
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective sensing of cyanide ions: impact of molecular design and assembly on the response of π-conjugated acylhydrazone compounds†
Harshal V. Barkale and
Nilanjan Dey *
*
Department of Chemistry, Birla Institute of Technology and Science Pilani, Hyderabad Campus, Hyderabad, Telangana 500078, India. E-mail: nilanjandey.iisc@gmail.com; nilanjan@hyderabad.bits-pilani.ac.in
First published on 12th August 2024
Abstract
This study investigates the sensing properties of two distinct compounds, denoted as 1 and 2, featuring acylhydrazone units. Spectroscopic analyses reveal the disruption of the supramolecular assembly upon binding with cyanide ions, consequently due to the hydrogen bonding interaction with acylhydrazone units. This leads to a ratiometric, color-changing response of both the compounds specifically towards cyanide ions. The investigation sheds light on the reversible nature of the cyanide-probe interaction and highlights the potential for reusability in cyanide ion detection. Moreover, compound 1, distinguished by its long alkyl chains, displays a superior response to CN− ions (∼4-fold larger signal), in contrast to compound 2. However, interference was observed from other basic anions, such as F− and AcO−. The research suggests the dominating role of supramolecular assembly, intermolecular interaction, and local hydrophobic environment around the binding sites on the analytical performance of the probe molecules. The findings underscore the significance of structural design and molecular assembly in dictating the selectivity and sensitivity of compounds, offering valuable insights for the development of efficient sensor systems in diverse real-world applications.
1. Introduction
The inclusion of lengthy alkyl chains within dye molecules can significantly impact their optical characteristics and self-assembly tendencies. These modifications are pivotal in various applications, including sensors, organic electronics, and other fields requiring precise control over absorption properties.1 The hydrophobic nature of alkyl chains influences the solubility, stability, and overall behavior of dye molecules in bulk mediums. Moreover, these chains can adjust the electronic structure of dye molecules, causing shifts in their absorption spectrum through effects on charge separation and conformational planarity.2 Longer alkyl chains can enhance the solubility of dye molecules in non-polar solvents while limiting it in polar solvents.3 As a result, dye molecules with lengthy alkyl chains tend to extensively self-assemble in polar solvents due to π–π stacking and hydrophobic interactions. Additionally, these chains can create steric hindrance, affecting the spatial orientation of the dye molecule and influencing its electronic transitions and optoelectronic properties.4 Furthermore, the presence of alkyl chains might render the dye molecule sensitive to environmental changes such as pH or polarity, further impacting its absorption characteristics.5The presence of lengthy alkyl chains can modify both the structure and electronic properties of dye molecules, thereby influencing their response to specific metal ions or anions. The hydrophobic environment created by the alkyl chains may facilitate responses to ionic analytes through non-covalent interactions like charge pairing and hydrogen bonding.6 The bulky alkyl chains may modify the arrangement of dye molecules in the aggregated state, affecting the binding of certain ions, and altering the ion-sensing properties within these aggregates compared to individual dye molecules.7 Consequently, the modifications induced by lengthy alkyl chains on dye molecules can significantly affect their selectivity, sensitivity, and response to various metal ions or anions.
In light of these observations, we compared the anion sensing properties of two acylhydrazone-based chromogenic probes (1 and 2) (Fig. 1d). Compound 1, unlike 2, featured two lengthy hexadecyl alkyl chains attached to the core aryl unit. Both compounds exhibited a very similar response (a red-shifted absorption band with ratiometric color changing response) to CN− ions in the CH3CN–water (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) medium. Mechanistic investigations indicated the formation of a 1:2 hydrogen-bonded complex with CN− ions involving the acylhydrazone units. However, the presence of alkyl chains predominantly influenced the selectivity and sensitivity of these compounds. Compound 1 displayed approximately a 4-fold larger response to CN− ions compared to compound 2. Conversely, interference from other basic anions, such as F− and AcO−, was more significant with compound 1. Hence, this study demonstrates that by altering the aggregation properties of probe molecules, we can modify their sensing efficacy in terms of both selectivity and sensitivity. This finding holds promise for the development of sensors across various real-life sample analyses.
:3) medium. Mechanistic investigations indicated the formation of a 1:2 hydrogen-bonded complex with CN− ions involving the acylhydrazone units. However, the presence of alkyl chains predominantly influenced the selectivity and sensitivity of these compounds. Compound 1 displayed approximately a 4-fold larger response to CN− ions compared to compound 2. Conversely, interference from other basic anions, such as F− and AcO−, was more significant with compound 1. Hence, this study demonstrates that by altering the aggregation properties of probe molecules, we can modify their sensing efficacy in terms of both selectivity and sensitivity. This finding holds promise for the development of sensors across various real-life sample analyses.
