DOI:
10.1039/D4RA01834F
(Review Article)
RSC Adv., 2024,
14, 13100-13128
Exploring the synthetic potential of epoxide ring opening reactions toward the synthesis of alkaloids and terpenoids: a review
Received
10th March 2024
, Accepted 15th April 2024
First published on 23rd April 2024
Abstract
Epoxides are oxygen containing heterocycles which are significantly employed as crucial intermediates in various organic transformations. They are considered highly reactive three-membered heterocycles due to ring strain and they undergo epoxide ring opening reactions with diverse range of nucleophiles. Epoxide ring-opening reactions have gained prominence as flexible and effective means to obtain various functionalized molecules. These reactions have garnered substantial attention in organic synthesis, driven by the need to comprehend the synthesis of biologically and structurally important organic compounds. They have also found applications in the synthesis of complex natural products. In this review article, we have summarized the implementation of epoxide ring opening reactions in the synthesis of alkaloids and terpenoids based natural products reported within the last decade (2014–2023).
Introduction
Epoxides are 3-membered oxygen containing heterocycles that play an important role in the synthesis of natural products. Epoxides are considered highly reactive molecules due to ring strain1 and they undergo enantioselective ring opening reaction with a diverse range of reagents (Gilman reagent2) and nucleophiles3 such as carbon4 and oxygen nucleophiles i.e., trimethylsilyl cyanides,5 aryllithiums,6 trialkylsilyl halides,7 alkylamines,8 carboxylic acids9 and phenols10 in the presence of numerous catalysts. The general reaction of epoxide ring opening reaction in the presence of nucleophile is shown in the Fig. 1.
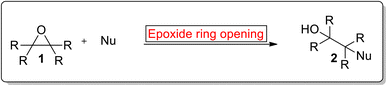 |
| | Fig. 1 General reaction of epoxide ring opening reaction.11 | |
Similarly, π-nucleophiles i.e., allylsilanes, olefins,12 and arenes13 have also been used for the ring opening reactions of epoxides. Sulfur nucleophiles14 also play an important role towards the epoxide ring opening reactions i.e., alkanes and arene-thiols can also be used for the ring opening reaction of epoxides to synthesize β-hydroxy mercaptans or regioselective products i.e., β-hydroxysulfides.15 Similarly, α-azido ketone is prepared by the reaction of chloroepoxide with sodium azide.16
The epoxide ring opening reactions are highly employed in the synthesis of various natural products such as alkaloids, steroids, terpenoids, biopolymers, carotenoids, polyketides and flavonoids etc. which exhibit medicinal value. In 1963, an epoxide was first time reacted with heteroaromatic π-nucleophile to synthesize a natural product named as Indolmycin (an antibiotic).17 Vinyl epoxides are important synthetic precursors in organic synthesis18 as they lead towards the synthesis of (−)-(4R,5R)-Muricatacin (a natural acetogenin) via regio or stereoselective ring opening reaction of epoxide.19 Syntheses of numerous biological active compounds such as Sphingofungin,20 Zoanthenol,21 marine ladder polyethers,22 Zampanolide,23 epi-Muscarine,24 Molestin E,25 (−)-Epicoccin G and (−)-Rostratin A,26 homoisoflavonoids,27 Chlorotonils,28 Lingzhiol,29 Phomarol,30 Isoandrographolide,31 Citrinadins,32 Isosilybin,33 polyester,34 Mupirocin35 and Bisfuranoxide36 etc. have been accomplished by involving epoxide chemistry.
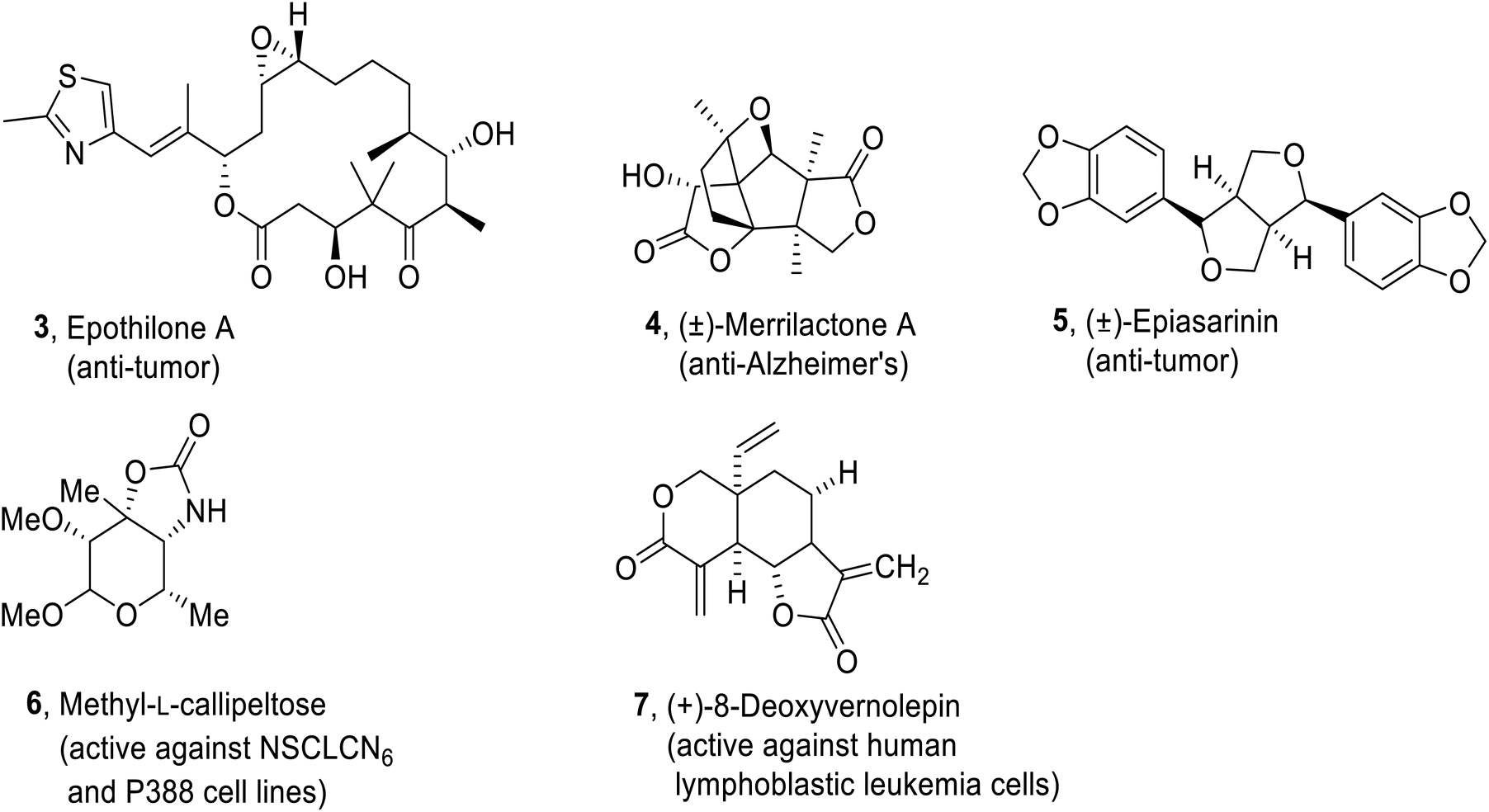
Predominantly, biologically active natural products i.e., Epothilone A37 3 (antitumor activity), methyl-L-Callipeltose38 6 (cytotoxic activity against NSCLCN6 and P388 cell lines), (−)-Dactylolide,39 Petuniasterone D40 (insecticidal), (±)-Merrilactone A41 4 (active against Alzheimer's disease), (−)-Preussomerin,42, (+)-8-Deoxyvernolepin43 7 (active against human lymphoblastic leukemia cells), (±)-Epiasarinin44 5 (antitumour activity), Laulimalide,45, (−)-Nakamurol A,46 Prenylbisabolane,47, (−)- Delobanone and (−)-epi-Delobanone47 are generated by employing epoxide ring opening reactions (Fig. 2).
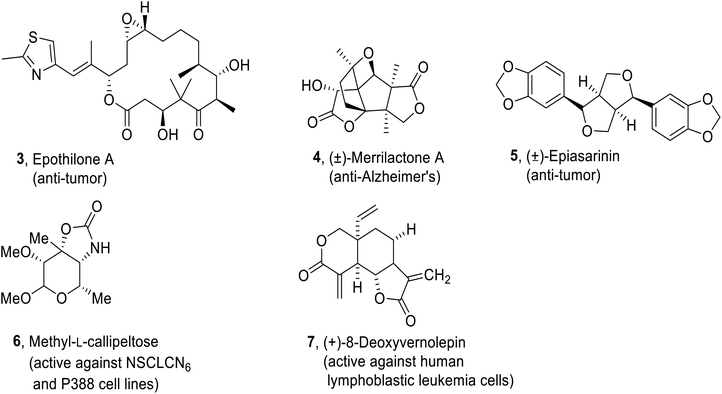 |
| | Fig. 2 Structures of some biologically active natural products (Epothilone A 3, (±)-Merrilactone A 4, (±)-Epiasarinin 5, Methyl-L-callipeltose 6, (+)-8-Deoxyvernolepin 7). | |
In 2006, research group of Oltra48 reviewed the synthesis of natural products by involving epoxide opening reaction in the presence of titanocene (III). Vilotijevic and Jamison overviewed the syntheses of polycyclic polyether natural products and marine polycyclic polyethers by employing epoxide-opening reactions and endo-selective epoxide-opening pathways in 2009 (ref. 49) and 2010 (ref. 50) respectively. Similarly, Bugarin and allies, provided a review on the syntheses of polypropionates and other bioactive natural products by using epoxide chemistry in 2023.51 Our review focuses on the formation of alkaloids and terpenoids due to the comprehensive work reported on the syntheses of these natural products by involving epoxide ring opening reaction as one of the key steps, reported within the past decade (2014–2023).
Review of literature
Syntheses of alkaloid-based natural products
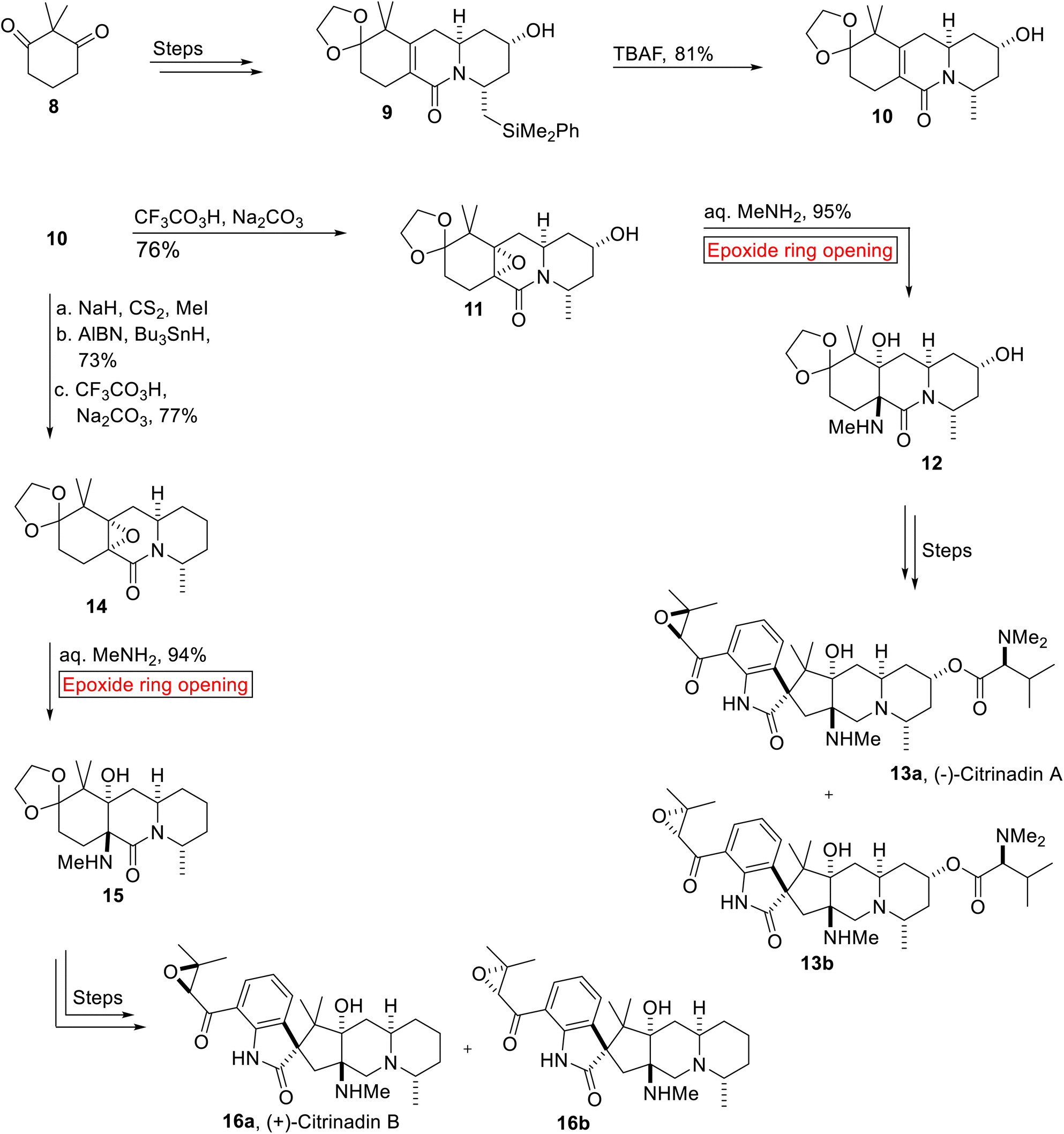
Spirooxindole alkaloid. (−)-Citrinadin A 13a and (+)-Citrinadin B 16a are spirooxindole alkaloids which were isolated and reported by research group of Kobayashi in 2004 and 2005. They were found to demonstrate significant efficacy against human epidermoid carcinoma KB (13a, IC50 = 10 μg mL−1) and murine leukemia L1210 (13a, IC50 = 6.2 μg mL−1; 16a, IC50 = 10 μg mL−1)52 cells. In 2014, Martin and co-workers disclosed the concise, enantioselective syntheses of Citrinadins A 13a and B 16a that resulted in the revision of their stereochemical structures. Their strategy minimized protection/deprotection processes, refunctionalization and showed high diastereoselectivity. The stereochemistry of Citrinadins was based on the substrate-controlled reactions involving stereoselective epoxidation and epoxide ring opening reaction as key steps.53 Total synthesis of (−)-Citrinadin A 13a and (+)-Citrinadin B 16b was accomplished from common intermediate lactam 10. The synthetic scheme initiated with the preparation of 9 from 2,2-dimethylcyclohexane-1,3-dione 8 over few steps. Then, 9 experienced tetra-n-butylammonium fluoride (TBAF)-mediated desilylation to produce unsaturated lactam 10 (81%) which was further subjected to peroxytrifluoroacetic acid-mediated diastereoselective epoxidation in order to furnish epoxide 11 (76%). In the next step, 11 underwent base-catalyzed diastereoselective epoxide ring opening reaction to generate amino alcohol 12 (95%) which forged (−)-Citrinadins A 13a over few steps. Similarly, synthesis of (+)-Citrinadin B 16a commenced with the formation of epoxide 14 (77%) from lactam 10 upon Barton deoxygenation followed by CF3CO3H-mediated epoxidation. Then, epoxide 14 was exposed to similar base (Me2NH2)-catalyzed epoxide ring opening reaction to generate another amino alcohol 15 (94%) which was further converted into a mixture of (+)-Citrinadin B (dr = 2.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) 16a and 16b over few steps (Scheme 1).
:1) 16a and 16b over few steps (Scheme 1).
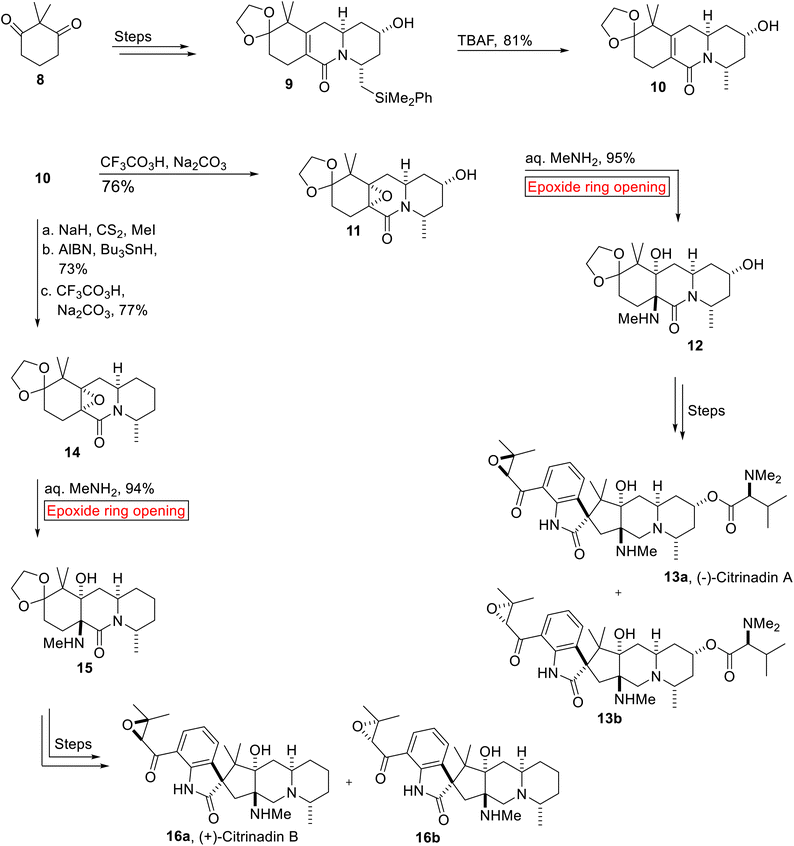 |
| | Scheme 1 Synthesis of (−)-Citrinadin A 13a and (+)-Citrinadin B 16b. | |
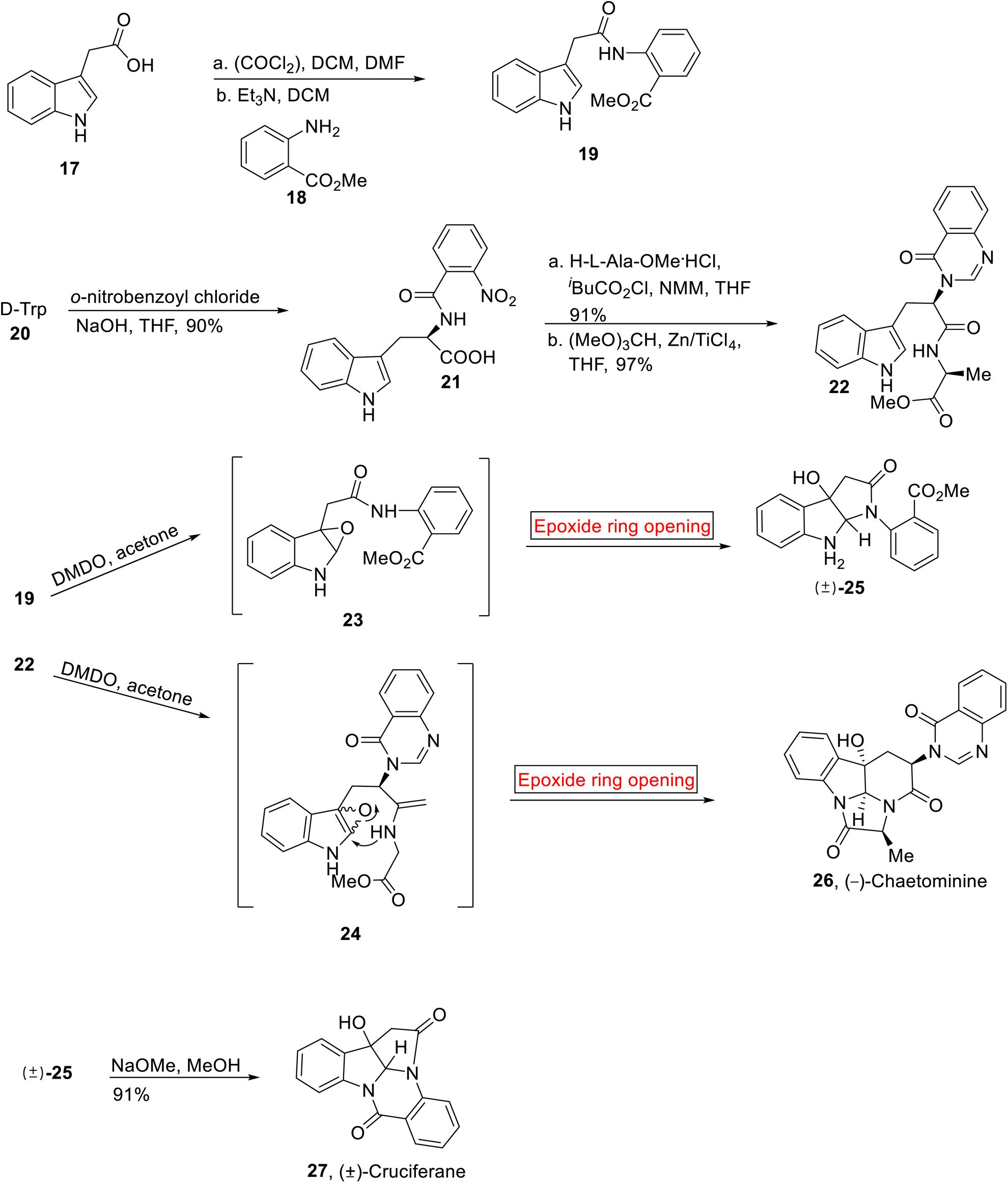
Cruciferane 27 (alkaloid) was extracted from the roots of plant (Isatis indigotica54) by the research group of Shi in 2012. It is used in traditional medicines to treat encephalitis B, carbuncles, erysipelas, influenza, and epidermic hepatitis. It has antipyretic properties as well. Total synthesis of Cruciferane was first reported in 2013 by research groups of Nair55 and then by Argade.56 Further, Ji57 has developed the synthesis of Cruciferane from complex and expensive starting materials and reagents. In the present work, Ghosh and Nagarajan in 2014 have reported the synthesis of (±)-Cruciferane 31 from simple route and economical starting materials by employing epoxide ring opening reaction as a key step (60.3%, overall yield).58 Similarly, (−)-Chaetominine 36 is an alkaloid which was isolated by research group of Tan from Chaetomium sp. IFB-E015 (fungus present on the leaves of Adenophora axilliflora) in 2006.59 It exhibits strong activity opposite to human colon cancer SW1116 (28 nM)59 and human leukemia K562 (21 nM) cell lines. The first total synthesis of (−)-Chaetominine 26 has been previously reported by research group of Snider,60 second by Evano,61 third by Papeo.62 Then, the fourth approach was reported by Evano and colleagues in nine steps (14% overall yield).63 In 2014, Huang et al., reported the asymmetric, protecting group free total synthesis of (−)-Chaetominine 26 that features excellent overall yield (33.4%) as compared to other approaches, in one-pot sequence reactions. They devised the facile and shorter synthetic route by employing epoxide ring opening reaction as one of the key steps.64 Total synthesis of Cruciferane 27 was initiated by the reaction between indole 3-acetic acid 17 and oxalyl chloride, followed by base (Et3N)-catalyzed amidation in the presence of methyl anthranilate 18 to afford amidation product 19 (78%). The synthesis towards (−)-Chaetominine 36 began with the preparation of aroylated product 21. The aroylated product 21 (90%) was obtained by the treatment of D-tryptophan 20 with o-nitrobenzoyl chloride which was further treated with L-alanine methyl ester hydrochloride and iBuCO2Cl/NMM (N-methylmorpholine) followed by reaction with HC(OMe)3 and in situ generated TiCl4 to provide quinazolinone 22 (97%). Then, compound 19 and 22 were individually subjected to DMDO-mediated epoxidation to provide epoxides 23 and 24 (96%) respectively which further experienced intramolecular epoxide ring opening reaction to furnish (±)-25 (85%) and (−)-Chaetominine 26 (42%) respectively. Then, the compound (±)-25 was reacted with NaOMe (MeOH) to render racemic (±)-Cruciferane 27 in 91% yield (Scheme 2).
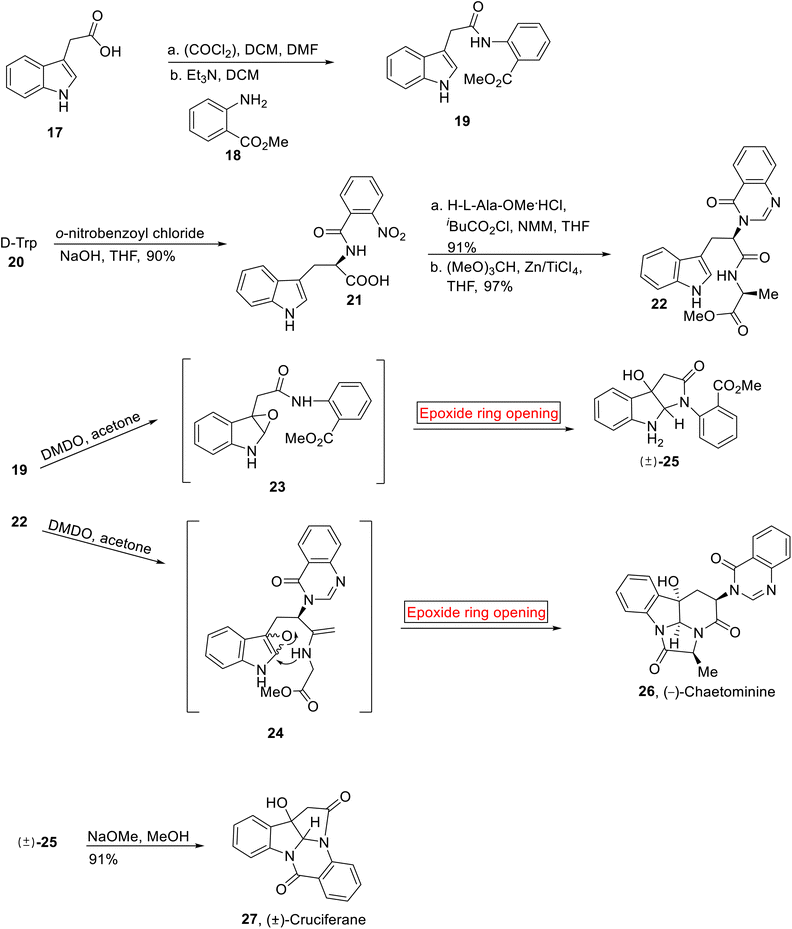 |
| | Scheme 2 Synthesis of (−)-Chaetominine 26 and (±)-Cruciferane 27. | |
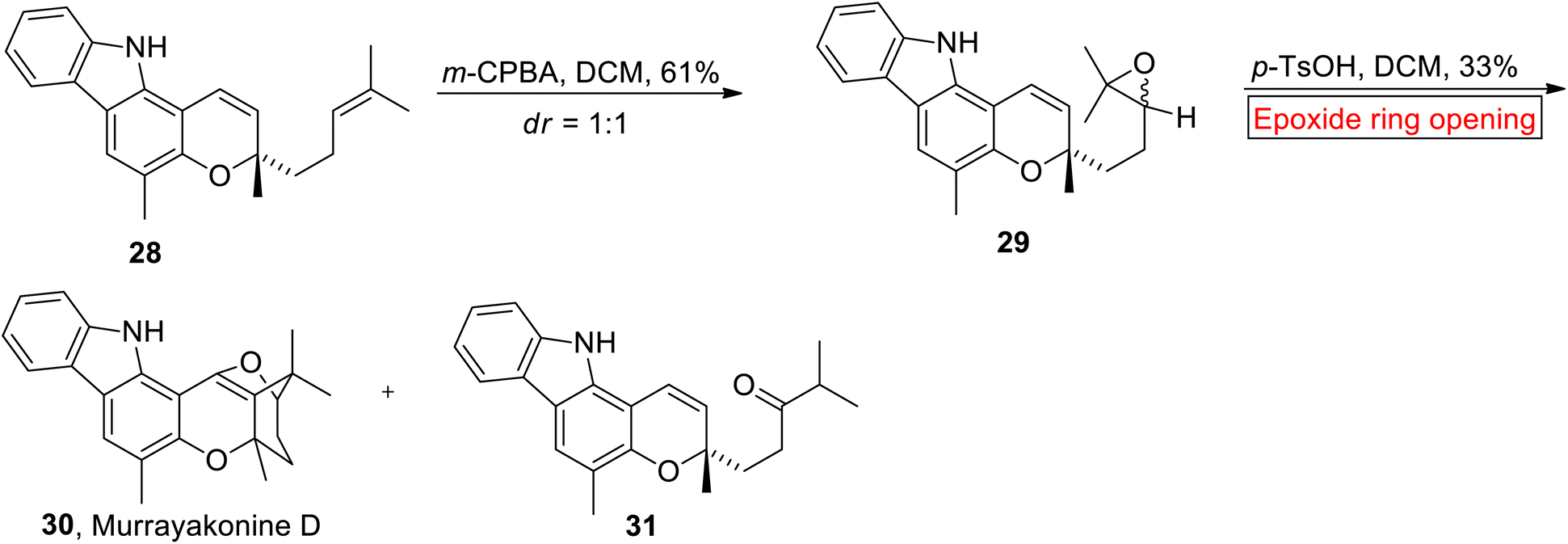
Carbazole alkaloids. Carbazole alkaloids (with a pyrrole moiety) are prevalent natural substances derived from multiple plant species as well as bacteria and fungi.65 Murrayakonine D 30 (carbazole alkaloid) has 6/6/6/5 ring system which was extracted from Murraya koenigi (medicinal plant).66 Research group of George has reported the synthesis of Murrayakonine D 30 (33% overall yield) in 2017 via epoxide ring opening reaction.67 In the first step, Mahanimbine 28 (which was prepared according to research group of Knölker68) went through meta-chloroperoxybenzoic acid (m-CPBA)-mediated epoxidation to generate 29 (61%). The compound 29 was further subjected to acid (p-TsOH)-catalyzed epoxide ring opening reaction in DCM to provide inseparable mixture of 30 and undesired ketone 31. The mixture was washed with cold MeOH to separate Murrayakonine D 30 (Scheme 3).
