
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectronic, magnetic and optical properties of the charge-disproportionated YNiO3 compound calculated using the GGA+U method
Edjan Alves da Silvaa,
Samuel Domenech de Candidob and
Miguel Abbate *c
*c
aInstituto de Fisica, Universidade Federal do Rio Grande do Sul, Av. Bento Gonçalves 9500, 91501-970 Porto Alegre, RS, Brazil
bInstituto Federal de Educação, Ciência e Tecnologia, Campus Campo Grande, Rua Taquari 831, 79100-510 Campo Grande, MS, Brazil
cDepartamento de Fisica, Universidade Federal do Parana, Caixa Postal 19044, 81531-990 Curitiba, PR, Brazil. E-mail: miguel@fisica.ufpr.br
First published on 7th June 2024
Abstract
The electronic, magnetic and optical properties of YNiO3 were studied using the GGA+U method. This compound presents charge disproportionation with two nonequivalent Ni ion sites, namely, Ni1 with Ni(3−δ)+ and Ni2 with Ni(3+δ)+. A band-structure calculation was performed for the observed P21/n monoclinic phase; a 2 × 1 × 2 supercell with 80 atoms was used to reproduce the observed magnetic ordering. The density of states (DOS) calculation clearly showed the bonding differences between the Ni1 and the Ni2 sites; the Ni1 site has a more ionic character, whereas the Ni2 site presents a more covalent character. The band-structure results correspond to an insulator phase with a band gap of about 0.5 eV. The magnetic ordering is antiferromagnetic and the magnetic moments are about 1.24 μB for Ni1 and around 0.00 μB for Ni2. The calculated magnetic ordering results are in good agreement with neutron diffraction results. The calculated optical conductivity is similar to previous experimental data.
1 Introduction
The rare-earth nickelates present a broad range of very interesting physical properties. These include, for instance, a metal–insulator transition, diverse structural and magnetic transitions, as well as charge disproportionation.1–3 This variety is partly related to the interplay between the electronic, magnetic and structural degrees of freedom in these compounds. In addition, the highly correlated electronic structures of the rare-earth nickelates adds to the complexity of their physical properties. To this end, the study of the relationship between their electronic structures and their physical properties is highly desirable. The aim of this work is to study the electronic structure of the YNiO3 compound using the GGA+U method. The theoretical results are compared to the experimental electronic, magnetic and optical properties of this compound.The YNiO3 oxide presents a metal–insulator phase transition around 582 K, which also involves structural and magnetic ordering changes.4–6 In the low-temperature phase, the YNiO3 compound is an antiferromagnetic insulator (AFM-I); the electrical conductivity and the magnetic susceptibility results reflect electron-correlation effects.7 The crystal structure in this phase is monoclinic with space group P21/n;5 the schematic representation of the crystalline structure of YNiO3 is shown in Fig. 1. The AFM-I phase is characterized by a distortion of the NiO6 octahedra, which gives rise to two nonequivalent Ni sites: Ni1 sites with larger Ni–O bonds and Ni2 sites with shorter Ni–O bonds. This difference was attributed to charge disproportionation with charge ordering leading to Ni(3−δ)+ and Ni(3+δ)+ ionic states.5,8,9 Hereafter, all the calculations and comparisons will be only concerned with the low-temperature AFM-I phase.