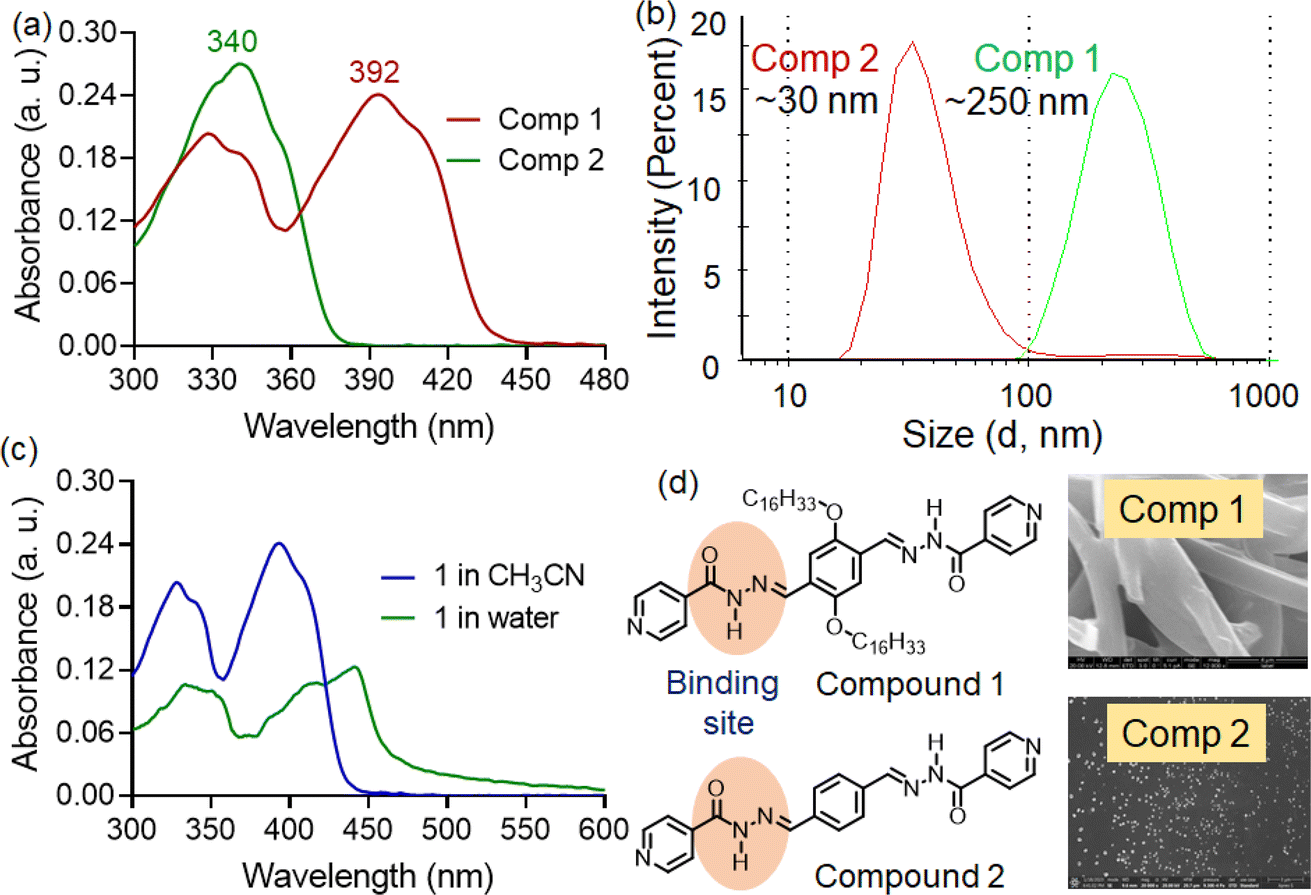 | ||
| Fig. 1 (a) UV-visible spectra of 1 and 2 (10 μM) in CH3CN–water (7:3) medium. (b) Dynamic light scattering experiment of 1 and 2 (10 μM) in CH3CN–water (7:3) medium. (c) UV-visible spectra of 1 (10 μM) in water and CH3CN medium. (d) FESEM images and structure of compound 1 and 2. | ||
2. Experimental section
2.1 Materials and methods
All chemicals (reagents, solvents, and chemicals) were bought from the best-known local chemical suppliers, such as Spectrochem, Avra, Alfa Acer, etc., and used without further purification. FTIR spectra were recorded on a PerkinElmer FTIR Spectrum BX system and were reported in wave numbers (cm−1). 1H NMR and 13C NMR spectra were recorded with a Bruker Advance DRX 400 spectrometer operating at 400 and 100 MHz respectively. Chemical shifts were reported in ppm downfield from the internal standard TMS. Mass spectra were recorded on Micromass Q-TOF Micro TM spectrometer.2.2 Sample preparation for spectroscopic studies
The UV-vis spectroscopic studies were recorded on a Shimadzu model 2100 spectrometer. For spectroscopic studies, 10 μL DMSO solutions of either 1 or 2 from a stock (1 mM) were added to CH3CN–water (7:3) medium and make the final volume of 1 mL. Thus, the final concentrations of both 1 and 2 were 1 × 10−5 M. The stock solutions of anions (tetrabutyl ammonium salts of F−, Cl−, Br−, I−, CN−, AcO−, ClO4−, NO3−, H2PO4−, PF6−) were prepared in DMSO medium. The concentration range of anions in spectroscopic studies was kept between 0–100 μM. Each experiment was performed three times (independently) to ensure statistical reliability. The spectroscopic data was plotted using graphical softwares, such as Origin 8.5, Graph Pad Prism 5.0 etc.