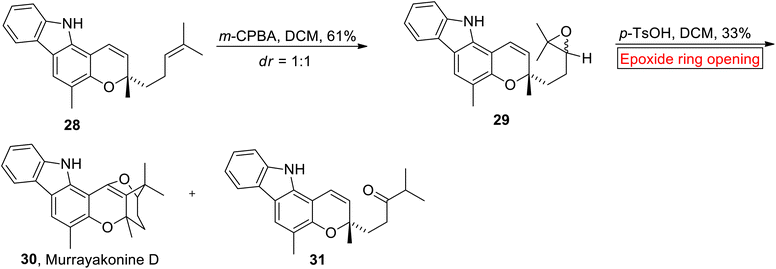 |
| | Scheme 3 Synthesis of Murrayakonine D 30. | |
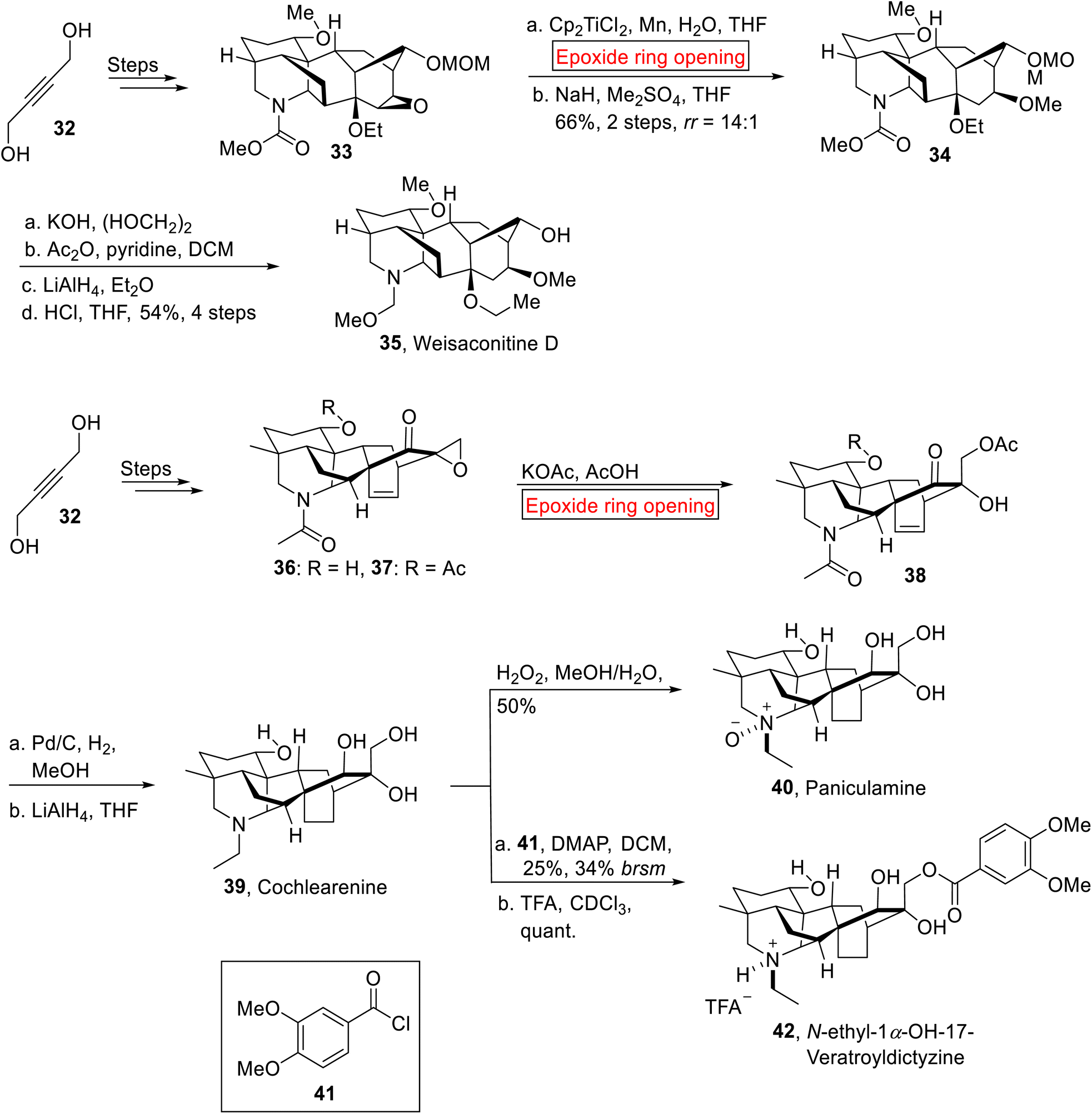
Diterpenoid alkaloids. Weisaconitine D 35, Cochlearenine 39, Paniculamine 40 and N-ethyl-1α-hydroxy-17-Veratroyldictyzine 42 are polycyclic diterpenoid alkaloids which are extracted from Aconitum, Delphinium, and Consolidum plants. These diterpenoid alkaloids exhibit anti-arrhythmic, anti-inflammatory, analgesic, and other medicinal properties.69–71 Owing to the structural complexity and pharmacological activities of diterpenoid alkaloids with varying oxygenation and substitution patterns, research group of Sarpong in 2017 reported the first syntheses of C-18, C-19 and C-20 diterpenoid alkaloids by involving ring opening reaction of epoxide.72 The synthetic route towards C-18 diterpenoid alkaloid (Weisaconitine D) 35 initiated with the formation of epoxy ether 33 from allylic alcohol 32 over few steps. The epoxy ether 33 underwent titanium-mediated epoxide ring opening reaction regioselectivity, followed by methylation, which rendered 34 (66%, 2 steps, rr = 14:1). The compound 34 was subjected to KOH-catalyzed hydrolysis, acetylation, LiAlH4-mediated reduction and subsequent deprotection using aq. HCl to furnish Weisaconitine D 35 (54%, 4 steps). Similarly, the synthetic endeavor towards C-20 diterpenoid alkaloids (Cochlearenine 39, Paniculamine 40, Veratroyldictyzine 42) was commenced with the preparation of demethylated products 36 and 37 (51%) from allylic alcohol 32 over few steps. Then, compound 36 and 37 were subjected to KOAc-catalyzed epoxide ring opening reaction to render α-hydroxyketone 38 which was further exposed to Pd/C hydrogenation accompanied by reduction (LiAlH4) to forge Cochlearenine 39 (63–77%, 3 steps). Then, further treatment of 39 with H2O2 furnished Paniculamine 40. Finally, N-ethyl-1α-OH-17-Veratroyldictyzine 42 (quant.) was attained by veratroylation of 39 with 41 in DMAP followed by titration with trifluoroacetic acid (TFA) (Scheme 4).
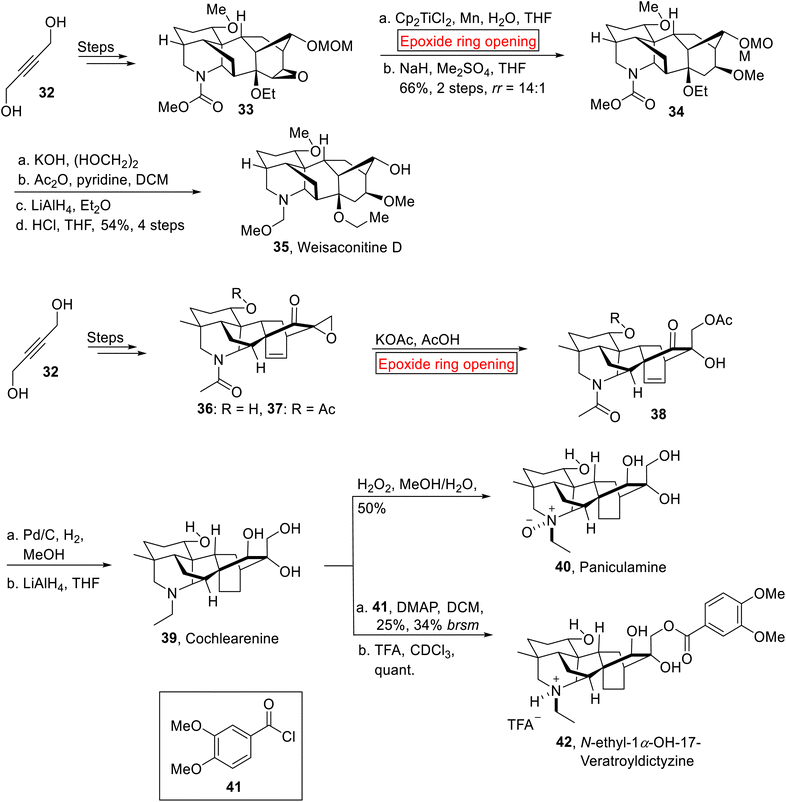 |
| | Scheme 4 Synthesis of Weisaconitine D 35, Cochlearenine 39, Paniculamine 40, Veratroyldictyzine 42. | |
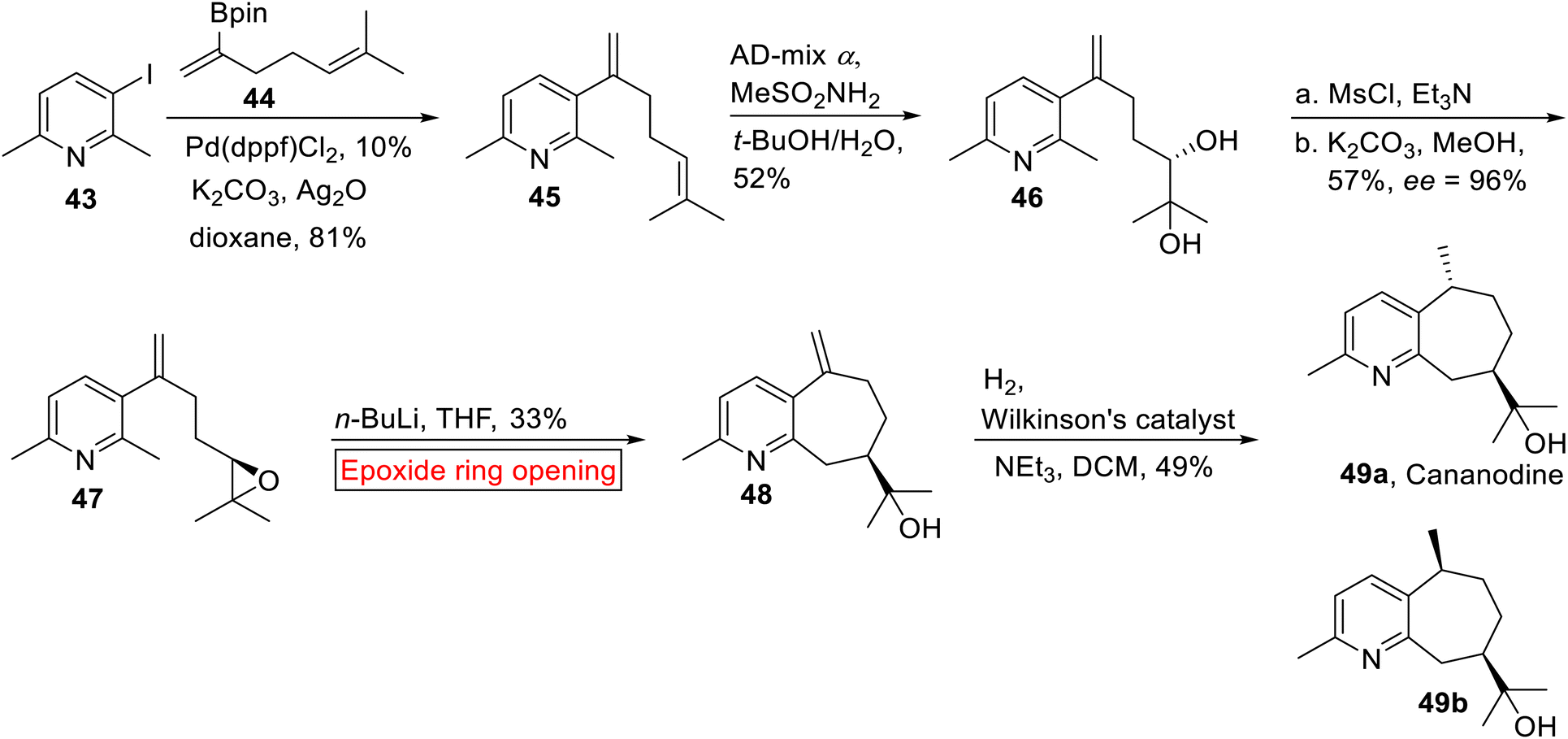
Guaipyridine alkaloid. Cananodine 49a is a rare73 guaipyridine alkaloid which was isolated from Cananga odorata74 (ylang–ylang). In South Asia, C. odorata has been used in folk medicines. Recently, Cananodine 49a was isolated from Cyperus scariosus.75 It is biologically potent opposite to Hep 2,2,15 hepatocarcinoma (IC50 = 3.8 μg mL−1) and Hep G2 (IC50 = 0.22 μg mL−1) cell lines.76 Various transition metals-mediated cross-coupling reactions have been employed to carry out the synthesis of several intricate organic compounds.77,78 In 2017, Shelton et al. reported the synthesis of optically active Cananodine 49a by utilizing palladium-mediated cross coupling and epoxide ring opening reaction as significant steps.79 In the first step, pyridyl iodide 43 and dienylboronate 44 were reacted under Pd-catalyzed cross coupling reaction in the presence of Ag2O to afford 45 in excellent yield (81%). Next, 45 underwent asymmetric dihydroxylation to furnish diol 46 (52%). In the next step, diol 46 was converted into epoxide 47 (57%) which was further subjected to n-butyllithium-mediated intramolecular epoxide ring opening reaction as a key step to provide 48 (33%). Lastly, compound 48 underwent hydrogenation of methylene group in the presence of Wilkinson's catalyst to synthesize Cananodine 49a and its epimer 49b as a 1:1 inseparable mixture of diastereomers in 49% yield (Scheme 5).
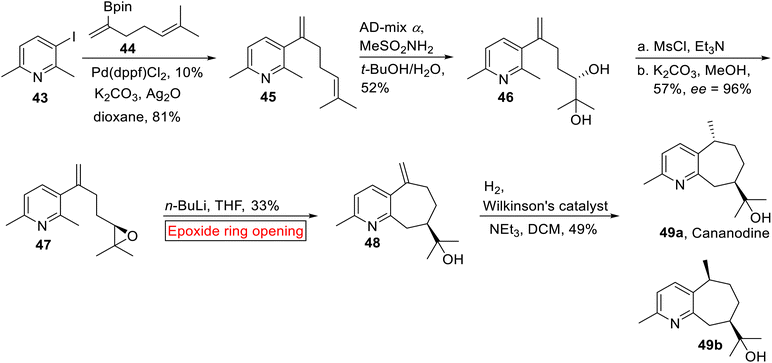 |
| | Scheme 5 Synthesis of optically active Cananodine 49a. | |
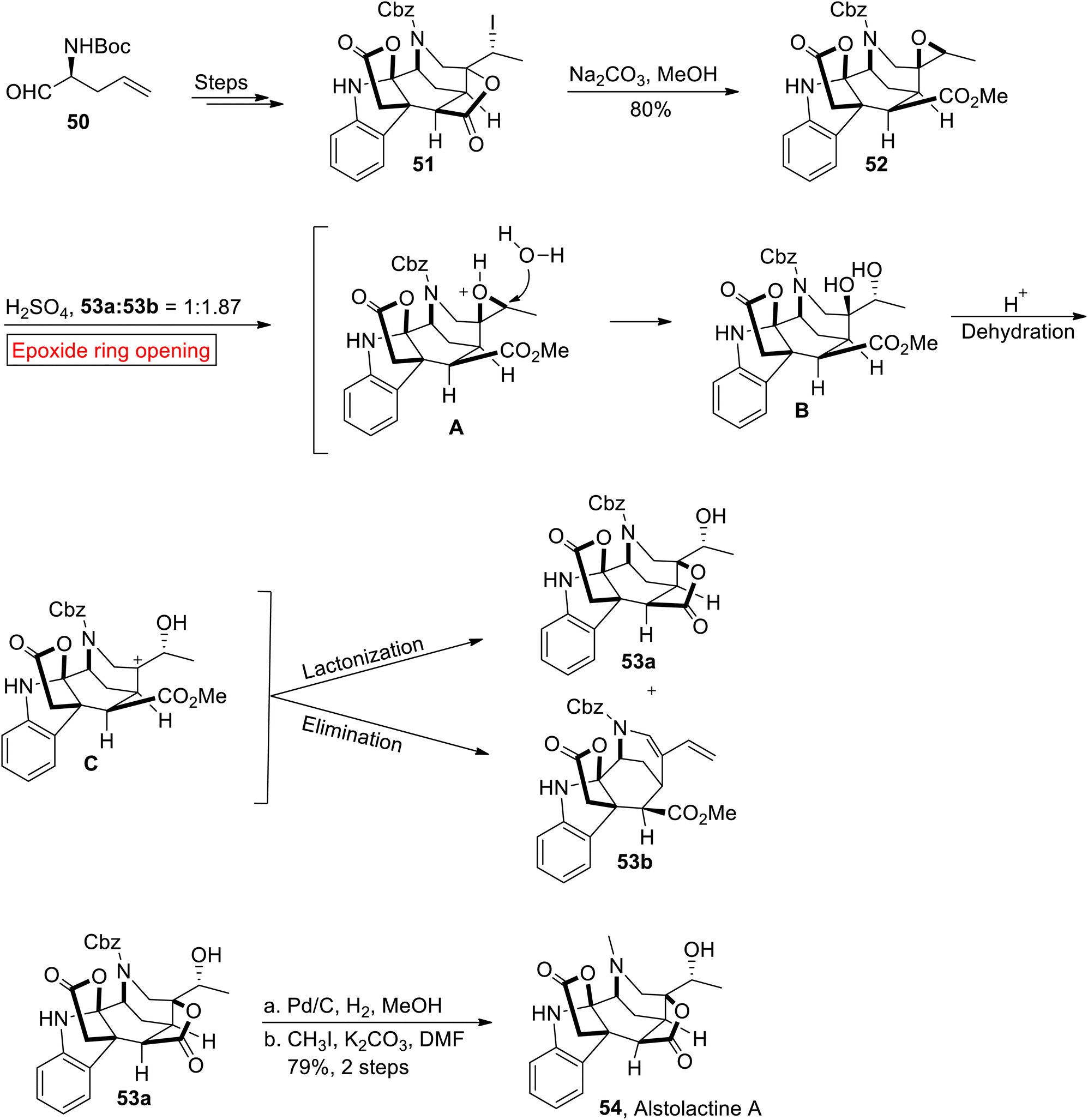
Akuammiline alkaloid. Alstolactine A 54 belongs to a family of akuammiline alkaloids which is genetically related to indole monoterpenoids. It was isolated by Liu and research group of Luo, from leaves of Alstonia scholaris.80,81 In the present research, Gao and co-workers have used an epoxide ring opening reaction for the first total synthesis of Alstolactine A 54 in 2017.82 The strategies described by research group of Gao may offer a unique perspective in the synthesis of akuammilines and their derivatives for future medicinal studies. The synthetic endeavor commenced with the synthesis of 51 from L-2-allylglycine 50 over several steps, which further experienced base-mediated epoxidation in MeOH to forge epoxide 52 (80%). In the next step, epoxide 52 underwent acid-catalyzed epoxide ring opening reaction to afford compounds 53a (31%) and 53b (42%). Finally, compound 53a was subjected to Pd/C-mediated hydrogenation followed by methylation to furnish asymmetric Alstolactine 54 in 79% yield (Scheme 6).
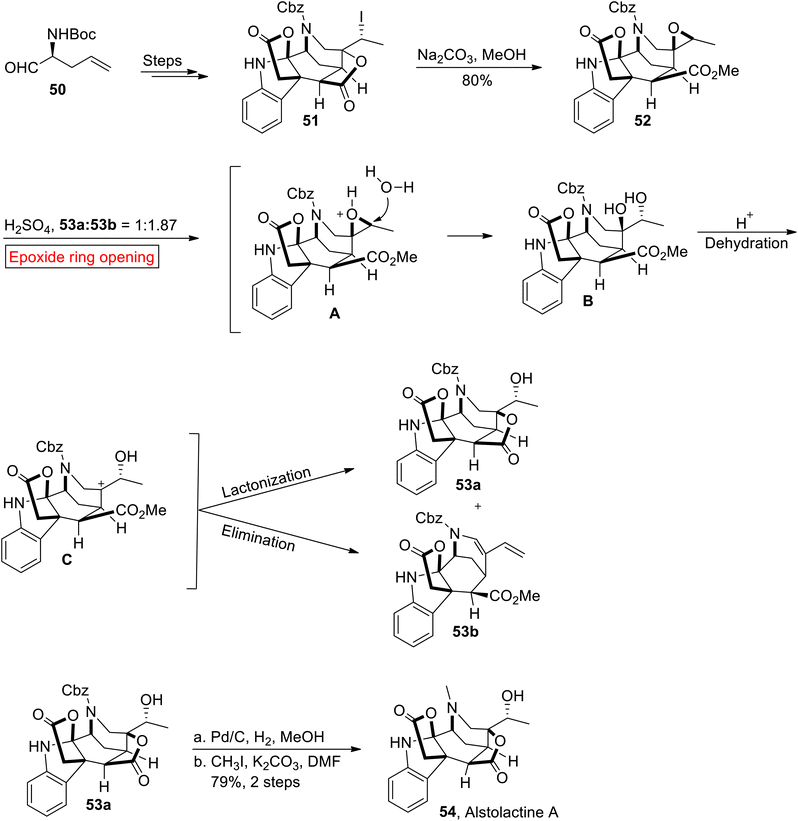 |
| | Scheme 6 Total synthesis of Alstolactine A 54. | |
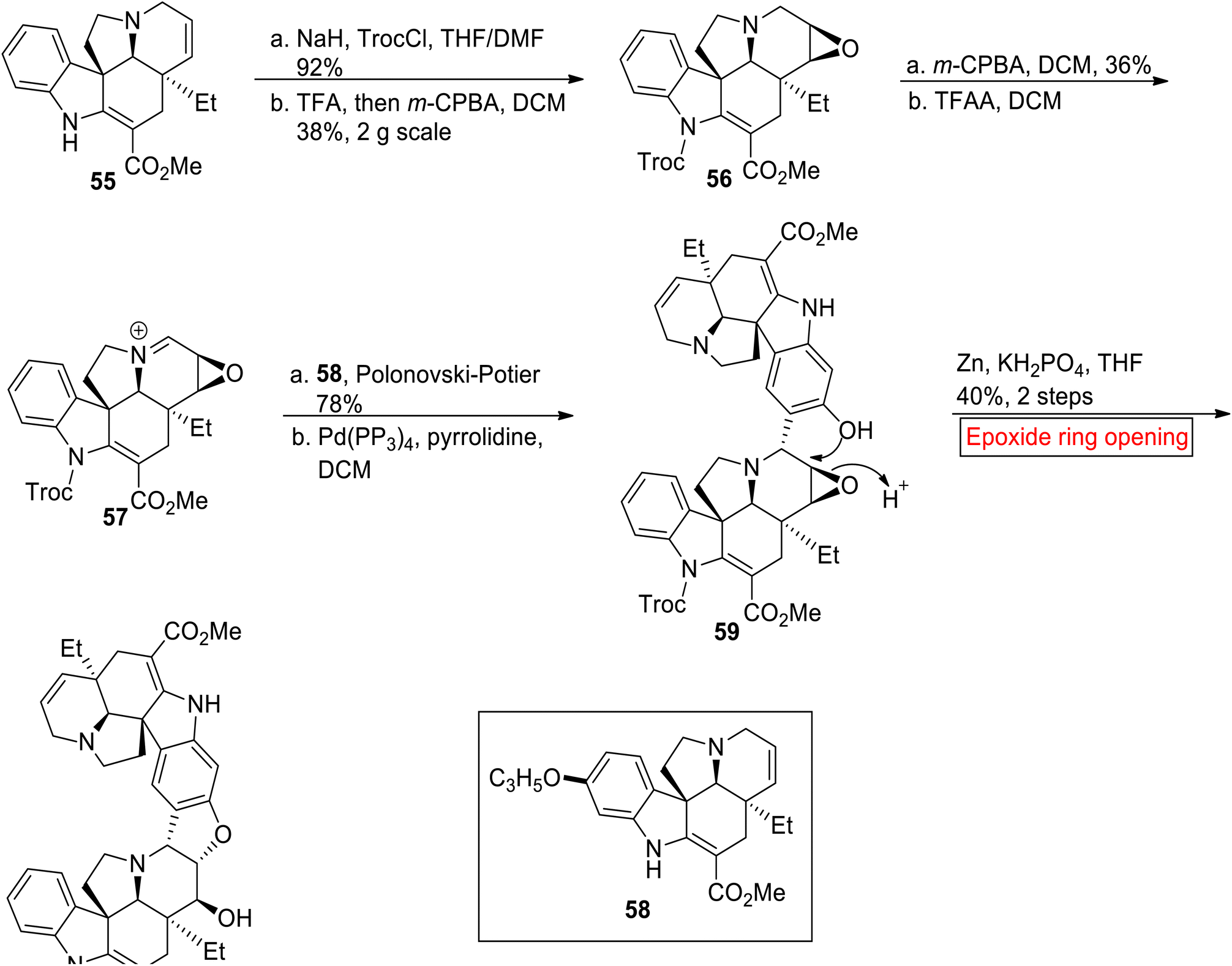
Bis-indole alkaloid. (−)-Melodinine K 60 is a bis-indole alkaloid which belongs to the class of Aspidosperma–Aspidosperma which was isolated from Melodinus tenuicaudatus (plant) by Luo and colleagues in 2010.83 It displays strong cytocidal activity opposite to cell line of breast cancer (IC50 = 2.7 μM) as compared to cisplatin (IC50 = 18.7 μM) and vinorelbine (IC50 = 17.2 μM). In 2020, Andrade and allies disclosed the first synthesis of (−)-Melodinine K 60 by employing epoxide ring opening reaction as one of the key steps.84 Due to structural complexity and low isolation yield from natural source, they developed an efficient method for the synthesis of (−)-Melodinine K 60 for future research on its biological functions. In the first step, (−)-tabersonine 55 was treated with NaH and TrocCl (2,2,2-trichloroethoxycarbonyl chloride) followed by stereo divergent oxidation in the presence of TFA and m-CPBA to afford N-Troc pachysiphine 56 (38%). In the next step, compound 56 was treated with m-CPBA and then with trifluoroacetic anhydride (TFAA) to afford iminium ion 57. Then, the stereo divergent attack of iminium ion 57 at the C-15 of (−)-16-allyloxytabersonine 58 followed by Pd (PPh3)4-catalyzed deprotection afforded alcohol 59. Finally, alcohol 59 went through epoxide ring opening reaction with subsequent deprotection reaction to forge (−)-Melodinine K 60 in 40% yield (Scheme 7).