 | ||
| Fig. 1 Schematic VESTA representation of the crystalline structure of YNiO3 with shorter and larger Ni–O bonds indicated by orange and dark green octahedra, respectively.10 | ||
The electronic structure of YNiO3 was studied using soft X-ray resonant magnetic powder diffraction,11 X-ray absorption spectroscopy (XAS)12 and optical absorption.7 The magnetic and structural properties of this oxide were investigated using synchrotron radiation diffraction,13 neutron and synchrotron radiation diffraction,5,14 and electron spin resonance (ESR) and susceptibility.15,16 The electronic structure of YNiO3 was calculated using several band-structure methods, including: LDA,17 LDA+U,18 LSDA+U,19 GGA+U,20 and the Hartree–Fock approach.21 Recent results include studies of the optical spectra,22 and additional band-structure calculations.23
Considerable insight on the properties of YNiO3 was obtained from the above band structure and by modeling Hamiltonian models. But a comprehensive comparison of the calculations and the experimental results is still lacking and would be desirable. This work presents the electronic structure of YNiO3 calculated by the GGA+U method. The results are in reasonable agreement with the available experimental data, and help to explain the electronic, magnetic and optical properties of this compound. The results are also interpreted in terms of the configuration interaction (CI) scheme of highly correlated pd models. Finally, the interplay between the structural, charge and spin degrees of freedom is discussed using the CI language.
2 Calculation details
Calculations for the AFM-I phase at room temperature were performed using the WIEN2k package,24 mainly because most of the available experimental results were obtained under this condition. This program is based in the full-potential linearized augmented plane wave (LAPW) method. The GGA exchange–correlation potential was obtained using the PBEsol scheme,25 and the on-site Hubbard parameter U was set to 1.5 eV. This value was chosen because it yields the best overall agreement with the experimental results. We also note that this value is in line with those previously used for this compound.19,20,26The spin–orbit splitting of the Ni 3d orbitals was included to determine the magnetic ordering. The spin–orbit coupling determines the orientation of the spin, so it is crucial for the calculation of the magnetic ordering. But the value of the coupling constant (about 0.04 eV for Ni) is relatively small, and does not affect much the electronic structure or the physical properties. The calculation of the magnetic ordering was unconstrained, and all the magnetic moments were allowed to rotate, until the criterion of total energy convergence was achieved.
The crystal structure was monoclinic and the space group was P21/n. The lattice parameters are listed in Table 1; the experimental values were retrieved from Alonso et al.,5 and the theoretical parameters were obtained by a total energy calculation. The cell volume was reduced about 2.8% from 211.85 Å3 (experimental) to 205.98 Å3 (calculated).
| Experimental | Calculated | |
|---|---|---|
| a | 5.17932 | 5.13105 |
| b | 5.51529 | 5.46389 |
| c | 7.41656 | 7.34744 |
The nonequivalent atomic positions are given in Table 2; the experimental results were retrieved from Alonso et al.5 and the theoretical positions were obtained by an atomic force calculation (the convergence criteria for the force was 1 mRy Bohr−1). The experimental and calculated average Ni–O bond lengths are in excellent agreement: 1.994 Å (exper.) vs. 1.993 Å (theor.) at the Ni1 site, and 1.923 Å (exper.) vs. 1.922 Å (theor.) at the Ni2 site.
| Atom | Experimental | Calculated | ||||
|---|---|---|---|---|---|---|
| x | y | z | x | y | z | |
| Y | 0.9816 | 0.0729 | 0.2502 | 0.9796 | 0.0735 | 0.2505 |
| Ni1 | 0.5000 | 0.5000 | 0.0000 | 0.5000 | 0.5000 | 0.0000 |
| Ni2 | 0.5000 | 0.0000 | 0.5000 | 0.5000 | 0.0000 | 0.5000 |
| O1 | 0.0998 | 0.4705 | 0.2457 | 0.0977 | 0.4729 | 0.2504 |
| O2 | 0.6973 | 0.3080 | 0.0467 | 0.6942 | 0.3011 | 0.0472 |
| O3 | 0.1882 | 0.2038 | 0.9465 | 0.1952 | 0.2002 | 0.9501 |
The maximum wavevector of the plane-wave basis set was given by RMTKMAX = 7.0, the integration was performed using 500 k-points in the irreducible part of the Brillouin zone, the energy convergence criteria was set to 10−5 Ry, and the 2 × 1 × 2 super-cell was formed by 80 atoms (please note that the monoclinic cell contains four chemical formulas with 20 atoms).