2.3 DLS and SEM measurement
The samples (1 and 2) were made under dust-free conditions and drop-cast over double-sided tapes attached to the brass stubs. Then the stubs were air-dried for 48 h. The coatings with gold vapor were done before analysing the samples on a Quanta 200 SEM operated at 15 kV. DLS measurements were done using a Malvern Zetasizer Nano ZS particle sizer (Malvern Instruments Inc., MA) instrument.3. Results and discussion
3.1 Aggregation studies in solution phase
The compounds 1, 2 were prepared through a carbonyl-nucleophile addition protocol using isoniazid and the corresponding dialdehyde as reported earlier.8 At first, the UV-visible spectra compounds 1 and 2 were recorded in organic solvents of varying micropolarity, where not much polarity-dependent shift in absorption maxima was witnessed (Fig. S1†). The UV-visible spectra of compound 1 exhibited multiple absorption bands at approximately 328 nm (ε = 2.02 × 105 M−1 cm−1), 340 nm (ε = 1.87 × 105 M−1 cm−1), and around 392 nm (ε = 2.45 × 105 M−1 cm−1) in CH3CN–water (7:3) medium. The absorption bands at the higher energy region (328 and 340 nm) were associated with π−π* transitions, while the red-shifted band around 392 nm was likely due to charge transfer interactions (Fig. 1a).9 Interestingly, compound 2, lacking hexadecyl chains, only showed absorption bands at 328 and 340 nm under similar condition. The charge transfer band observed in compound 1 was absent in compound 2. In general, acylhydrazones often exhibit either photo or thermally-induced geometric isomerism (E/Z isomerization) due to restricted rotation around the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond. This is often influenced by factors such as steric hindrance, electronic effects, and solvent interactions. Moreover, these compounds can form self-assembled nanostructures via alternative donor–acceptor type hydrogen bonding interactions. Thus, we suspect that due to the absence of long alkyl chains, the likelihood of self-assembled aggregate formation in the CH3CN–water (7:3) medium is lower for compound 2. Such aggregates often promote intermolecular charge transfer interactions. The aggregation of compound 1 was also supported by dynamic light scattering experiments (DLS) and field-effect scanning electron microscopic (FE-SEM) analysis. DLS experiments indicated that compound 1 formed tape-like structures (Fig. 1d), suggesting the formation of extended supramolecular assemblies. In contrast, compound 2 exhibited small spherical aggregates, significantly smaller in size than those observed for compound 1. The DLS experiment revealed aggregate sizes with average hydrodynamic diameters of 250 ± 15.8 nm and 30 ± 2.6 nm for compounds 1 and 2, respectively (Fig. 1b).
N bond. This is often influenced by factors such as steric hindrance, electronic effects, and solvent interactions. Moreover, these compounds can form self-assembled nanostructures via alternative donor–acceptor type hydrogen bonding interactions. Thus, we suspect that due to the absence of long alkyl chains, the likelihood of self-assembled aggregate formation in the CH3CN–water (7:3) medium is lower for compound 2. Such aggregates often promote intermolecular charge transfer interactions. The aggregation of compound 1 was also supported by dynamic light scattering experiments (DLS) and field-effect scanning electron microscopic (FE-SEM) analysis. DLS experiments indicated that compound 1 formed tape-like structures (Fig. 1d), suggesting the formation of extended supramolecular assemblies. In contrast, compound 2 exhibited small spherical aggregates, significantly smaller in size than those observed for compound 1. The DLS experiment revealed aggregate sizes with average hydrodynamic diameters of 250 ± 15.8 nm and 30 ± 2.6 nm for compounds 1 and 2, respectively (Fig. 1b).
Additionally, we recorded the absorption spectra of compound 1 in the aqueous medium. Here, we observed a more red-shifted absorption band (λabs = 440 nm) with notable hypochromism, indicating the formation of aggregated structures with significant charge-transfer characteristics (Fig. 1c). Moreover, we noticed substantial tailing in the longer wavelength region. This residual absorbance at lower energy levels likely originated from Mie scattering effects, owing to the presence of colloidal nano aggregates in the solution.10
3.2 Anion sensing in solution phase
The acylhydrazone based probes can form hydrogen bonded stable complex with basic anions, leading to changes in optical signals. These complexes are less likely to dissociate under typical conditions, providing a reliable means of capturing and retaining anions. Moreover, the binding of anions to the acylhydrazone unit is reversible, suggesting the reusability of such probe molecules. Considering these, both compounds 1 and 2 were employed to screen various anions in a CH3CN–water (7:3) medium. Among the different anions tested, both compounds exhibited alterations in solution color exclusively upon the addition of cyanide ions. Compound 1 resulted in a deep yellow solution upon the addition of CN− ions, while compound 2 produced a pale yellow color. The comparatively higher nucleophilicity and lower hydration energy for the cyanide ion (ΔHhyd = −67 kJ mol−1) than that of the fluoride (ΔHhyd = −505 kJ mol−1) and acetate ion (ΔHhyd = −375 kJ mol−1) explains such selectivity of probe molecules towards CN− ions. To understand the differences in color response, a detailed UV-visible spectroscopic analysis was conducted. The UV-visible titration studies with compound 1 revealed the gradual formation of a new red-shifted charge transfer band around the ∼440 nm region upon the addition of CN− ions (Fig. 2a). Consequently, the absorbance at the 392 and 328 nm bands decreased during the titration, displaying prominent isosbestic points at 402 nm. The presence of clear isosbestic points during concentration-varied studies indicated a one-to-one equilibrium between compound 1 and the corresponding cyanide-mediated hydrogen bonding adduct. Meanwhile, titration of compound 2 with CN− ions under similar conditions led to an increase in absorbance at the ∼410 nm band at the expense of the absorbance at the 340 nm band (Fig. 2c). Here as well, an isosbestic point was detected at 358 nm. Additionally, we plotted the changes in absorbance (A/A0) at the 440 and 410 nm bands for compound 1 and 2, respectively, against the concentrations of added cyanide ions (Fig. 2b).
