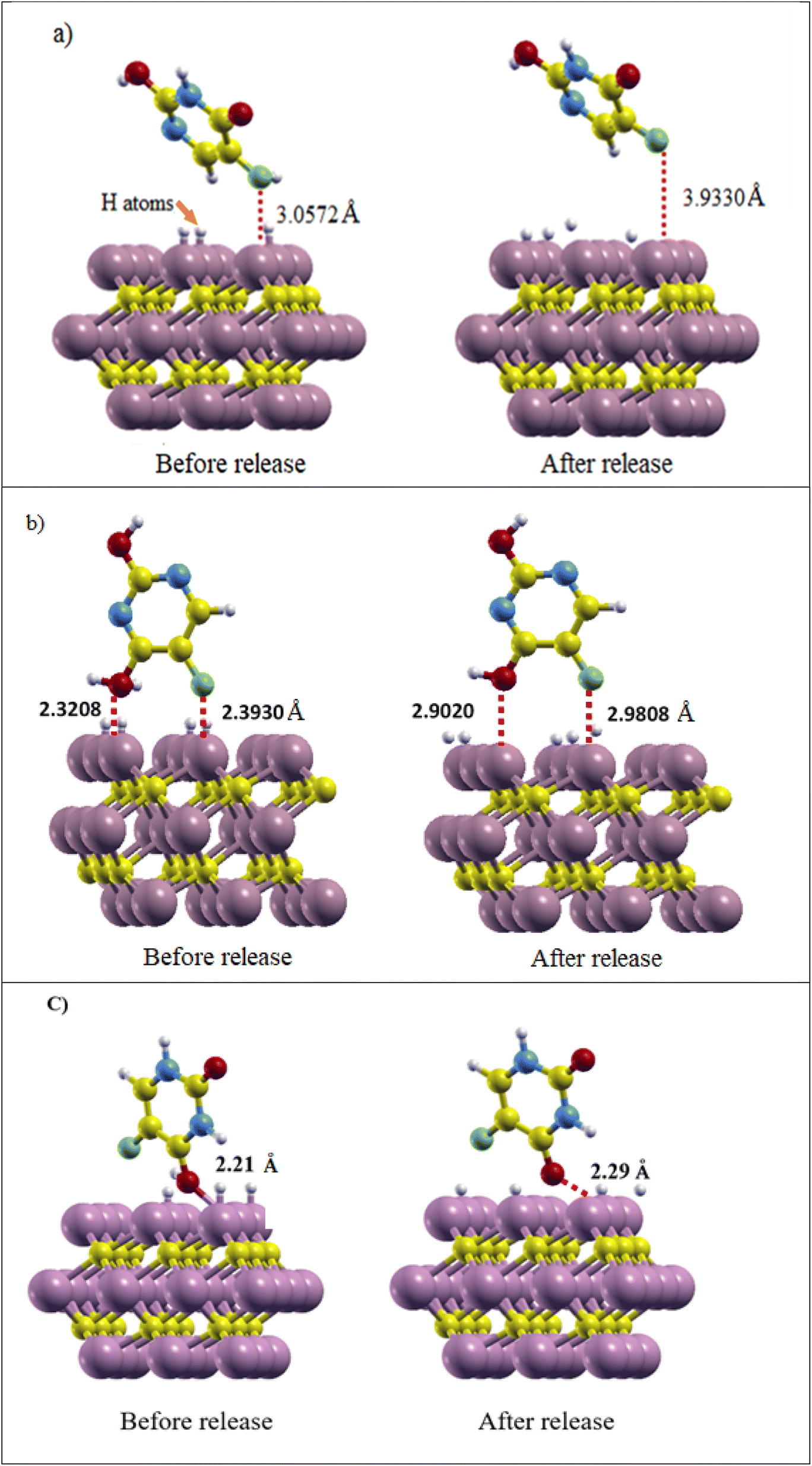 | ||
| Fig. 7 (a) Ti3C2@Ketonel, (b) Ti3C2@Dienol, and (c) Ti3C2@Diketo before and after drug release. The distance of the drug from the surface and its amount before and after release is shown. | ||
In Dienol in the ⊥Ti2 state, due to the fact that the O atom is closer to the surface than the F atom, a hydrogen atom was added to the oxygen of the drug and the Ti atoms around it so the following release energy was obtained: +1.47 eV. The distance of the O atom from the Ti3C2 MXene surface increased from 2.32 to 2.90 Å and the F atom from 2.39 to 2.98 Å. Therefore, Dienol starts to be released in the slightly acidic environment of the tumor tissue, which reduces the toxins and side effects of this drug. In the Diketo form in the ⊥C state, the distance of drug from Ti3C2 surface change about 0.08 Å with the change of the environment pH and release energy was obtained +1.57 eV, and it can be said that the release process has been started.