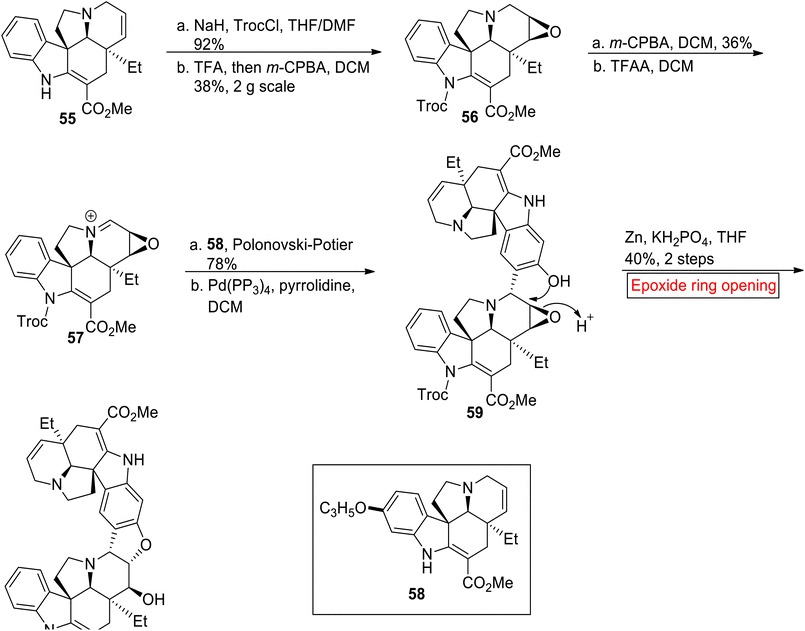 |
| | Scheme 7 Synthesis of (−)-Melodinine K 60. | |
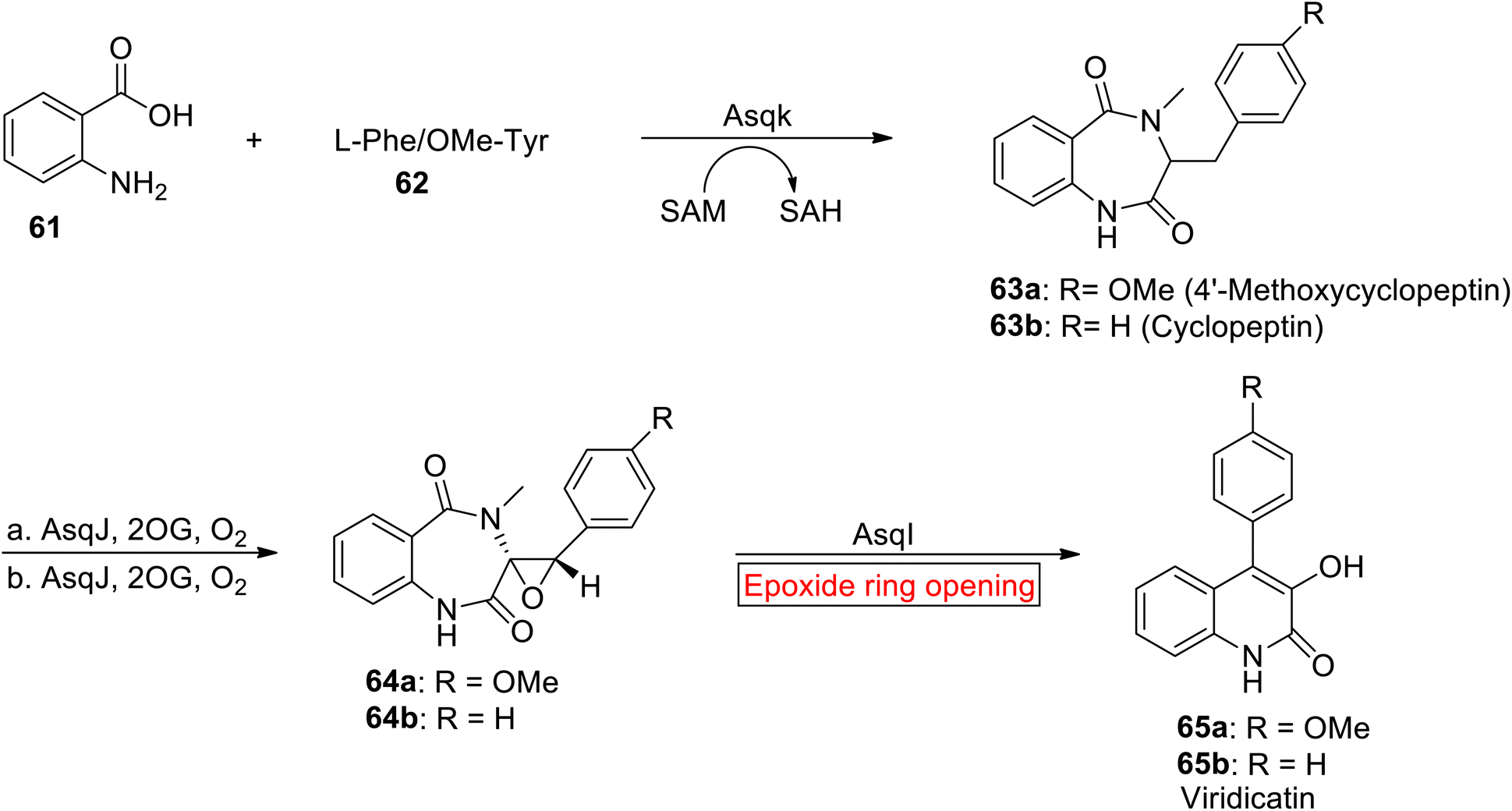
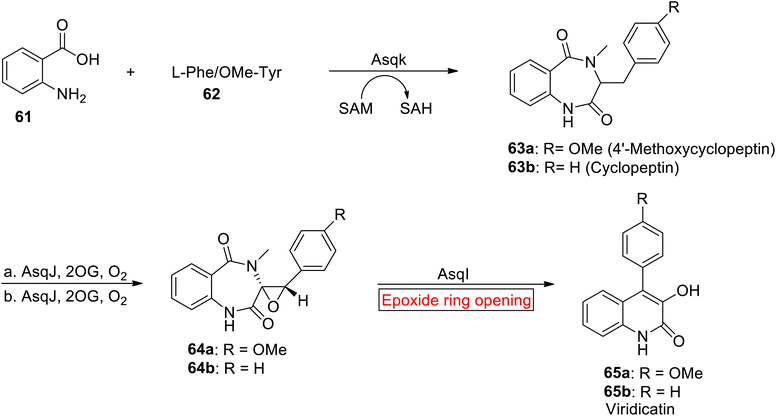
Quinolone alkaloid. Virdicatin 65 is a quinolone alkaloid (secondary metabolites) which exhibits antiviral, antibacterial, antiallergic, and antitumor activities.85,86 Quinolone core containing Virdicatin 65 acts as a scaffold for synthesis of natural and synthetic compounds.87 In 2021, Chang et al. reported the synthesis of Viridicatin 97 by involving epoxide ring opening reaction as one of the key steps.88 Their research work demonstrates the utilization of natural and synthetic processes for the efficient synthesis of quinolone alkaloids that would suggest promising directions for exploring their biological activities in future studies. In the first step, cyclopeptin 63 was obtained by Asqk (methyl transferase)-mediated cyclization with subsequent treatment (methylation) of 61 with 62. Then, cyclopeptin 63 was subjected to desaturation accompanied by asymmetric epoxidation in the presence of 2-oxoglutarate (Fe/2OG) dependent oxygenase and AsqJ to synthesize cyclopenin 64. In the next step, cyclopenin 64 experienced AsqI-catalyzed epoxide ring opening reaction to render Virdicatin 65 (Scheme 8).
 |
| | Scheme 8 Synthesis of Virdicatin 65. | |
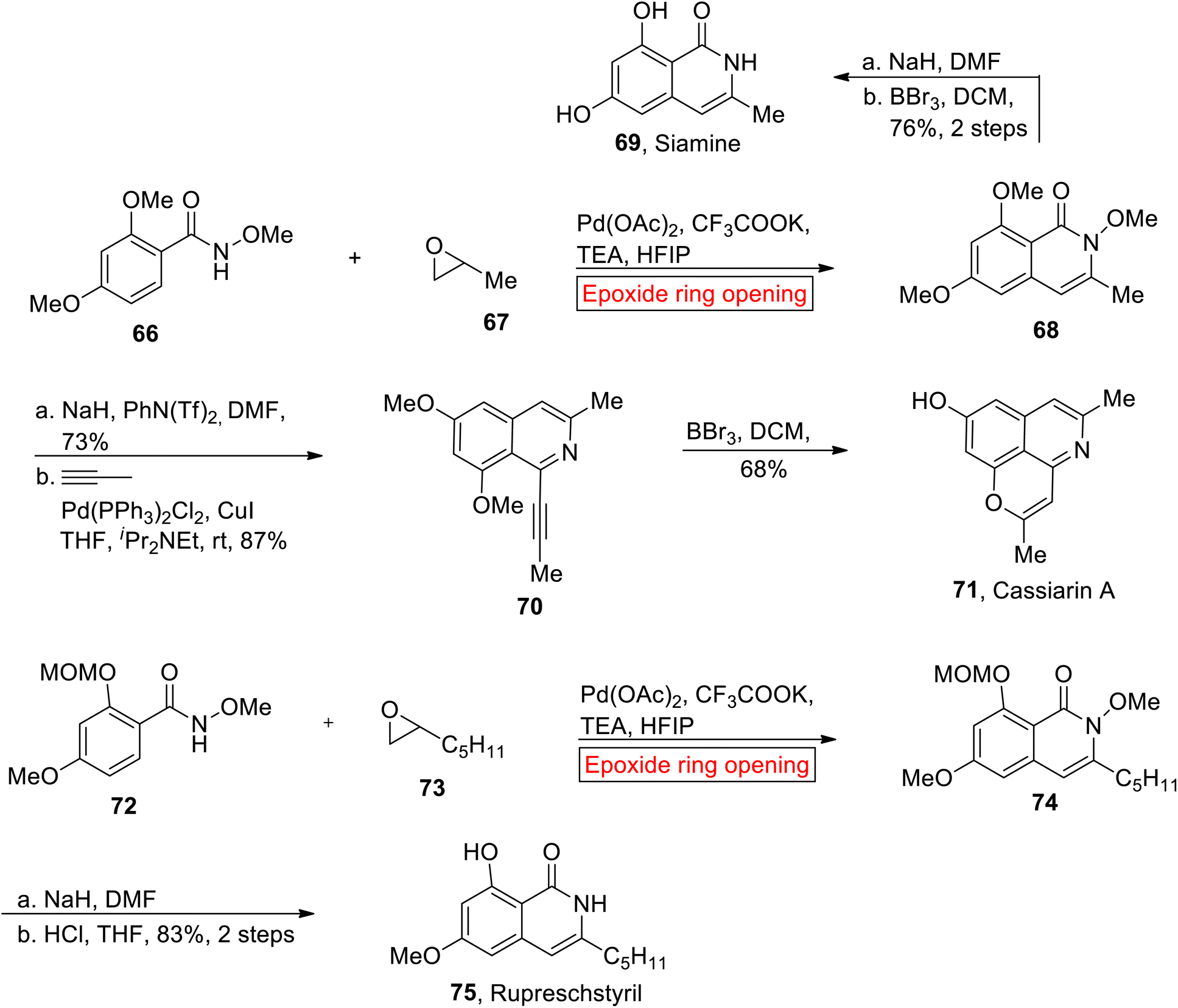
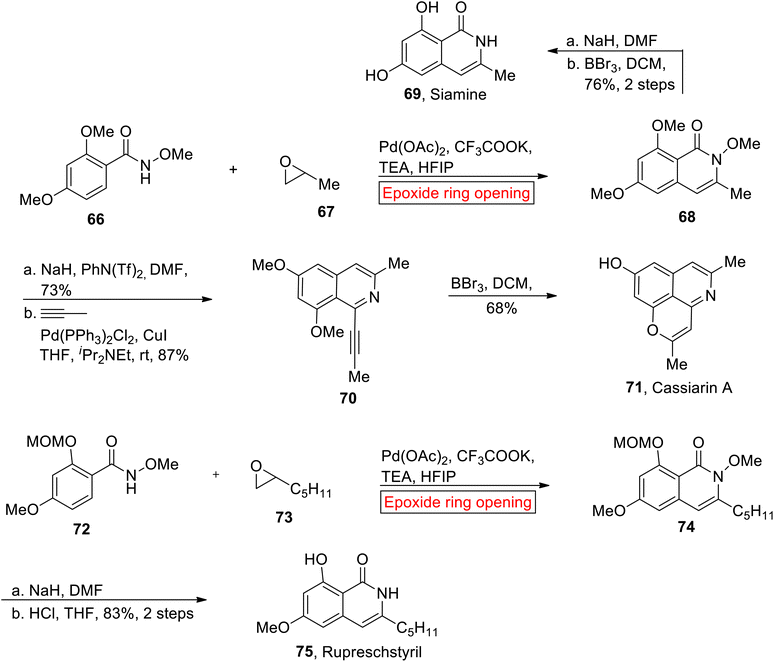
Isoquinolones such as Siamine 69, Cassiarin A 71 and Rupreschstyril 75 are biological active compounds. Research groups of Glorius89 (2014), Huang90 (2017) and Kou91 described the synthesis of isoquinolones in different ways but their synthetic route faced several drawbacks due to coupling partners. In 2021, Wang and coworkers reported the syntheses of isoquinolones by employing epoxide ring opening reaction as one of the key steps for the first time in efficient yield within fewer steps.92 In the first step, Pd-catalyzed ring opening of epoxide 67 occurred with N-methoxybenzamide 98 to render N-methoxyisoquinolone 68 in the presence of hexafluoroisopropanol (HFIP) and appropriate base i.e., triethylamine (TEA). In the next step, isoquinolone 68 was converted into Siamine 69 (76%, 2 steps) via sequential reduction and demethylation. Similarly, isoquinolone 68 underwent reduction (NaH) followed by coupling with in situ prepared propyne to afford alkyne 70 (87%). Finally, Cassiarin A 71 (37%, overall yield) was obtained from alkyne 70 via demethylation (BBr3) with subsequent 6-endo-dig cyclization. The synthetic endeavor towards Rupreschstyril 75 was initiated with the formation of isoquinolin-1-one 74 from 72 and epoxide 73 through epoxide ring opening reaction under optimized conditions. Lastly, compound isoquinolin-1-one 74 experienced reduction followed by deprotection (MOM) reaction to afford Rupreschstyril 75 in 83% yield over 2 steps (Scheme 9).
 |
| | Scheme 9 Synthesis of Siamine 69, Cassiarin A 71, and Rupreschstyril 75. | |
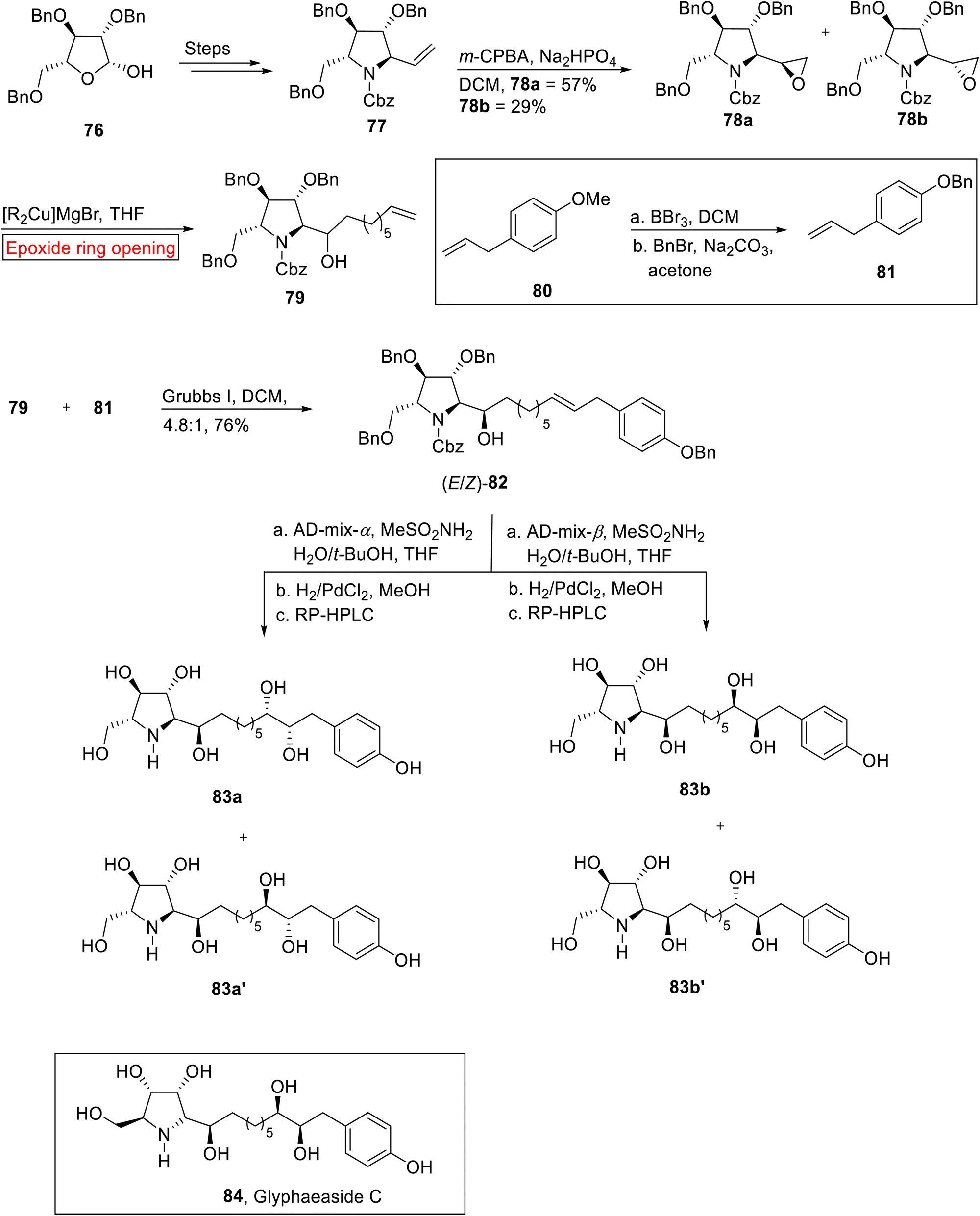
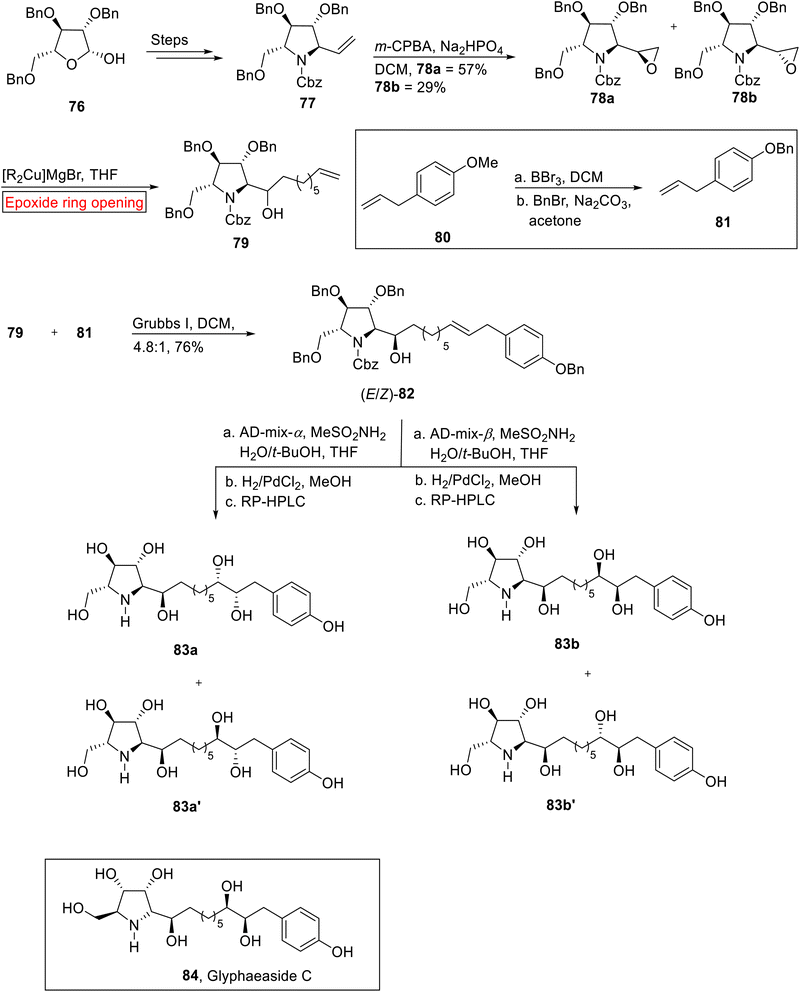
Broussonetine alkaloid. The Glyphaeaside C 84 (iminosugars) having polyhydroxylated phenylalkyl chain belongs to broussonetine alkaloid family which was extracted from Glyphaea brevis.93 Because of side-chain character of Glyphaeaside C 84, it inhibits almond β-glucosidase, rice α-glucosidase and snail β-mannosidase. In 2021, Pyne et al. reported the synthesis of Glyphaeaside C 84 by utilizing epoxide ring opening reaction.94 For stereochemical reasons, they synthesized 79 by utilizing epoxide ring opening reaction instead of carbonyl addition. Firstly, β-D-arabinofuranose 76 rendered vinylpyrrolidine 77 (28%) via treatment over few steps which upon epoxidation (m-CPBA) furnished epoxides 78a (57%) and 78b (29%). In the next step, 78a underwent epoxide ring opening reaction in the presence of Gilman reagent in THF to synthesize alcohol 79 in a good yield (84%). Secondly, 4-allylanisole 80 was subjected to O-demethylation with BBr3 (in DCM) accompanied by benzylation (acetone) to yield 81 (44%). In the next step, alkene 82 was prepared by the Grrubs' I-catalyzed reaction of 79 with 81. Finally, alkene 82 was reacted with AD-mix-α/AD-mix-β (Sharpless asymmetric dihydroxylation) with subsequent deprotection reaction to furnish four enantiomers 83a/83a′, and 83b/83b′ respectively. According to the specific rotation data, it was suggested that 83a was the enantiomer of Glyphaeaside C 84 (Scheme 10).
 |
| | Scheme 10 Synthesis of Glyphaeaside C 84. | |
Syntheses of terpenoid-based natural products
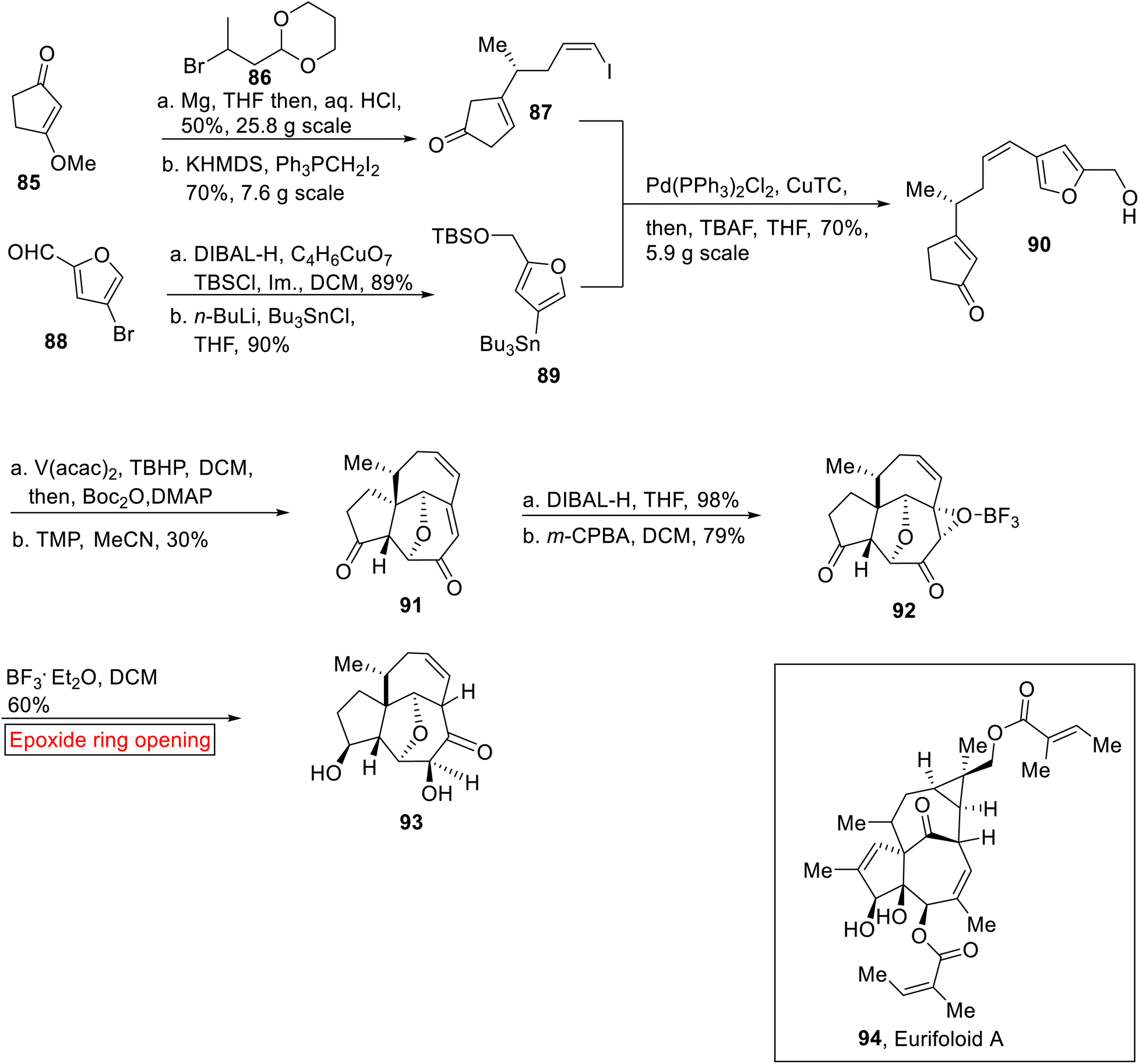
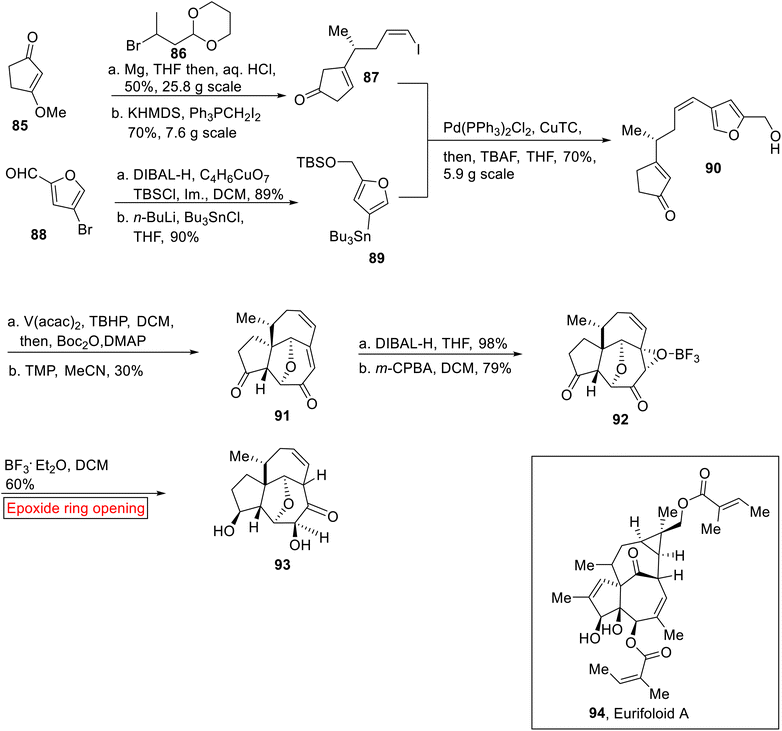
Diterpenoid. Eurifoloid A 94 is a diterpenoid, belongs to a phorboid family of natural product which was extracted from Euphorbia neriifolia.95 It exhibits biological activities96 such as antitumor and anti-HIV. Synthesis of Eurifoloid A 94 has remained a challenging task97 for chemists due to the presence of congested stereogenic centers such as quaternary stereocenter, angeloyloxy and tigloyloxy groups in its structure. Liu98 and colleagues, first time reported the synthesis of tricyclic core 93 of Eurifoloid A 94 by employing ring opening reaction of epoxide as one of the key steps in 2017. The synthetic route initiated by the reaction of compound 85 with alkyl bromide 86 using Mg, THF (in situ Grignard reagent) followed by Wittig reaction to generate Z-vinyl iodide 87 (70%). In the next step, bromofuran carbaldeyde 88 underwent reduction (DIBAL-H), followed by protection and then Br–Sn exchange yielded tin reagent 89 (90%). Further, Z-vinyl iodide 87 and tin reagent 89 were reacted under Stille coupling conditions. Later, TBS group deprotection rendered coupled product 90 (70%). The coupled product 90 further experienced oxidative rearrangement and then type II intramolecular [5 + 2] cycloaddition reaction in the presence of tetramethylpiperidine (TMP) to provide the compound 91 (30%) having [5-7-7] tricyclic core. Compound 91 was then subjected to sequential diastereoselective reduction and m-CPBA-mediated oxidation (DCM) to furnish the epoxide 92 (79%). Finally, the ring opening reaction of epoxide 92 in the presence of BF3·Et2O using DCM as solvent, resulted in the core structure 93 (that contains reduced strain bicyclo[4.4.1]undecane ring system featuring cis-bridgehead) of Eurifoloid A 94 but with opposite stereochemistry at C8 (Scheme 11).
