
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Robust leishmanicidal upshot of some new diphenyl triazine-based molecules†
Anju Singha,
Mirza Adil Begb,
Samra Jamalc,
Arif Khand,
Abdur Rahmana,
Angamuthu Selvapandiyanb,
Syed Shafid and
Nasimul Hoda *a
*a
aDepartment of Chemistry, Drug Design and Synthesis Lab., Jamia Millia Islamia, Jamia Nagar, New Delhi 110025, India. E-mail: nhoda@jmi.ac.in; Fax: +0091-11-26985507; Tel: +0091-9910200655
bDepartment of Molecular Medicine, Jamia Hamdard, New Delhi 110062, India
cDepartment of Biotechnology, Jamia Hamdard, New Delhi 110062, India
dDepartment of Chemistry, SCLS, Jamia Hamdard, New Delhi 110062, India
First published on 17th July 2024
Abstract
Amongst the neglected tropical diseases, leishmaniasis alone causes 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths annually due to the protozoan parasite genus Leishmania. Existing therapies have serious drawbacks in safety, drug resistance, field-adapted application and cost. Therefore, new safer and shorter treatments are an urgent need of the time. Herein, we report the synthesis of fifteen novel diphenyl triazine and diphenyl triazine pyrimidine derivatives and their antileishmanial properties against Leishmania donovani, that causes fatal visceral leishmaniasis. Most of the synthesized analogues exhibited more than 90% inhibition against the promastigote stage of the parasite. Moreover, compounds T4 and T7 showed potent activity against extracellular promastigote (IC50 = 1.074 μM and IC50 = 1.158 μM) as compared to miltefosine (IC50 = 1.477 μM) and is nontoxic towards the host THP-1 macrophage cell line. Interestingly, compound T4 exhibited significant activity against amastigotes (7.186 μM) and induced the macrophages to prevent the survival of the parasite. Our results indicate that T4 represents a new structural lead for this serious and neglected disease.
000 deaths annually due to the protozoan parasite genus Leishmania. Existing therapies have serious drawbacks in safety, drug resistance, field-adapted application and cost. Therefore, new safer and shorter treatments are an urgent need of the time. Herein, we report the synthesis of fifteen novel diphenyl triazine and diphenyl triazine pyrimidine derivatives and their antileishmanial properties against Leishmania donovani, that causes fatal visceral leishmaniasis. Most of the synthesized analogues exhibited more than 90% inhibition against the promastigote stage of the parasite. Moreover, compounds T4 and T7 showed potent activity against extracellular promastigote (IC50 = 1.074 μM and IC50 = 1.158 μM) as compared to miltefosine (IC50 = 1.477 μM) and is nontoxic towards the host THP-1 macrophage cell line. Interestingly, compound T4 exhibited significant activity against amastigotes (7.186 μM) and induced the macrophages to prevent the survival of the parasite. Our results indicate that T4 represents a new structural lead for this serious and neglected disease.
1. Introduction
Leishmaniasis is an endemic protozoan disease and is one of the major health problems worldwide. It is caused by the genus Leishmania transmitted from infected persons to normal ones by the bite of female phlebotomine sand flies.1,2 It appears in three clinical forms: cutaneous (CL), mucocutaneous (MCL), and visceral leishmaniasis (VL).3,4 VL5 is the life threatening form of the disease due to the failure of the host immune system.6 As per reports, 200 million people reside in leishmaniasis endemic areas and at least 2 million cases are reported every year with leishmaniasis causing 20000–30000 deaths.7,8
An absolute treatment for this ailment is still lacking, which makes the quest for the discovery of new potential antileishmanial agents. Amphotericin B (Ampho B), a well-known antifungal drug works as an essential medication for leishmaniasis.1 In addition to it, other available antileishmanial drugs comprise: miltefosine (MTF), paromomycin (P) and antimonials (Sb(III)).9 Due to the increasing resistance and prolonged treatment of existing drugs, there is an immense need for the development of safer antileishmanial agents.
Among the scaffolds explored, quinoline derivatives have shown promising biological activities against various disease-causing parasites. Compound 1 (Fig. 1) passed in the Drugs for Neglected Diseases initiative (DNDi) pipeline for in vivo testing due to their better selectivity index as compared to MTF.10 Recently, the complex quinaldine derivative 2 (Fig. 1) was designed through virtual screening methods to inhibit infantum type 2 NADH dehydrogenase (NDH2). It experimentally proved the inhibition of the enzyme and displayed notable activity against L. infantum axenic amastigotes and promastigotes.11
 | ||
| Fig. 1 Structure of some known antileishmanial drugs.10 | ||
The potentiality of 1,3,5-triazine derivatives against leishmaniasis has been reported by several research groups.12–16 Triazine core moiety linked through an ether linkage of pentamidine resulted in the development of two antileishmanial compounds 2 (Fig. 2) with good activity against intracellular L. donovani amastigotes.16 Analogues of β-carboline-1,3,5-triazine have been reported to act against promastigote and amastigote forms of Leishmania amazonensis.17 1,3,4-thiadiazole analogues like compound 4 (Fig. 2) also showed very capable antileishmanial effects even at lower concentrations.18 1,2,4-triazino[5,6-b]indol-3-ylthio-1,3,5-triazine 1 (Fig. 2) displayed more than 90% inhibition against promastigote form of L. donovani.17 It was found to be the most active and least toxic with 20 and 10-fold more selectivity (S.I. = 56.61) in comparison to the standard drugs pentamidine and sodium stibogluconate, respectively.15 In addition, the compound 5 exhibited significant activity IC50 < 25 μM against promastigote as well as amastigote.19 A triazine dimer 3,3′-(((ethane-1,2-diylbis(azanediyl)) bis(4-(mesityloxy)-1,3,5-triazine-6,2-diyl)) bis(azanediyl))di benzonitrile, 3 (Fig. 2) was observed to display very potent in vitro and moderate in vivo antitrypanosomal activity.20 Interestingly, some natural products Meridianin G, Annomontine (Fig. 2) are potent antileishmanial agents.21
 | ||
| Fig. 2 Designing of diphenyl triazine, pyrimidine-based hybrids showing antileishmanial activity. | ||
Leishmaniasis is an infectious disease caused by protozoa and is the second most critical parasitic disease after malaria. The suggested drugs for leishmaniasis consist of triazole, chalcone, chromone, thiazole, thiosemicarbazone, indole, quinoline etc.22 Chloroquine and its derivatives, whose nuclear structure is composed of quinoline, have always fascinated chemists and biologists due to the diversity of their chemical and medicinal properties, for example antiparasitic, anticancer, antibacterial, antiviral, antifungal, antioxidant, Anti-asthmatic, antipsychotic, antiglaucoma and cardiovascular agents.23–25 The majority of anti-leishmanial agents act by interacting with key regulators including PTR-I, DHFR, LdMetAP1, MAPK, 14 α-demethylase and pteridine reductase-I, etc. Also, these tend to induce the production of ROS, which causes damage to parasites. In the present study the in vitro and ex vivo antileishmanial activity of a chloroquinolin inhibitors, namely 7-chloro-N, N-dimethylquinolin-4-amine was evaluated against L. infantum and L. amazonensis.26 The Results showed that the compound was highly effective against L. infantum and L. amazonensis, presenting a selectivity index of 154.6 and 86.4 against the promastigotes and of 137.6 and 74.3 against the axenic amastigotes respectively. Keeping these things into consideration, we attempted to incorporate different heterocyclic moieties, the synthesis and antileishmanial evaluation of new fifteen 1,2,4 Triazine, pyrimidine hybrid molecules.