3 Results and discussion
3.1 Band structure and band gap
Fig. 2 shows the projected density of states (DOS) of the YNiO3 compound, separated into the Ni1 (upper panel) and the Ni2 (lower panel) nonequivalent sites.5,13,14 These results correspond to a stable and fully converged antiferromagnetic insulating (AFM-I) phase. The DOS for each nonequivalent site are split into the majority (above) and minority (below) spin bands. The Fermi level (EF = 0 eV) separates the occupied states in the valence band and the unoccupied states in the conduction band. The Ni 3d and the O 2p bands present a strong covalent mixing from −7 to +3 eV, whereas the Y 4d bands exhibit a more ionic character from +5 to +9 eV.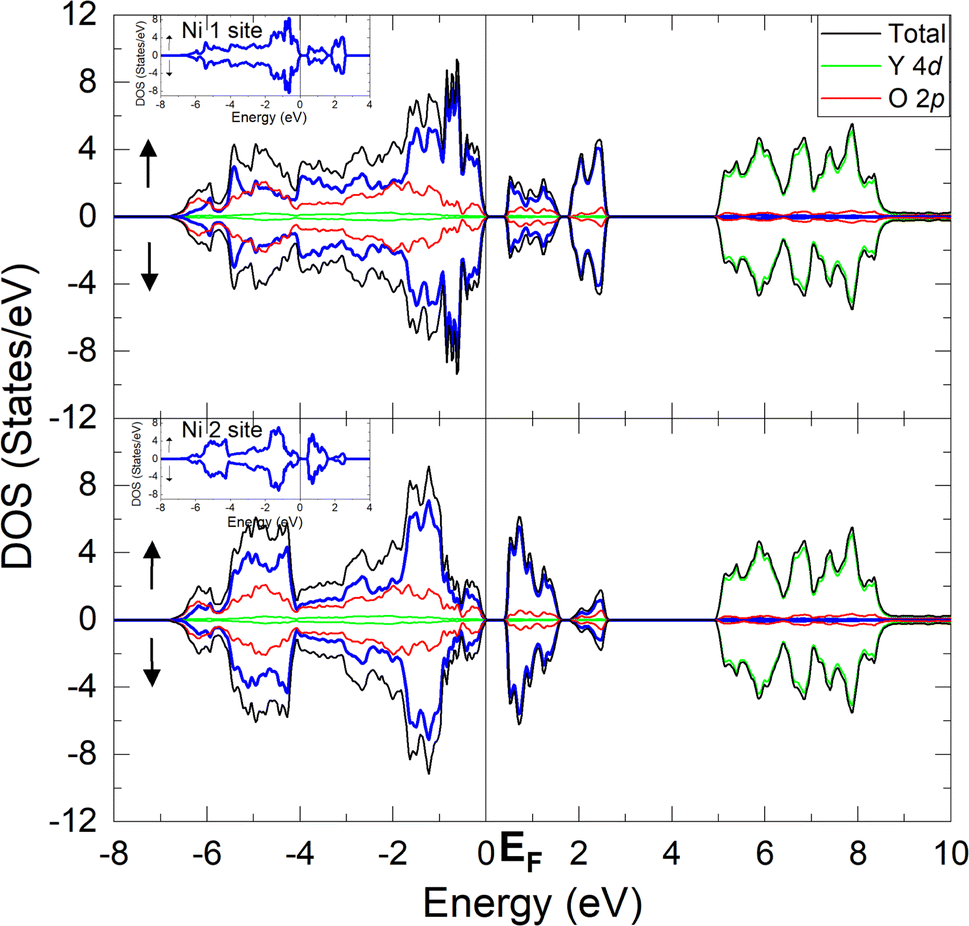 | ||
| Fig. 2 Projected density of states (DOS) of the YNiO3 compound separated into the Ni1 (upper panel) and the Ni2 (lower panel) nonequivalent sites. The DOS in each panel are split in the majority (above) and minority (below) spin bands. The Fermi level separates the occupied states in the valence band from the unoccupied states in the conduction band. | ||
The Ni 3d bands of the Ni1 and Ni2 nonequivalent sites show clear differences; see the inset in Fig. 2. In particular, the Ni 3d states contribute more to the bonding part of the Ni 3d–O 2p bands, from −7 to −4 eV, in the Ni2 site (lower panel). This means that, as expected, the Ni1 site is slightly more ionic, whereas the Ni2 site is more covalent; see below.