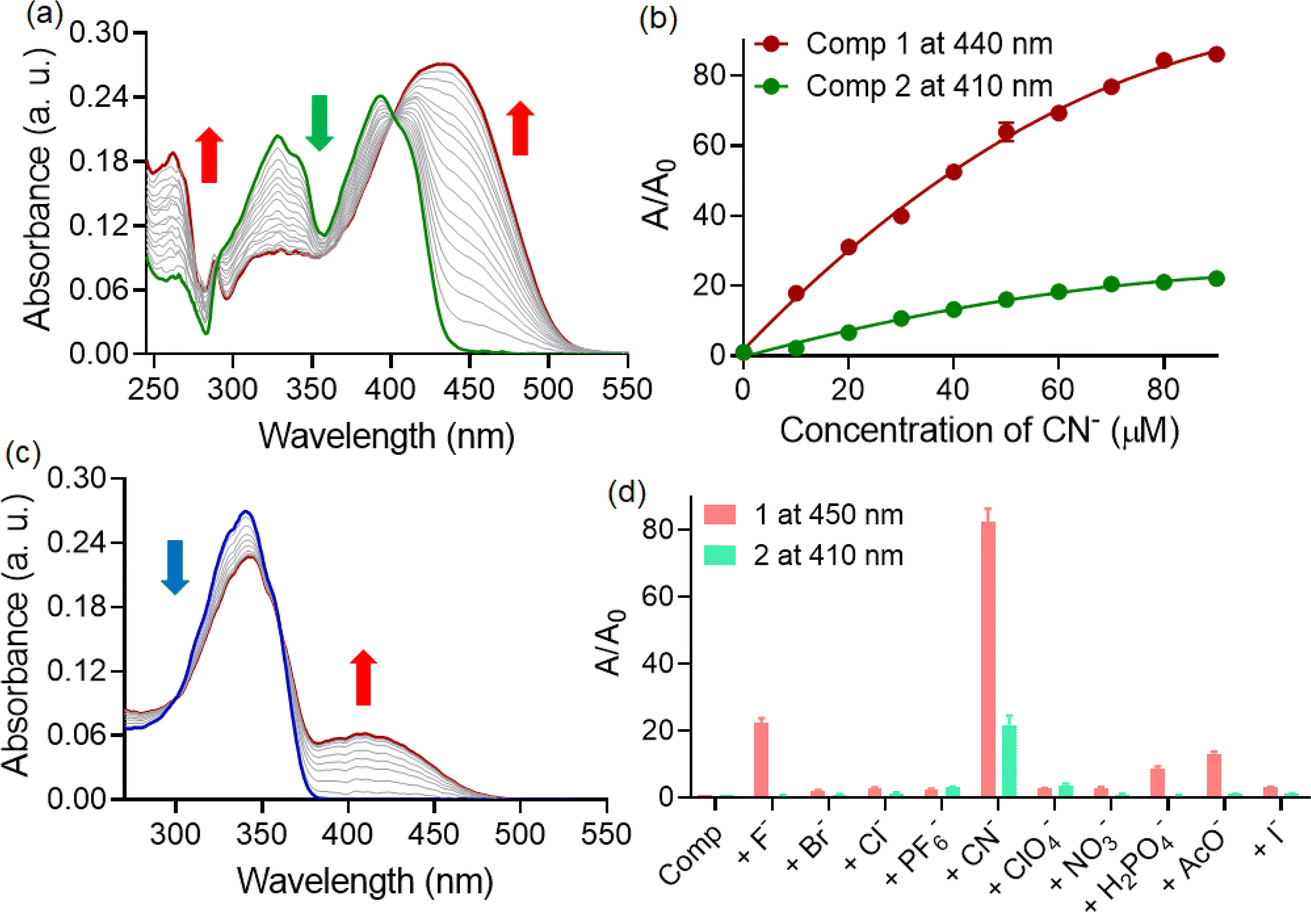 | ||
| Fig. 2 UV-visible titrations of (a) 1 (c) 2 (10 μM) with CN− ion (100 μM) in CH3CN–water (7:3) medium. (b) Change in absorbance of compounds 1 and 2 (10 μM) with CN− ion (0–100 μM) in CH3CN–water (7:3) medium. (d) Change in absorbance of 1 and 2 (10 μM) with different anions (100 μM) in CH3CN–water (7:3) medium. | ||
Compound 1 exhibited approximately an 85-fold change in the absorption signal, whereas the changes were approximately 20-fold for compound 2. This observation indicated that despite extensive self-assembly, compound 1 demonstrated a better response (higher sensitivity) towards CN− ions. We also investigated the interaction of cyanide ions with 1 in CH3CN–H2O mixture medium with varying water contents (Fig. S4†). It was observed that the degree of response, as defined by A/A0, decreased drastically with increasing the fraction of water in the mixture, presumably due to high hydration energy and competitive hydrogen bonding interactions of CN− ions with solvent molecules. Additionally, the kinetics of cyanide interaction was studied for both probes 1 and 2 under identical conditions. Both 1 and 2 exhibited immediate changes in the solution color upon exposure to CN− with no further time-delayed response (Fig. S2†).