2. Results and discussions
2.1. Chemistry
The target molecules were synthesized via multiple steps as represented in Scheme 1. In the first step benzil (1) was made to react with thiosemicarbazide (hydrazinecarbothioamide) 2 in water and ethanol in equal proportions to get 3 according to the reported literature.27 The compound 3 was treated with substituted chloro containing complex to get target compounds T1–T5. On the other hand, compounds 3 was treated with 4,6-dichloropyrimidine to get compound 4 in good yields. Further compound 4 was reacted with different nitrogen containing nucleophiles in presence of DMF as a reaction solvent, triethylamine as base to obtain the target molecules T6–T15. Column chromatography technique was used for the purification of all the molecules and 1H-NMR, 13C-NMR, ESI MS and elemental analysis for characterisation purposes. | ||
| Scheme 1 Synthesis of diphenyl triazine based compounds. Reagents and conditions: (i) EtOH, water 1:1, 80 °C, reflux, 16 h, (ii) [R1H {4,7 dichloroquinoline}] Et3N Acetone, reflux, 60 °C, 8 h. (iii) K2CO3, DMF, reflux, 100 °C, 10 h (iv) [R2H{Morpholine}] Et3N, DMF, reflux, 100 °C, 10 h. | ||
2.2. Biological evaluation
Each candidate was evaluated for its in vitro activity against reference strain (MHOM/IN/83/AG83) of extracellular promastigotes and intracellular amastigotes of L. donovani. The in vitro cytotoxicity assay was performed using human macrophage (cell line THP-1). Compounds, which showed good inhibitory activity against the promastigotes, standard antileishmanial, miltefosine and Ampho B were included in the study. We were very pleased to find that among the synthesised analogues T4 and T7 more potent against promastigotes and axenic amastigotes.
| Entity | Compound | % Inhibition of L. donovani promastigotes |
|---|---|---|
| 1 | T1 | 66.78 |
| 2 | T2 | 71.02 |
| 3 | T3 | 83.43 |
| 4 | T4 | 99.67 |
| 5 | T5 | 67.93 |
| 6 | T6 | 43.86 |
| 7 | T7 | 99.89 |
| 8 | T8 | 73.70 |
| 9 | T9 | 29.81 |
| 10 | T10 | 22.41 |
| 11 | T11 | 32.15 |
| 12 | T12 | 21.86 |
| 13 | T13 | 18.54 |
| 14 | T14 | 22.76 |
| 15 | T15 | 28.82 |
| Compound | L. donovani promastigotes | L. donovani amastigotes |
|---|---|---|
| IC50 (μM) | IC50 (μM) | |
| T4 | 1.074 | 7.186 |
| T7 | 1.158 | nt |
| MTF | 1.477 | |
| Ampho B | 1.038 |
 | ||
| Scheme 2 Reagents and conditions: K2CO3, DMF, reflux 100 °C, overnight. | ||
| Compounds | Percentage reduction of parasites | ||||
|---|---|---|---|---|---|
| 100 μM | 50 μM | 25 μM | 12.5 μM | 6.25 μM | |
| a T4 and T7 reduced the infection of macrophages, with highest effect observed at 100 μM. Antiamastigote activity of test molecules against Leishmania (donovani) decreasing gradually with decrease in concentrations. This effect can be correlated to the anti-promastigote activity of molecules. | |||||
| T4 | 78.0 | 74 | 70 | 68.80 | 66 |
| T7 | 72 | 70.6 | 66 | 62 | 56 |
| Ampho B | 84.2 | 82.5 | 79.44 | 76 | 74 |
| Milte | 76 | 74 | 73 | 72 | 68 |
 | ||
| Scheme 3 Reagents and conditions: (i) 3-chloropropanoyl chloride, AlCl3, CH2Cl2, 24 h, room temperature. | ||
 | ||
| Scheme 4 Reagents and conditions. (i) Benzene, 83 °C reflux, 1.5 h (ii) acetic acid, 110 °C reflux 4–5 h; (iii) POCl3, 110 °C,18 h. | ||
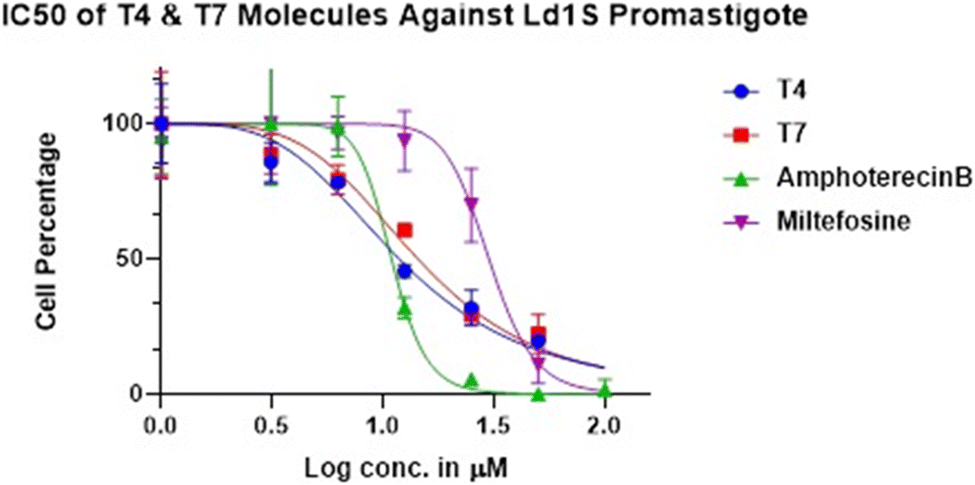 | ||
| Fig. 3 IC50 of T4 and T7 molecules against the promastigotes. | ||
