DOI:
10.1039/D4RA02090A
(Paper)
RSC Adv., 2024,
14, 13095-13099
Regioselective C(sp2)–H halogenation of pyrazolo[1,5-a]pyrimidines facilitated by hypervalent iodine(III) under aqueous and ambient conditions†
Received
19th March 2024
, Accepted 15th April 2024
First published on 23rd April 2024
Abstract
An efficient and mild approach has been developed for the regio-selective direct C3 halogenation of pyrazolo[1,5-a]pyrimidines employing readily available potassium halide salts and a hypervalent iodine(III) reagent at ambient temperature. The protocol is both practical and environmentally friendly, utilizing water as a green solvent, potassium halides as an inexpensive and bench stable halogen source and PIDA as a non-toxic reagent, enabling clean and efficient halogenation at room temperature. The procedure yields a range of C3 halogenated pyrazolo[1,5-a]pyrimidines in good to excellent yields. Mechanistic studies suggest the involvement of electrophilic substitution mechanism in the halogenation process.
Introduction
Organohalogen compounds are prevalent in nature and are recognized for their remarkable biological activities, rendering them intriguing candidates for drug development.1 Numerous naturally existing metabolites containing halogens hold medicinal and therapeutic significance.2 Additionally, these compounds find diverse applications in the realm of material science.3 Halogenated heteroaromatic compounds hold significant importance as foundational components for synthesizing complex natural products and drugs. Their pivotal role in transition metal-catalysed cross-coupling reactions further underscores their importance in these processes.4
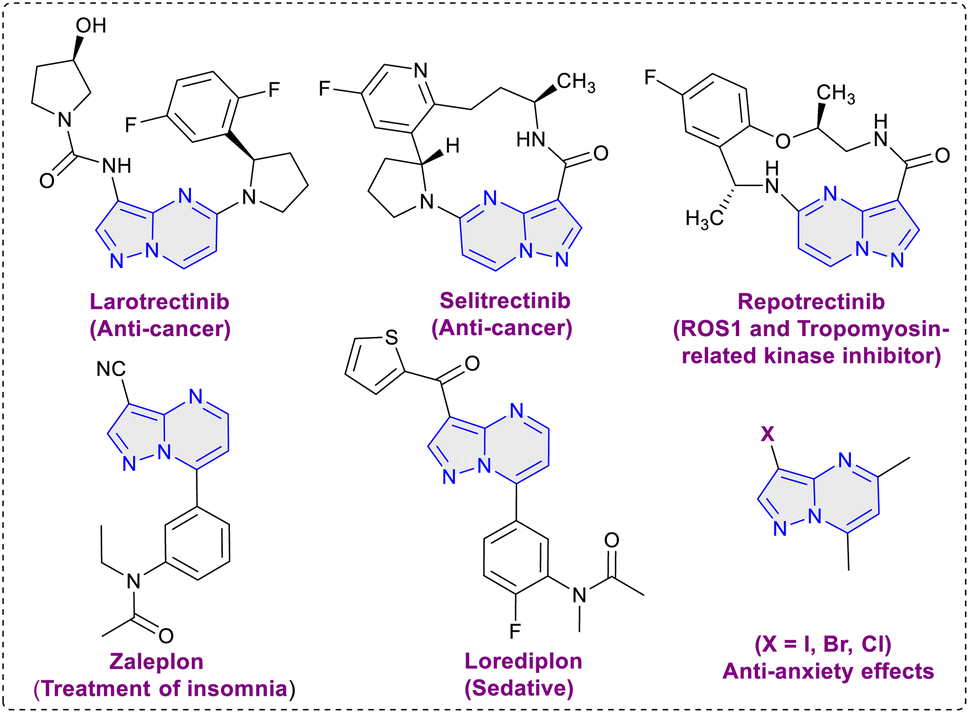
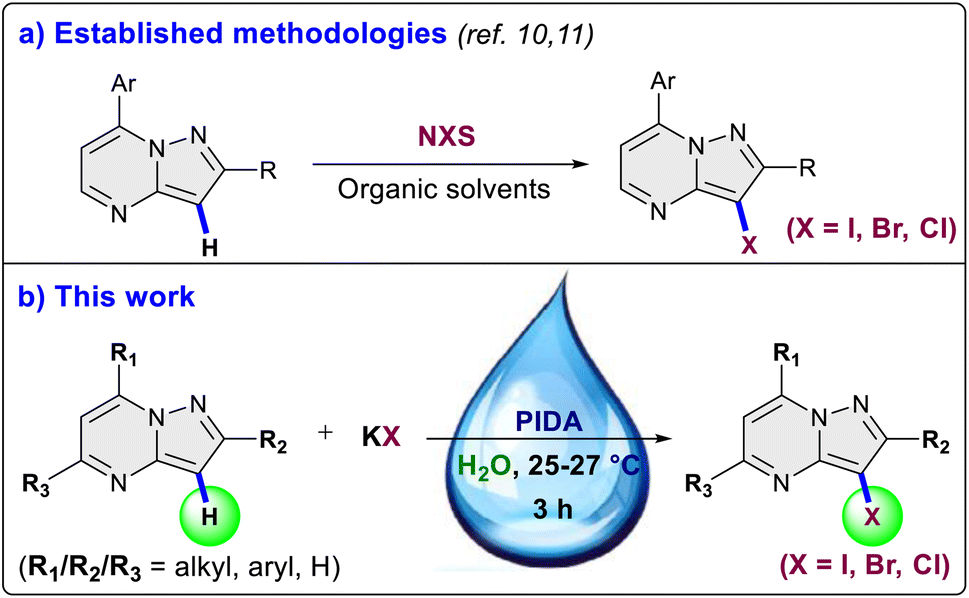
The pyrazolo[1,5-a]pyrimidine scaffold serves as a central structure for a diverse range of pharmacologically and biologically active compounds.5 In the field of medicinal chemistry, this scaffold is renowned for its anti-tumor, antiviral, anticancer, anti-malarial, and anti-inflammatory properties.6 Notably, the pyrazolo[1,5-a]pyrimidine core is a crucial element present in various anti-cancer medications like selitrectinib, repotrectinib, larotrectinib and is also a constituent of insomnia medications such as zaleplon and lorediplon7 (Fig. 1). Beyond its therapeutic relevance, the pyrazolo[1,5-a]pyrimidine core bears significant