During the release of the drug from the carrier, the added protons approach the Ti3C2 nanosheets, which shows that the hydrogen atoms tend to interact with Ti atoms of the surface so the binding strength of these atoms with the drug could be weakened, respectively.
In order to ensure that the structure of the drug has not changed after release, the bond lengths of the drugs are compared with its the optimal model. The results are represented in row 6 of Tables 2–4. Our results showed that the structures of the drugs were not changed, so the properties of the drugs were preserved.
4. Conclusion
We modeled a drug delivery system consisting of Ti3C2 MXene nanosheets as a carrier and FU as a selected drug molecule using the quantum espresso computing package. In this study, the adsorption energies of different forms of the FU drug on the Ti3C2 carrier surface were calculated.Our calculations confirm the stability of the Ti3C2@FU systems and exothermic interaction between FU and the Ti3C2 surface. Furthermore, charge flows from the Ti3C2 surface to drugs during the adsorption process. The amount of Ti3C2 surface DOS at the Fermi surface increases slightly after drug adsorption. The total magnetic moment of the 3 × 3 × 1 supercell of the Ti3C2 surface was calculated to be 20.28 μB per cell. For the Ti3C2@Ketonel and Ti3C2@Dienol systems, the total magnetic moment did not change significantly with respect to that of Ti3C2, so drug adsorption did not destroyed the magnetic properties of these systems. However, for the Ti3C2@Diketo case, the total magnetic moment value decreased to 17.82 μB per cell.
Drug release can be achieved in the low pH environment of cancerous cells; the release energy was obtained at +1.75, +1.47 and +1.57 eV for Ketoenol, Dienol and Diketo, respectively. Results of the calculations provide a deep understanding of the interaction mechanism of 2D MXene materials with drugs at the atomistic scale.
Author contributions
M. Sadeghi: (PhD student) performed research, modeling and calculations, writing draft version. B. Khoshnevisan: (supervisor) designed research, grant holder, modeling, data analysis.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.References
- P. Boisseau and B. Loubaton, “Nanomedicine, nanotechnology in medicine”, C. R. Phys., 2011, 12(7), 620–636 CrossRef CAS.
- H. Zhang, T. Fan, W. Chen, Y. Li and B. Wang, “Recent advances of two-dimensional materials in smart drug delivery nano-systems”, Bioact. Mater., 2020, 5(4), 1071–1086 Search PubMed.
- J. Damodharan, “Nanomaterials in medicine–An overview”, Mater. Today: Proc., 2021, 37, 383–385 CAS.
- R. Davis Jr, R. A. Urbanowski Jr and A. K. Gaharwar, “2D layered nanomaterials for therapeutics delivery”, Curr. Opin. Biomed. Eng., 2021, 20, 100319 CrossRef PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. E. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, “Electric field effect in atomically thin carbon films”, Science, 2004, 306(5696), 666–669 CrossRef CAS PubMed.
- F. Nasrollahi, J. Varshosaz, A. A. Khodadadi, S. Lim and A. Jahanian-Najafabadi, “Targeted delivery of docetaxel by use of transferrin/poly (allylamine hydrochloride)-functionalized graphene oxide nanocarrier”, ACS Appl. Mater. Interfaces, 2016, 8(21), 13282–13293 CrossRef CAS PubMed.
- M. Vatanparast and Z. Shariatinia, “Hexagonal boron nitride nanosheet as novel drug delivery system for anticancer drugs: Insights from DFT calculations and molecular dynamics simulations”, J. Mol. Graphics Modell., 2019, 89, 50–59 CrossRef CAS PubMed.
- X. Zhou, H. Sun and X. Bai, “Two-dimensional transition metal dichalcogenides: synthesis, biomedical applications and biosafety evaluation”, Front. bioeng. biotechnol., 2020, 8, 236 CrossRef PubMed.
- D. Zeng, L. Wang, L. Tian, S. Zhao, X. Zhang and H. Li, “Synergistic photothermal/photodynamic suppression of prostatic carcinoma by targeted biodegradable MnO2 nanosheets”, Drug Delivery, 2019, 26(1), 661–672 CrossRef CAS PubMed.