The calculated DOS presented here are in good agreement with previous results in the literature.8,18,19,27 The calculated band gap between the valence and the conduction band, about 0.5 eV, agrees with the optical band gap measured by Arima and Tokura,28 around 0.3 eV, although the present calculation slightly overestimated the value of the experimental band gap. We note that the value of the band gap found here is similar to those reported in recent studies.19,20,26
3.2 Magnetic moments and magnetic ordering
The calculated magnetic moments for the Ni ions at the Ni1 sites are about 1.24 μB and for those at the Ni2 sites they are around 0.00 μB. A comparison with the experimental values obtained by Alonso et al.5 using neutron diffraction is shown in Table 3. The results at the Ni1 site, 1.4 (exper.) vs. 1.24 μB (theor.), are in reasonable agreement, but the values at the Ni2 site, 0.7 (exper.) vs. 0.00 μB (theor.), are in clear disagreement. It is worth noting that recent calculations by other groups also yield a negligible magnetic moment at the Ni2 site.29,30 Additional work, both theoretical and experimental, would be desirable to reconcile this discrepancy.| Magnetic moments (μB) | ||
|---|---|---|
| Ni sites | Experimental5 | This work |
| Ni1 | 1.4 | 1.24 |
| Ni2 | 0.7 | 0.00 |
Fig. 3 compares the antiferromagnetic ordering of the YNiO3 oxide at room temperature obtained by Alonso et al.5 (top) with the result of the present calculation produced with the VESTA program10 (bottom). The small arrows at the Ni2 sites represent the orientation of the residual 0.0083 μB magnetic moments given by the calculation. Despite the discrepancy in the value of the magnetic moment at the Ni2 site, there is an overall good agreement in the relative orientation of these moments. Please note that the use of the 2 × 1 × 2 supercell structure with 80 atoms and the inclusion of the spin–orbit coupling of the Ni 3d orbitals were crucial to reproduce the experimental magnetic ordering.
 | ||
| Fig. 3 Antiferromagnetic ordering of the YNiO3 perovskite at room temperature obtained by Alonso et al.5 (top) compared to the present calculated result produced with the VESTA program10 (bottom). | ||
3.3 Optical conductivity
Fig. 4 compares the optical conductivity obtained by Arima and Tokura28 at room temperature and the calculated value of σ(ω) for the YNiO3 compound. The experimental optical conductivity of the YNiO3 compound presents a narrow band gap of about 0.3 eV,28 whereas the calculated result yields a slightly larger band gap of around 0.5 eV. Both the experimental and calculated optical conductivity values reflect the insulating character of the YNiO3 compound at room temperature. Please note the absence of the Drude peak in the spectra, which is a characteristic of a metallic phase. The increase of σ(ω) at higher energies, above 5 eV, is attributed to the onset of interband O 2p to Ni 3d transitions. | ||
| Fig. 4 Experimental optical conductivity extracted from Arima and Tokura (ref. 28) compared to the calculation of the YNiO3 compound. | ||
3.4 Configuration interaction description