Considering that selectivity is a crucial parameter alongside sensitivity for an optimal sensory system, we also investigated the changes in absorbance at 440 nm for compound 1 and at 410 nm for compound 2 upon the addition of other relevant anions (Fig. 2d). Although both compounds displayed a notably high response to CN− ions, the interference from other competing anions was not identical. Besides CN−, compound 1 exhibited slight yet noticeable changes with F− ions (∼21-fold) and AcO− ions (∼12-fold). However, interference from these two anions was significantly less pronounced for compound 2. Therefore, we can infer that despite possessing a similar binding site, compound 2 demonstrated greater specificity for cyanide ions in a CH3CN–water (7:3) medium. We believe that the interaction with CN− ion could influence the distribution of electron density within the acylhydrazone (modify the effective conjugation length), leading to a change in its electronic transitions and resulting in a red-shifted absorption. Not only this, hydrogen bonding interaction could also alter the molecular geometry and affect the electronic environment around the chromophoric groups, leading to a shift in the absorption spectrum towards longer wavelengths. We have compared the results obtained from previous published studies. The table is attached in the ESI file (Table S1).†
3.3 Mechanistic investigation with anions
The Job's plot analysis revealed that despite differing responses, both compounds 1 and 2 exhibited a 1:2 binding stoichiometry with CN− ions (Fig. 3d). In the investigation of compound 1, FT-IR and 1H-NMR spectra were recorded both in the presence and absence of CN− ions. The FT-IR spectra displayed a shift in the carbonyl stretching frequency from 1640 to 1628 cm−1 in the presence of CN− ions, while the –NH stretching band in the ∼3345 cm−1 region became broad and almost unrecognizable (Fig. 3a). This indicated the involvement of acyl hydrazone units in interaction with cyanide ions.11 Furthermore, 1H-NMR titration studies with 1 were conducted in a CDCl3 medium due to limited solubility compared to CD3CN. The peaks corresponding to aromatic protons (a, b, c, and d) were shifted to a more shielded region upon the addition of CN− ions (Fig. 4). Notably, a significant shift was observed for the protons close to the acyl hydrazone unit, denoted by ‘b’. This shift was likely due to the accumulation of negative charges resulting from hydrogen bonding interactions with acyl hydrazone units. Additionally, the introduction of a stoichiometric amount of Hg2+ to the solution of 1. CN− led to a complete reversal of the UV-visible spectrum (Fig. 3b). The restoration of the characteristic absorption spectrum of the probe suggested that CN− ions could be removed by Hg2+. These observations indicated the reversible nature of the interaction between the probe and CN− ions, implying that the same probe solution could