- F. Wu, M. Zhang, X. Chu, Q. Zhang, Y. Su, B. Sun, T. Lu, N. Zhou, J. Zhang, J. Wang and X. Yi, “Black phosphorus nanosheets-based nanocarriers for enhancing chemotherapy drug sensitiveness via depleting mutant p53 and resistant cancer multimodal therapy”, Chem. Eng. J., 2019, 370, 387–399 CrossRef CAS.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, “Two-dimensional nanocrystals produced by exfoliation of Ti3AlC2”, Adv. Mater., 2011, 23(37), 4248–4253 CrossRef CAS PubMed.
- M. Alhabeb, K. Maleski, B. Anasori, P. Lelyukh, L. Clark, S. Sin and Y. Gogotsi, “Guidelines for synthesis and processing of two-dimensional titanium carbide (Ti3C2T x MXene)”, Chem. Mater., 2017, 29(18), 7633–7644 CrossRef CAS.
- X. Zhang, Z. Zhang and Z. Zhou, “MXene-based materials for electrochemical energy storage”, J. Energy Chem., 2018, 27(1), 73–85 CrossRef.
- E. M. Siriwardane, I. Demiroglu, C. Sevik, F. M. Peeters and D. Çakır, “Assessment of Sulfur-functionalized MXenes for Li-ion Battery applications”, J. Phys. Chem. C, 2020, 124(39), 21293–21304 CrossRef CAS.
- Z. W. Seh, K. D. Fredrickson, B. Anasori, J. Kibsgaard, A. L. Strickler, M. R. Lukatskaya, Y. Gogotsi, T. F. Jaramillo and A. Vojvodic, “Two-dimensional molybdenum carbide (MXene) as an efficient electrocatalyst for hydrogen evolution”, ACS Energy Lett., 2016, 1(3), 589–594 CrossRef CAS.
- S. J. Kim, H. J. Koh, C. E. Ren, O. Kwon, K. Maleski, S. Y. Cho, B. Anasori, C. K. Kim, Y. K. Choi, J. Kim and Y. Gogotsi, “Metallic Ti3C2T x MXene gas sensors with ultrahigh signal-to-noise ratio”, ACS Nano, 2018, 12(2), 986–993 CrossRef CAS PubMed.
- D. H. Ho, Y. Y. Choi, S. B. Jo, J. M. Myoung and J. H. Cho, “Sensing with MXenes: progress and prospects”, Adv. Mater., 2021, 33(47), 2005846 CrossRef CAS PubMed.
- H. Lin, Y. Chen and J. Shi, “Insights into 2D MXenes for versatile biomedical applications: current advances and challenges ahead”, Adv. Sci., 2018, 5(10), 1800518 CrossRef PubMed.
- W. Tang, Z. Dong, R. Zhang, X. Yi, K. Yang, M. Jin, C. Yuan, Z. Xiao, Z. Liu and L. Cheng, “Multifunctional two-dimensional core–shell mxene@ gold nanocomposites for enhanced photo–radio combined therapy in the second biological window”, ACS Nano, 2018, 13(1), 284–294 CrossRef PubMed.
- H. Lin, X. Wang, L. Yu, Y. Chen and J. Shi, “Two-dimensional ultrathin MXene ceramic nanosheets for photothermal conversion”, Nano Lett., 2017, 17(1), 384–391 CrossRef CAS PubMed.
- M. Naguib, O. Mashtalir, J. Carle, V. Presser, J. Lu, L. Hultman, Y. Gogotsi and M. W. Barsoum, “Two-dimensional transition metal carbides”, ACS Nano, 2012, 6(2), 1322–1331 CrossRef CAS PubMed.
- A. M. Jastrzębska, A. Szuplewska, T. Wojciechowski, M. Chudy, W. Ziemkowska, L. Chlubny, A. Rozmysłowska and A. Olszyna, “In vitro studies on cytotoxicity of delaminated Ti3C2 Mxene”, J. Hazard. Mater., 2017, 339, 1–8 CrossRef PubMed.
- A. M. Jastrzębska, A. Szuplewska, A. Rozmysłowska-Wojciechowska, M. Chudy, A. Olszyna, M. Birowska, M. Popielski, J. A. Majewski, B. Scheibe, V. Natu and M. W. Barsoum, “On tuning the cytotoxicity of Ti3C2 (MXene) flakes to cancerous and benign cells by post-delamination surface modifications”, 2D Mater., 2020, 7(2), 025018 CrossRef.