The electronic state of the divalent Ni2+ ions in NiO oxide is mainly represented by an ionic 3d8 configuration.31,32 However, the trivalent Ni3+ ions in the LaNiO3 compound are not dominated by the ionic 3d7 configuration, but rather by the covalent 3d8![[L with combining low line]](https://www.rsc.org/images/entities/char_004c_0332.gif) configuration, where corresponds to a ligand (O 2p) hole.33,34 This means that the third hole does not go to the Ni 3d states, as expected in an ionic approximation, but rather to the O 2p orbitals, which reflects the larger covalent contribution to the bonding in this material. In terms of the highly correlated pd models used to study these materials, this corresponds to the so-called negative charge-transfer regime.35
configuration, where corresponds to a ligand (O 2p) hole.33,34 This means that the third hole does not go to the Ni 3d states, as expected in an ionic approximation, but rather to the O 2p orbitals, which reflects the larger covalent contribution to the bonding in this material. In terms of the highly correlated pd models used to study these materials, this corresponds to the so-called negative charge-transfer regime.35
In the LaNiO3 compound, the oxygen octahedra are similar and the 3d8 configuration is uniformly spread throughout the crystal. In the YNiO3 oxide, there is a structural distortion in the octahedra, which produces two nonequivalent Ni sites. The ensuing charge disproportionation is the result of a charge redistribution between the Ni(3−δ)+ site and the Ni(3+δ)+ site. The charge redistribution does not involve the strongly bound Ni 3d states, but rather the loosely bound hole in the O 2p orbitals. In particular, the Ni1 site (with a larger octahedron and weaker Ni 3d–O 2p interactions) is better described by a 3d8 configuration, whereas the Ni2 sites (with a smaller octahedron and stronger Ni 3d–O 2p interactions) is mainly given by a 3d82 configuration.26,36
The DFT band-structure method is an independent particle approach, but it is a first-principles method and takes into account translational invariance. On the other hand, the highly correlated pd models consider many-body effects, but they are based on adjustable parameters and are often applied to small systems. This discussion illustrates that they have complementary advantages and limitations. In the present work, the first method is mostly used because it provides straightforward results that can be compared with the experimental results.
3.5 Structural, charge and spin interactions
The YNiO3 material presents a clear interaction between the structural, charge and spin degrees of freedom. The structural distortion produces a Ni1 site at the larger octahedra with larger Ni–O bonds, and a Ni2 site at the smaller octahedra with smaller Ni–O bonds. This results in charge disproportionation with a more ionic Ni1 site with a 3d8 configuration, and a more covalent Ni2 site with a 3d82 configuration. The spin at the Ni1 site, with the 3d8 configuration, corresponds to a S = 1 high-spin state, whereas the spin at the Ni2 site, with the 3d82 configuration, is given by a S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O low-spin state.26,36 In turn, this helps to explain the calculated 1.24 μB magnetic moment at the Ni1 site, and the 0.00 μB magnetic moment at the Ni2 site. For an easier reference, the relationships between the Ni–O bonds, the electronic configuration and the spin state for each nonequivalent site are listed in Table 4.
O low-spin state.26,36 In turn, this helps to explain the calculated 1.24 μB magnetic moment at the Ni1 site, and the 0.00 μB magnetic moment at the Ni2 site. For an easier reference, the relationships between the Ni–O bonds, the electronic configuration and the spin state for each nonequivalent site are listed in Table 4.