 |
| | Scheme 11 Synthesis of tricyclic core 93 of Eurifoloid A 94. | |
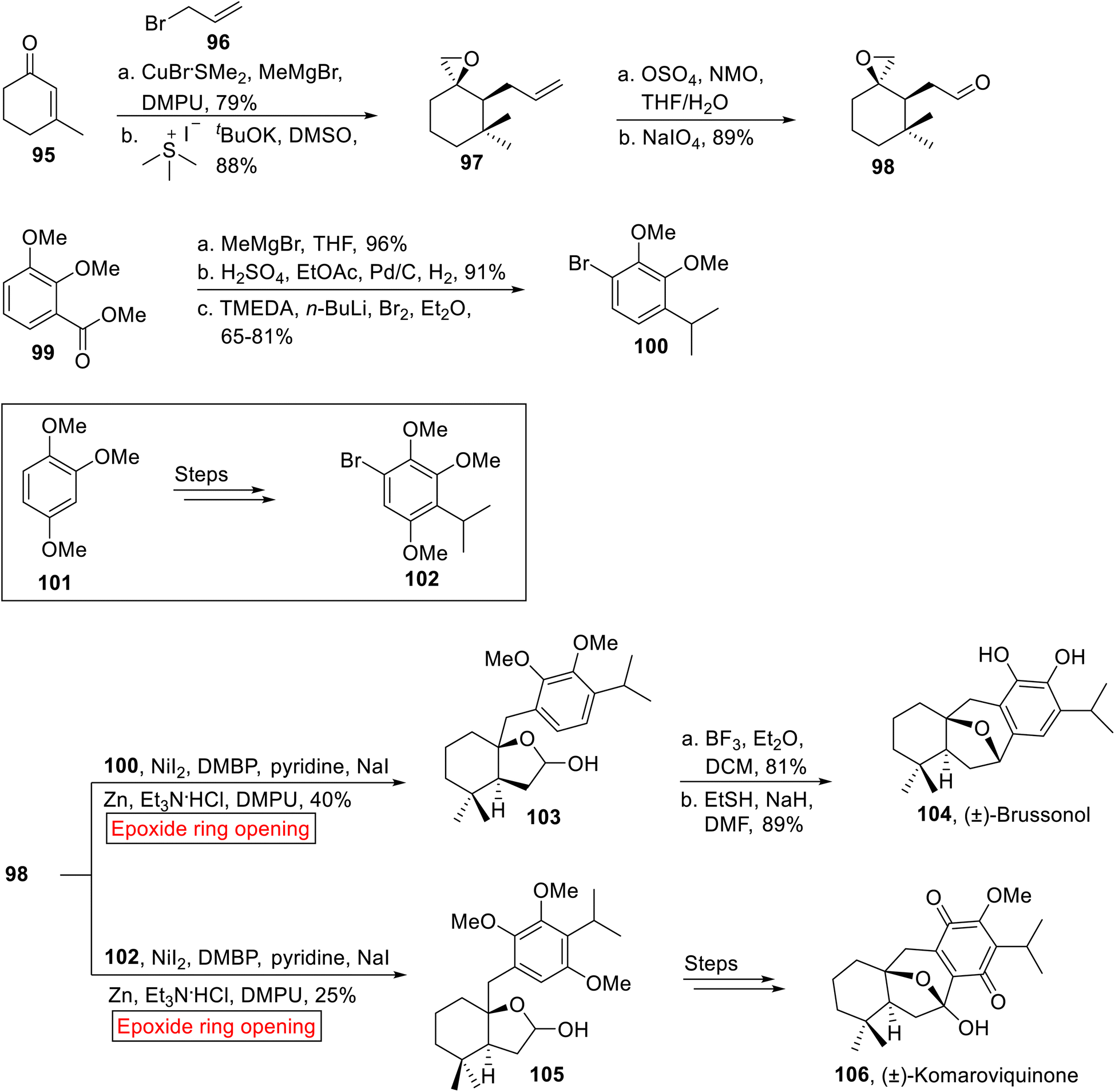
(±)-Brussonol 104 and (±)-Komaroviquinone 106 belong to a family of ictexane diterpenes, isolated from terrestrial plants containing fused tricyclic skeleton (6-7-6). (±)-Brussonol 104 and (±)-Komaroviquinone 106 display antiprotozoal, anticancer and cytotoxic activities. These natural products were synthesized by a number of reactions such as Ga(III)-catalyzed cycloisomerization,99 tandem C–H oxidation/cyclization/rearrangement,100 intramolecular Marson-type cyclization,101 Pd-catalyzed intramolecular Heck reaction,102 intramolecular nucleophilic cyclization,103 cationic ring expansion,104 TiCl4-catalyzed Friedel–Crafts cyclialkylation105,106 and Pt-mediated hydrative cyclization.107 In 2019, Burtoloso and Ahmad108 disclosed the syntheses of (±)-Brussonol 104 (18.4%, overall yield) and (±)-Komaroviquinone 106 (7% overall yield) by utilizing regioselective epoxide ring-opening reaction. The method developed by Burtoloso and Ahmad facilitated the efficient synthesis of these compounds till date, in order to achieve high yields in less steps as compared to previous approaches by employing similar starting materials. The synthetic approach towards (±)-Brussonol 104 and Komaroviquinone 106 was initiated with the formation of coupling fragment 98. In the first step, α, β-unsaturated ketone 95 experienced Michael addition reaction with MeMgBr in the presence of DPMU and CuBr·SMe2, followed by enolate quenching with allyl bromide 96 and Corey–Chaykovsky epoxidation to forge single diastereomer of epoxide 97 (88%). In the next step, epoxy-aldehyde 98 (89%) was obtained by Lemieux–Johnson oxidation of epoxide 97. Then, aryl ester 99 underwent nucleophilic addition reaction (MeMgBr) followed by dehydration/hydrogenation reaction and selective bromination in the presence of tetramethylethylenediamine (TMEDA) and Et2O to afford aryl bromide 100 (65–81%). Similarly, the coupling partner 1-bromo-4-isopropyl-2,3,5-trimethoxybenzene 102 was synthesized from 1,2,4-trimethoxybenzene 101 over few steps. Then, the epoxide 98 was coupled with aryl bromide 100 and trimethoxybenzene 102 individually via Zn-catalyzed epoxide ring opening reaction in the presence of NaI, Et3N·HCl and 4,4′-dimethoxy-2,2′-bipyridine (DMBP) to yield acetals 103 (40%) and 105 (25%) respectively. Acetal 103 was then subjected to Friedel–Crafts cyclization followed by sodium thiolate-mediated demethylation to furnish (±)-Brussonol 104 (89%). Similarly, diastereoselective (±)-Komaroviquinone 106 was obtained from acetal 105 over few steps (Scheme 12).
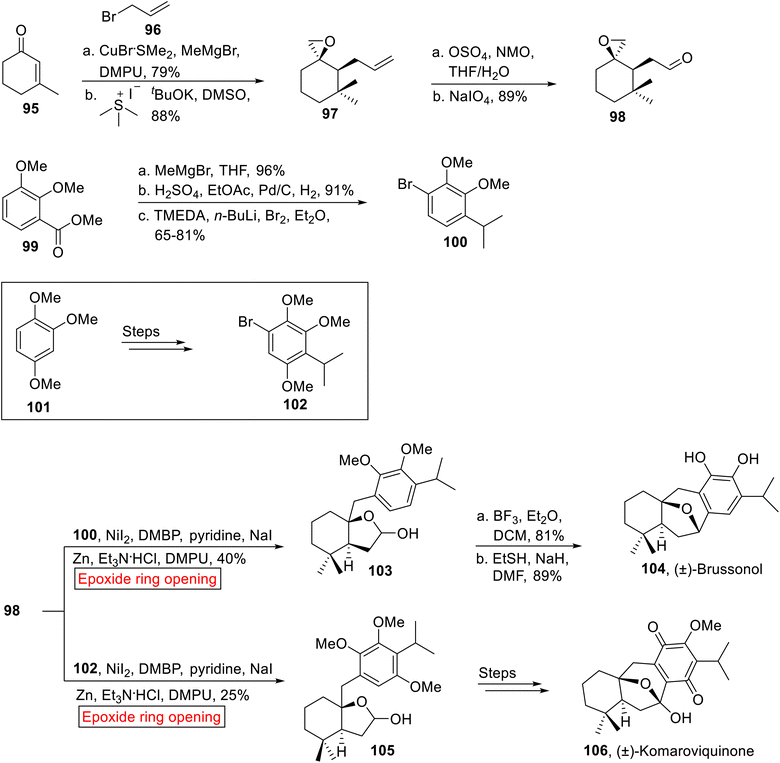 |
| | Scheme 12 Synthesis of (±)-Brussonol 104 (±)-Komaroviquinone 106. | |
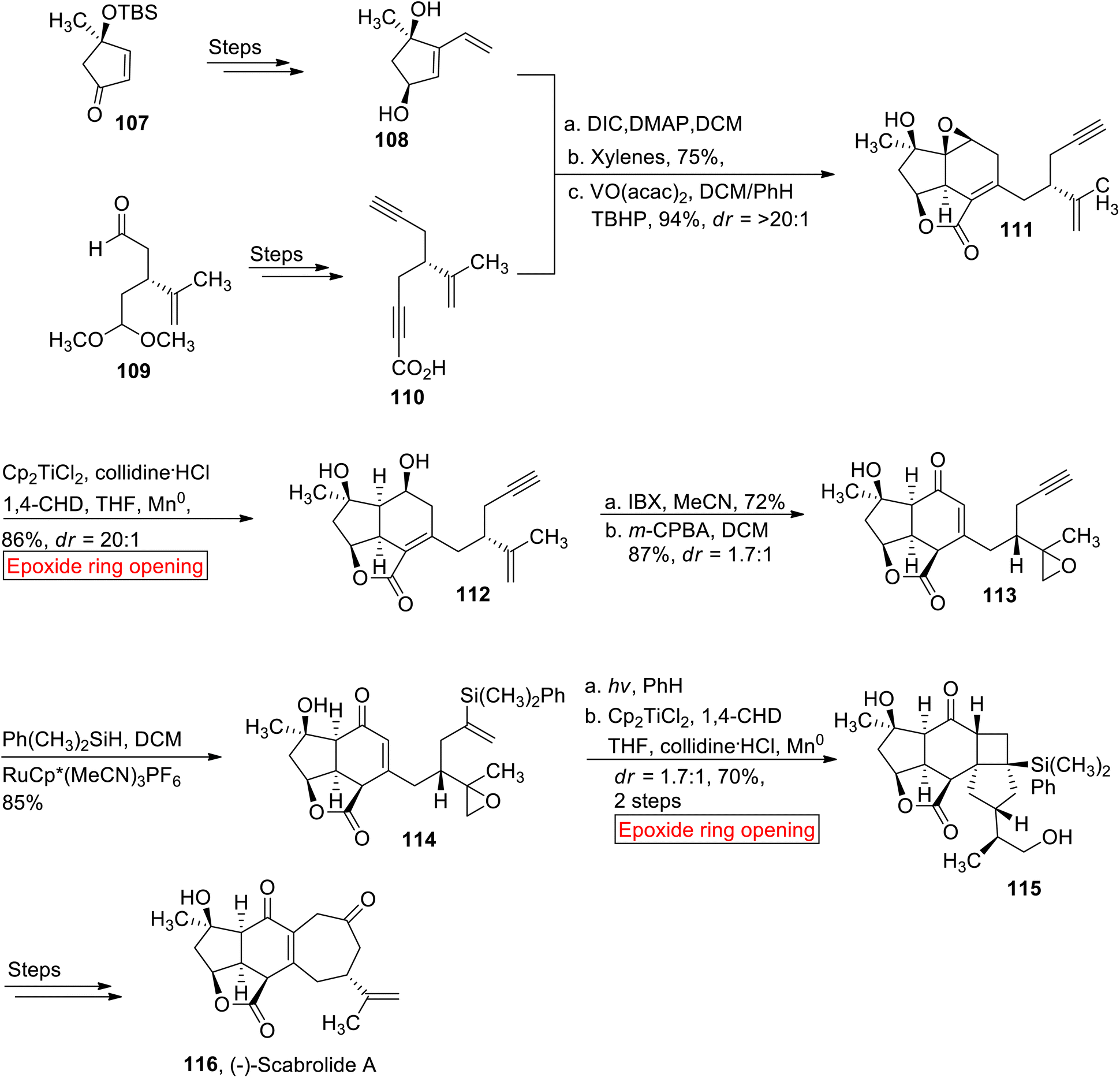
(−)-Scabrolide A 116 is a norcembranoid diterpenoid which belongs to the family of marine natural products.109 In 2002, it was isolated for the first time from Sinularia scabra (soft coral) by the research group of Sheu.110 (−)-Scabrolide A 116 acts as an anti-inflammatory agent111 as it inhibits the production of IL-6 and IL-12. In 2020, Stoltz112 reported the first total synthesis of (−)-Scabrolide A 116 by employing epoxide ring opening reaction as one of the key steps. This research report represented the first synthesis of any member of polycyclic furanobutenolide-derived norcembranoid diterpenoid family that have remained elusive to synthetic efforts for over two decades since their original discovery. The synthetic route began with the preparation of dihydroxyvinyl cyclopentene 108 and ynoic acid 110 which were utilized as the starting materials for the synthesis of (−)-Scabrolide A 116. In the first step, cyclopentene 108 and ynoic acid 110 were synthesized individually from enone 107 and monoprotected dialdehyde 109 respectively over few steps. Compounds 108 and 110 were reacted under Steglich conditions followed by Diels's reaction to yield Diels's alder adduct with subsequent epoxidation to furnish intermediate 111 (94%). In the next step, intermediated 111 was exposed to Cp2TiCl2-mediated epoxide ring opening reaction in the presence of collidine·HCl, 1,4-cyclohexadiene (1,4-CHD), THF and Mn0 to render diol 112 (86%, dr = >20:1). The diol 112 was further treated with 2-iodoxybenzoic acid (IBX) accompanied by selective epoxidation to afford mixture of epimers in good yield 113 (87%, 1.7:1). Then, the mixture 113 experienced Ru-mediated hydrosilylation to generate diastereomeric mixture of 114 (85%). In the next step, compound 114 was subjected to irradiation in the presence of PhH followed by Cp2TiCl2-mediated reductive epoxide ring opening reaction by utilizing earlier mentioned conditions (1,4-CHD, collidine·HCl, THF) to produce diol 115 (dr = 1.7:1, 70%). The resulting diol 115 was converted to (−)-Scabrolide A 116 over few steps (Scheme 13).
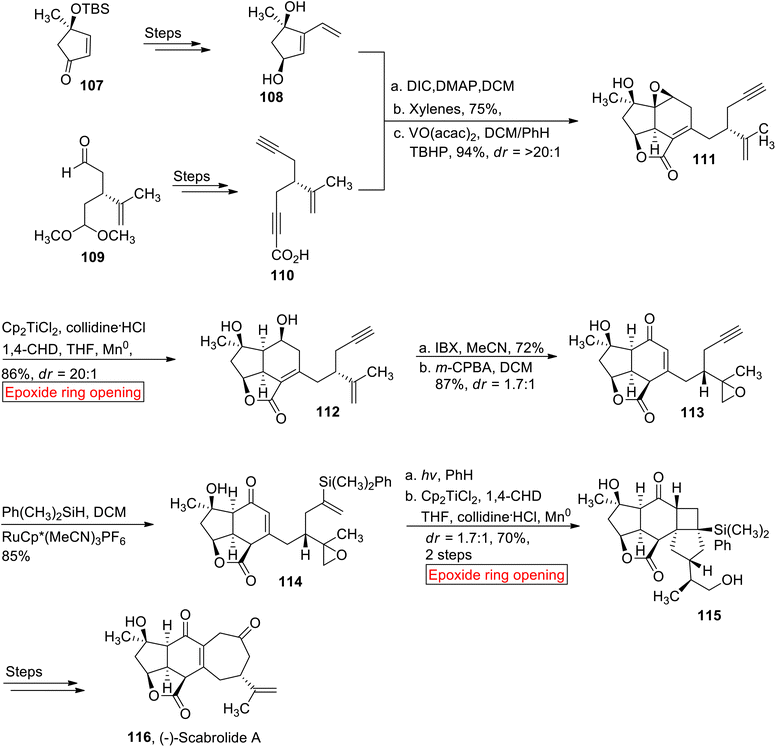 |
| | Scheme 13 Synthesis of (−)-Scabrolide A 116. | |
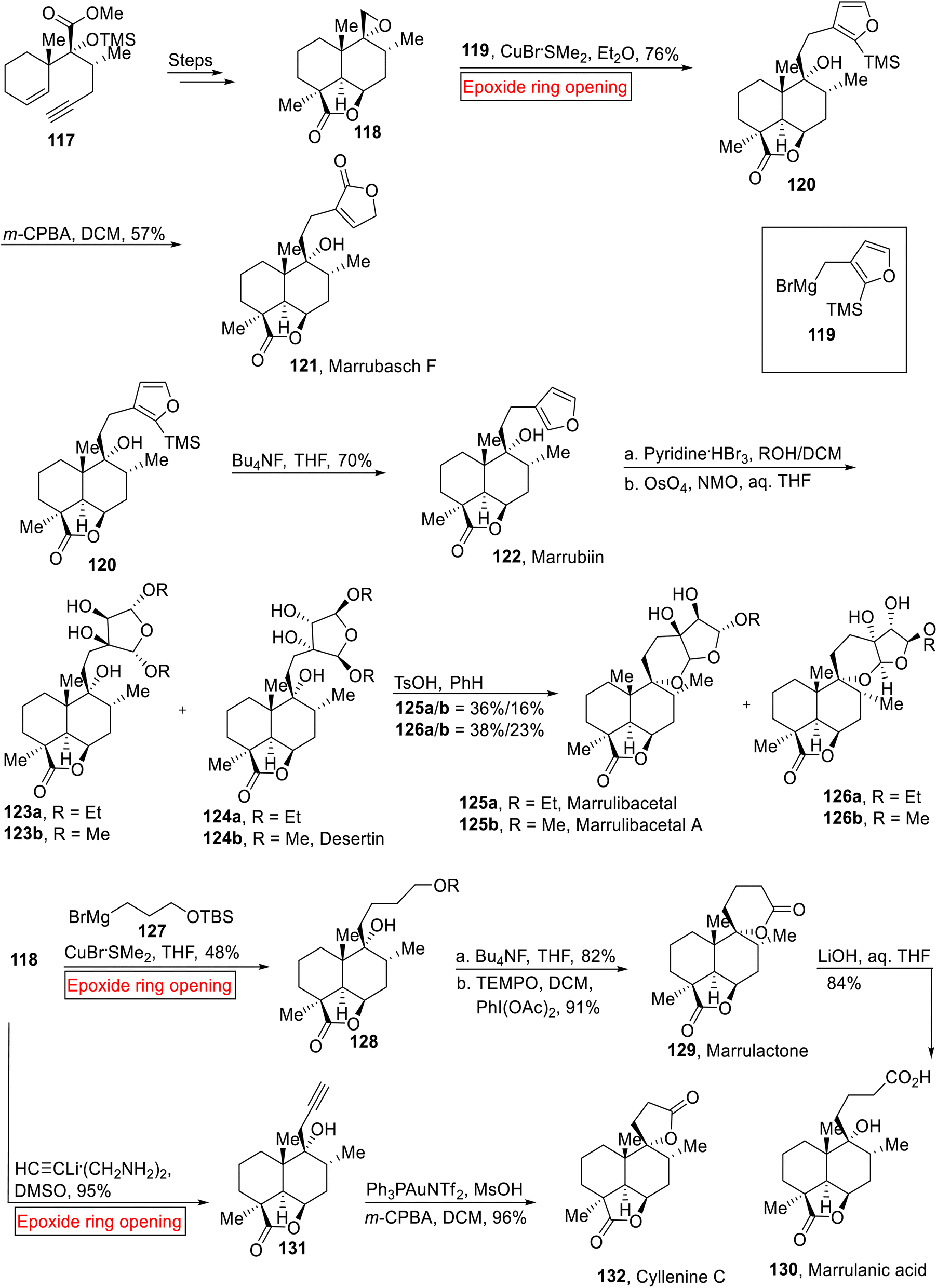
Flowering plants that belong to genus Marrubium are found in temperate and Mediterranean region.113 These plants are used as a local medicine in Eurasion zone due to the presence of Marrubiin 122 (furanoid) compound as it acts as analgesic, vasorelaxant, antispasmodic, anti-diabetic, hypoglycemic and anti-inflammatory agent.114 Marrubiin 122 is a labdane diterpene lactone which was extracted from Marrubium vulgare in 1842 for the first time and later its effect on rat aorta115 was also explored. The phytochemical analysis of this family (mints) also led to the isolation of various labdane diterpene lactones.116 Previously, research group of Mangoni117 described the total synthesis of Marrubasch F, Marrulibanoside, Premarrubiin (semi-syntheses) and Marrubiin (racemic form). Nakamura et al. disclosed the stereoselective total syntheses of Marrubasch F 121, Marrubiin 122, Desertine 124b, Marrulibacetal 159a, Marrulibacetal A 125b, Marrulactone 129, Marrulanic acid 130 and Cyllenine C 132 in 2020.118 Their synthetic approach features the extention of C-9 side chain of 118 via nucleophilic epoxide ring opening reaction with high level of malleability and convergency. The synthetic route initiated with the preparation of epoxide intermediate 118 from the enyne 117 over few steps. In the next step, intermediate 118 underwent epoxide ring opening reaction with Grignard reagent 119 in the presence of CuBr·SMe2 (Et2O) to afford 120 (76%) which was then exposed to oxidation (m-CPBA, DCM) to furnish Marrubasch F 121 (57%). In the same way, Marrubiin 122 (70%) was obtained by the deprotection (BuNF, THF) of TMS group of 120. Marrubiin 122 went through oxidative acetalization (pyridine·HBr) followed by reaction with NMO (4-methylmorpholine N-oxide) in aq. THF to provide Desertine 124b along other isomers. Mixture of diols 123a and 124a were treated with TsOH and PhH to generate Marrulibacetal 125a and Marrulibacetal A 125b respectively. Some other isomers and diastereomers were also formed with them. In accordance, intermediate 118 experienced Cu-catalyzed epoxide ring opening reaction with Grignard reagent 127 to render alcohol 128 (48%), which was further exposed to desilylation with subsequent oxidation to forge Marrulactone 129 (91%). Then, the Marrulactone 129 was converted to Marrulanic acid 130 (84%) via LiOH-mediated regioselective saponification. Synthesis of Cyllenine C 132 (96%) was achieved by nucleophilic epoxide ring opening reaction of intermediate 118 in the presence of lithium acetylide ethylenediamine in dimethyl sulfoxide (DMSO) to afford alcohol 131 (95%) followed by gold(I)-mediated cycloisomerization/oxidation sequence (Scheme 14).