- C. Xing, S. Chen, X. Liang, Q. Liu, M. Qu, Q. Zou, J. Li, H. Tan, L. Liu, D. Fan and H. Zhang, “Two-dimensional Mxene (Ti3C2)-integrated cellulose hydrogels: toward smart three-dimensional network nanoplatforms exhibiting light-induced swelling and bimodal photothermal/chemotherapy anticancer activity”, ACS Appl. Mater. Interfaces, 2018, 10(33), 27631–27643 CrossRef CAS PubMed.
- X. Han, J. Huang, H. Lin, Z. Wang, P. Li and Y. Chen, “2D ultrathin Mxene-based drug-delivery nanoplatform for synergistic photothermal ablation and chemotherapy of cancer”, Adv. Healthcare Mater., 2018, 7(9), 1701394 CrossRef PubMed.
- G. Liu, J. Zou, Q. Tang, X. Yang, Y. Zhang, Q. Zhang, W. Huang, P. Chen, J. Shao and X. Dong, “Surface modified Ti3C2 MXene nanosheets for tumor targeting photothermal/photodynamic/chemo synergistic therapy”, ACS Appl. Mater. Interfaces, 2017, 9(46), 40077–40086 CrossRef CAS PubMed.
- B. Zhu, J. Shi, C. Liu, J. Li and S. Cao, “In-situ self-assembly of sandwich-like Ti3C2 MXene/gold nanorods nanosheets for synergistically enhanced near-infrared responsive drug delivery”, Ceram. Int., 2021, 47(17), 24252–24261 CrossRef CAS.
- Y. Liu, Q. Han, W. Yang, X. Gan, Y. Yang, K. Xie, L. Xie and Y. Deng, “Two-dimensional MXene/cobalt nanowire heterojunction for controlled drug delivery and chemo-photothermal therapy”, Mater. Sci. Eng. C., 2020, 116, 111212 CrossRef CAS PubMed.
- Z. Bai, L. Zhao, H. Feng, H. Xu, N. Zhang, Y. Li, J. Song, Y. Bai, R. Yang and F. Feng, ”Fabricating Aptamer-functionalized Ti3C2 therapeutic nanoplatform for targeted chemo-photothermal therapy of cancer”, Mater. Des., 2023, 226, 111656 CrossRef CAS.
- D. B. Longley, D. P. Harkin and P. G. Johnston, “5-fluorouracil: mechanisms of action and clinical strategies”, Nat. Rev. Cancer, 2003, 3(5), 330–338 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, “From ultrasoft pseudopotentials to the projector augmented-wave method”, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59(3), 1758 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, “Generalized gradient approximation made simple”, Phys. Rev. Lett., 1996, 77(18), 3865 CrossRef CAS PubMed.
- S. Grimme, “Accurate description of van der Waals complexes by density functional theory including empirical corrections”, J. Comput. Chem., 2004, 25(12), 1463–1473 CrossRef CAS PubMed.
- S. Grimme, “Semiempirical GGA-type density functional constructed with a long-range dispersion correction”, J. Comput. Chem., 2006, 27(15), 1787–1799 CrossRef CAS PubMed.
- F. Wu, K. Luo, C. Huang, W. Wu, P. Meng, Y. Liu and E. Kan, “Theoretical understanding of magnetic and electronic structures of Ti3C2 monolayer and its derivatives”, Solid State Commun., 2015, 222, 9–13 CrossRef CAS.
- M. Faraji, A. Bafekry, M. M. Fadlallah, F. Molaei, N. N. Hieu, P. Qian, M. Ghergherehchi and D. Gogova, “Surface modification of titanium carbide MXene monolayers (Ti 2 C and Ti 3 C 2) via chalcogenide and halogenide atoms”, Phys. Chem. Chem. Phys., 2021, 23(28), 15319–15328 RSC.
- Q. Meng, J. Ma, Y. Zhang, Z. Li, C. Zhi, A. Hu and J. Fan, “The S-functionalized Ti 3 C 2 Mxene as a high capacity electrode material for Na-ion batteries: a DFT study”, Nanoscale, 2018, 10(7), 3385–3392 RSC.
- M. Iqbal, J. Fatheema, Q. Noor, M. Rani, M. Mumtaz, R. K. Zheng, S. A. Khan and S. Rizwan, “Co-existence of magnetic phases in two-dimensional MXene”, Mater. Today Chem., 2020, 16, 100271 CrossRef CAS.