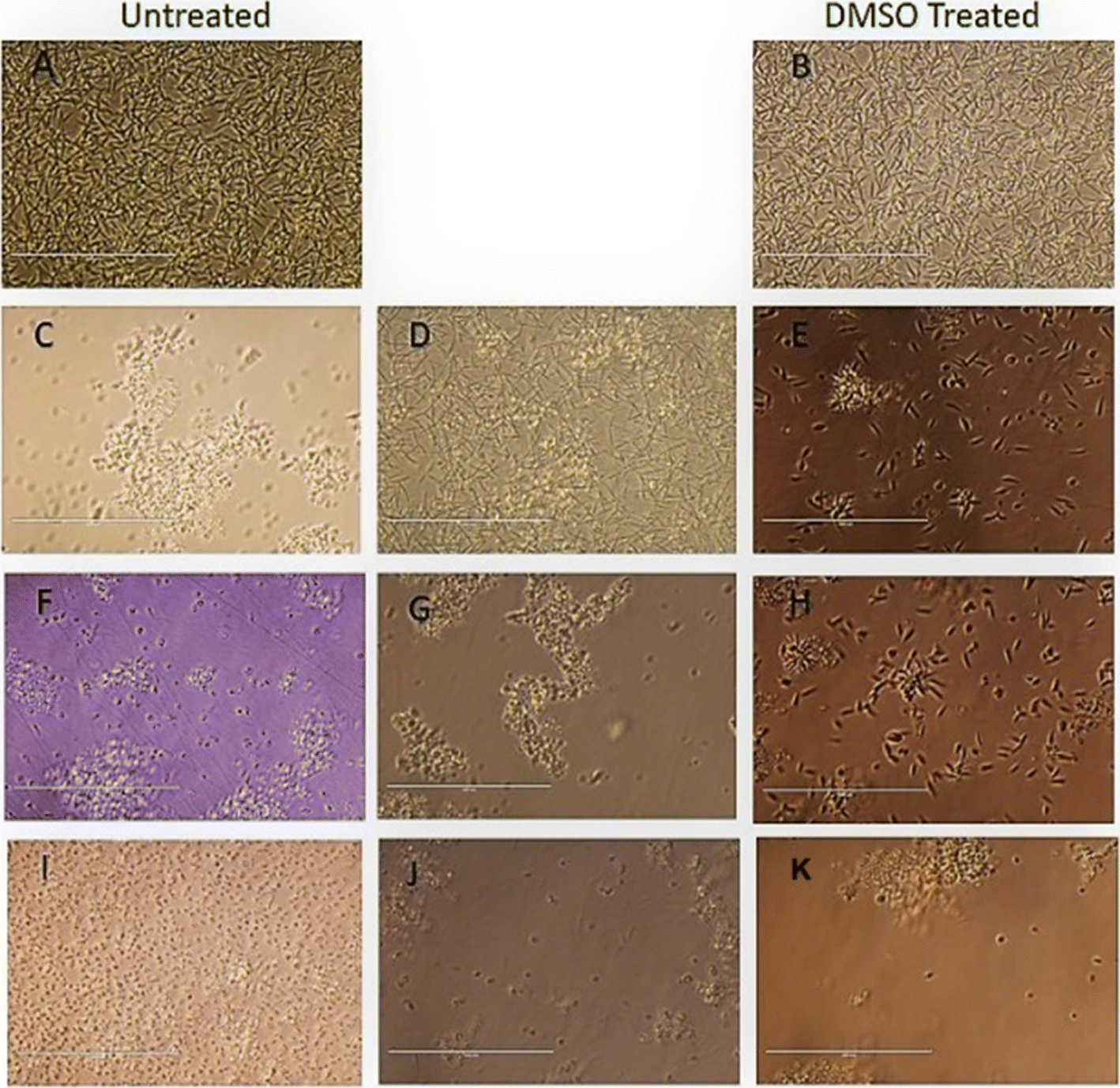 | ||
| Fig. 4 Morphological Analysis of promastigote stage cells treated with compound T4 and MTF, compared to the untreated control (A) and DMSO treated control (B). Exponential-phase promastigotes (1 × 106 cells per mL) were incubated with 25 μm, 50 μm and 100 μm and analysed by light microscopy (40× magnification). (A) Untreated control (B) DMSO Treated cells (C–E) Ampho B, (F–H) T4 and T7 (I–K) at 25, 50, and 100 μM respectively. Scale bar in all is 10 μm. | ||
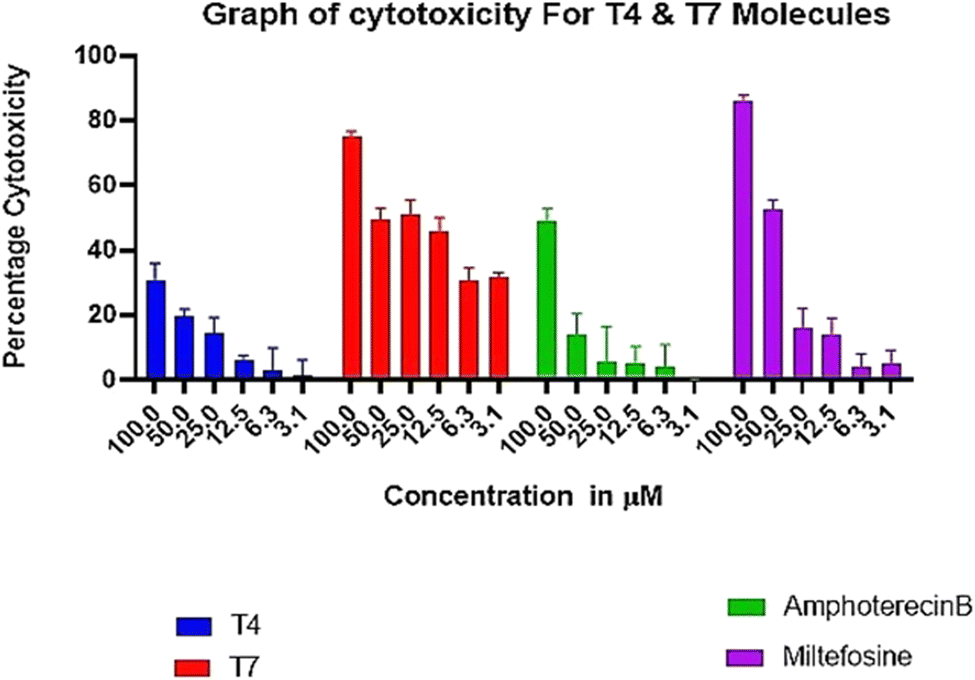 | ||
| Fig. 5 Absence of adverse toxicity of test compounds T4 on THP-1 cells up to 3.125–100 μM in comparison to MTF and Ampho B. | ||
3. Micrographs of L. donovani infected macrophages treated with compound T4 and standard drugs Ampho B and miltefosine
We find that the diphenyl triazine and diphenyl-triazine-pyrimidine showed better activity against promastigotes and amastigotes. Unfortunately, compound T7 displayed cytotoxicity on THP-1 cell line. After that we evaluated the compound T4 on the infected THP-1cells, which was compared with standard antileishmanials, Miltefosine and Ampho B at different concentrations (Fig. 6). Interestingly, we found the compound T4 showed great effect against the promastigotes better than that of well-established antileishmanials. Diphenyl-triazine analogues showing robust potency against the amastigotes of the parasite as well as L donovani promastigotes (IC50 = 1.074 μM), (IC50 = 7.186 μM). Compound T4 can serve as lead for the development of more active antileishmanial agents.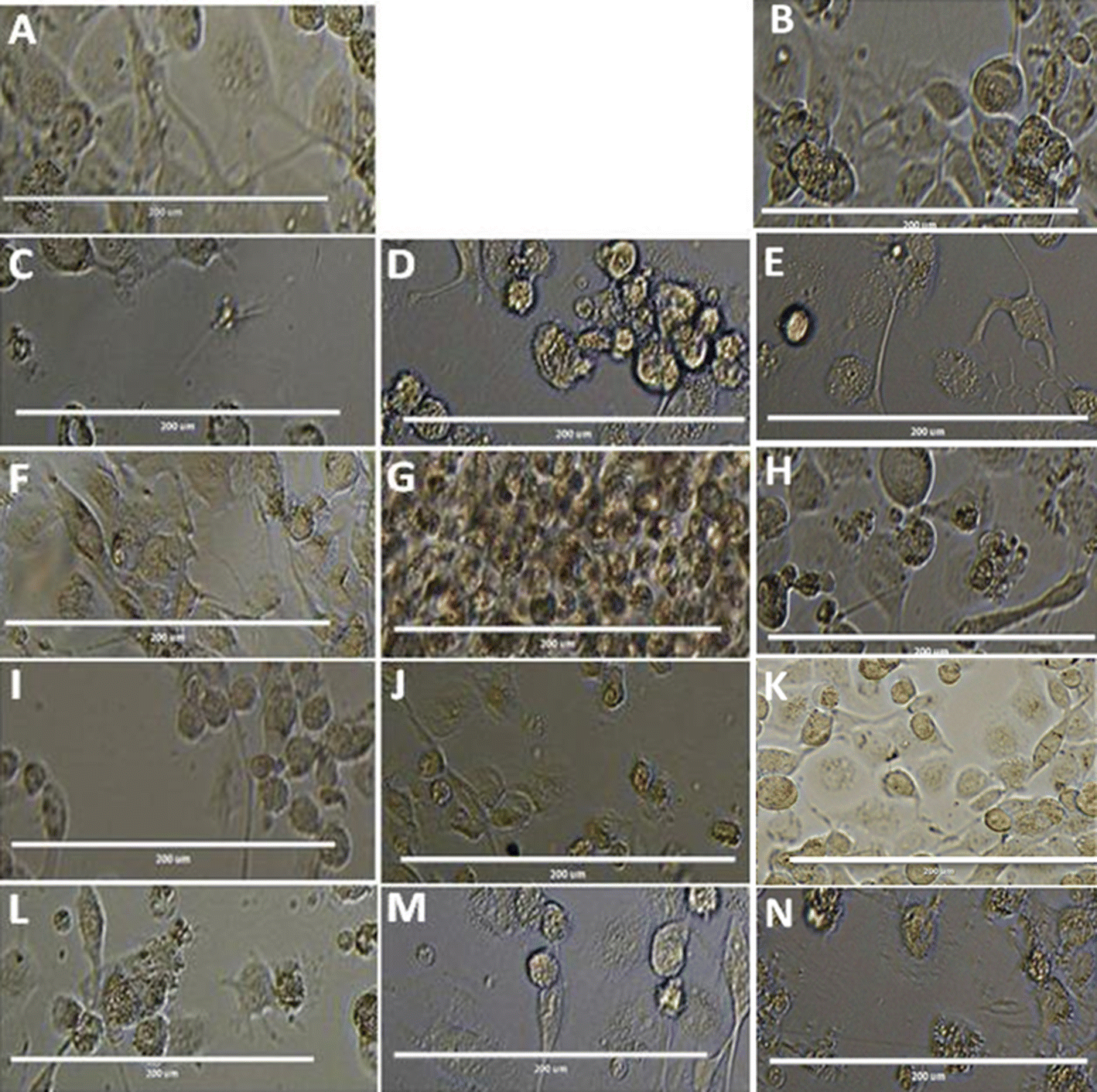 | ||
| Fig. 6 Morphological analysis of compound T4 on THP-1 cells along with controls AmphoB and Milte with different concentrations of Test molecules where (A) untreated control (B) DMSO treated cells (C–H) T4 treated in 100, 50, 25, 12.5, 6.25, 3.125 μM conc. (I–K) Ampho treated cells in 100, 50, 25 μM (L–N) Milte treated cells in 100, 50, 25 μM concentrations respectively. Scale bars in all is 10 μm. | ||
3.1. Infection and treatment of L. donovani
To evaluate the anti-leishmanial effect of the compound T4 and T7 on intracellular L. donovani parasites, macrophage was infected with stationary -phase L. donovani for 6 h. The cells were then washed with saline to remove any non-internalised parasites. To assess the effect of compounds T4 and T7 on intracellular parasites at early stages of infection, macrophages were subsequently treated with T4 and T7 at concentrations of 100, 50, 25, 12.5, or 6.25 μM (refer to Table 3 and Fig. 7) for 6, 24, or 48 hours. Observation of treated macrophages revealed that both T4 and T7 were capable of inhibiting the growth of Leishmania L. donovani species, indicating the potential of these compounds as chemotherapeutic agents for the treatment of L. donovani leishmaniasis. | ||
| Fig. 7 Treatment with internalized parasites. (A) Untreated (B) DMSO, (C) Ampho B, (D) MTF and (E and F) T4 at 100, 50 μM respectively. The scale bar in all is100 μm. | ||
4. Conclusions