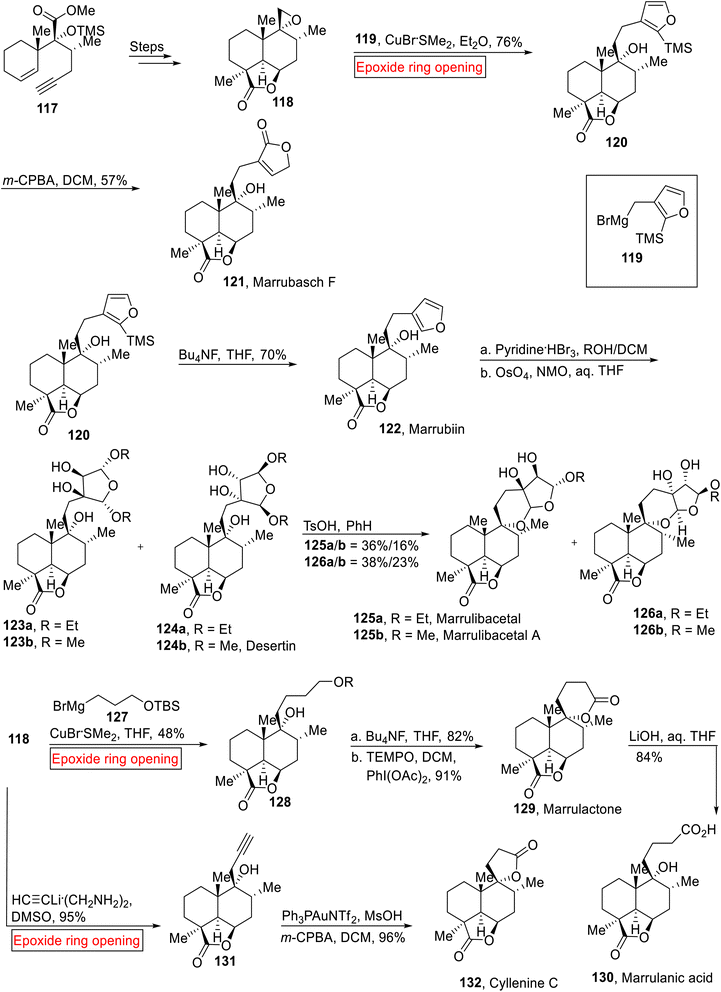 |
| | Scheme 14 Total syntheses of Marrubasch F 121, Marrubiin 121, Desertine 124b, Marrulibacetal 125a, Marrulibacetal A 125b, Marrulactone 129, Marrulanic acid 130 and Cyllenine C 132. | |
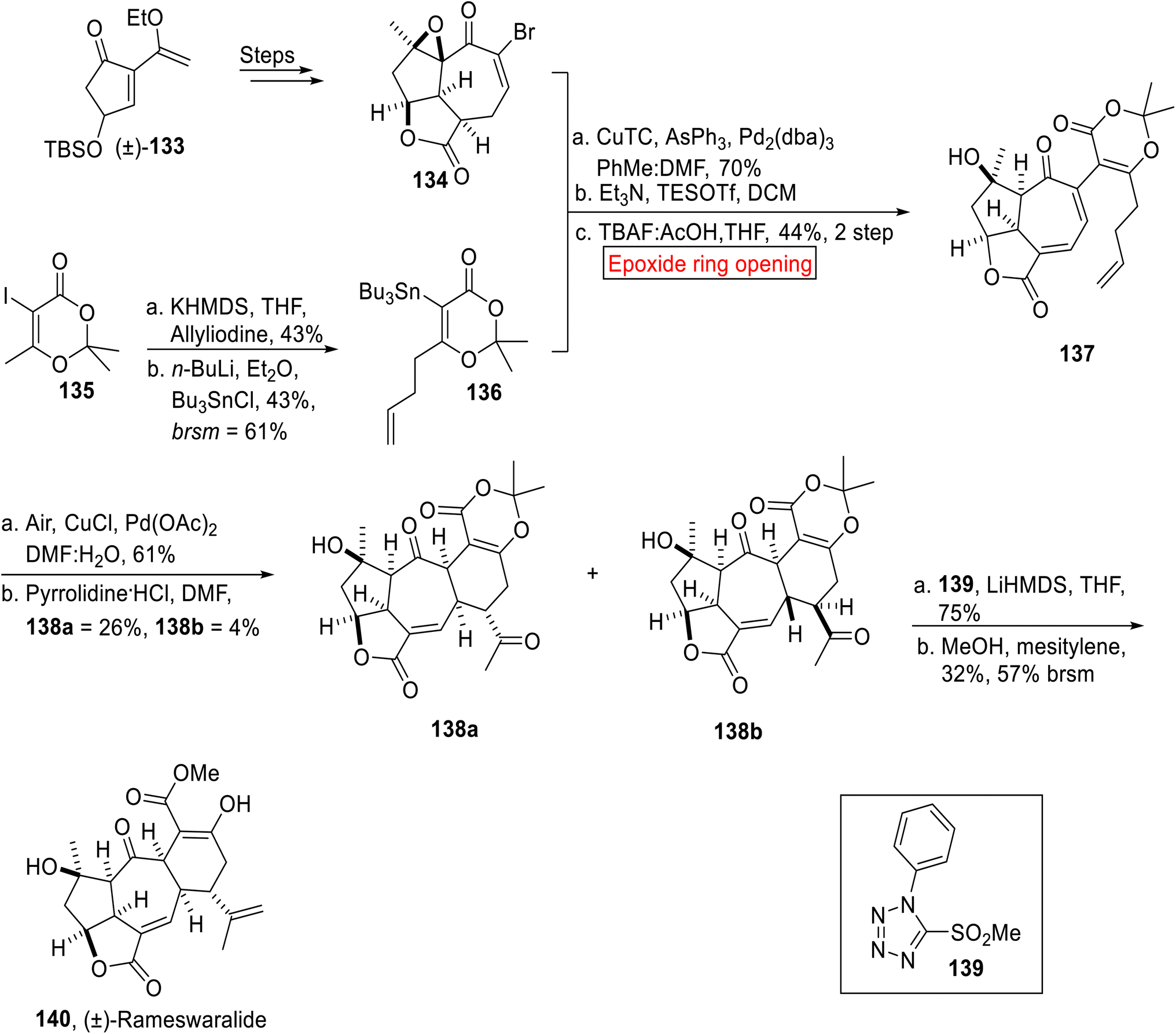
(±)-Rameswaralide 140 is a diterpene related to the class of secondary metabolites119 (a marine cembranoid), which was extracted in 1998 (ref. 120) from Sinularia dissecta (soft coral) featuring a tricyclic skeleton. It exhibits anti-inflammatory and anti-cancer activity.121 In this regard, development of various anticancer agents has been the focus of researchers.122 In 2022, research group of Romo developed an elegant approach toward the first total synthesis of (±)-Rameswaralide 140 by employing ring opening reaction of epoxide as one of the key steps.123 The synthetic route initiated with the syntheses of epoxy α-bromo enone 134 from 133 over few steps. The stannane 136 was obtained from vinyl iodide 135 via sequential alkylation, treatment with n-BuLi and tributyl tin chloride. In the next step, epoxy α-bromo enone 134 and stannane 136 upon Stille coupling followed by base-catalyzed oxidation with subsequent ring opening reaction of epoxide under desilylation conditions afforded α-methelyene butyrolactone 137 (44%, 2 steps). The compound 137 was further subjected to Pd(OAc)2-catalyzed oxidation followed by Stork enamine cyclization to render 138a (26%) and 138b (4%) diastereomers. In the next step, major diastereomeric ketone 138a was treated with 139 under Julia–Kocienski olefination by utilizing optimized conditions to forge (±)-Rameswawalide 140 in 32% yield (Scheme 15).
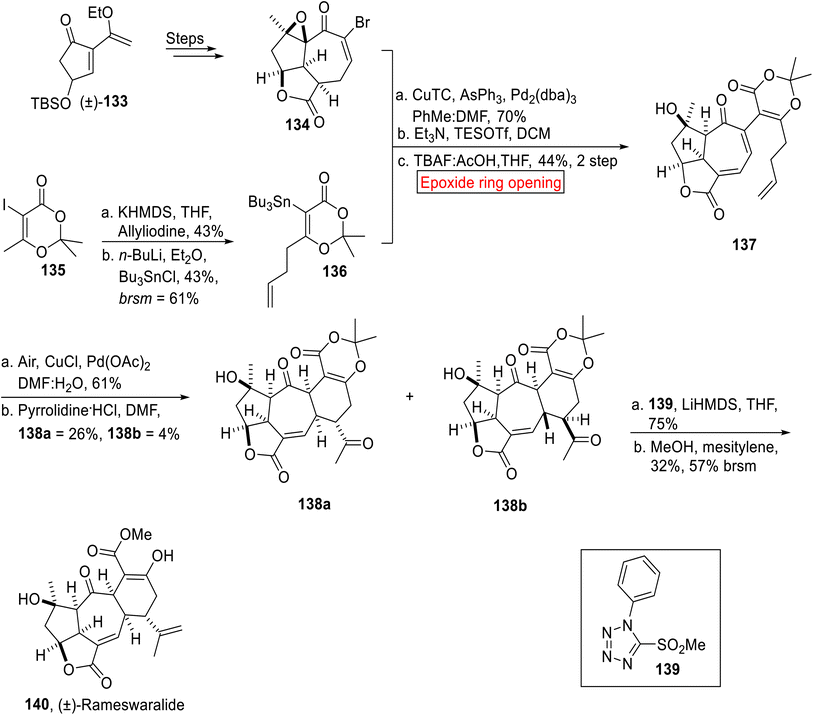 |
| | Scheme 15 First total synthesis of (±)-Rameswaralide 140. | |
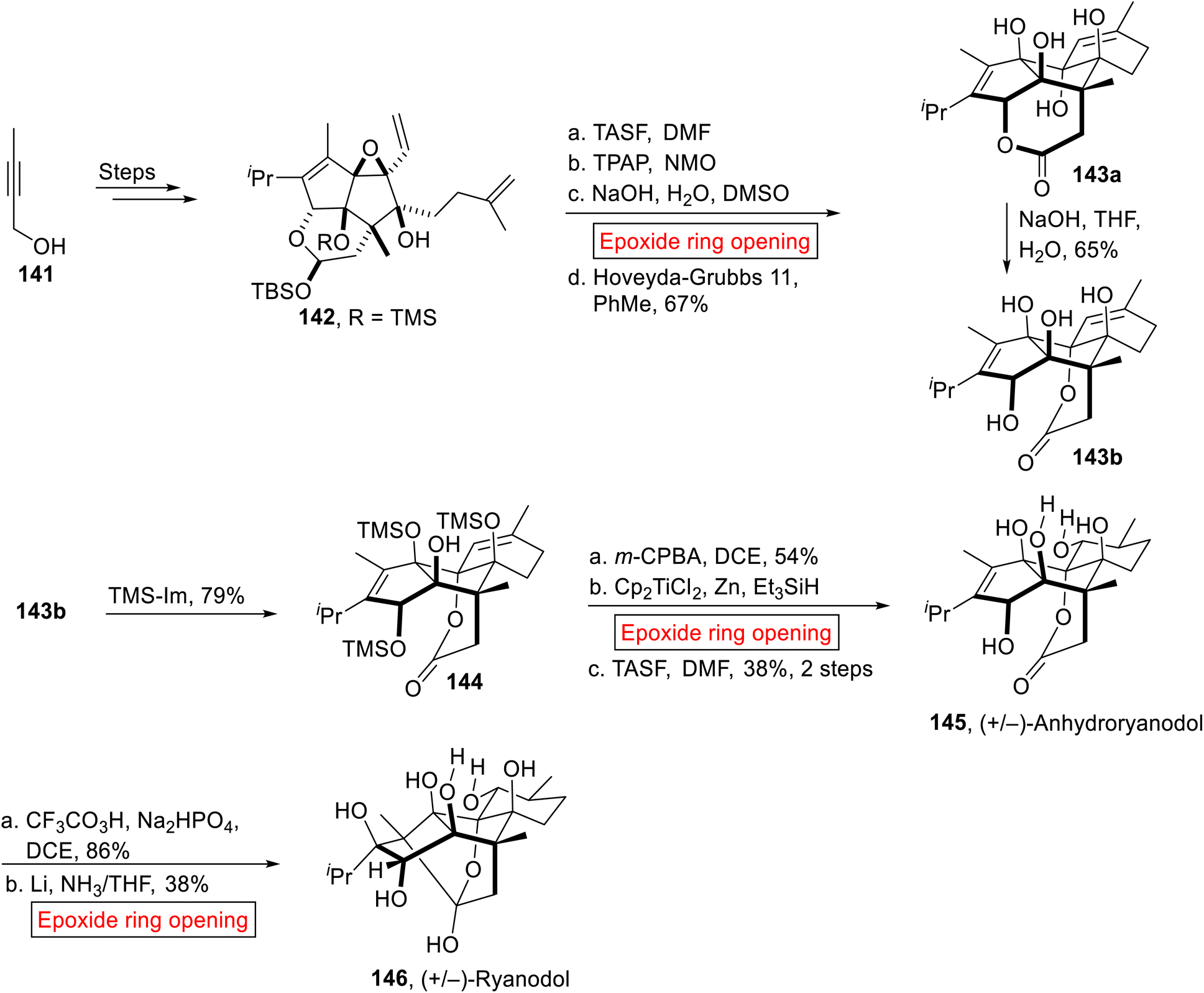
(+)-Ryanodine was extracted from Ryana speciosa Vahl (plant) found in South America.124 In 1967, ryanodine's structure was disclosed by Wiesner125 and afterward reported by Deslongchamps.126 The de novo synthesis of Ryanodine and Ryanodol (hydrolysis product of Ryanodine) was very difficult because they are composed of eleven stereogenic centers. Previously different efforts have been reported on the preparation of these targets, Ryanodol by Deslongchamps,126 Ryanoid by Inoue127 and recently Ryanodol by Reisman.128 In 2020, Micalizio et al. reported the synthetic approach towards the novel synthesis of Ryanodol 146 that was achieved by the synthesis of its degradation product (+/−)-Anhydroryanodol 145.129 They utilized carbocyclic approach for the synthesis of (+/−)-Anhydroryanodol 145 by employing epoxide ring opening reaction as of the key steps. Initially, divinyl epoxide 142 was obtained from alkyne 141 over few steps. In the next step, divinyl epoxide 142 went through sequential deprotection (TASF = trissulfonium difluorotrimethylsilicate), oxidation (TPAP = tetrapropylammonium perruthenate, NMO), base (NaOH)-catalyzed epoxide ring opening reaction in DCM. Then, subsequent ring-closing metathesis afforded a mixture of lactones 143a and 143b in equal amount. Compound 143a was also converted into 143b (65%) in the presence of base (NaOH). Then, 143b was silylated selectively by treating it with neat TMS-Im (imidazole) to forge 144. In the next step, (+/−)-Anhydroryanodol 145 (38%, 2 steps) was obtained by m-CPBA-mediated epoxidation, Cp2TiCl2-catalyzed epoxide ring opening reaction in the presence of Et3SiH followed by TASF-mediated deprotection. Finally, (+/−)-Anhydroryanodol 145 was converted into (+/−)-Ryanodol 146 (38%) via site-specific epoxidation (CF3CO3H) followed by Li/NH3-catalyzed epoxide ring opening reaction reported by Deslongchamps126 and Reisman128 (Scheme 16).
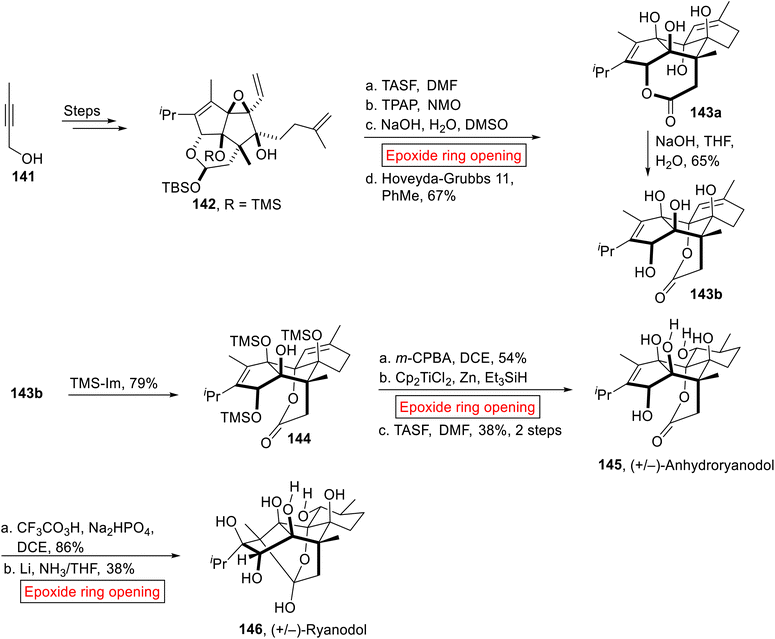 |
| | Scheme 16 Synthesis of (+/−)-Anhydroryanodol 145. | |
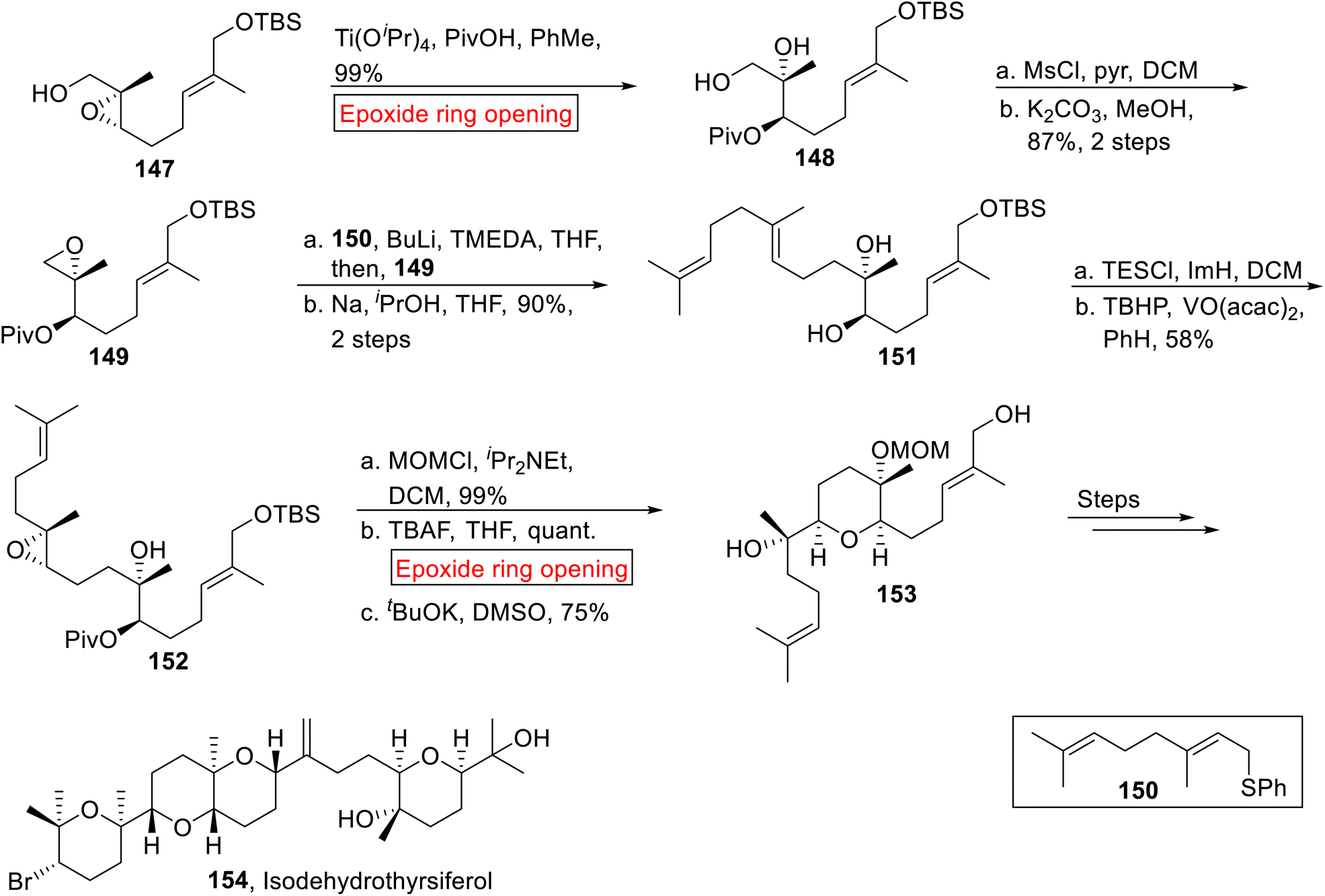
Triterpenoid. Isodehydrothyrsiferol 154 belongs to the family of triterpenoid130 which was drawn from Laurencia viridis (red alga) by Munro and colleagues in 1978.131 It depicts potent cytocidal activity opposite to P388 cell lines (IC50 = 17 nm).130 It has unique structural features because it displays partial enantiodivergent as a novel phenomenon in the biosynthesis of natural products. In 2017, research group of Morimoto disclosed the synthesis of Isodehydrothyrsiferol 154 by employing epoxide ring opening reaction as a key step.132 In the first step, 2,3-epoxy alcohol 147 experienced Ti(OiPr)4-mediated epoxide ring opening reaction in the presence of PivOH and toulene to generate pivalate 148 (99%). The resulting pivalate 148 was further converted into another epoxide 149 (87%, 2 steps) by sequential protection and epoxidation. Next, geranyl phenyl sulfide 150 was added to 149 followed by reaction with Na, iPrOH to furnish compound 151 (90%, over 2 steps). The tertiary alcohol moiety of compound 151 was exposed to protection with TESCl (Triethylchlorosilane) and then diastereoselective epoxidation provided epoxide 152 (58%). In the next step, the –OH group of compound 152 underwent protection followed by TBAF-mediated epoxide ring opening reaction in THF with subsequent deprotection (tBuOK) to attain 153 (75%). The synthesized compound 153 was finally converted to Isodehydrothyrsiferol 154 over few steps (Scheme 17).
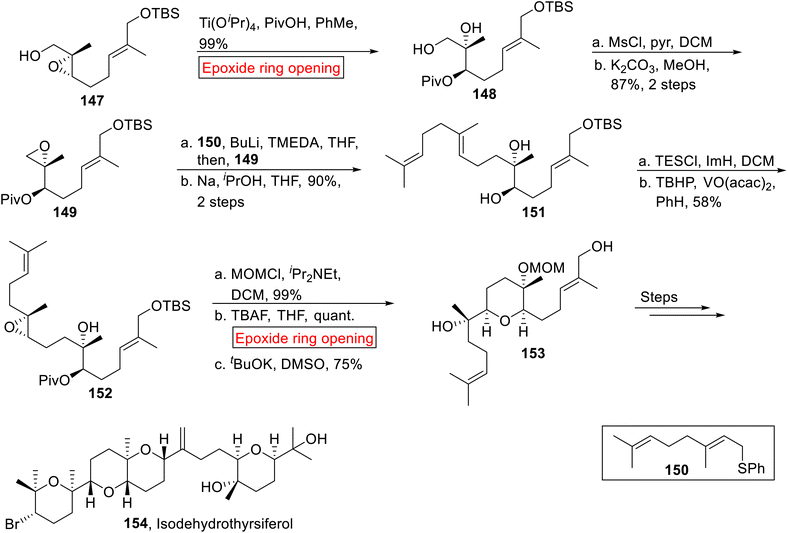 |
| | Scheme 17 Synthesis of Isodehydrothyrsiferol 154. | |
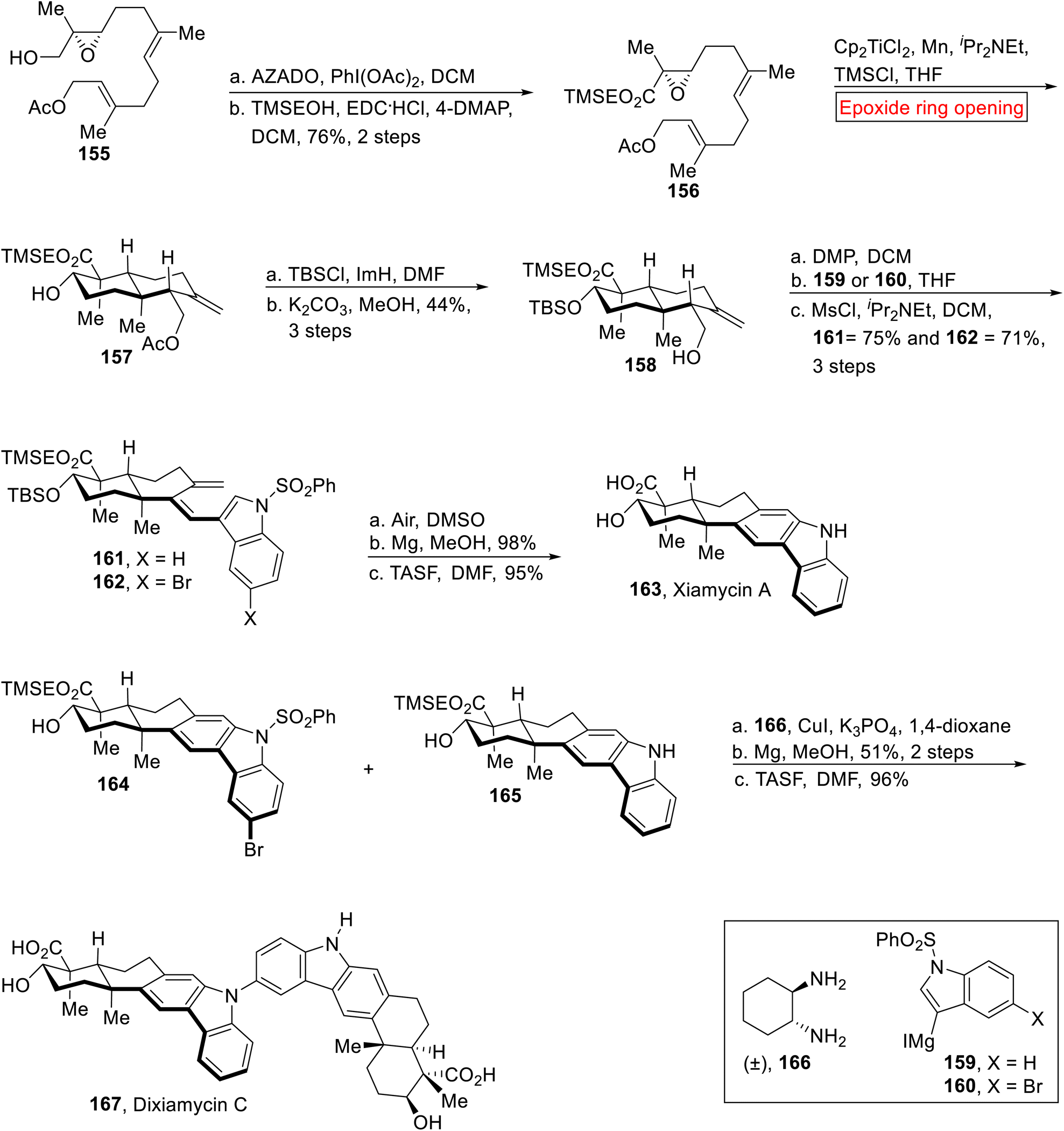
Indole sequequiterpenenoids. Xiamycin A 163 and Dixiamycin C 167 are indole sequequiterpenenoids which were extracted by Hertweck133–136 and Zhang137–139 from Streptomyces species. Xiamycin 163 and Dixiamycin C 167 demonstrate substantial inhibitory activity opposite to herpes simplex virus-1 (HSV-1). Recently, work on Xiamycin A 163 and Dixiamycin B was reported by research group of Baran140 for the first. In 2015, Meng et al. described the total synthesis of Xiamycin 163 and Dixiamycin C 167 (for the first time) by employing epoxide ring opening reaction as one of the key steps.141 The synthetic route started with the preparation of α, β-epoxy ester 156 (76%, 2 steps) from optically active epoxide 155 through AZADO (2-azaadamantane N-oxyl)-mediated oxidation and esterification. Next, the compound 156 underwent Cp2TiCl2-catalyzed epoxide ring opening reaction in the presence of iPr2Net, TMSCl, Mn and THF to furnish trans-decalin 157. In the next step, compound 158 (44%, 3 steps) was obtained from decalin 157 via TBS protection accompanied by acetyl deprotection. Then, compound 158 was subjected to oxidation (using DMP) followed by Grignard addition (159 or 160) with subsequent dehydration in the presence of MsCl/iPr2Net to afford triene 161 or 162. Next, triene 161 or 162 experienced thermal 6π-electrocyclization accompanied by aromatization, desulfonation and TMSE deprotection to achieve Xiamycin A 163 in excellent yield (95%). Similarly, compounds 164 and 165 were reacted with compound 166 in the presence of CuI followed by deprotection with TSAF to render Dixiamycin C 167 (96%, over 2 steps) in a good yield (Scheme 18).