be used multiple times (reusability) for CN− ion detection. Conversely, a similar experiment conducted with compound 3, featuring a styryl linkage instead of acyl hydrazone, demonstrated no detectable response to CN− ions (Fig. 3c).12 Hence, it was inferred that the acyl hydrazone unit was likely the primary binding site for the cyanide ion. Further, we investigated the reversibility of cyanide coordination using HCl as a proton source. The regeneration of the free probe, as evident by changes in absorbance, was noticed upon treatment of 1. CN− mixture with equimolar amounts of HCl (Fig. S3†).
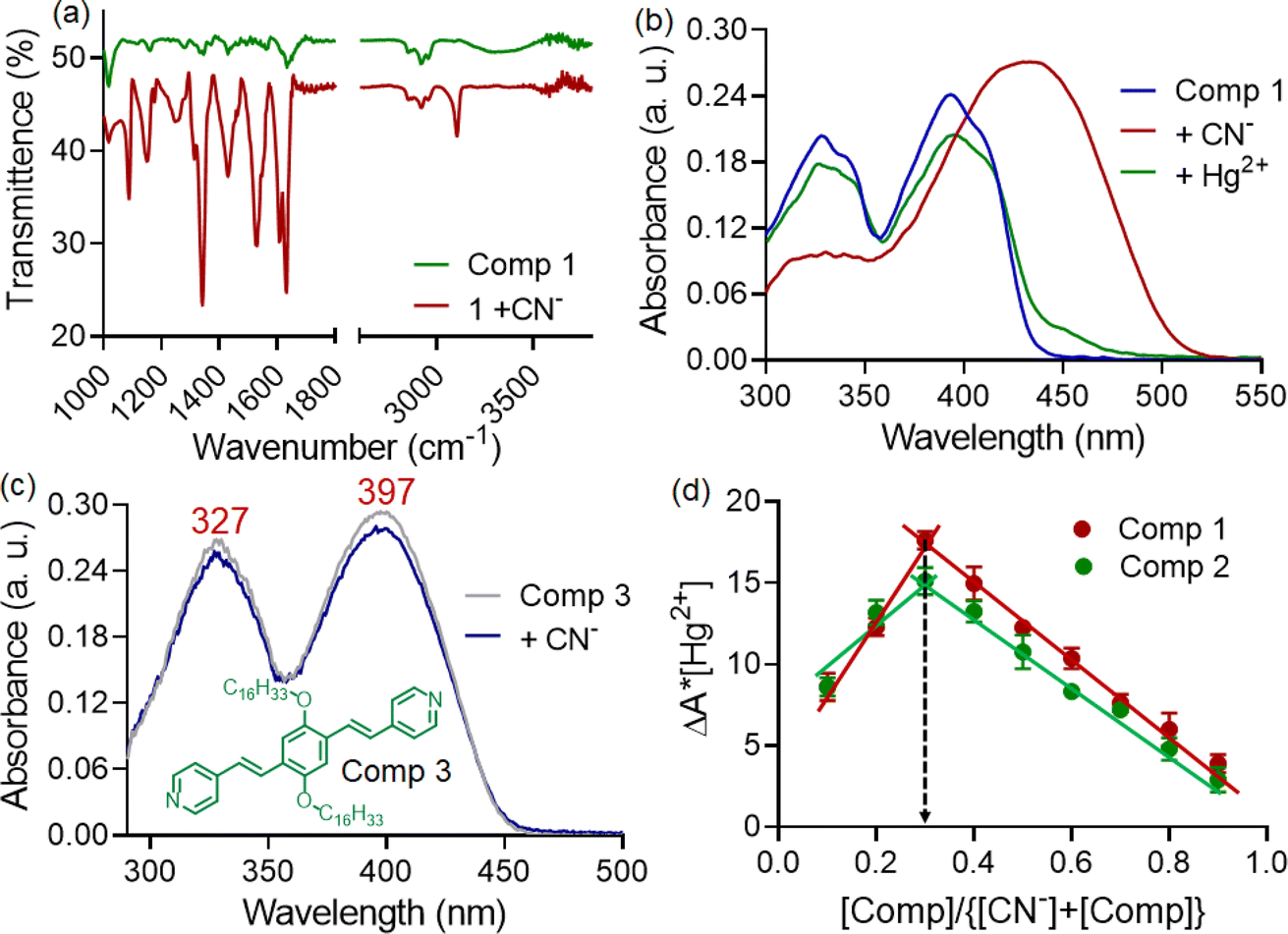 | ||
| Fig. 3 (a) FT-IR spectra of 1 with CN− ion. (b) UV-visible spectra of 1 (10 μM) with CN− ion (100 μM) and subsequently treated with Hg2+ (50 μM). (c) UV-visible spectra of 3 (10 μM) with CN− ion (100 μM) in CH3CN–water (7:3) medium. (d) Job's plot analysis shows 1:2 binding stoichiometry between probe molecules (1, 2) and CN− ion. | ||
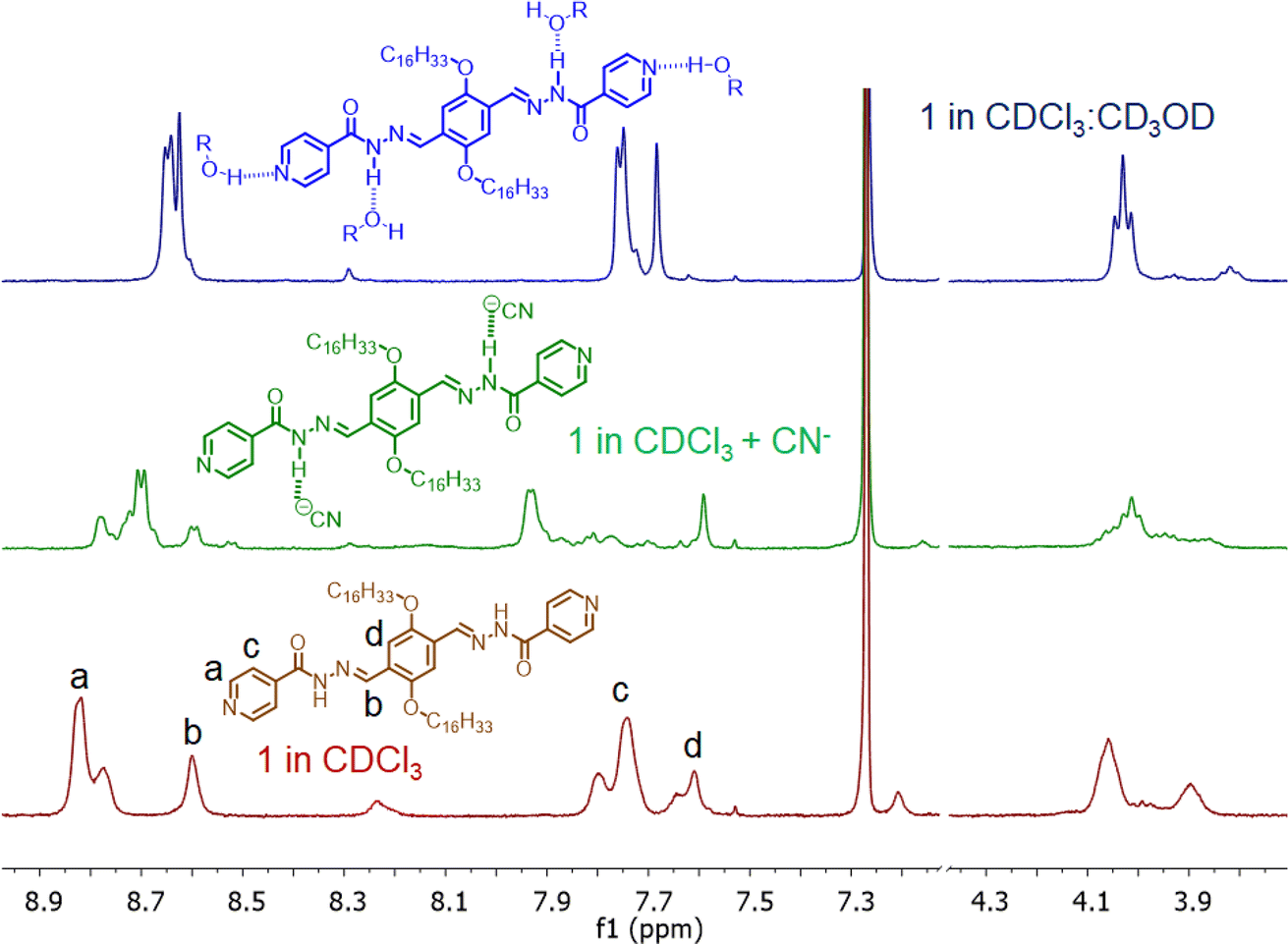 | ||
| Fig. 4 Partial 1H-NMR spectra of 1 with CN− ion in CDCl3 medium and also in presence of CD3OD (CDCl3:CD3OD = 5:1). | ||
3.4 Effect of alkyl chains on analytical performance
It is hypothesized that compound 1, due to the presence of long alkyl chains, is likely to form a supramolecular assembly in a bulk medium, supported by donor–acceptor hydrogen bonding interactions involving the amide functional groups. In contrast, cyanide is anticipated to form hydrogen bonds with the acidic –NH groups of hydrazone units. This cyanide-mediated hydrogen bonding interaction is expected to facilitate intramolecular charge transfer from the core aryl moiety to the terminal pyridine units. Moreover, the binding with cyanide ions is assumed to disrupt the supramolecular assembly structure, reducing the possibility of π–π stacking interaction and intermolecular charge transfer. In the aggregated state, CN− ions could bind simultaneously with multiple molecules, leading to a greater extent of change (measured by changes in absorption signal) for compound 1 compared to compound 2. Additionally, because of the extensive self-assembly, the microenvironment near the hydrazone functional groups might be relatively hydrophobic. This hydrophobicity could result in interactions with other basic anions, such as F− and AcO−.3.5 Real-life applications
Contamination of natural water samples with toxic anions, such as F−, CN− etc. is one of the major health-care challenges. Therefore, we utilized compound 1 for the detection of excess CN− in the natural water samples (CH3CN–H2O, 7:3 v/v). For spectroscopic studies, water samples were collected water from a laboratory tap, a local pond (located near the industrial region of Medchal area), and the Arabian Sea (near Mangalore, India). The samples were filtered to remove the insoluble dirt particles before analysis. However, no additional purification was carried