A series of diphenyl-triazine core moieties was synthesized and evaluated for their antileishmanial efficacy. The compounds were synthesized using organic chemistry methods and their structures were evaluated using NMR and mass spectrometry techniques. These diphenyl-triazine hybrids were screened against the promastigote stage of L. donovani. Compounds T4 and T7 demonstrated significant inhibition of the promastigote stage of the parasites with >90% inhibition and IC50 values of 1.074 and 1.158 μM, respectively. Conversely, compounds T12, T13, T14 and T15 exhibited minimal activity against the parasite. Compound T4, which incorporates (3,4-dimethoxyphenyl) propanone attached to the diphenyl-triazine core moiety, also effectively inhibited the parasite at intracellular amastigotes, with an IC50 of 5.18 μM. Assessment of the toxicity profile of T4 on the human macrophage cell line THP-1 indicated its non-toxic nature compared to the marketed MTF. In conclusion, compounds such as T4 show promise as lead candidates for the development of more potent antileishmanial agents.5. Experimental section
5.1. Chemistry
The chemicals, reagents and solvents used during the synthesis and characterization of the compounds were from Sigma-Aldrich. 1H NMR and 13CNMR spectra were observed on Bruker 126, 75 and 500, 399, 300 MHz spectrophotometer using deuterated solvents (CDCl3, δ 7.26; DMSO-d6 δ 2.54) and multiplicities of NMR signals were designated as s (singlet), d (doublet), dd (double doublet), t (triplet), q (quartet), m (multiplet, for unresolved lines). Chemical shifts were showed in δ (ppm) and tetramethylsilane was used as the internal standard. Mass spectra were recorded on UPLC XEVO G2-XS QTOF Spectrometer (HRMS) instrument. Thin layer chromatography was performed with Merck silica gel (60–120 and F254) aluminum-coated sheets of 0.25 mm thickness. Spots on these were observed in short (254 nm) with ultraviolet light and long (365 nm) wavelengths. Elemental analyses were obtained on Elementar Vario analyser. Elemental analyses of the compounds were found to be within ±0.4% of the theoretical values. The purity of tested compounds was >95%.15.1.2.1. 7-Chloro-4-((5,6-diphenyl-1,2,4-triazin-3-yl)thio)quinoline (T1). Yellow, m.p. 250–252 °C, 60% yield was obtained after column chromatography. HRMS (ESI+): calcd for [C24H15ClN4S + H]+: m/z = 426.07 found: 427.0834. 1H NMR (400 MHz) δ 8.96 (d, J = 4.4 Hz 1H), 8.29 (d, J = 9.0 Hz, 1H), 8.19 (d, J = 2.1 Hz, 1H), 7.89 (d, J = 4.4 Hz, 1H), 7.54 (dd, J = 9.0, 2.1 Hz, 1H), 7.48 (d, J = 8.0 Hz, 2H), 7.41–7.34 (m, 3H), 7.32 (dd, J = 7.8, 1.4 Hz, 3H), 7.25–7.22 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 169.01, 155.89, 154.81, 150.80, 134.78, 134.52, 131.31, 129.80, 129.74, 129.31, 128.96, 128.82, 128.68, 128.56, 128.38, 127.91, 126.89. Anal. calcd for C24H15ClN4S C, 67.52; H, 3.54; N, 13.12 found C, 66.51; H, 4.01; N, 13.02.
5.1.2.2. Ethyl 4-((5,6-diphenyl-1,2,4-triazin-3-yl)thio)-6-methoxyquinoline-3-carboxylate (T2). Light yellow, m.p. 200–202 °C, 55% yield was obtained after column chromatography. HRMS (ESI+): calcd for [C28H22N4O3S + H]+: m/z = 494.14 found: 495.1500 1H NMR (500 MHz, CDCl3) δ 9.23 (s, 1H), 8.14 (d, J = 10.0 Hz, 1H), 7.90 (d, J = 2.6 Hz, 1H), 7.50 (d, J = 10.0 Hz, 3H), 7.44–7.32 (m, 5H), 7.26 (dd, J = 15.4, 7.7 Hz, 2H), 4.32 (q, J = 7.1 Hz, 2H), 3.91 (s, 3H), 1.27 (t, J = 5.0 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 169.53, 165.79, 159.41, 155.57, 154.38, 147.19, 145.68, 135.85, 134.93, 134.60, 131.58, 131.16, 130.73, 129.89, 129.80, 129.67, 129.62, 129.29, 128.65, 128.48, 124.54, 104.06, 61.94, 55.79, 14.04. Anal. calcd for C28H22N4O3S C, 68.00; H, 4.48; N, 11.33; found C, 67.50; H, 4.07; N, 11.03.