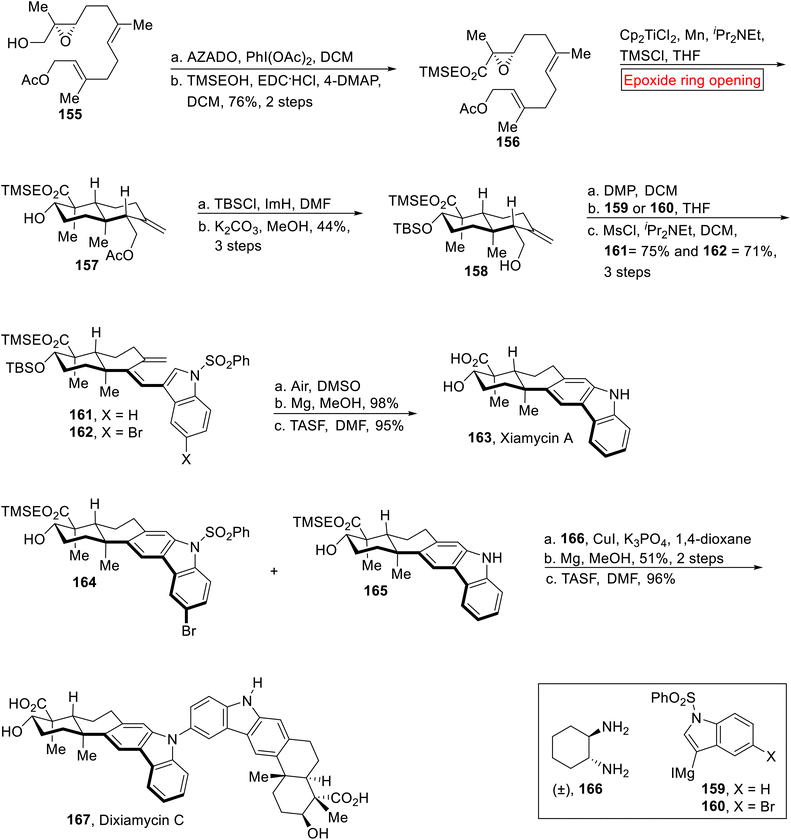 |
| | Scheme 18 Total synthesis of Xiamycin 163 and Dixiamycin C 167. | |
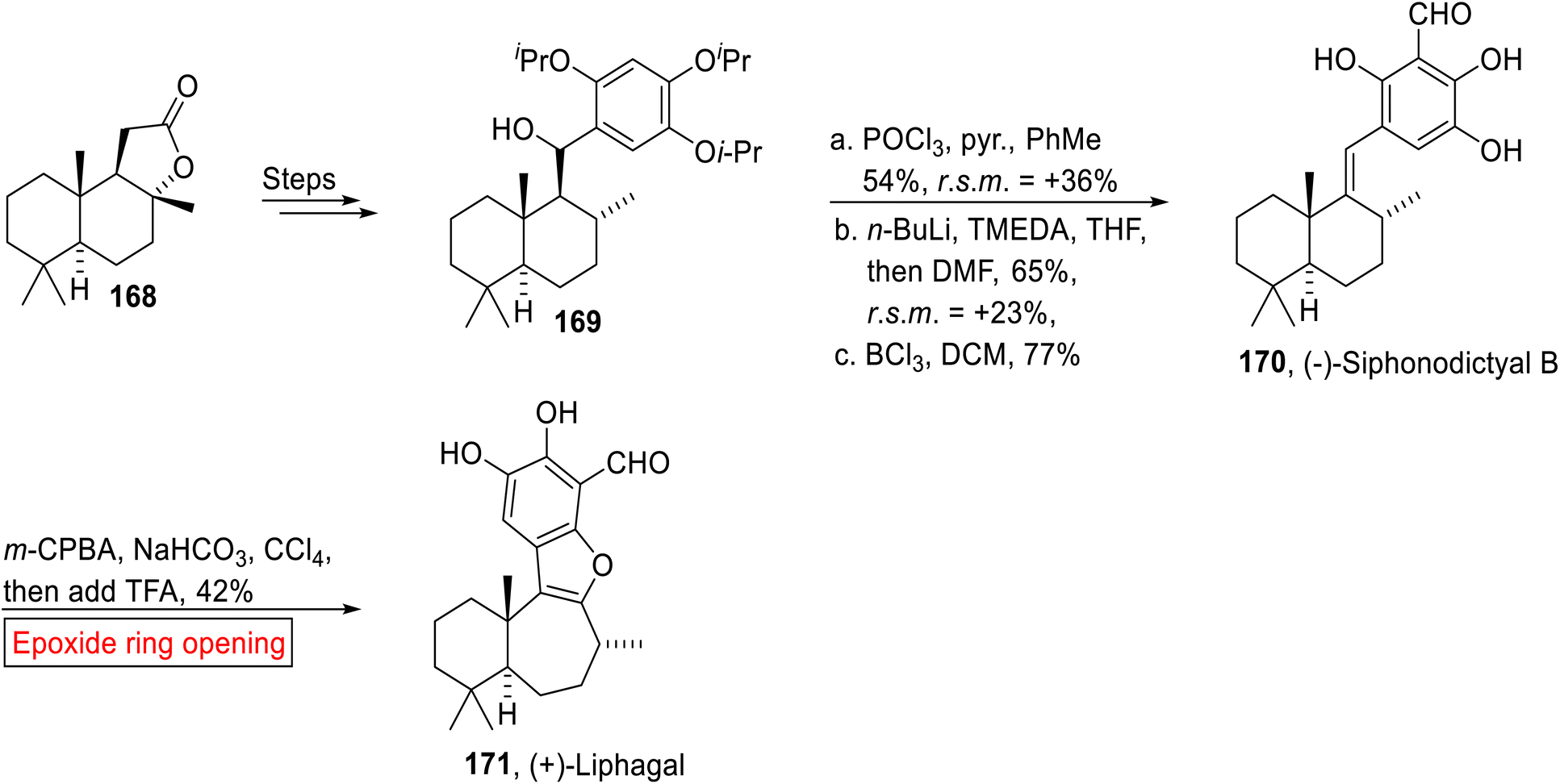
Meroterpenoid. (−)-Siphonodictyal B 170 (marine sponge) and (+)-Liphagal 171 belong to the meroterpenoid family of natural products. (+)-Liphagal 171 was extracted by Andersen and colleagues from Aka coralliphaga142 while Siphonodictyal B was also isolated from Aka coralliphaga by the research groups of Faulkner143 and Köck.144 Its structure was first reported in 1981 by Faulkner and colleagues and then was revised by Clardy and coworkers145 (1986). In 2015, George et al. reported the correct revised structure of (−)-Siphonodictyal B 170 which was then converted into (+)-Liphagal 171 by employing epoxide ring opening reaction as one of the key steps.146 Their research work highlights the significance of biosynthetic suppositions which help in the development of innovative sequential reactions and structural reassignment of natural products. The synthetic route commenced with the preparation of benzylic alcohol 169 from (+)-sclareolide 168 over few steps. In the next step, alcohol 169 underwent POCl3-mediated dehydration followed by ortho-lithiation, then quenching by utilizing DMF with subsequent deprotection (BCl3) of isopropyl ether to afford (−)-Siphonodictyal B 170 (77%). Finally, synthesis of (+)-Liphagal 171 (42%) was achieved by m-CPBA-mediated epoxidation in the presence of NaHCO3 and CCl4 accompanied by TFA-catalyzed epoxide ring opening reaction (Scheme 19).
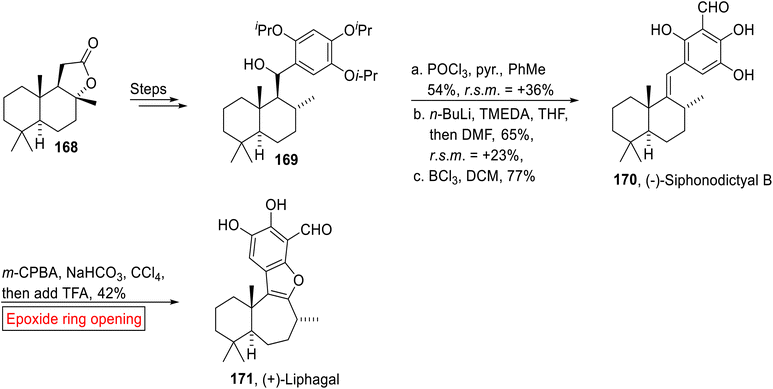 |
| | Scheme 19 Synthesis of (−)-Siphonodictyal B 170 and (+)-Liphagal 171. | |
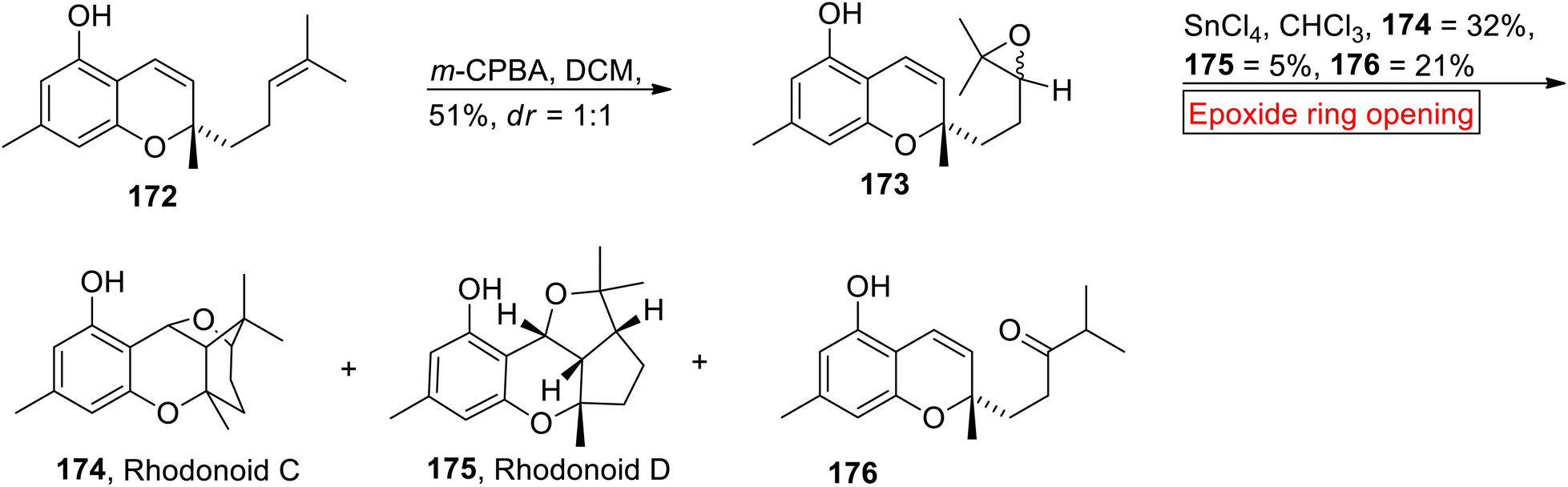
Rhodonoids C 174 and D 175 belong to the class of meroterpenoids extracted from Rhododendron capitatum.147 Rhodonoids C 174 and D 175 have 6/6/6/5 and 6/6/5/5 ring systems respectively. In 2017, research group of Day67 synthesized Rhodonoids C 174 and D 175 by using Chromene 172 (meroterpenoid) which was prepared according to Hsung and colleagues148 from orcinol and citral. In the first step, prenyl side chain of Chromene 172 underwent m-CPBA-mediated epoxidation to furnish diastereomer 173 (51%, dr = 1:1). Then, 173 experienced SnCl4-catalyzed epoxide ring opening reaction with subsequent cyclization in the presence of CHCl3 to synthesize Rhodonoids C 174 (32%), D 175 (5%) and 176 (Scheme 20).
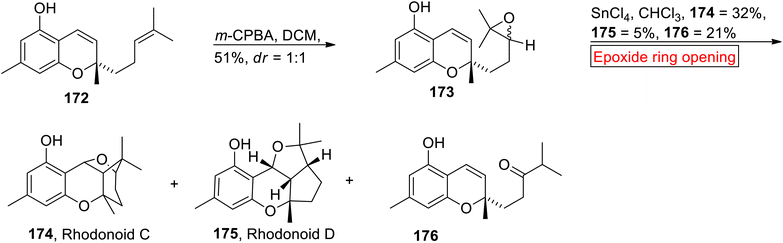 |
| | Scheme 20 Synthesis of Rhodonoids C 174, D 175 and 176. | |
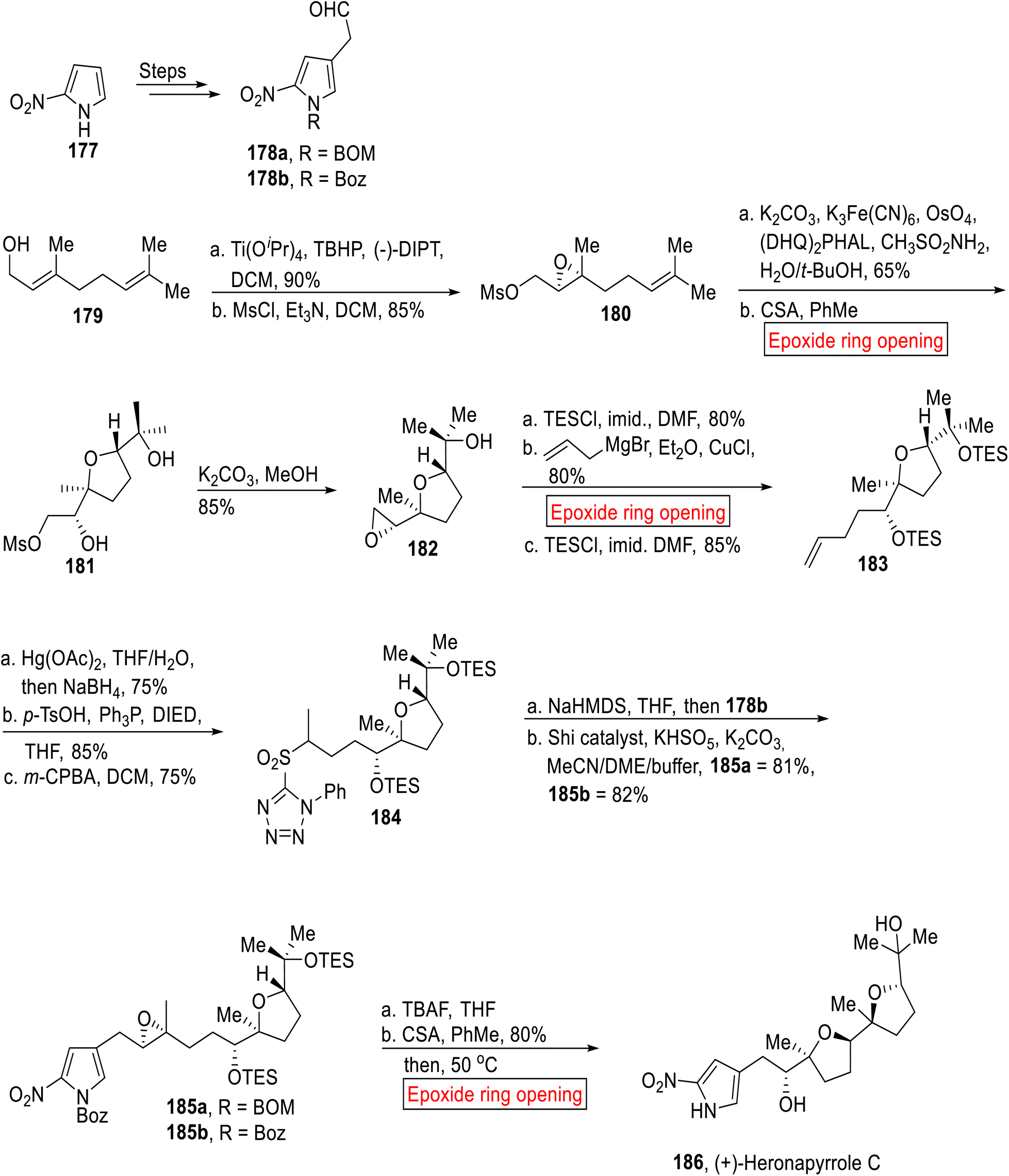
Sesquiterpenoids. Heronapyrrole C 186 belongs to a sesquiterpene (rare nitropyrrole) family of natural product which was isolated from the culture of Streptomyces sp. (CMB-M0423) in 2010 (Australia).149 Considering the widespread microbial diseases, researchers are continuously making efforts to develop efficient anti-microbial agents.150 Nitropyrrole natural products exhibit activity opposite to Gram-positive bacteria without exhibiting cytocidal activity towards mammalian cell lines. In 2012, Stark disclosed the synthesis of Heronapyrrole C with less yield.151 In 2014, Brimble et al. reported the regioselective synthesis of (+)-Heronapyrrole C 186 by employing epoxide ring opening reaction as one of the key steps.152 Their strategy was based on the synthesis of regiocontrolled 2-nitropyrrole building blocks that have received limited attention until now. The synthetic scheme towards (+)-Heronapyrrole C 186 began with the syntheses of aldehyde fragments 178a and 178b from 2-nitropyrrole 177 over few steps. The synthesis of second fragment 184 was initiated with the preparation of epoxy mesylate 180 from geraniol 179 by Sharpless asymmetric epoxidation followed by mesylation. Then, epoxy mesylate 180 went through Sharpless asymmetric dihydroxylation accompanied by acid-catalyzed (CSA = Camphorsulfonic acid) epoxide ring opening reaction to afford 181. In the next step, 181 went through epoxidation to render terminal epoxide 182 (85%). Then, compound 182 was subjected to protection (TESCl) of tertiary alcohol moiety followed by copper(I) chloride-catalyzed epoxide ring opening reaction with allyl magnesium bromide in Et2O and again protection with TESCl to furnish alkene 183 (85%). Next, alkene 183 upon Markovnikov oxymercuration reduction followed by Mitsunobu inversion (p-TsOH) with m-CPBA-mediated oxidation rendered olefination partner 184 (75%). Then, the coupling partner 178b and 184 were coupled through Julia-Kocienski olefination followed by (−)-Shi ketone-mediated asymmetric epoxidation to give epoxides 185a (81%) and 185b (82%). Finally, 185b was converted into (+)-Heronapyrrole C 186 via TBAF-mediated deprotection accompanied by acid (CSA)-catalyzed epoxide ring opening reaction in toluene with subsequent heating at 50 °C (Scheme 21).
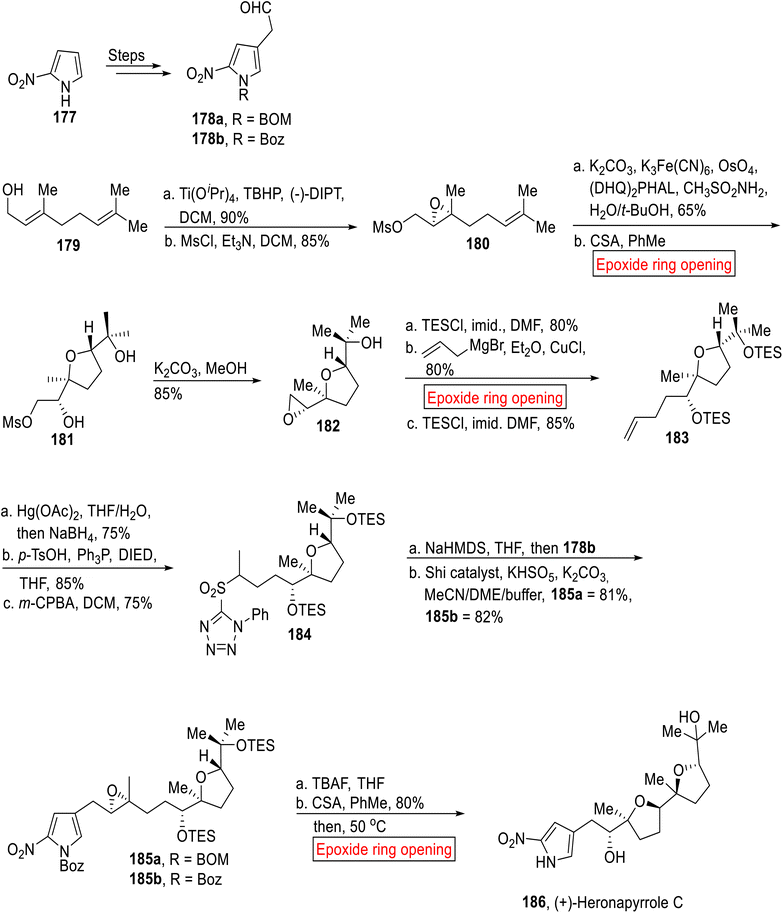 |
| | Scheme 21 Regioselective synthesis of (+)-Heronapyrrole C 186. | |
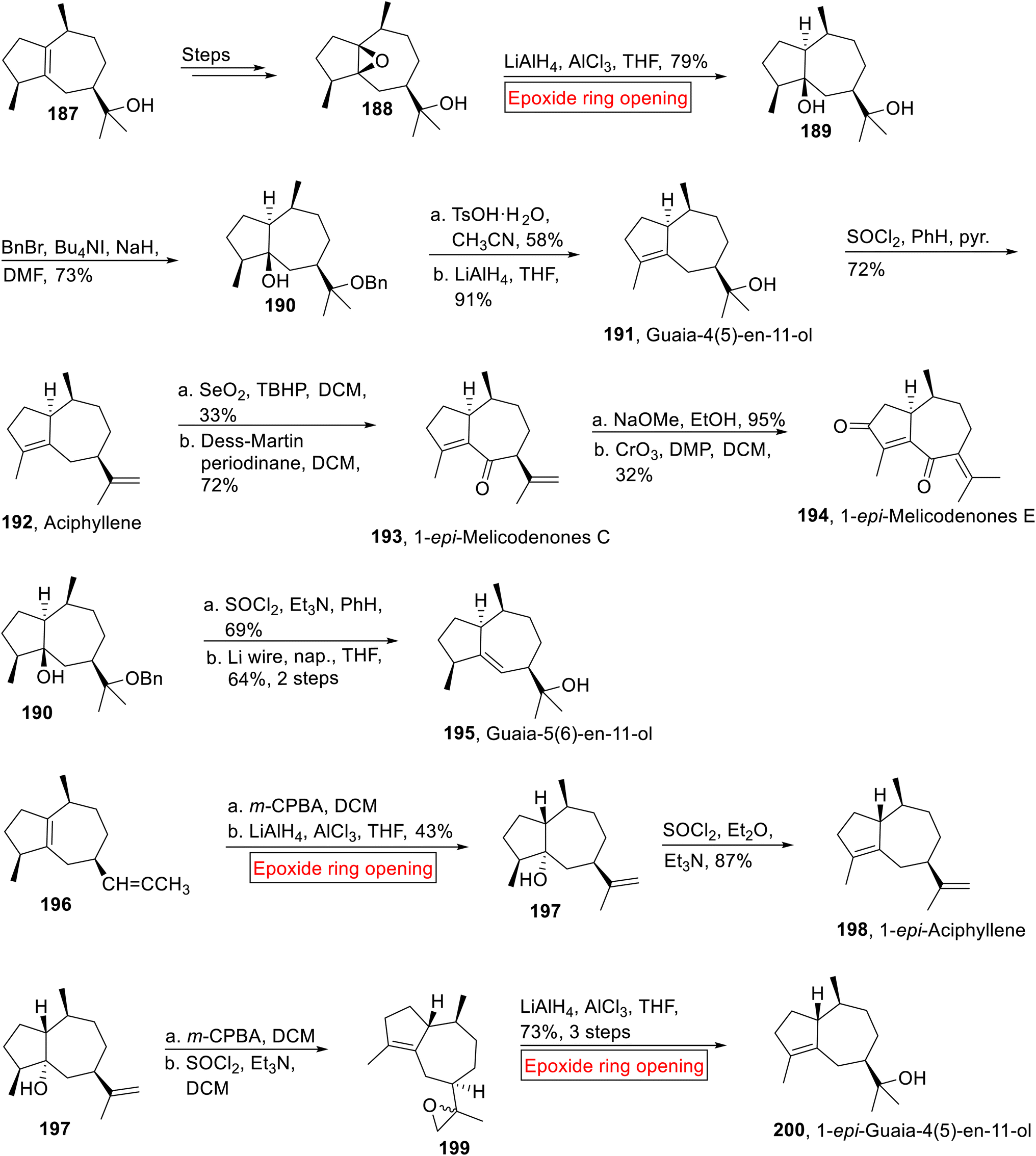
Guaiane-type sesquiterpenoids (Guaia-4(5)-en-11-ol 191, Aciphyllene 192, 1-epi-Melicodenones C 193, 1-epi-Melicodenones E 194, Guaia-5(6)-en-11-ol 195, 1-epi-Aciphyllene 198, 1-epi-Guaia-4(5)-en-11-ol 200) were isolated from fungi plants and marine life.153,154 Previously synthesized Guaiane-type sesquiterpenoids have low overall yield155,156 and experienced difficulties in installing the required stereochemistry while synthesizing the [5.3.0]-bicyclic ring cores. The problem was then solved by Taylor and coworkers by using inexpensive naturally occurring Guaiol 187 as a starting material for the synthesis of Guaiane-type sesquiterpenoids. In 2014, research group of Taylor157 reported the synthesis of these sesquiterpenoids via epoxide ring opening reaction as one of the key steps. The synthetic route initiated with the formation of β-epoxyguaiol 188 from 187 over few steps. Then, 188 was subjected to LiAlH4-catalyzed epoxide ring opening reaction in the presence of AlCl3 in THF to render 189 (79%). Then, compound 189 was exposed to selective benzylation under Ogawa's method in the presence of in situ generated benzyl iodide to afford compound 190 (73%). Treatment of compound 190 with TsOH·H2O (MeCN) followed by reduction (LiAlH4) yielded Guaia-4(5)-en-11-ol 191 with 26% overall yield (91%). In the next step, alcohol moiety of 191 experienced dehydration (SOCl2) followed by SNIS (silver nitrate impregnated upon silica gel) chromatography purification to synthesize Aciphyllene 192 in 18% overall yield (72%). Then, Aciphyllene 192 upon sequential allylic and Dess-Martin periodinane oxidation provided 1-epi-Melicodenones C 193 (72%) which was further treated with base (NaOMe) followed by CrO3-mediated oxidation to generate the 1-epi-Melicodenones E 194 (32%).
Guaia-5(6)-en-11-ol 195 (31%, overall yield) was obtained by the dehydration (SOCl2) of 190 accompanied by treatment with Li wire and naphthalene in THF. Similarly, compound 196 went through m-CPBA-mediated epoxidation accompanied by LiAlH4-mediated epoxide ring opening reaction in the presence of AlCl3 in THF to afford alcohol 197 (43%) which was subjected to dehydration to afford 1-epi-Aciphyllene 198 (87%). Then, alcohol 197 was subjected to sequential epoxidation and dehydration to generate epoxide 199. In the next step, compound 199 went through LiAlH4-catalyzed epoxide ring opening reaction reductively by using AlCl3 and THF to accomplish 1-epi-Guaia-4(5)-en-11-ol 200 in 73% yield over 3 steps (Scheme 22).