out to eliminate the remaining soluble impurities, and the filtrates were used directly for the spectroscopic studies. In all instances, the absorption spectra of compound 1 were recorded in the water samples collected from the mentioned sources. The absorption spectra of compound 1, obtained under these conditions, were consistent with those observed in Milli-Q water, thereby ruling out the possibility of interference from other analytes.
Herein, we observed a concentration-dependent linear change in absorbance, which indicated that the present method could achieve the quantitative estimation of CN− even in natural water samples without any pretreatment (Fig. 5a). In all cases, the percentage recovery values varied between 95.2 and 103.4%, with relative standard deviation (RSD) values less than 5% (Fig. 5b). These results suggested the quantitative nature of the present protocol. Additionally, the minimum detectable concentrations (LODs) for CN− in all cases were found to be less than the permissible limit.
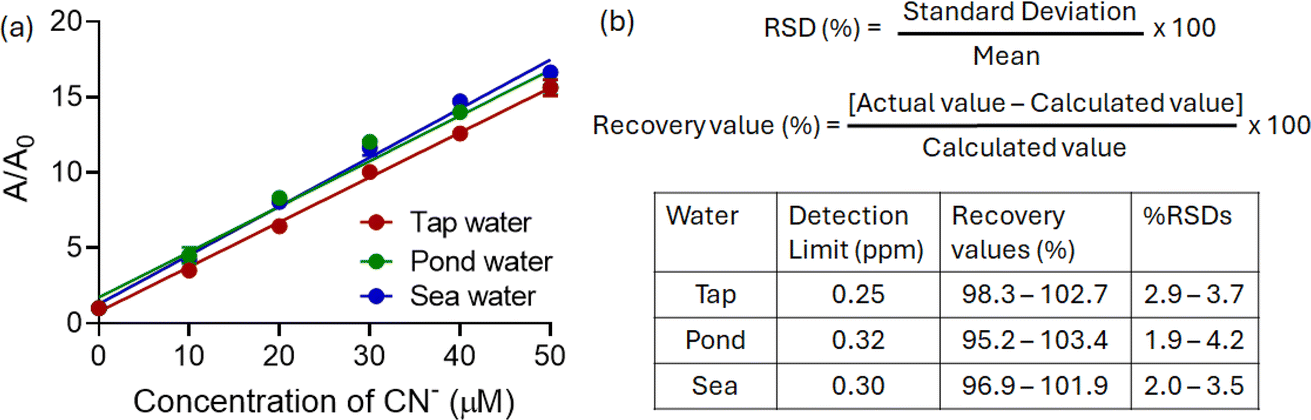 | ||
| Fig. 5 (a) Changes in the absorbance of 1 at 440 nm upon addition of CN− ion in real-life samples (CH3CN–H2O, 7:3 v/v). (b) Quantitative analysis of CN− ion in real-life water samples. | ||
4. Conclusion
In summary, the study elucidated the distinct behavior of compounds 1 and 2 in response to various anions in CH3CN–water (7:3) medium. Both compounds exhibited a color-changing response towards cyanide ions, leading to ratiometric change in absorption signal. The spectroscopic analyses provided insights into the binding interactions, affirming the acylhydrazone units as the primary binding site for cyanide ions for both 1 and 2. Additionally, such competitive hydrogen bonding interaction with CN− ions led to a disruption in the supramolecular assembly of probe molecules. The reversible nature of the cyanide-probe interaction suggested the potential for the reusability of the probe solution in detecting CN− ions. Interestingly, compound 1, characterized by its long alkyl chains, demonstrated a stronger affinity for cyanide ions than that observed with 2. However, the selectivity index for compound 1 appeared to be not so promising, as small yet perceptible interferences were observed with other basic anions. Despite having the same interaction sites, the distinct spectral changes observed in compound 1, attributed to self-assembly, intermolecular interactions, and the relatively hydrophobic microenvironment near the hydrazone functional groups provided a comprehensive understanding of the superior interaction of 1 with cyanide ion. Overall, the findings underline the significance of molecular design and structural composition in dictating the sensing properties of compounds, thereby contributing to the development of efficient and selective sensor systems for the detection of specific ions, a crucial aspect in various real-life applications.