5.1.2.3. Ethyl 4-((5,6-diphenyl-1,2,4-triazin-3-yl)thio)-6-methylquinoline-3-carboxylate (T3). White, m.p. 210–212 °C, 62% yield was obtained after column chromatography. HRMS (ESI+): calcd for [C28H22N4O2S + H]+: m/z = 478.15 found: 479.1561. 1H NMR (500 MHz, CDCl3) δ 9.32 (s, 1H), 8.44 (s, 1H), 8.17 (d, J = 8.5 Hz, 1H), 7.71 (d, J = 8.6 Hz, 1H), 7.50 (d, J = 8.2 Hz, 1H), 7.43–7.33 (m, 1H), 7.27 (dd, J = 14.2, 6.3 Hz, 1H), 4.32 (q, J = 7.1 Hz, 1H), 2.58 (s, 1H), 1.27 (t, J = 7.1 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 169.55, 165.58, 155.57, 154.40, 148.60, 138.95, 134.96, 134.63, 134.09, 131.13, 129.80, 129.65, 129.51, 129.30, 129.26, 128.64, 128.47, 125.43, 61.99, 22.02, 14.03. Anal. calcd for C28H22N4O2S C, 70.27; H, 4.63; N, 11.71; found C, 71.03; H, 4.23; N, 12.31.
5.1.2.4. 1-(3,4-Dimethoxyphenyl)-3-((5,6-diphenyl-1,2,4-triazin-3-yl)thio)propan-1-one (T4). Pale yellow, m.p. 210–212 °C, 71% yield was obtained after column chromatography. HRMS (ESI+): calcd for [C26H23N3O3S] m/z = 457.15 found: 480.1518 and 496.1119 [C26H23N3O3S + Na]+: [C26H23N3O3S + K]+. 1H NMR (500 MHz, CDCl3) δ 7.63 (dd, J = 8.3, 1.4 Hz, 2H), 7.58 (d, J = 10.5 Hz, 1H), 7.55 (d, J = 3.7 Hz, 1H), 7.53 (s, 1H), 7.46–7.41 (m, 2H), 7.39 (d, J = 7.7 Hz, 2H), 7.34 (t, J = 7.7 Hz, 2H), 7.29 (s, 1H), 6.87 (d, J = 10.0 Hz, 1H), 3.96 (s, 6H), 3.76 (t, J = 6.9 Hz, 3H), 3.62 (t, J = 6.9 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 196.68, 170.63, 155.71, 153.92, 153.48, 149.09, 135.26, 135.14, 130.93, 129.86, 129.82, 129.47, 129.31, 128.62, 128.52, 122.86, 110.15, 110.07, 56.09, 56.04, 38.14, 25.54. Anal. calcd for C26H23N3O3S C, 68.25; H, 5.07; N, 9.18; found C, 67.65; H, 5.02; N, 10.00.
5.1.2.5. 2-((5,6-Diphenyl-1,2,4-triazin-3-yl)thio)-N-(2-oxo-2H-chromen-4-yl)acetamide (T5). Light yellow, m.p. 200–202 °C, 52% yield was obtained after column chromatography. HRMS (ESI+): calcd for [C26H18N4O3S + H]+: m/z = 466.11 found: 467.1179 and [C26H18N4O3S + Na]+: 489.1132. 1H NMR (500 MHz, CDCl3) δ 9.61 (s, 1H), 8.70 (s, 1H), 7.57 (t, J = 6.5 Hz, 4H), 7.51 (d, J = 10.0 Hz, 1H), 7.47–7.42 (m, 3H), 7.39 (d, J = 5.0 Hz, 1H), 7.36 (s, 1H), 7.34 (s, 1H), 7.32 (s, 1H), 7.30 (s, 1H), 7.29 (s, 1H), 4.21 (s, 2H). 13C NMR (126 MHz, CDCl3) δ 168.83, 167.79, 158.36, 156.30, 154.84, 150.11, 134.87, 134.73, 131.24, 129.91, 129.75, 129.70, 129.47, 128.65, 128.57, 127.84, 125.09, 124.18, 123.88, 119.73, 116.35, 35.41. Anal. calcd for C26H18N4O3S C, 66.94; H, 3.89; N, 12.01; found C, 67.01; H, 3.59; N, 11.89.
5.1.4.1. 4-(6-((5,6-Diphenyl-1,2,4-triazin-3-yl)thio)pyrimidin-4-yl)morpholine (T6). Yellow, m.p. 198–200 °C, 50% yield was obtained after column chromatography. ESI m/z [M + H]+ calcd 428.14 found 428.9. 1H NMR (399 MHz, cdcl3) δ 8.55 (s, 1H), 7.54 (d, J = 0.7 Hz, 1H), 7.52 (d, J = 1.2 Hz, 2H), 7.43 (d, J = 3.2 Hz, 1H), 7.41 (d, J = 1.5 Hz, 1H), 7.39 (s, 2H), 7.37 (d, J = 1.6 Hz, 1H), 7.34 (s, 1H), 7.32 (s, 1H), 7.26 (s, 1H), 3.752 (t, J = 4.8 Hz,4H), 3.66 (t, J = 5.2 Hz,4H). 13C NMR (126 MHz, cdcl3) δ 170.67, 163.97, 159.94, 157.83, 156.98, 133.19, 131.85, 131.79, 131.37, 130.72, 130.57, 106.68, 68.43, 46.18. Anal. calcd for C23H20N6OS C, 64.47; H, 4.70; N, 19.61; found C, 64.42; H, 5.20; N, 19.42.
5.1.4.2 3-((6-(4-Methylpiperidin-1-yl)pyrimidin-4-yl)thio)-5,6-diphenyl-1,2,4-triazine (T7). Chock white, m.p. 250–252 °C, 53% yield was obtained after column chromatography. ESI m/z [M + H]+ calcd 440.18 found 442.0. 1H NMR (399 MHz, cdcl3) δ 8.52 (s, 1H), 7.53 (s, 1H), 7.51 (d, J = 0.8 Hz, 2H), 7.41 (d, J = 2.1 Hz, 1H), 7.40 (d, J = 1.2 Hz, 1H), 7.37 (s, 1H), 7.35 (d, J = 0.8 Hz, 2H), 7.32 (s, 1H), 7.30 (s, 1H), 7.28 (s, 1H), 3.67 (s, 4H), 2.56 (t, J = 5.0 Hz, 1H), 2.45 (t, J = 5.2 Hz,4H), 2.32 (s, 3H).13C NMR (100 MHz, cdcl3) δ 168.75, 161.69, 161.63, 157.96, 155.75, 154.88, 134.90, 134.82, 131.12, 130.16, 129.83, 129.72, 129.34, 129.07, 128.67, 128.53, 128.24, 128.20, 104.85, 54.47, 46.02, 43.78, 43.41. Anal. calcd for C25H24N6S C, 68.16; H, 5.49; N, 19.08; found C, 67.76; H, 5.14; N, 18.78.