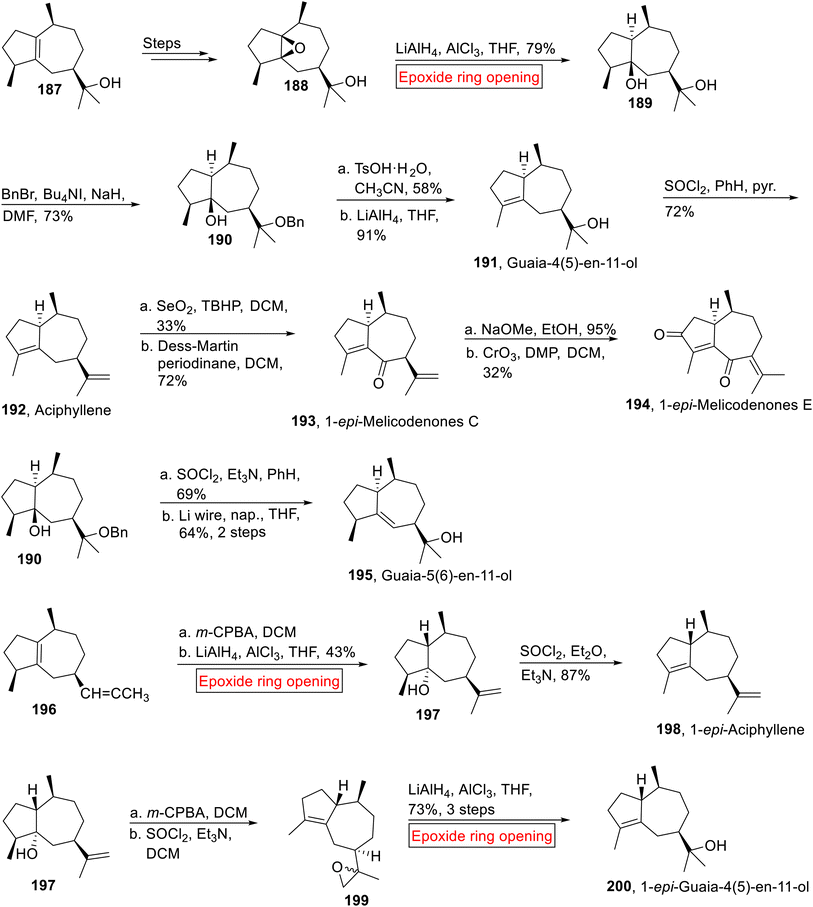 |
| | Scheme 22 Syntheses of Guaiane-type sesquiterpenoids (Guaia-4(5)-en-11-ol 191, Aciphyllene 192, 1-epi-Melicodenones C 193, 1-epi-Melicodenones E 194, Guaia-5(6)-en-11-ol 195, 1-epi- Aciphyllene 198, 1-epi-Guaia-4(5)-en-11-ol 200). | |
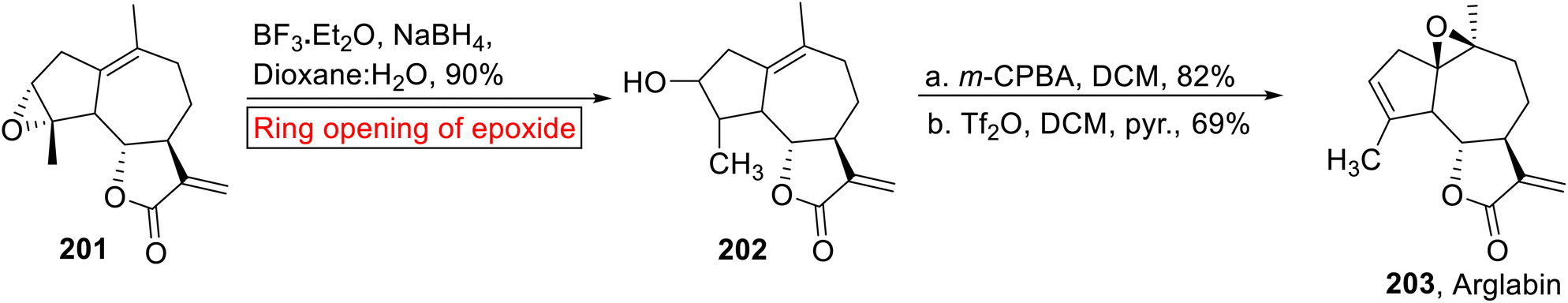
Arglabin 203 (guaianolide) belongs to the family of sesquiterpene lactones which was isolated from Artemisia glabella158 specie. The dimethyl amino adduct of Arglabin exhibits antitumor activity against lung, breast, colon and ovarian cancer.159 Total synthesis and hemisynthesis of Arglabin 203 from parthenolide have been reported by the research groups of Reiser160 and Chen161 respectively. In 2015, Lone and Bhat have accomplished the hemisynthesis of Arglabin 203 (51%, overall yield) from ludartin 201 (ref. 162) via short and efficient reaction process, by taking into account the less side effects of Arglabin as compared to other chemotherapeutics. They utilized epoxide ring opening reaction as one of the key steps in their strategy for the synthesis of Arglabin. In the first step, ludartin 201 underwent Lewis acid (BF3·Et2O)-catalyzed regioselective ring opening of epoxide using NaBH4, which resulted in compound 202 (90%). Stereoselective epoxidation of compound 202 in DCM followed by selective reduction synthesized Arglabin 203 in excellent yield (Scheme 23).
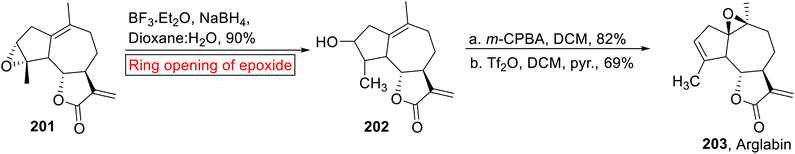 |
| | Scheme 23 Hemisynthesis of Arglabin 203. | |
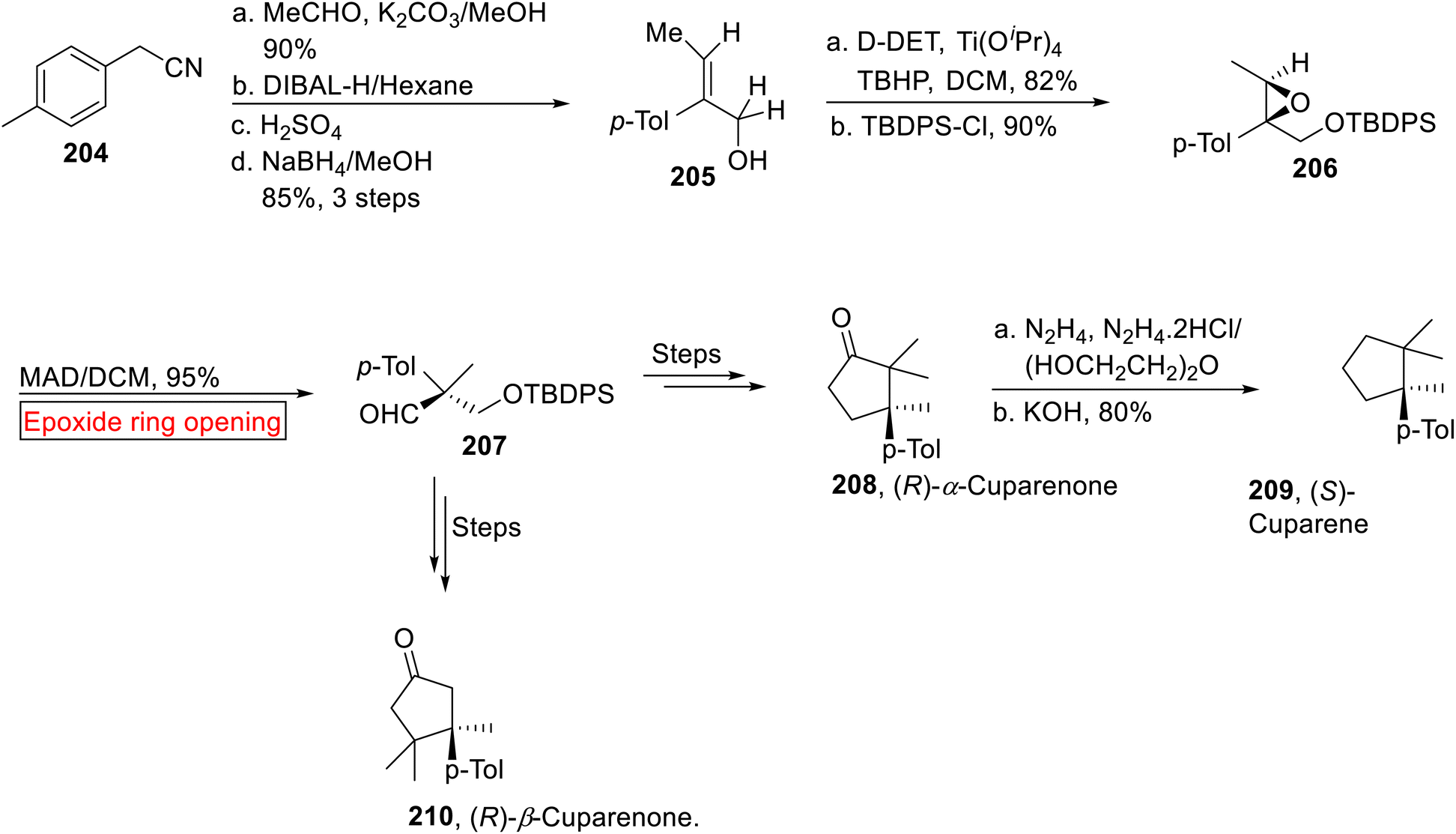
In 1964, Chetty and Dev163 extracted α-Cuparenones 208 and β-Cuparenones 210, sesquiterpenes from the oil of Thuja orientallis leaves (Family – Cupressaceae) which display a number of biological activities. Then in 1976, Benesova164 and co-workers isolated α and β-Cuparenones from Mania fragrans (liverworts). Similarly, (S)-Cuparene 209, a sesquiterpene was isolated in 1958 by Erdtman and colleagues.165 Nanda and allies reported the optically pure synthesis of (R)-α-Cuparenone 208, (R)-β-Cuparenone 210 and (S)-Cuparene 209 by employing epoxide ring opening reaction as one of the key steps.166 In the first step, 4-methylbenzyl cyanide 204 went through aldol reaction (Me-CHO) followed by DIBAL-H-mediated reduction, acid-catalyzed hydrolysis with subsequent reduction (NaBH4) to furnish E-allylic alcohol 205 (85%, 3 steps). Then, E-allylic alcohol 205 was subjected to asymmetric epoxidation followed by tert-butyldiphenyl chlorosilane (TBDPSCl)-mediated protection to provide protected epoxide 206 (90%). In the next step, compound 206 experienced Lewis acid-mediated epoxide ring opening reaction which caused Meinwald rearrangement to forge aldehyde intermediate 207 (95%). The compound 207 was converted to (R)-α-Cuparenone 208 and (R)-β-Cuparenone 210 via several steps. In the next step, (S)-cuparene 209 was obtained (7.5%, overall yield) from (R)-α-Cuparenone 208 (9.4%, overall yield) via reductive deoxygenation under Huang-Minlon conditions (Scheme 24).
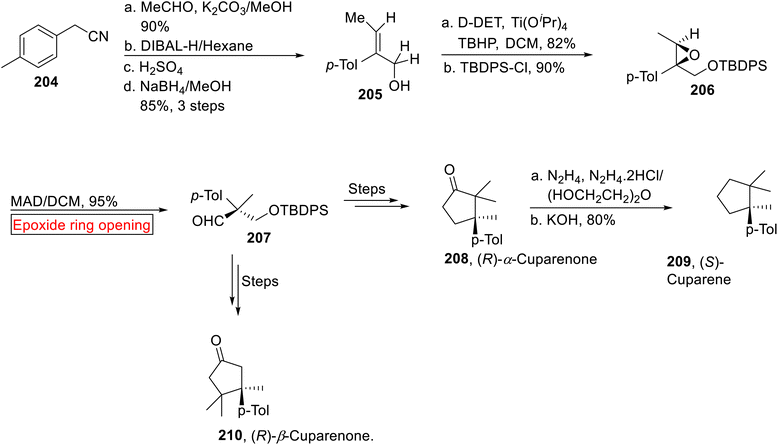 |
| | Scheme 24 Synthesis of (R)-α-Cuparenone 208, (R)-β-Cuparenone 210 and (S)-Cuparene 209. | |
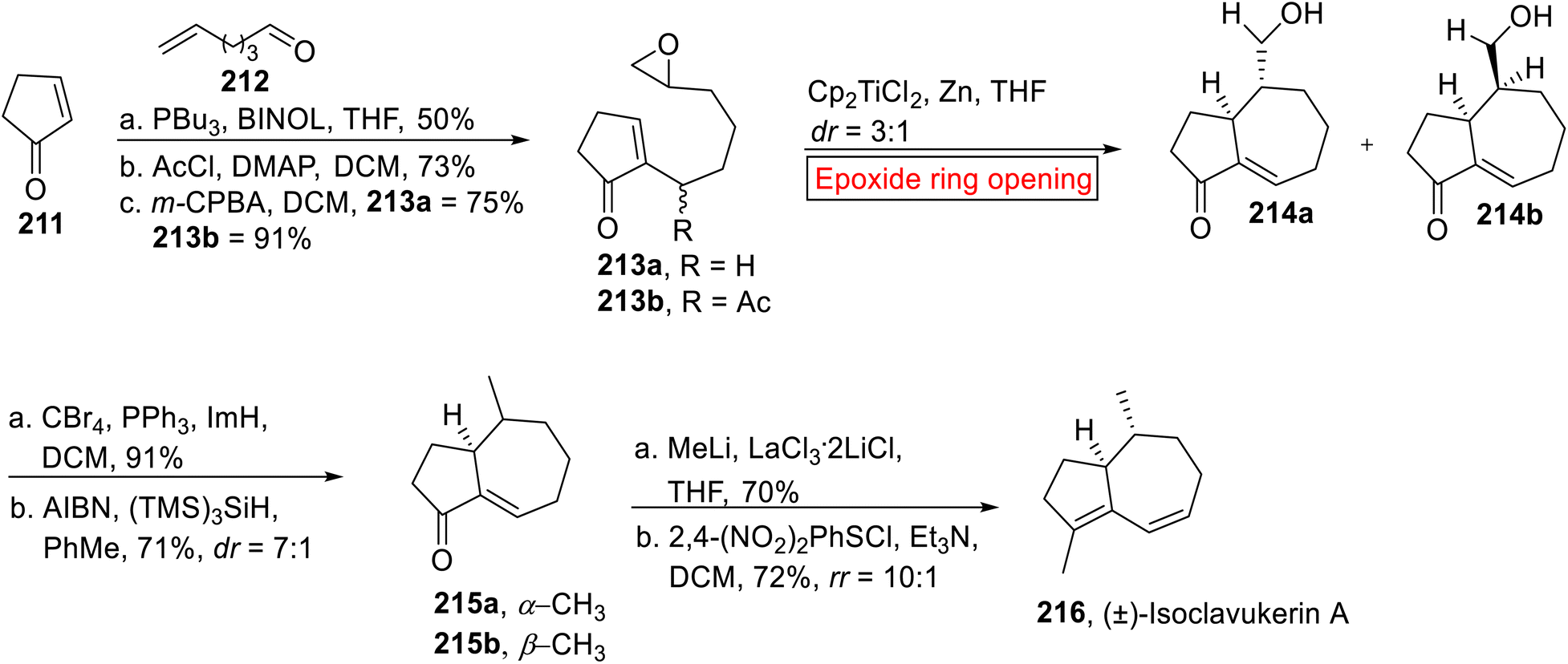
(±)-Isoclavukerin A 216 belongs to sesquiterpene (tri-nor-guaiane) family of natural products. It was isolated by research groups of Kitagawa167,168 and Bowden169 from Clavularia koellikeri (Okinawan soft coral) and Cespitularia sp. (Australian soft coral) respectively. In 2023, Chakraborty170 et al. reported the synthesis of (±)-Isoclavukerin A 216 by employing epoxide ring opening reaction as one of the key steps. The method used by research group of Chakraborty is an attractive approach for the synthesis of sesquiterpenoids owing to high diastereoselective route, simple pathway focusing on step economy and mild reaction conditions. The synthetic endeavor started with the preparation of epoxides 213a (75%) and 213b (91%) from cyclopentenone 211 and hex-6-enal 212 in the presence of PBu3 followed by treatment with AcCl with subsequent epoxidation (m-CPBA). In the next step, compound 213a and 213b were subjected to Cp2TiCl-catalyzed epoxide ring opening reaction in THF to afford corresponding cyclized products 214a and 214b. Then, the mixture of 214a and 214b experienced sequential bromination and debromination to afford enones 215a and 215b (dr = 7:1). In the last step, mixture of enones 215a and 215b upon treatment with MeLi and additive LaCl3·2LiCl accompanied by 1,4-conjugate elimination in the presence of 2,4-dinitrobenzenesulfinylchloride and Et3N forged (±)-Isoclavukerin A 216 in 72% yield (Scheme 25).
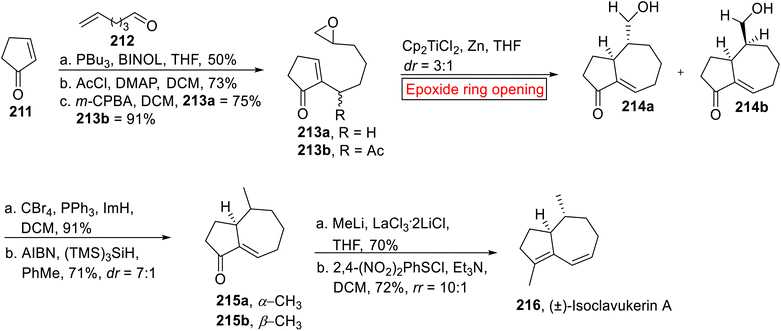 |
| | Scheme 25 Synthesis of (±)-Isoclavukerin A 216. | |
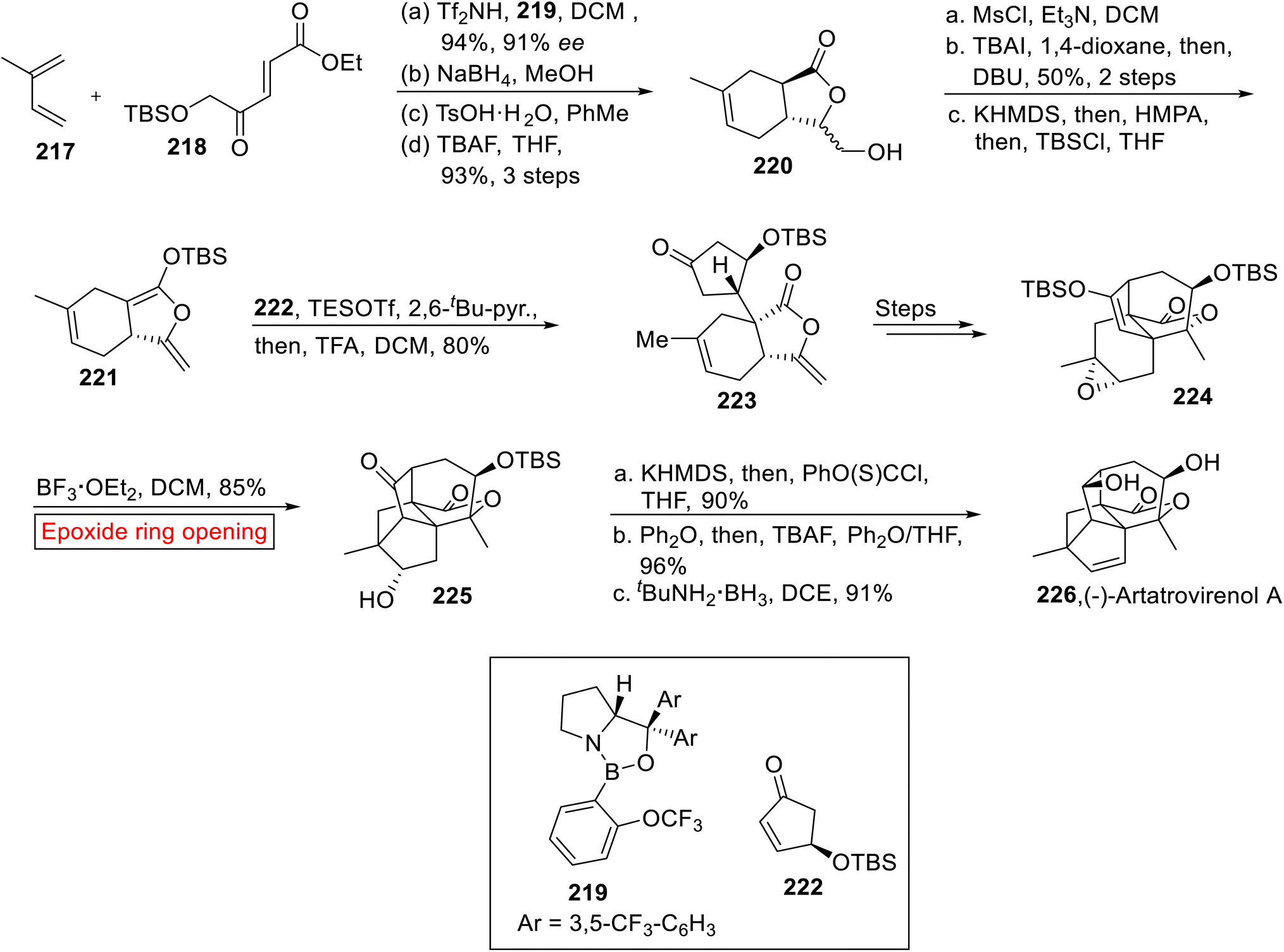
Taking into account the deadly aspects of cancer, several research groups have focused their attention towards the synthesis of efficacious anti-proliferative agents.171–173 (−)-Artatrovirenol A 226 was isolated from Artemisia atrovirens in 2020 (ref. 174) which exhibits cytocidal activity opposite to hepatocellular carcinoma (SMMC-7721, HepG2, and Huh7). Bridged, fused, and spirocyclic ring systems make a cage-like structure of (−)-Artatrovirenol A 226. In 2023,175 Zhu et al. disclosed the first total synthesis of (−)-Artatrovirenol A 226 by employing epoxide ring opening reaction as one of the significant steps. The enantioselective synthesis of (−)-Artatrovirenol A 226 initiated with the formation of TBS silyl ketene acetal 221. In the first step, iso-prene 217 was reacted with enolate 218 using oxazaborolidine 219 and bistriflimide (Tf2NH) followed by chemoselective reduction (NaBH4), acid-mediated lactonization (TsOH) and deprotection (TBAF) of silyl ether to provide γ-lactone 220 (93%, 3 steps). In the next step, –OH group of lactone 220 was protected with MsCl accompanied by TBAI-mediated Finkelstein iodination and elimination with subsequent deprotection reaction (by using KHMDS = potassium bis(trimethylsilyl)amide). The deprotection strategy was then followed by HMPA (hexamethylphosphoramide) and TBSCl (tert-butyldimethylsilyl chloride) addition to furnish silyl ketene acetal 221. Then, acetal 221 and enone 222 were reacted in the presence of TESOTf (trimethylsilyl trifluoromethanesulfonate) followed by TFA-mediated hydrolysis to render 5,6-cis-fused bicyclic lactone 223 (80%) which was further converted into 224 over several steps. In the next step, compound 224 experienced BF3·OEt2-catalyzed epoxide ring opening reaction in DCM to afford the product 225 (85%). In the next step, the compound 225 went through sequential thioacylation of alcohol, TBS deprotection (TBAF) and treatment with tBuNH2·BH3 to furnish (−)-Artatrovirenol 226 in 91% yield (Scheme 26).
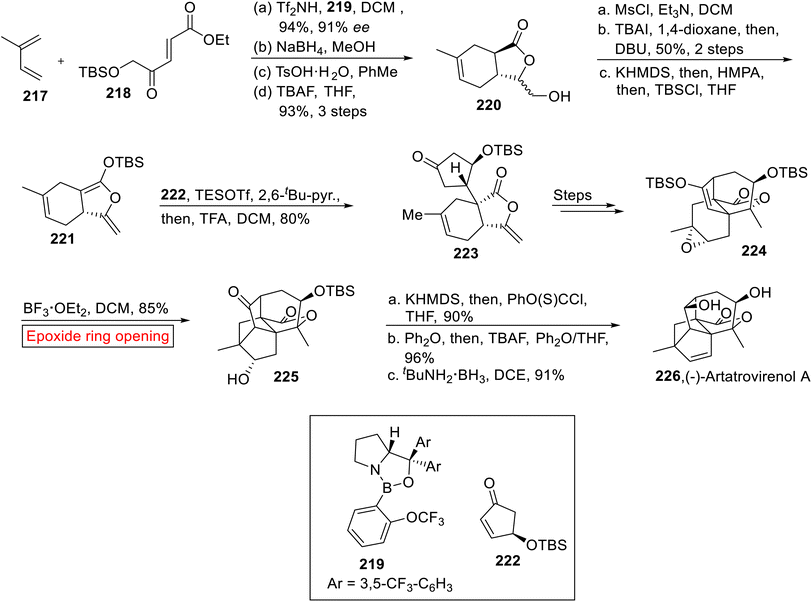 |
| | Scheme 26 Total synthesis of (−)-Artatrovirenol A 226. | |
Conclusion
This article provides an overview of the previous ten years of research on the epoxide ring opening reactions that have been employed towards the synthesis of natural products. Epoxides are frequently used as building blocks in synthetic organic chemistry by undergoing epoxide ring opening reaction. A number of reagents, catalysts, and solvents are used in an epoxide ring opening process which determine its outcome. Moreover, the total synthesis of various natural products also utilizes epoxide ring opening reaction as one of the key steps. An updated summation of synthetic methods involving the utilization of epoxide ring opening for the production of interesting natural products with important biological activities is presented in this review article. These natural products include alkaloids and terpenoids. This review will motivate organic chemists to focus their efforts on epoxide ring opening reactions and their chemistry to devise more effective pathways for further advancements towards the synthesis of natural products in the future.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
A. Irfan extends his appreciation to the Deanship of Research and Graduate Studies at King Khalid University for funding this work through Large Groups Research Project under grant number (RGP2/156/45). A. R. Chaudhry is thankful to the Deanship of Graduate Studies and Scientific Research at the University of Bisha, for supporting this work through the Fast-Track Research Support Program.
References
- G. Sabitha, R. S. Babu, M. Rajkumar, C. S. Reddy and J. Yadav, Tetrahedron Lett., 2001, 42, 3955–3958 CrossRef CAS.
- R. Munir, A. F. Zahoor, U. Nazeer, M. A. Saeed, A. Mansha, A. Irfan and M. U. Tariq, RSC Adv., 2023, 13, 35172–35208 RSC.
- S. E. Schaus and E. N. Jacobsen, Org. Lett., 2000, 2, 1001–1004 CrossRef CAS PubMed.
- U. Nazeer, A. Mushtaq, A. F. Zahoor, F. Hafeez, I. Shahzadi and R. Akhtar, RSC Adv., 2023, 13, 35695–35732 RSC.
- B. M. Cole, K. D. Shimizu, C. A. Krueger, J. P. Harrity, M. L. Snapper and A. H. Hoveyda, Angew. Chem., Int. Ed. Engl., 1996, 35, 1668–1671 CrossRef CAS.
- S. Mori, E. Nakamura and K. Morokuma, J. Am. Chem. Soc., 2000, 122, 7294–7307 CrossRef CAS.
- W. A. Nugent, J. Am. Chem. Soc., 1998, 120, 7139–7140 CrossRef CAS.
- S. Sagawa, H. Abe, Y. Hase and T. Inaba, J. Org. Chem., 1999, 64, 4962–4965 CrossRef CAS PubMed.
- Z. Yan, Z. Ma, J. Deng and G. Luo, Chem. Eng. Sci., 2021, 242, 116746 CrossRef CAS.
- S. Matsunaga, J. Das, J. Roels, E. M. Vogl, N. Yamamoto, T. Iida, K. Yamaguchi and M. Shibasaki, J. Am. Chem. Soc., 2000, 122, 2252–2260 CrossRef CAS.
- R.-E. Parker and N. Isaacs, Chem. Rev., 1959, 59, 737–799 CrossRef CAS.
- D. J. Goldsmith, J. Am. Chem. Soc., 1962, 84, 3913–3918 CrossRef CAS.
- R. A. Smith and S. Natelson, J. Am. Chem. Soc., 1931, 53, 3476–3479 CrossRef CAS.
- S. Ahmad, A. F. Zahoor, S. A. R. Naqvi and M. Akash, Mol. Diversity, 2018, 22, 191–205 CrossRef CAS PubMed.
- Z. Li, Z. Zhou, K. Li, L. Wang, Q. Zhou and C. Tang, Tetrahedron Lett., 2002, 43, 7609–7611 CrossRef CAS.
- S. Faiz, A. F. Zahoor, N. Rasool, M. Yousaf, A. Mansha, M. Zia-Ul-Haq and H. Z. Jaafar, Molecules, 2015, 20, 14699–14745 CrossRef CAS PubMed.
- M. S. Von Wittenau and H. Els, J. Am. Chem. Soc., 1963, 85, 3425–3431 CrossRef.
- K. Fagnou and M. Lautens, Org. Lett., 2000, 2, 2319–2321 CrossRef CAS PubMed.
- C. Baylon, G. Prestat, M.-P. Heck and C. Mioskowski, Tetrahedron Lett., 2000, 41, 3833–3835 CrossRef CAS.
- M. Inai, T. Asakawa and T. Kan, Tetrahedron Lett., 2018, 59, 1343–1347 CrossRef CAS.
- J. T. Bagdanoff, D. C. Behenna, J. L. Stockdill and B. M. Stoltz, Eur. J. Org. Chem., 2016, 12, 2101–2104 CrossRef PubMed.
- F. X. Li, S. J. Ren, P. F. Li, P. Yang and J. Qu, Angew. Chem., 2020, 132, 18631–18636 CrossRef.
- C. P. Bold, M. Gut, J. Schürmann, D. Lucena-Agell, J. Gertsch, J. F. Diaz and K. H. Altmann, Chem.–Eur. J., 2021, 27, 5936–5943 CrossRef CAS PubMed.
- A. Gehlawat, R. Prakash and S. Kumar Pandey, ChemistrySelect, 2020, 5, 6373–6375 CrossRef CAS.