- I. R. Shein and A. L. Ivanovskii, “Graphene-like titanium carbides and nitrides Tin+ 1Cn, Tin+ 1Nn (n= 1, 2, and 3) from de-intercalated MAX phases: First-principles probing of their structural, electronic properties and relative stability”, Comput. Mater. Sci., 2012, 65, 104–114 CrossRef CAS.
- H. Gholivand, S. Fuladi, Z. Hemmat, A. Salehi-Khojin and F. Khalili-Araghi, “Effect of surface termination on the lattice thermal conductivity of monolayer Ti3C2Tz MXenes”, J. Appl. Phys., 2019, 126(6), 065101 CrossRef.
- M. Kurtoglu, M. Naguib, Y. Gogotsi and M. W. Barsoum, “First principles study of two-dimensional early transition metal carbides”, MRS Commun., 2012, 2, 133–137 CrossRef CAS.
- S. Rafiq, S. Awan, R. K. Zheng, Z. Wen, M. Rani, D. Akinwande and S. Rizwan, “Novel room-temperature ferromagnetism in Gd-doped 2-dimensional Ti3C2Tx MXene semiconductor for spintronics”, J. Magn. Magn. Mater., 2020, 497, 165954 CrossRef CAS.
- O. Mashtalir, M. Naguib, V. N. Mochalin, Y. Dall'Agnese, M. Heon, M. W. Barsoum and Y. Gogotsi, “Intercalation and delamination of layered carbides and carbonitrides”, Nat. Commun., 2013, 4(1), 1716 CrossRef PubMed.
- K. L. Firestein, J. E. von Treifeldt, D. G. Kvashnin, J. F. Fernando, C. Zhang, A. G. Kvashnin, E. V. Podryabinkin, A. V. Shapeev, D. P. Siriwardena, P. B. Sorokin and D. Golberg, “Young's modulus and tensile strength of Ti3C2 MXene nanosheets as revealed by in situ TEM probing, AFM nanomechanical mapping, and theoretical calculations”, Nano Lett., 2020, 20(8), 5900–5908 CrossRef CAS PubMed.
- R. G. Parr and R. G. Pearson, “Absolute hardness: companion parameter to absolute electronegativity”, J. Am. Chem. Soc., 1983, 105(26), 7512–7516 CrossRef CAS.
- N. Zhang, Y. Hong, S. Yazdanparast and M. A. Zaeem, “Superior structural, elastic and electronic properties of 2D titanium nitride MXenes over carbide MXenes: a comprehensive first principles study”, 2D Mater., 2018, 5(4), 045004 CrossRef CAS.
- Q. Tang, Z. Zhou and P. Shen, “Are MXenes promising anode materials for Li ion batteries? Computational studies on electronic properties and Li storage capability of Ti3C2 and Ti3C2X2 (X= F, OH) monolayer”, J. Am. Chem. Soc., 2012, 134(40), 16909–16916 CrossRef CAS PubMed.
- G. R. Berdiyorov, “Optical properties of functionalized Ti3C2T2 (T= F, O, OH) MXene: First-principles calculations”, AIP Adv., 2016, 6(5), 055105 CrossRef.
- H. Yekeler and D. Özbakır, “Concerning the solvent effect in the tautomerism of uracil, 5-fluorouracil, and thymine by density-functional theory and ab initio calculations”, J. Mol. Model., 2001, 7, 103–111 CrossRef CAS.
- A. Yaraghi, O. M. Ozkendir and M. Mirzaei, “DFT studies of 5-fluorouracil tautomers on a silicon graphene nanosheet”, Superlattices Microstruct., 2015, 85, 784–788 CrossRef CAS.
- B. Blicharska and T. Kupka, “Theoretical DFT and experimental NMR studies on uracil and 5-fluorouracil”, J. Mol. Struct., 2002, 613(1–3), 153–166 CrossRef CAS.
- M. B. Javan, A. Soltani, Z. Azmoodeh, N. Abdolahi and N. Gholami, “A DFT study on the interaction between 5-fluorouracil and B 12 N 12 nanocluster”, RSC Adv., 2016, 6(106), 104513–104521 RSC.
- H. Othmani, R. Ben Said, N. Terzi, N. E. Jaidane, M. Mogren Al Mogren, A. Elmarghany and M. Hochlaf, “Structural, energetic and spectroscopic characterisation of 5-fluorouracil anticarcinogenic drug isomers, tautomers and ions”, Mol. Phys., 2019, 117(13), 1589–1603 CrossRef CAS.