5.1.4.3 5,6-Diphenyl-3-((6-(piperidin-1-yl)pyrimidin-4-yl)thio)-1,2,4-triazine (T8). Pale yellow, m.p. 240–242 °C, 63% yield was obtained after column chromatography. ESI m/z [M + H]+ calcd 426.16 found 427.15. Anal. calcd for C24H22N6S C, 67.58; H, 5.20; N, 19.70; found C, 67.58; H, 5.20; N, 19.70; 1H NMR (500 MHz, cdcl3) δ 8.53 (s, 1H), 8.38 (s, 1H), 7.54 (dd, J = 6.9, 1.3 Hz, 1H), 7.43 (d, J = 8.0 Hz, 1H), 7.39 (s, 2H), 7.38 (s, 1H), 7.32 (t, J = 7.6 Hz, 1H), 7.30 (s, 1H),7.29 (s,1H), 6.78 (s, 2H), 3.61 (d, J = 25.6 Hz, 4H), 1.69 (dd, J = 11.2, 5.1 Hz, 2H), 1.60 (dd, J = 10.2, 4.3 Hz, 4H). 13C NMR (126 MHz, cdcl3) δ 166.34, 161.51, 158.11, 157.50, 131.06, 129.86, 129.68, 129.38, 128.66, 128.52, 105.11, 96.20, 45.15, 25.43, 24.45.
5.1.4.4 2-(4-(6-((5,6-Diphenyl-1,2,4-triazin-3-yl)thio)pyrimidin-4-yl)piperazin-1-yl)nicotinonitrile (T9). Light yellow, m.p. 250–252 °C, 59% yield was obtained after column chromatography. ESI m/z [M + H]+ calcd 529.18 found 530.12 1H NMR (500 MHz, cdcl3) δ 8.58 (s, 1H), 8.40–8.36 (m, 1H), 7.82 (d, J = 7.6 Hz, 1H), 7.56 (s, 2H), 7.55 (s, 2H), 7.45 (s, 2H), 7.43 (d, J = 8.3 Hz, 1H), 7.39 (d, J = 6.8 Hz, 2H), 7.35 (d, J = 7.3 Hz, 2H), 6.83 (dd, J = 7.5, 4.9 Hz, 1H), 3.83 (d, J = 8.0 Hz, 8H). 13C NMR (126 MHz, cdcl3) δ 168.54, 162.19, 161.52, 157.88, 155.92, 155.07, 154.12, 138.90, 134.82, 134.76, 131.24, 129.87, 129.35, 128.75, 128.59, 126.03, 112.46, 104.65, 46.17, 43.07. Anal. calcd for C29H23N9S C, 65.77; H, 4.38; N, 23.80; found C, 66.01; H, 4.53; N, 23.43.
5.1.4.5 7-Chloro-4-(4-(6-((5,6-diphenyl-1,2,4-triazin-3-yl)thio)pyrimidin-4-yl)piperazin-1-yl) quinoline (T10). Yellow, m.p. 260–262 °C, 57% yield was obtained after column chromatography. ESI m/z [M + H]+ calcd 588.16 found 589.15. 1H NMR (399 MHz, cdcl3) δ 8.75 (d, J = 5.0 Hz, 1H), 8.60 (s, 1H), 8.10 (d, J = 2.0 Hz, 1H), 7.98 (d, J = 9.0 Hz, 1H), 7.56 (s, 1H), 7.55–7.54 (m, 2H), 7.53 (d, J = 2.0 Hz, 2H), 7.48 (dd, J = 9.0, 2.1 Hz, 1H), 7.43 (d, J = 2.7 Hz, 1H), 7.42–7.41 (m, 1H), 7.40 (s, 1H), 7.38 (s, 1H), 7.33 (d, J = 7.8 Hz, 1H), 7.26 (s, 1H), 6.86 (d, J = 5.0 Hz, 1H), 3.96 (s, 4H), 3.44 (t, J = 5.2 Hz, 4H). 13C NMR (126 MHz, cdcl3) δ 168.96, 162.38, 158.01, 156.51, 155.90, 155.05, 151.71, 134.83, 134.78, 131.27, 131.22, 129.87, 129.84, 129.35, 128.91, 128.88, 128.75, 128.58, 128.54, 126.68, 126.66, 124.80, 109.99, 109.13, 104.83, 51.76, 43.96. Anal. calcd for C32H25ClN8S C, 65.24; H, 4.28; Cl, 6.02; N, 19.02; found C, 65.32; H, 4.45; Cl, 5.88; N, 19.12.
5.1.4.6 7-(4-(6-((5,6-Diphenyl-1,2,4-triazin-3-yl)thio)pyrimidin-4-yl)piperazin-1-yl)-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidine (T11). Yellow cotton, m.p. 280–285 °C, 68% yield was obtained after column chromatography. ESI m/z [M + H]+ calcd 559.20 found 559.8. 1H NMR (300 MHz, CDCl3) δ 8.58 (s, 1H), 8.32 (s, 1H), 8.01 (s, 1H), 7.56–7.54 (m, 2H), 7.54–7.50 (m, 3H), 7.41 (d, J = 3.3 Hz, 2H), 7.38–7.28 (m, 3H), 6.19 (s, 1H), 2.96 (s, 4H), 2.88 (s, 4H), 2.60 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 168.46, 165.09, 162.52, 162.45, 161.64, 157.91, 157.11, 155.93, 155.05, 154.27, 149.77, 134.72, 131.26, 129.83, 129.33, 128.73, 128.60, 104.69, 94.57, 47.29, 43.20, 36.50. Anal. calcd for C29H25N11S C, 62.24; H, 4.50; N, 27.53; found C, 61.89; H, 3.95; N, 27.33.
5.1.4.7. 103-((6-(4-(2-methoxyphenyl)piperazin-1-yl)pyrimidin-4-yl)thio)-5,6-diphenyl-1,2,4-triazine (T12). White m.p. 250–255 °C, 64% yield was obtained after column chromatography. ESI m/z [M + H]+ calcd 533.20 found 534.20. 1H NMR (399 MHz, cdcl3) δ 8.56 (d, J = 0.9 Hz, 1H), 7.53 (dd, J = 7.6, 0.8 Hz, 3H), 7.42 (s, 2H), 7.40 (s, 1H), 7.38 (s, 1H), 7.37 (d, J = 1.6 Hz, 1H), 7.35 (t, J = 1.6 Hz, 1H), 7.32 (d, J = 7.8 Hz, 1H), 7.26 (s, 1H), 6.92 (s, 1H), 6.90 (s, 1H), 6.86 (s, 1H), 6.84 (s, 1H), 3.81 (s, 3H), 3.76 (d, J = 3.9 Hz, 4H), 3.16 (t, J = 5.5 4H). 13C NMR (126 MHz, cdcl3) δ 163.61, 159.86, 157.66, 156.78, 156.25, 147.04, 136.73, 136.67, 133.01, 131.71, 131.62, 131.22, 130.55, 130.41, 120.71, 120.65, 116.40, 106.76, 57.41, 52.49, 45.90. Anal. calcd for C30H27N7OS C, 67.52; H, 5.10; N, 18.37; found C, 67.32; H, 4.89; N, 18.17.
5.1.4.8. 113-((6-(4-Ethylpiperazin-1-yl)pyrimidin-4-yl) thio)-5,6-diphenyl-1,2,4-triazine (T13). Light yellow, m.p. 280–285 °C, 55% yield was obtained after column chromatography. ESI m/z [M + H]+ calcd 455.19 found 456.0. 1H NMR (399 MHz, cdcl3) δ 7.54 (s, 1H), 7.52 (s, 1H), 7.48 (d, J = 1.4 Hz, 2H), 7.43 (dd, J = 5.0, 2.8 Hz, 2H), 7.38 (dd, J = 7.4, 1.9 Hz, 2H), 7.32–7.29 (m, 4H), 7.26 (s, 1H), 4.14 (s, 4H), 2.70 (s, 4H), 2.60 (q, J = 7.1 Hz, 2H), 1.21 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, cdcl3) δ 138.19, 132.19, 131.81, 131.61, 131.60, 131.31, 131.04, 130.65, 130.51, 130.23, 130.19, 54.43, 54.27, 45.03, 13.40. Anal. calcd for C25H25N7S C, 65.91; H, 5.53; N, 21.52; found C, 66.11; H, 5.44; N, 21.34.