| Degrees of freedom | |||
|---|---|---|---|
| Ni sites | Ni–O bonds | Configuration | Spin |
| Ni1 | Larger | 3d8 | S = 1 |
| Ni2 | Shorter | 3d82 |
S = 0 |
3.6 Charge disproportionation
Alonso et al.5 used the phenomenological Brown bond-valence model to estimate the Ni valence; they obtained two different values of the delta parameter: around δ = 0.38 at the Ni1 site and about δ′ = 0.17 at the Ni2 site. Later on, in the analysis of the different models used to interpret the neutron diffraction results, they concluded that δ = δ′ = 0.35.5 The present band-structure calculation gives a smaller estimate of around δ = 0.05. But, as discussed above, a double-cluster method predicts small changes at the Ni sites and larger changes at the ligand (O 2p) band36 (this is reasonable because O 2p–O 2p charge fluctuations cost less energy than Ni 3d–Ni 3d charge variations). Using this method, we estimate that the ligand contribution is about δ = 0.3.36 The combination of δ = 0.05 from the band structure and δ = 0.3 from the double-cluster calculations is similar to the value δ = 0.35 obtained from neutron diffraction data by Alonso et al.54 Summary and conclusions
In summary, we studied the electronic structure of the YNiO3 compound using the GGA+U method with U = 1.5 eV. In particular, we investigated the charge disproportionation between the nonequivalent Ni1 and Ni2 sites. The stable phase at room temperature is an antiferromagnetic insulating (AFM-I) state. The density of states (DOS) at the nonequivalent Ni1 and the Ni2 sites present clear differences; in particular, the Ni 3d–O 2p bonds are more ionic at the Ni1 site and more covalent at the Ni2 site. The calculated band-gap energy is in reasonable agreement with the experimental value. The calculated magnetic moment at the Ni1 site is in agreement with the experimental value, but there is a discrepancy in the calculated moment at the Ni2 site. Despite this disagreement, the calculated magnetic ordering agrees with the neutron diffraction results. We note that the use of a 2 × 1 × 2 supercell structure with 80 atoms and the inclusion of the Ni 3d spin–orbit coupling were crucial to obtain the orientation of the magnetic moments. The calculated optical conductivity is in reasonable agreement with the experimental result. The results are also interpreted in terms of the configuration interaction (CI) scheme of highly correlated pd models. Finally, the interactions between the structural, charge and spin degrees of freedom in YNiO3 are also discussed.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was partially supported by the Brazilian funding agencies Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES).References
- J. B. Torrance, P. Lacorre, A. I. Nazzal, E. J. Ansaldo and C. Niedermayer, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 8209–8212 CrossRef CAS PubMed.
- G. Catalan, Phase Transitions, 2008, 81, 729–749 CrossRef CAS.
- M. L. Medarde, J. Phys.: Condens. Matter, 1997, 9, 1679–1707 CrossRef CAS.
- J. L. García-Muñoz, J. Rodriguez-Carvajal, P. Lacorre and J. B. Torrance, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 4414 CrossRef PubMed.
- J. A. Alonso, J. L. García-Muñoz, M. T. Fernández-Díaz, M. A. G. Aranda, M. J. Martínez-Lope and M. T. Casais, Phys. Rev. Lett., 1999, 82, 3871–3874 CrossRef CAS.
- J.-S. Zhou and J. B. Goodenough, Phys. Rev. Lett., 2005, 94, 065501 CrossRef PubMed.
- T. Arima, Y. Tokura and J. B. Torrance, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 17006–17009 CrossRef CAS PubMed.
- I. I. Mazin, D. I. Khomskii, R. Lengsdorf, J. A. Alonso, W. G. Marshall, R. M. Ibberson, A. Podlesnyak, M. J. Martínez-Lope and M. M. Abd-Elmeguid, Phys. Rev. Lett., 2007, 98, 176406 CrossRef.
- U. Staub, G. I. Meijer, F. Fauth, R. Allenspach, J. G. Bednorz, J. Karpinski, S. M. Kazakov, L. Paolasini and F. d'Acapito, Phys. Rev. Lett., 2002, 88, 126402 CrossRef CAS PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- Y. Bodenthin, U. Staub, C. Piamonteze, M. García-Fernández, M. Martínez-Lope and J. Alonso, J. Phys.: Condens. Matter, 2011, 23, 036002 CrossRef CAS PubMed.
- A. Y. Ramos, C. Piamonteze, H. C. Tolentino, N. M. Souza-Neto, O. Bunau, Y. Joly, S. Grenier, J.-P. Itié, N. E. Massa and J. A. Alonso, et al., Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 045102 CrossRef.