Data availability
Data will be made available on request.Author contributions
The author (Harshal V. Barkale) confirms the responsibility for the following: synthesis of compounds, characterizations, investigation, data collection and validation. The author (Nilanjan Dey) confirms the responsibility for the following: conceptualization, formal analysis, project administration, resources, software, supervision, visualization, writing – original draft, writing – review & editing.Conflicts of interest
The author herewith declares no conflict of interest for the manuscript.Acknowledgements
We acknowledge the Indian Council of Medical Research for ITR grant (2021-8350) for financial support and DST for SYST grant (SP/YO/2021/1632). Also, authors thank BITS Pilani, Hyderabad for technical help and support.References
- (a) D. V. Talapin, M. Engel and P. V. Braun, MRS Bull., 2020, 45, 799–806 CrossRef CAS; (b) A. Pal and N. Dey, Soft Matter, 2024, 20, 3044–3052 RSC.
- (a) I. Robayo-Molina, A. F. Molina-Osorio, L. Guinane, S. A. M. Tofail and M. D. Scanlon, J. Am. Chem. Soc., 2021, 143, 9060–9069 CrossRef CAS PubMed; (b) H. Zhang, F. Tao, Y. Cui and Z. Xu, J. Mol. Liq., 2020, 302, 112550 CrossRef CAS; (c) D. Biswakarma, N. Dey and S. Bhattacharya, Soft Matter, 2020, 16, 9882–9889 RSC.
- (a) T. Lee, C. E. Song, S. K. Lee, W. S. Shin and E. Lim, ACS Omega, 2021, 6, 4562–4573 CrossRef CAS PubMed; (b) S. Ogi, V. Stepanenko, J. Thein and F. Würthner, J. Am. Chem. Soc., 2016, 138, 670–678 CrossRef CAS PubMed.
- (a) T. Shimoaka, Y. Yamaguchi, N. Shioya, A. Ajayaghosh, T. Mori, K. Ariga and T. Hasegawa, J. Phys. Chem. C, 2023, 127, 9336–9343 CrossRef CAS; (b) S. Banerjee, A. Akhuli and M. Sarkar, Chem. Phys., 2023, 565, 111762 CrossRef CAS.
- (a) X. Wang and Y. Gao, Food Chem., 2018, 246, 242–248 CrossRef CAS; (b) Y. Zhang, M. Li, H. Feng, W. Ni, H. Zhang, F. Liu, X. Wan and Y. Chen, Dyes Pigm., 2017, 141, 262–268 CrossRef CAS.
- (a) X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed; (b) Y. Hu, D. X. Cao, A. T. Lill, L. Jiang, C.-A. Di, X. Gao, H. Sirringhaus and T.-Q. Nguyen, Adv. Electron. Mater., 2018, 4, 1800175 CrossRef.
- (a) L. Xue, E. Gurung, G. Tamas, Y. P. Koh, M. Shadeck, S. L. Simon, M. Maroncelli and E. L. Quitevis, J. Chem. Eng. Data, 2016, 61, 1078–1091 CrossRef CAS; (b) R. S. Fernandes and N. Dey, ACS Appl. Nano Mater., 2023, 6, 5168–5176 CrossRef CAS.
- (a) B. Maiti, N. Dey and S. Bhattacharya, ACS Appl. Bio Mater., 2019, 2, 2365–2373 CrossRef CAS PubMed; (b) C. Rao, Z. Wang, Z. Li, L. Chen, C. Fu, T. Zhu, X. Chen, Z. Wang and C. Liu, Analyst, 2020, 145, 1062–1068 RSC.
- (a) S. K. Samanta and S. Bhattacharya, Chem. Commun., 2013, 49, 1425–1427 RSC; (b) S. M. Landge, E. Tkatchouk, D. Benítez, D. A. Lanfranchi, M. Elhabiri, W. A. Goddard III and I. Aprahamian, J. Am. Chem. Soc., 2011, 133, 9812–9823 CrossRef CAS PubMed.
- (a) B. Chettri, S. Jha and N. Dey, J. Photochem. Photobiol., A, 2023, 435, 114210 CrossRef CAS; (b) N. Dey, New J. Chem., 2022, 46, 8105–8111 RSC; (c) R. S. Fernandes and N. Dey, Talanta, 2022, 250, 123703 CrossRef CAS PubMed.
- (a) P. Anzenbacher, D. S. Tyson, K. Jursíkova and F. N. Castellano, J. Am. Chem. Soc., 2002, 124, 6232–6233 CrossRef CAS PubMed; (b) R. S. Fernandes and N. Dey, J. Mol. Struct., 2022, 132968 CrossRef CAS.
- (a) S. K. Samanta, N. Dey, N. Kumari, D. Biswakarma and S. Bhattacharya, ACS Sustainable Chem. Eng., 2019, 7, 12304–12314 CAS; (b) S. Mondal, H. V. Barkale and N. Dey, Colloids Surf., A, 2023, 677, 132322 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra01884b |
| This journal is © The Royal Society of Chemistry 2024 |