5.1.4.9. 123-((6-(4-(4-Nitrophenyl)piperazin-1-yl)pyrimidin-4-yl)thio)-5,6-diphenyl-1,2,4-triazine (T14). Yellow, m.p. 260–265 °C, 73% yield was obtained after column chromatography. ESI m/z [M + H]+ calcd 548.17 found 548.9 1H NMR (300 MHz, CDCl3) δ 8.58 (s, 1H), 8.15 (d, J = 9.3 Hz, 2H), 7.56–7.54 (m, 2H), 7.53 (dd, J = 2.9, 1.4 Hz, 2H), 7.53 (s, 2H), 7.41 (d, J = 4.8 Hz, 2H), 7.34 (dd, J = 15.4, 7.6 Hz, 3H), 6.80 (d, J = 9.3 Hz, 2H), 3.88 (t, J = 4.8 Hz, 4H), 3.75 (t, J = 5.7 Hz, 4H). 13C NMR (75 MHz, CDCl3) δ 168.56, 162.27, 161.52, 157.95, 155.92, 155.05, 138.83, 134.76, 131.25, 129.87, 129.35, 128.76, 128.60, 126.03, 112.43, 104.65, 46.14, 43.03. Anal. calcd for C29H24N8O2S C, 63.49; H, 4.41; N, 20.42; found C, 62.89; H, 4.10; N, 21.01.
5.1.4.10. 3-((6-(4-(4-Fluorophenyl)piperazin-1-yl)pyrimidin-4-yl)thio)-5,6-diphenyl-1,2,4-triazine (T15). Light yellow, m.p. 270–272 °C, 57% yield was obtained after column chromatography. ESI m/z [M + H]+ calcd 521.18 found 522.0. 1H NMR (300 MHz, CDCl3) δ 8.56 (s, 1H), 7.54 (d, J = 7.2 Hz, 4H), 7.43 (s, 2H), 7.41 (d, J = 3.3 Hz, 2H), 7.33 (dd, J = 15.6, 7.8 Hz, 3H), 6.99 (t, J = 8.4 Hz, 2H), 6.90 (dd, J = 7.8, 3.6 Hz, 2H), 3.82 (s, 4H), 3.15 (t, J = 4.5 Hz, 4H). 13C NMR (75 MHz, CDCl3) δ 168.73, 161.94, 161.74, 158.04, 156.03, 155.84, 154.97, 147.55, 134.89, 131.19, 129.88, 129.38, 128.73, 128.59, 118.55, 118.44, 115.89, 115.60, 104.90, 50.13, 43.94. Anal. calcd for C29H24FN7S C, 66.78; H, 4.64; N, 18.80; found C, 65.99; H, 5.21; N, 19.01.
:75.6. Biological assays
6.1. Leishmanial parasite culture and maintenance
Promastigotes were routinely cultured at 26 °C in medium M199 supplemented with 10% heat inactivated fetal bovine serum (FBS, Gibco Laboratories, Mumbai, India), penicillin 100 (IU mL−1), streptomycin (100 μg mL−1). Log phase promastigotes were sub-cultured every 72–96 h, the inoculum being 2 × 106 cells per mL.296.2. Cell line culture
THP-1 human monocytic cells were grown at 37 °C in medium RPMI-1640 (pH 7.4, Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% heat-inactivated FBS for 48 to 72 h in a humidified atmosphere of 5% CO2 and sub-cultured in fresh RPMI-1640 medium at an average density 2 × 105 cells per mL (Sharma et al. 2023).306.3. Anti-leishmanial activity
The triazin hybrids series was evaluated for their antileishmanial ability against L. donovani promastigotes in vitro. Promastigotes of L. donovani (1× 106 cells per mL) in M199 medium were incubated at 26 °C for 72 h with each compound at a concentration of 100 μM. Miltefosine (MTF) was used as a reference drug, 0.2% DMSO as solvent control. Parasites with media alone were taken as control. Parasite viability was achieved by MTT assay after 72 h (Sharma et al. 2023).306.4. Cytotoxicity assay
Human macrophage THP-1 cell line of 0.5 × 106 cells per mL seeded in RPMI-1640 medium (Sigma-Aldrich, USA) supplemented with 10% FBS (GIBCO) in presence of 5% CO2 at 37 °C in triplicate in 96 well culture plate. The cells were treated with two-fold serially diluted concentrations (100–3.125 μM) of the candidate molecules for 72 h. The IC50 was calculated using MTT assay (Sharma et al. 2023).306.5. Dose-dependent anti-promastigote activity and determination of IC50
Promastigotes at density of 1 × 106 cells per mL were incubated in the absence and or presence of most active compounds T1–T15 of the series at serial six-fold dilutions starting at 3.125 μM for 3 days at 26 °C. MTF was used as a standard antileishmanial drug control. The cell viability was evaluated (by MTT Assay) and the mean percentage viability was calculated as follows: Mean cell number of treated parasites/mean cell number of untreated parasites × 100. The 50 and 90% inhibitory concentration (IC50) i.e., the concentration of drugs that decreased the cell growth by 50 and 90% respectively, was determined by graphical extrapolation after plotting the graph of percentage viability vs. concentration of the drug.316.6. Determination of promastigote cellular morphology
Variations in the cellular morphology of Leishmania parasites due to the treatment with compounds T4 and T7 was detected microscopically. Briefly, the promastigotes (1× 106 cells per mL) were incubated in the absence or presence of test compounds and MTF and AmphoB for 72 h with different concentrations and observed under 40× objective of a phase-contrast microscope. At least 20 microscopic fields were observed for each sample. Data were recorded by using NIS-Elements imaging software.32The 50% inhibitory concentration of the highly potent compounds T4 and T7 is represented in (Table 2). The test compounds showed a similar trend like that of MTF and Ampho B in dose dependent parasite killing at promastigote stage with IC50 values at 1.074 ± 0.09403 μM and 1.158 ± 0.92568 μM respectively. Parasite viability was not affected by DMSO (0.2%, data not shown) used as solvent control.33
6.7. Anti-amastigote assay
THP-1 cells (5 × 105 cells per mL) were cultivated in RPMI-1640 media (Roswell Park Memorial Institute 1640) (Sigma-Aldrich, USA), supplemented with 10% fetal calf serum and maintained at 37 °C with 5% CO2 using phorbol ester as the inducer. These cells were added on circular coverslips (1 × 106 cells per mL). Promastigotes of L. donovani were introduced at 1:20 ratio. Treatment of the samples was carried out at doses of 100, 50, 25, 12.5, 6.25 and 3.125 μM/72 h. After being taken off, the coverslips were stained with Giemsa. The rate of macrophage infection was then ascertained.
Author contributions
A. S. and A. R. were involved in the design and synthesis of the compounds under the guidance of NH. A. S. analyzed all chemical data. M. A. B., S. J., and A. K. performed in vitro biological studies under the supervision A. S., S. S. and M. A. B. also analyzed all biological data.Conflicts of interest
There are no conflicts of interest to declare for any of the authors.Acknowledgements
A. S. would like to acknowledge University Grants Commission, Government of India for RGNF Senior Research Fellowship. Abdur Rahman thanks to UGC, Govt. of India for the research fellowship [No. f.82-27/2019(SA-III), UGC-Ref. No.; 191620050863].References
- WHO, Leishmaniasis.
- A. S. Nagle, S. Khare, A. B. Kumar, F. Supek, A. Buchynskyy, C. J. N. Mathison, N. K. Chennamaneni, N. Pendem, F. S. Buckner, M. H. Gelb and V. Molteni, Chem. Rev., 2014, 114, 11305–11347 CrossRef CAS PubMed.