- J. A. Alonso, M. J. Martinez-Lope, M. T. Casais, M. A. G. Aranda and M. T. Fernandez-Diaz, J. Am. Chem. Soc., 1999, 121, 4754–4762 CrossRef CAS.
- J. A. Alonso, M. J. Martínez-Lope, M. T. Casais, J. L. García-Muñoz and M. T. Fernández-Díaz, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 1756 CrossRef CAS.
- M. Causa, R. Sánchez, M. Tovar, J. Alonso and M. Martinez-Lope, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 024429 CrossRef.
- R. Sánchez, M. Causa, M. Tovar, J. Alonso and M. Martinez-Lope, J. Magn. Magn. Mater., 2004, 272–276, 390–391 CrossRef.
- X. Xu, X. Meng, C. Wang, F. Wu and G. Chen, J. Phys. Chem. B, 2004, 108, 1165–1167 CrossRef CAS.
- Y. Quan, V. Pardo and W. E. Pickett, Phys. Rev. Lett., 2012, 109, 216401 CrossRef PubMed.
- S. Yamamoto and T. Fujiwara, J. Phys. Chem. Solids, 2002, 63, 1347–1351 CrossRef CAS.
- A. Mercy, J. Bieder, J. Íñiguez and P. Ghosez, Nat. Commun., 2017, 8, 1677 CrossRef PubMed.
- T. Mizokawa, D. I. Khomskii and G. A. Sawatzky, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 11263–11266 CrossRef CAS.
- J. Bieder, A. Mercy, W.-Y. Tong and P. Ghosez, Phys. Rev. B, 2020, 102, 081111 CrossRef CAS.
- L. Binci, M. Kotiuga, I. Timrov and N. Marzari, Phys. Rev. Res., 2023, 5, 033146 CrossRef CAS.
- P. Blaha, K. Schwarz, G. Madsen, D. Kvasnicka and J. Luitz, WIEN2k: An Augmented Plane Wave Plus Local Orbitals Program for Calculating Crystal Properties, Technische Universität Wien, Wien, 2001, vol. 28 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. Varignon, M. N. Grisolia, J. Íñiguez, A. Barthélémy and M. Bibes, npj Quantum Mater., 2017, 2, 21 CrossRef.
- A. Hampel and C. Ederer, Phys. Rev. B, 2017, 96, 165130 CrossRef.
- T. h. Arima and Y. Tokura, J. Phys. Soc. Jpn., 1995, 64, 2488–2501 CrossRef CAS.
- J. Du, S.-L. Shang, Y. Wang, A. Zhang, S. Xiong, F. Liu and Z.-K. Liu, AIP Adv., 2021, 11, 015028 CrossRef CAS.
- J. Du, O. Malyi, S.-L. Shang, Y. Wang, X.-G. Zhao, F. Liu, A. Zunger and Z.-K. Liu, Mater. Today Phys., 2022, 27, 100805 CrossRef CAS.
- G. A. Sawatzky and J. W. Allen, Phys. Rev. Lett., 1984, 53, 2339–2342 CrossRef CAS.
- A. Fujimori and F. Minami, Phys. Rev. B: Condens. Matter Mater. Phys., 1984, 30, 957–971 CrossRef CAS.
- M. Abbate, G. Zampieri, F. Prado, A. Caneiro, J. M. Gonzalez-Calbet and M. Vallet-Regi, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 155101 CrossRef.
- E. Alves, H. Martins, S. Domenech and M. Abbate, Phys. Lett. A, 2019, 383, 2952–2956 CrossRef CAS.
- P. Kuiper, G. Kruizinga, J. Ghijsen, G. A. Sawatzky and H. Verweij, Phys. Rev. Lett., 1989, 62, 221–224 CrossRef CAS PubMed.
- R. J. Green, M. W. Haverkort and G. A. Sawatzky, Phys. Rev. B, 2016, 94, 195127 CrossRef.
| This journal is © The Royal Society of Chemistry 2024 |