- H. Li, A. Li, D. Zhang, Q. Wu, P. Mao, Y. Qiu, Z. Zhao, P. Yu, X. Su and M. Bai, “First-Principles Study on the Structural, Electronic, and Lithium Storage Properties of Ti3C2T2 (T= O, F, H, OH) MXene”, ACS Omega, 2022, 7(44), 40578–40585 CrossRef CAS PubMed.
- T. Ahmed, M. A. Rahman, R. Islam, A. A. Piya and S. U. D. Shamim, “Unravelling the adsorption performance of BN, AlN, GaN and InN 2D nanosheets towards the ciclopirox, 5-fluorouracil and nitrosourea for anticancer drug delivery motive: A DFT-D with QTAIM, PCM and COSMO investigations”, Comput. Theor. Chem., 2022, 1214, 113797 CrossRef CAS.
- M. K. Hazrati, Z. Javanshir and Z. Bagheri, “B24N24 fullerene as a carrier for 5-fluorouracil anti-cancer drug delivery: DFT studies”, J. Mol. Graphics Modell., 2017, 77, 17–24 CrossRef CAS PubMed.
- M. H. Mohammed and F. H. Hanoon, “Application of zinc oxide nanosheet in various anticancer drugs delivery: quantum chemical study”, Inorg. Chem. Commun., 2021, 127, 108522 CrossRef CAS.
- N. Ershadi, R. Safaiee and M. M. Golshan, “Functionalized (4, 0) or (8, 0) SWCNT as novel carriers of the anticancer drug 5-FU; a first-principle investigation”, Appl. Surf. Sci., 2021, 536, 147718 CrossRef CAS.
- K. Shayan and A. Nowroozi, “Boron nitride nanotubes for delivery of 5-fluorouracil as anticancer drug: a theoretical study”, Appl. Surf. Sci., 2018, 428, 500–513 CrossRef CAS.
- A. Soltani, M. T. Baei, E. T. Lemeski, S. Kaveh and H. Balakheyli, “A DFT study of 5-fluorouracil adsorption on the pure and doped BN nanotubes”, J. Phys. Chem. Solids, 2015, 86, 57–64 CrossRef CAS.
- F. Safdari, H. Raissi, M. Shahabi and M. Zaboli, “DFT calculations and molecular dynamics simulation study on the adsorption of 5-fluorouracil anticancer drug on graphene oxide nanosheet as a drug delivery vehicle”, J. Inorg. Organomet. Polym. Mater., 2017, 27, 805–817 CrossRef CAS.
- N. Wazzan, K. A. Soliman and W. A. Halim, “Theoretical study of gallium nitride nanocage as a carrier for 5-fluorouracil anticancer drug”, J. Mol. Model., 2019, 25, 1–19 CrossRef CAS PubMed.
- M. H. Mohammed and F. H. Hanoon, “Theoretical prediction of delivery and adsorption of various anticancer drugs into pristine and metal-doped graphene nanosheet”, Chin. J. Phys., 2020, 68, 578–595 CrossRef CAS.
- R. G. Parr, R. A. Donnelly, M. Levy and W. E. Palke, “Electronegativity: the density functional viewpoint”, J. Chem. Phys., 1978, 68(8), 3801–3807 CrossRef CAS.
- S. Zhao, W. Kang and J. Xue, “MXene nanoribbons”, J. Mater. Chem. C, 2015, 3(4), 879–888 RSC.
- J. Fatheema, M. Fatima, N. B. Monir, S. A. Khan and S. Rizwan, “A comprehensive computational and experimental analysis of stable ferromagnetism in layered 2D Nb-doped Ti3C2 MXene”, Phys. E, 2020, 124, 114253 CrossRef CAS.
- A. Adnan, R. Lam, H. Chen, J. Lee, D. J. Schaffer, A. S. Barnard, G. C. Schatz, D. Ho and W. K. Liu, “Atomistic simulation and measurement of pH dependent cancer therapeutic interactions with nanodiamond carrier”, Mol. Pharmaceutics, 2011, 8(2), 368–374 CrossRef CAS PubMed.
- M. Rouhani, “Density functional theory study towards capability of Ga-doped boron nitride nanosheet as a nanocarrier for 3-allyl-2 selenohydantoin anticancer drug delivery”, Phys. E, 2021, 126, 114437 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra02399d |
| This journal is © The Royal Society of Chemistry 2024 |