- F. Chappuis, S. Sundar, A. Hailu, H. Ghalib, S. Rijal, R. W. Peeling, J. Alvar and M. Boelaert, Nat. Rev. Microbiol., 2007, 5, 873–882 CrossRef CAS PubMed.
- H. W. Murray, J. D. Berman, C. R. Davies and N. G. Saravia, Lancet, 2005, 366, 1561–1577 CrossRef CAS PubMed.
- A. Selvapandiyan, S. L. Croft, S. Rijal, H. L. Nakhasi and N. K. Ganguly, PLoS Neglected Trop. Dis., 2019, 13, e0007616 CrossRef PubMed.
- A. C. Pinheiro, M. N. Rocha, P. M. Nogueira, T. C. M. Nogueira, L. F. Jasmim, M. V. N. de Souza and R. P. Soares, Diagn. Microbiol. Infect. Dis., 2011, 71, 273–278 CrossRef CAS PubMed.
- D. Liu and J. E. Uzonna, Front. Cell. Infect. Microbiol., 2012, 2 DOI:10.3389/fcimb.2012.00083.
- J. L. Santos, A. A. Andrade, A. A. M. Dias, C. A. Bonjardim, L. F. L. Reis, S. M. R. Teixeira and M. F. Horta, J. Interferon Cytokine Res., 2006, 26, 682–688 CrossRef CAS PubMed.
- J. Chakravarty and S. Sundar, Expert Opin. Pharmacother., 2019, 20, 1251–1265 CrossRef CAS PubMed.
- P. M. Loiseau, S. Gupta, A. Verma, S. Srivastava, S. K. Puri, F. Sliman, M. Normand-Bayle and D. Desmaele, Antimicrob. Agents Chemother., 2011, 55, 1777–1780 CrossRef CAS PubMed.
- S. Stevanović, A. Perdih, M. Senćanski, S. Glišić, M. Duarte, A. Tomás, F. Sena, F. Sousa, M. Pereira and T. Solmajer, Molecules, 2018, 23, 772 CrossRef.
- N. Sunduru, A. Agarwal, S. B. Katiyar, Nishi, N. Goyal, S. Gupta and P. M. S. Chauhan, Bioorg. Med. Chem., 2006, 14, 7706–7715 CrossRef CAS PubMed.
- R. G. Booth, C. D. Selassie, C. Hansch and D. V. Santi, J. Med. Chem., 1987, 30, 1218–1224 CrossRef CAS PubMed.
- N. Sunduru, Nishi, S. Palne, P. M. S. Chauhan and S. Gupta, Eur. J. Med. Chem., 2009, 44, 2473–2481 CrossRef CAS PubMed.
- L. Gupta, N. Sunduru, A. Verma, S. Srivastava, S. Gupta, N. Goyal and P. M. S. Chauhan, Eur. J. Med. Chem., 2010, 45, 2359–2365 CrossRef CAS.
- K. Chauhan, M. Sharma, R. Shivahare, U. Debnath, S. Gupta, Y. S. Prabhakar and P. M. S. Chauhan, ACS Med. Chem. Lett., 2013, 4, 1108–1113 CrossRef CAS PubMed.
- P. Baréa, V. A. Barbosa, D. L. Bidóia, J. C. de Paula, T. F. Stefanello, W. F. da Costa, C. V. Nakamura and M. H. Sarragiotto, Eur. J. Med. Chem., 2018, 150, 579–590 CrossRef.
- A. Al-Qahtani, Y. M. Siddiqui, A. A. Bekhit, O. A. El-Sayed, H. Y. Aboul-Enein and M. N. Al-Ahdal, Saudi Pharm. J., 2009, 17, 227–232 CrossRef.
- S. Chander, P. Ashok, R. M. Reguera, M. Y. Perez-Pertejo, R. Carbajo-Andres, R. Balana-Fouce, K. V. Gowri Chandra Sekhar and M. Sankaranarayanan, Exp. Parasitol., 2018, 189, 49–60 CrossRef CAS.
- M. Venkatraj, I. G. Salado, J. Heeres, J. Joossens, P. J. Lewi, G. Caljon, L. Maes, P. Van der Veken and K. Augustyns, Eur. J. Med. Chem., 2018, 143, 306–319 CrossRef CAS PubMed.
- E. V. Costa, M. L. B. Pinheiro, C. M. Xavier, J. R. A. Silva, A. C. F. Amaral, A. D. L. Souza, A. Barison, F. R. Campos, A. G. Ferreira, G. M. C. Machado and L. L. P. Leon, J. Nat. Prod., 2006, 69, 292–294 CrossRef CAS.
- S. Kumar, S. Bawa and H. Gupta, Mini-Rev. Med. Chem., 2009, 9, 1648–1654 CrossRef CAS.
- O. Afzal, S. Kumar, M. R. Haider, M. R. Ali, R. Kumar, M. Jaggi and S. Bawa, Eur. J. Med. Chem., 2015, 97, 871–910 CrossRef CAS.
- R. Kaur and K. Kumar, Eur. J. Med. Chem., 2021, 215, 113220 CrossRef CAS.
- J. E. Gutiérrez, H. Ramírez, E. Fernandez-Moreira, M. E. Acosta, M. R. Mijares, J. B. De Sanctis, S. Gurská, P. Džubák, M. Hajdúch, L. Labrador-Fagúndez, B. G. Stella, L. J. Díaz-Pérez, G. Benaim and J. E. Charris, Pharmaceuticals, 2023, 16, 1709 CrossRef.
- T. G. Soyer, D. V. C. Mendonça, G. S. V. Tavares, D. P. Lage, D. S. Dias, P. A. F. Ribeiro, L. Perin, F. Ludolf, V. T. S. Coelho, A. C. G. Ferreira, P. H. A. S. Neves, G. F. Matos, M. A. Chávez-Fumagalli, E. S. Coimbra, G. R. Pereira, E. A. F. Coelho and L. M. R. Antinarelli, Exp. Parasitol., 2019, 199, 30–37 CrossRef CAS.
- M. Khoshneviszadeh, M. H. Ghahremani, A. Foroumadi, R. Miri, O. Firuzi, A. Madadkar-Sobhani, N. Edraki, M. Parsa and A. Shafiee, Bioorg. Med. Chem., 2013, 21, 6708–6717 CrossRef CAS PubMed.
- J. Kumar, P. Meena, A. Singh, E. Jameel, M. Maqbool, M. Mobashir, A. Shandilya, M. Tiwari, N. Hoda and B. Jayaram, Eur. J. Med. Chem., 2016, 119, 260–277 CrossRef CAS PubMed.
- F. Afrin, R. Rajesh, K. Anam, M. Gopinath, S. Pal and N. Ali, Infect. Immun., 2002, 70, 6697–6706 CrossRef CAS.
- S. Sharma, P. A. Yakkala, M. A. Beg, S. Tanwar, I. Latief, A. Khan, K. Sharma, S. Rehman, A. Selvapandiyan and S. Shafi, ChemistrySelect, 2023, 8, e202302415 CrossRef CAS.
- M. Islamuddin, D. Sahal and F. Afrin, J. Med. Microbiol., 2014, 63, 74–85 CrossRef CAS.
- C. Paris, P. M. Loiseau, C. Bories and J. Breard, Antimicrob. Agents Chemother., 2004, 48, 852–859 CrossRef CAS.
- S. Shafi, F. Afrin, M. Islamuddin, G. Chouhan, I. Ali, F. Naaz, K. Sharma and M. S. Zaman, Front. Microbiol., 2016, 7 DOI:10.3389/fmicb.2016.01379.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra01904k |
| This journal is © The Royal Society of Chemistry 2024 |