- O. Hoff, N. Kratena, D. Aynetdinova, K. E. Christensen and T. J. Donohoe, Chem.–Eur. J., 2022, 28, e202202464 CrossRef CAS PubMed.
- P. Thesmar, S. Coomar, A. Prescimone, D. Häussinger, D. Gillingham and O. Baudoin, Chem.–Eur. J., 2020, 26, 15298–15312 CrossRef CAS PubMed.
- S. Meninno, L. Zullo, J. Overgaard and A. Lattanzi, Adv. Synth. Catal., 2017, 359, 913–918 CrossRef CAS.
- W. Hofer, E. Oueis, A. A. Fayad, F. Deschner, A. Andreas, L. P. de Carvalho, S. Hüttel, S. Bernecker, L. Pätzold and B. Morgenstern, Angew. Chem., Int. Ed., 2022, 61, e202202816 CrossRef CAS PubMed.
- R. Rengarasu and M. E. Maier, Asian J. Org. Chem., 2017, 6, 108–117 CrossRef CAS.
- X. Wang, G. Huang, Y. Wang and J. Gui, J. Am. Chem. Soc., 2023, 145, 9354–9363 CrossRef CAS PubMed.
- T. Kasemsuk, P. Piyachaturawat, R. Bunthawong, U. Sirion, K. Suksen, A. Suksamrarn and R. Saeeng, Eur. J. Med. Chem., 2017, 138, 952–963 CrossRef CAS PubMed.
- Z. Liu, F. Zhao, B. Zhao, J. Yang, J. Ferrara, B. Sankaran, B. Venkataram Prasad, B. B. Kundu, G. N. Phillips Jr and Y. Gao, Nat. Commun., 2021, 12, 4158 CrossRef CAS PubMed.
- B. R. McDonald and K. A. Scheidt, Acc. Chem. Res., 2015, 48, 1172–1183 CrossRef CAS PubMed.
- F. Isnard, M. Lamberti, C. Pellecchia and M. Mazzeo, ChemCatChem, 2017, 9, 2972–2979 CrossRef CAS.
- L. Wang, A. Parnell, C. Williams, N. A. Bakar, M. R. Challand, M. W. van der Kamp, T. J. Simpson, P. R. Race, M. P. Crump and C. L. Willis, Nat. Catal., 2018, 1, 968–976 CrossRef CAS.
- A. Rivas, M. Castiñeira, R. Álvarez, B. Vaz and A. R. de Lera, J. Nat. Prod., 2022, 85, 2302–2311 CrossRef CAS PubMed.
- Z. Y. Liu, Z. C. Chen, C. Z. Yu, R. F. Wang, R. Z. Zhang, C. S. Huang, Z. Yan, D. R. Cao, J. B. Sun and G. Li, Chem.–Eur. J., 2002, 8, 3747–3756 CrossRef CAS PubMed.
- H. Huang and J. S. Panek, Org. Lett., 2003, 5, 1991–1993 CrossRef CAS PubMed.
- T. R. Hoye and M. Hu, J. Am. Chem. Soc., 2003, 125, 9576–9577 CrossRef CAS PubMed.
- J. A. Faraldos and J.-L. Giner, J. Org. Chem., 2002, 67, 4659–4666 CrossRef CAS PubMed.
- L. Shi, K. Meyer and M. F. Greaney, Angew. Chem., 2010, 122, 9436–9439 CrossRef.
- K. Miyashita and T. Imanishi, Chem. Rev., 2005, 105, 4515–4536 CrossRef CAS PubMed.
- A. F. Barrero, J. E. Oltra, M. Alvarez and A. Rosales, J. Org. Chem., 2002, 67, 5461–5469 CrossRef CAS PubMed.
- D. J. Aldous, A. J. Dalençon and P. G. Steel, Org. Lett., 2002, 4, 1159–1162 CrossRef CAS PubMed.
- A. Ahmed, E. K. Hoegenauer, V. S. Enev, M. Hanbauer, H. Kaehlig, E. Öhler and J. Mulzer, J. Org. Chem., 2003, 68, 3026–3042 CrossRef CAS PubMed.
- S. Díaz, J. Cuesta, A. González and J. Bonjoch, J. Org. Chem., 2003, 68, 7400–7406 CrossRef PubMed.
- O. Smitt and H.-E. Högberg, Tetrahedron, 2002, 58, 7691–7700 CrossRef CAS.
- J. Cuerva, J. Justicia, J. Oller-López, B. Bazdi and J. Oltra, Mini-Rev. Org. Chem., 2006, 3, 23–35 CrossRef CAS.
- I. Vilotijevic and T. F. Jamison, Angew. Chem., Int. Ed., 2009, 48, 5250–5281 CrossRef CAS PubMed.
- I. Vilotijevic and T. F. Jamison, Mar. Drugs, 2010, 8, 763–809 CrossRef CAS PubMed.
- R. R. Rodríguez-Berríos, S. R. Isbel and A. Bugarin, Int. J. Mol. Sci., 2023, 24, 6195 CrossRef PubMed.
- M. Tsuda, Y. Kasai, K. Komatsu, T. Sone, M. Tanaka, Y. Mikami and J. I. Kobayashi, Org. Lett., 2004, 6, 3087–3089 CrossRef CAS PubMed.
- Z. Bian, C. C. Marvin, M. Pettersson and S. F. Martin, J. Am. Chem. Soc., 2014, 136, 14184–14192 CrossRef CAS PubMed.
- M. Chen, L. Gan, S. Lin, X. Wang, L. Li, Y. Li, C. Zhu, Y. Wang, B. Jiang, J. Jiang, J. Yang and J. Shi, J. Nat. Prod., 2012, 75, 1167–1176 CrossRef CAS PubMed.
- D. Gahtory, M. Chouhan, R. Sharma and V. A. Nair, Org. Lett., 2013, 15, 3942–3945 CrossRef CAS PubMed.
- S. D. Vaidya and N. P. Argade, Org. Lett., 2013, 15, 4006–4009 CrossRef CAS PubMed.
- Z.-J. Cai, S.-Y. Wang and S.-J. Ji, Org. Lett., 2013, 15, 5226–5229 CrossRef CAS PubMed.
- S. K. Ghosh and R. Nagarajan, RSC Adv., 2014, 4, 63147–63149 RSC.
- R. H. Jiao, S. Xu, J. Y. Liu, H. M. Ge, H. Ding, C. Xu, H. L. Zhu and R. X. Tan, Org. Lett., 2006, 8, 5709–5712 CrossRef CAS PubMed.
- B. B. Snider and X. Wu, Org. Lett., 2007, 9, 4913–4915 CrossRef CAS PubMed.
- A. Coste, M. Toumi, K. Wright, V. Raza, F. Couty, J. Marrot and G. Evano, Org. Lett., 2008, 10, 3841 CrossRef CAS PubMed.
- B. Malgesini, B. Forte, D. Borghi, F. Quartieri, C. Gennari and G. Papeo, Chem.–Eur. J., 2009, 15, 7922–7929 CrossRef CAS PubMed.
- A. Coste, G. Karthikeyan, F. Couty and G. Evano, Synthesis, 2009, 2927–2934 CAS.
- Q.-L. Peng, S.-P. Luo, X.-E. Xia, L.-X. Liu and P.-Q. Huang, Chem. Commun., 2014, 50, 1986–1988 RSC.
- S. Tabassum, A. F. Zahoor, S. Ahmad, R. Noreen, S. G. Khan and H. Ahmad, Mol. Diversity, 2021, 1–43 Search PubMed.
- Y. Nalli, V. Khajuria, S. Gupta, P. Arora, S. Riyaz-Ul-Hassan, Z. Ahmed and A. Ali, Org. Biomol. Chem., 2016, 14, 3322–3332 RSC.
- A. J. Day, H. C. Lam, C. J. Sumby and J. H. George, Org. Lett., 2017, 19, 2463–2465 CrossRef CAS PubMed.
- R. Hesse, K. K. Gruner, O. Kataeva, A. W. Schmidt and H. J. Knölker, Chem.–Eur. J., 2013, 19, 14098–14111 CrossRef CAS PubMed.
- M. Áiqbal Choudhary, Nat. Prod. Rep., 1999, 16, 619–635 RSC.
- F.-P. Wang, Q.-H. Chen and X.-Y. Liu, Nat. Prod. Rep., 2010, 27, 529–570 RSC.
- X.-W. Wang and H. Xie, Drugs Future, 1999, 24, 877–882 CrossRef CAS.
- K. G. Kou, S. Kulyk, C. J. Marth, J. C. Lee, N. A. Doering, B. X. Li, G. M. Gallego, T. P. Lebold and R. Sarpong, J. Am. Chem. Soc., 2017, 139, 13882–13896 CrossRef CAS PubMed.
- L. M. Liao, Alkaloids, Academic Press, New York, 2003, vol. 60, pp. 287–344 Search PubMed.
- T.-J. Hsieh, F.-R. Chang, Y.-C. Chia, C.-Y. Chen, H.-F. Chiu and Y.-C. Wu, J. Nat. Prod., 2001, 64, 616–619 CrossRef CAS PubMed.
- R. A. Clery, J. R. Cason and V. Zelenay, J. Agric. Food Chem., 2016, 64, 4566–4573 CrossRef CAS PubMed.
- L. Liu, Y. Cao, C. Chen, X. Zhang, A. McNabola, D. Wilkie, S. Wilhelm, M. Lynch and C. Carter, Cancer Res., 2006, 66, 11851–11858 CrossRef CAS PubMed.
- K. Zhao, L. Shen, Z. L. Shen and T. P. Loh, Chem. Soc. Rev., 2017, 46, 586–602 RSC.
- R. Akhtar and A. F. Zahoor, Synth. Commun., 2020, 50, 3337–3368 CrossRef CAS.
- P. Shelton, T. J. Ligon, J. M. Dell, L. Yarbrough and J. R. Vyvyan, Tetrahedron Lett., 2017, 58, 3478–3481 CrossRef CAS PubMed.
- X.-W. Yang, X.-D. Luo, P. K. Lunga, Y.-L. Zhao, X.-J. Qin, Y.-Y. Chen, L. Liu, X.-N. Li and Y.-P. Liu, Tetrahedron, 2015, 71, 3694–3698 CrossRef CAS.
- X.-W. Yang, X.-J. Qin, Y.-L. Zhao, P. K. Lunga, X.-N. Li, S.-Z. Jiang, G.-G. Cheng, Y.-P. Liu and X.-D. Luo, Tetrahedron Lett., 2014, 55, 4593–4596 CrossRef CAS.
- D. Wang, M. Hou, Y. Ji and S. Gao, Org. Lett., 2017, 19, 1922–1925 CrossRef CAS PubMed.
- T. Feng, Y. Li, Y.-Y. Wang, X.-H. Cai, Y.-P. Liu and X.-D. Luo, J. Nat. Prod., 2010, 73, 1075–1079 CrossRef CAS PubMed.
- M. Walia, C. N. Teijaro, A. Gardner, T. Tran, J. Kang, S. Zhao, S. E. O'Connor, V. Courdavault and R. B. Andrade, J. Nat. Prod., 2020, 83, 2425–2433 CrossRef CAS PubMed.
- N. Mizutani, Y. Aoki, T. Nabe, M. Ishiwara, S. Yoshino, H. Takagaki and S. Kohno, Eur. J. Pharmacol., 2009, 602, 138–142 CrossRef CAS PubMed.
- A. J. Duplantier, S. L. Becker, M. J. Bohanon, K. A. Borzilleri, B. A. Chrunyk, J. T. Downs, L.-Y. Hu, A. El-Kattan, L. C. James and S. Liu, J. Med. Chem., 2009, 52, 3576–3585 CrossRef CAS PubMed.
- J. Ramnauth, J. Speed, S. P. Maddaford, P. Dove, S. C. Annedi, P. Renton, S. Rakhit, J. Andrews, S. Silverman and G. Mladenova, J. Med. Chem., 2011, 54, 5562–5575 CrossRef CAS PubMed.
- H. Tang, Y. Tang, I. V. Kurnikov, H.-J. Liao, N.-L. Chan, M. G. Kurnikova, Y. Guo and W.-C. Chang, ACS Catal., 2021, 11, 7186–7192 CrossRef CAS PubMed.
- D. G. Yu, F. de Azambuja and F. Glorius, Angew. Chem., Int. Ed., 2014, 53, 2754–2758 CrossRef CAS PubMed.
- G.-D. Xu and Z.-Z. Huang, Org. Lett., 2017, 19, 6265–6267 CrossRef CAS PubMed.
- X. Kou and K. G. Kou, ACS Catal., 2020, 10, 3103–3109 CrossRef CAS.
- H. Wang, F. Cao, W. Gao, X. Wang, Y. Yang, T. Shi and Z. Wang, Org. Lett., 2021, 23, 863–868 CrossRef CAS PubMed.
- D. P. A. Gossan, A. A. Magid, P. A. Kouassi-Yao, J.-B. Behr, A. C. Ahibo, L. A. Djakouré, D. Harakat and L. Voutquenne-Nazabadioko, Phytochemistry, 2015, 109, 76–83 CrossRef CAS PubMed.
- B. J. Byatt, A. Kato and S. G. Pyne, Org. Lett., 2021, 23, 4029–4033 CrossRef CAS PubMed.
- J.-X. Zhao, C.-P. Liu, W.-Y. Qi, M.-L. Han, Y.-S. Han, M. A. Wainberg and J.-M. Yue, J. Nat. Prod., 2014, 77, 2224–2233 CrossRef CAS PubMed.
- C. M. Hasler, G. Acs and P. M. Blumberg, Cancer Res., 1992, 52, 202–208 CAS.
- E. Hecker, Pure Appl. Chem., 1977, 49, 1423–1431 CAS.
- X. Liu, J. Liu, J. Zhao, S. Li and C.-C. Li, Org. Lett., 2017, 19, 2742–2745 CrossRef CAS PubMed.
- E. M. Simmons, J. R. Yen and R. Sarpong, Org. Lett., 2007, 9, 2705–2708 CrossRef CAS PubMed.
- Z.-W. Jiao, Y.-Q. Tu, Q. Zhang, W.-X. Liu, S.-Y. Zhang, S.-H. Wang, F.-M. Zhang and S. Jiang, Nat. Commun., 2015, 6, 7332 CrossRef CAS PubMed.
- D. Martinez-Solorio and M. P. Jennings, Org. Lett., 2009, 11, 189–192 CrossRef CAS PubMed.
- S. Sengupta, M. G. Drew, R. Mukhopadhyay, B. Achari and A. K. Banerjee, J. Org. Chem., 2005, 70, 7694–7700 CrossRef CAS PubMed.
- Y. Suto, K. Kaneko, N. Yamagiwa and G. Iwasaki, Tetrahedron Lett., 2010, 51, 6329–6330 CrossRef CAS.
- C. Thommen, M. Neuburger and K. Gademann, Chem.–Eur. J., 2017, 23, 120–127 CrossRef CAS PubMed.
- G. Majetich, Y. Li and G. Zou, Heterocycles, 2007, 73, 217–225 CrossRef CAS PubMed.
- G. Majetich, Y. Li and G. Zou, Heterocycles: an International Journal for Reviews and Communications in Heterocyclic Chemistry, 2007, vol. 73, pp. 217–225 Search PubMed.
- C. H. Oh, L. Piao, J. Jung and J. Kim, Asian J. Org. Chem., 2016, 5, 1237–1241 CrossRef CAS.
- A. Ahmad and A. C. Burtoloso, Org. Lett., 2019, 21, 6079–6083 CrossRef CAS PubMed.
- Y. Li and G. Pattenden, Nat. Prod. Rep., 2011, 28, 429–440 RSC.
- J.-H. Sheu, A. F. Ahmed, R.-T. Shiue, C.-F. Dai and Y.-H. Kuo, J. Nat. Prod., 2002, 65, 1904–1908 CrossRef CAS PubMed.
- N. P. Thao, N. H. Nam, N. X. Cuong, T. H. Quang, P. T. Tung, D. Chae, S. Kim, Y.-S. Koh, P. Van Kiem and C. Van Minh, Bioorg. Med. Chem. Lett., 2013, 23, 228–231 CrossRef PubMed.
- N. J. Hafeman, S. A. Loskot, C. E. Reimann, B. P. Pritchett, S. C. Virgil and B. M. Stoltz, J. Am. Chem. Soc., 2020, 142, 8585–8590 CrossRef CAS PubMed.
- C. Meyre-Silva and V. Cechinel-Filho, Curr. Pharm. Des., 2010, 16, 3503–3518 CrossRef CAS PubMed.
- O. K. Popoola, A. M. Elbagory, F. Ameer and A. A. Hussein, Molecules, 2013, 18, 9049–9060 CrossRef CAS PubMed.
- S. El Bardai, N. Morel, M. Wibo, N. Fabre, G. Llabres, B. Lyoussi and J. Quetin-Leclercq, Planta Med., 2003, 69, 75–77 CrossRef CAS PubMed.
- F. Piozzi, M. Bruno, S. Rosselli and A. Maggio, Nat. Prod. Commun., 2006, 1, 1934578X0600100713 CrossRef.
- G. Laonigro, R. Lanzetta, M. Parrilli, M. Adinolfi and L. Mangoni, Gazz. Chim. Ital., 1979, 109, 145–150 CAS.
- Y. Sakagami, N. Kondo, Y. Sawayama, H. Yamakoshi and S. Nakamura, Molecules, 2020, 25, 1610 CrossRef CAS PubMed.
- R. A. Craig and B. M. Stoltz, Chem. Rev., 2017, 117, 7878–7909 CrossRef CAS PubMed.
- P. Ramesh, N. S. Reddy, Y. Venkateswarlu, M. V. R. Reddy and D. J. Faulkner, Tetrahedron Lett., 1998, 39, 8217–8220 CrossRef CAS.
- B. R. Chitturi, V. B. Tatipamula, C. B. Dokuburra, U. K. Mangamuri, V. R. Tuniki, S. V. Kalivendi, R. A. Bunce and V. Yenamandra, Tetrahedron, 2016, 72, 1933–1940 CrossRef CAS.
- I. Shahzadi, A. F. Zahoor, A. Rasul, N. Rasool, Z. Raza, S. Faisal, B. Parveen, S. Kamal, M. Zia-ur-Rehman and F. M. Zahid, J. Heterocycl. Chem., 2020, 57, 2782–2794 CrossRef CAS.
- N. J. Truax, S. Ayinde, J. O. Liu and D. Romo, J. Am. Chem. Soc., 2022, 144, 18575–18585 CrossRef CAS PubMed.
- E. F. Rogers, F. R. Koniuszy, J. Shavel Jr and K. Folkers, J. Am. Chem. Soc., 1948, 70, 3086–3088 CrossRef CAS PubMed.
- K. Wiesner, Pure Appl. Chem., 1963, 7, 285–296 CrossRef CAS.
- A. Bélanger, D. J. Berney, H.-J. Borschberg, R. Brousseau, A. Doutheau, R. Durand, H. Katayama, R. Lapalme, D. M. Leturc, C.-C. Liao, F. N. MacLachlan, J.-P. Maffrand, F. Marazza, R. Martino, C. Moreau, L. Saint-Laurent, R. Saintonge, P. Soucy, L. Ruest and P. Deslongchamps, Can. J. Chem., 1979, 57, 3348–3354 CrossRef.
- M. Nagatomo, M. Koshimizu, K. Masuda, T. Tabuchi, D. Urabe and M. Inoue, J. Am. Chem. Soc., 2014, 136, 5916–5919 CrossRef CAS PubMed.
- K. V. Chuang, C. Xu and S. E. Reisman, Science, 2016, 353, 912–915 CrossRef CAS PubMed.
- K. Du, M. J. Kier, Z. D. Stempel, V. Jeso, A. L. Rheingold and G. C. Micalizio, J. Am. Chem. Soc., 2020, 142, 12937–12941 CrossRef CAS PubMed.
- J. J. Fernández, M. L. Souto and M. Norte, Nat. Prod. Rep., 2000, 17, 235–246 RSC.
- J. Blunt, M. Hartshorn, T. McLennan, M. Munro, W. T. Robinson and S. Yorke, Tetrahedron Lett., 1978, 19, 69–72 CrossRef.
- A. Hoshino, H. Nakai, M. Morino, K. Nishikawa, T. Kodama, K. Nishikibe and Y. Morimoto, Angew. Chem., 2017, 129, 3110–3114 CrossRef.
- L. Ding, J. Münch, H. Goerls, A. Maier, H.-H. Fiebig, W.-H. Lin and C. Hertweck, Bioorg. Med. Chem. Lett., 2010, 20, 6685–6687 CrossRef CAS PubMed.
- L. Ding, A. Maier, H.-H. Fiebig, W.-H. Lin and C. Hertweck, Org. Biomol. Chem., 2011, 9, 4029–4031 RSC.
- Z. Xu, M. Baunach, L. Ding and C. Hertweck, Angew. Chem., 2012, 41, 10439–10443 CrossRef.
- M. Baunach, L. Ding, T. Bruhn, G. Bringmann and C. Hertweck, Angew. Chem., Int. Ed., 2013, 52, 9040–9043 CrossRef CAS PubMed.
- H. Li, Q. Zhang, S. Li, Y. Zhu, G. Zhang, H. Zhang, X. Tian, S. Zhang, J. Ju and C. Zhang, J. Am. Chem. Soc., 2012, 134, 8996–9005 CrossRef CAS PubMed.
- Q. Zhang, H. Li, S. Li, Y. Zhu, G. Zhang, H. Zhang, W. Zhang, R. Shi and C. Zhang, Org. Lett., 2012, 14, 6142–6145 CrossRef CAS PubMed.
- Q. Zhang, A. Mándi, S. Li, Y. Chen, W. Zhang, X. Tian, H. Zhang, H. Li, W. Zhang and S. Zhang, Eur. J. Org Chem., 2012, 2012, 5256–5262 CrossRef CAS.
- B. R. Rosen, E. W. Werner, A. G. O'Brien and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 5571–5574 CrossRef CAS PubMed.
- Z. Meng, H. Yu, L. Li, W. Tao, H. Chen, M. Wan, P. Yang, D. J. Edmonds, J. Zhong and A. Li, Nat. Commun., 2015, 6, 6096 CrossRef CAS PubMed.
- F. Marion, D. E. Williams, B. O. Patrick, I. Hollander, R. Mallon, S. C. Kim, D. M. Roll, L. Feldberg, R. Van Soest and R. J. Andersen, Org. Lett., 2006, 8, 321–324 CrossRef CAS PubMed.
- B. Sullivan, P. Djura, D. E. Mcintyre and D. J. Faulkner, Tetrahedron, 1981, 37, 979–982 CrossRef CAS.
- A. Grube, M. Assmann, E. Lichte, F. Sasse, J. R. Pawlik and M. Köck, J. Nat. Prod., 2007, 70, 504–509 CrossRef CAS PubMed.
- B. W. Sullivan, D. J. Faulkner, G. K. Matsumoto, C. H. He and J. Clardy, J. Org. Chem., 1986, 51, 4568–4573 CrossRef CAS.
- A. W. Markwell-Heys, K. K. Kuan and J. H. George, Org. Lett., 2015, 17, 4228–4231 CrossRef CAS PubMed.
- H.-B. Liao, G.-H. Huang, M.-H. Yu, C. Lei and A.-J. Hou, J. Org. Chem., 2017, 82, 1632–1637 CrossRef CAS PubMed.
- G.-Y. Luo, H. Wu, Y. Tang, H. Li, H.-S. Yeom, K. Yang and R. P. Hsung, Synthesis, 2015, 47, 2713–2720 CrossRef CAS.
- R. Raju, A. M. Piggott, L. X. Barrientos Diaz, Z. Khalil and R. J. Capon, Org. Lett., 2010, 12, 5158–5161 CrossRef CAS PubMed.
- S. Faiz, A. F. Zahoor, M. Ajmal, S. Kamal, S. Ahmad, A. M. Abdelgawad and M. E. Elnaggar, J. Heterocycl. Chem., 2019, 56, 2839–2852 CrossRef CAS.
- J. Schmidt and C. B. Stark, Org. Lett., 2012, 14, 4042–4045 CrossRef CAS PubMed.
- X.-B. Ding, D. P. Furkert, R. J. Capon and M. A. Brimble, Org. Lett., 2014, 16, 378–381 CrossRef CAS PubMed.
- B. M. Fraga, Nat. Prod. Rep., 2012, 29, 1334–1366 RSC.
- B. Fraga, Nat. Prod. Rep., 2013, 30, 1226 RSC.
- J. A. Marshall and A. E. Greene, J. Org. Chem., 1972, 37, 982–985 CrossRef CAS.
- A. Srikrishna and V. H. Pardeshi, Tetrahedron, 2010, 66, 8160–8168 CrossRef CAS.
- A.-C. Huang, C. J. Sumby, E. R. Tiekink and D. K. Taylor, J. Nat. Prod., 2014, 77, 2522–2536 CrossRef CAS PubMed.
- S. Adekenov, M. Mukhametzhanov, A. Kagarlitskii and A. Kupriyanov, Chem. Nat. Compd., 1982, 18, 623–624 CrossRef.
- N. Zhangabylov, L. Y. Dederer, L. Gorbacheva, S. Vasil'eva, A. Terekhov and S. Adekenov, Pharm. Chem. J., 2004, 38, 651–653 CrossRef CAS.
- S. Kalidindi, W. B. Jeong, A. Schall, R. Bandichhor, B. Nosse and O. Reiser, Angew. Chem., Int. Ed., 2007, 46, 6361–6363 CrossRef CAS PubMed.
- J.-D. Zhai, D. Li, J. Long, H.-L. Zhang, J.-P. Lin, C.-J. Qiu, Q. Zhang and Y. Chen, J. Org. Chem., 2012, 77, 7103–7107 CrossRef CAS PubMed.
- S. H. Lone and K. A. Bhat, Tetrahedron Lett., 2015, 56, 1908–1910 CrossRef CAS.
- G. L. Chetty and S. Dev, Tetrahedron Lett., 1964, 5, 73–77 CrossRef.
- V. Benesova, Collect. Czech. Chem. Commun., 1976, 41, 3812–3814 CrossRef CAS.
- H. Erdtman, B. Thomas, E. Stenhagen, L. G. Sillén, B. Zaar and E. Diczfalusy, Acta Chem. Scand., 1958, 12, 267–273 CrossRef CAS.
- R. Kumar, J. Halder and S. Nanda, Tetrahedron, 2017, 73, 809–818 CrossRef CAS.
- M. Kobayashi, B. Son, M. Kido, Y. Kyogoku and I. Kitagawa, Chem. Pharm. Bull., 1983, 31, 2160–2163 CrossRef CAS.
- M. Kobayashi, B. W. Son, Y. Kyogoku and I. Kitagawa, Chem. Pharm. Bull., 1984, 32, 1667–1670 CrossRef CAS.
- B. F. Bowden, J. C. Coll and D. M. Tapiolas, Aust. J. Chem., 1983, 36, 211–214 CrossRef CAS.
- S. Begum, R. Bhattacharya, S. Paul and T. K. Chakraborty, J. Org. Chem., 2023, 88, 12677–12697 CrossRef CAS PubMed.
- Z. Ma, G. Gao, K. Fang and H. Sun, ACS Med. Chem. Lett., 2019, 10, 191–195 CrossRef CAS PubMed.
- I. Shahzadi, A. F. Zahoor, A. Rasul, A. Mansha, S. Ahmad and Z. Raza, ACS Omega, 2021, 6, 11943–11953 CrossRef CAS PubMed.
- R. Akhtar, A. F. Zahoor, A. Rasul, M. Ahmad, M. N. Anjum, M. Ajmal, Z. Raza and Z. Pak, J. Pharm. Sci., 2019, 32, 2215–2222 Search PubMed.
- L.-H. Su, C.-A. Geng, T.-Z. Li, Y.-B. Ma, X.-Y. Huang, X.-M. Zhang and J.-J. Chen, J. Org. Chem., 2020, 85, 13466–13471 CrossRef CAS PubMed.
- R. Lavernhe, P. Domke, Q. Wang and J. Zhu, J. Am. Chem. Soc., 2023, 145, 24408–24415 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Muhammad Jawwad Saif
*a,
Muhammad Jawwad Saif