importance for material scientists, particularly in the realms of intriguing optical applications and chemosensor functionality.8 Halogenated pyrazolo[1,5-a]pyrimidines have been found to possess anxiolytic properties, their synthesis has been explored with the aim of assessing their potential effectiveness in addressing anxiety disorders.9 In 2004, Liebscher et al. introduced a protocol for the direct halogenation of pyrazolo[1,5-a]pyrimidines using corresponding N-halosuccinimides (Scheme 1a).10 However, this method requires elevated temperatures and the use of organic solvents. Similar approaches using N-halosuccinimides were later employed by Martins et al., and Portilla et al.11 (Scheme 1a). Hence, the development of an environmentally friendly and efficient methodology for the direct halogenation of pyrazolo[1,5-a]pyrimidines is of crucial significance. In this work, we disclose a method for the direct and regioselective C3 halogenation of pyrazolo[1,5-a]pyrimidines at room temperature. This approach employs potassium halides as a safe, bench stable, cheap and readily available halogenation source (Scheme 1b). The key advantage of this methodology is its operational feasibility at room temperature, demonstrating its efficiency and practicality. Additionally, it utilizes water as solvent, contributing to the overall sustainability of the process.
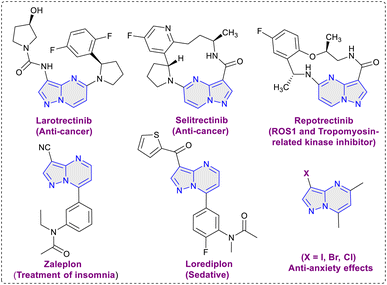 |
| | Fig. 1 Bioactive compounds with pyrazolo[1,5-a]pyrimidine scaffold. | |
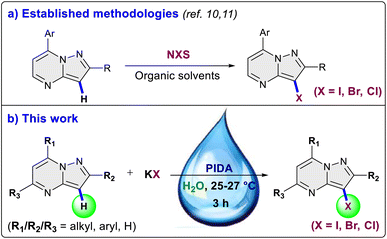 |
| | Scheme 1 Previous work on halogenation of pyrazolo[1,5-a]pyrimidines. | |
Results and discussion
The investigation into the iodination reaction commenced with the selection of 2-methyl-7-phenylpyrazolo[1,5-a]pyrimidine (1a) as the preferred substrate and potassium iodide (2a) as the halogen source. 2-Methyl-7-phenylpyrazolo[1,5-a]pyrimidine (1a) was reacted with 1.5 equivalents of KI and K2S2O8 (1.0 equiv.) for 3 hours at room temperature (25–27 °C) in water as a solvent, yielding desired 3-iodo-2-methyl-7-phenylpyrazolo[1,5-a]pyrimidine (3aa) in a 30% yield (Table 1, entry 1). Encouraged by the initial success, alternative oxidants for the iodination reaction were explored keeping other parameters constant. In the presence of Na2S2O8 and oxone the yield increased to 35% and 38% respectively (entry 2 and 3). However, the use of tert-butyl peroxybenzoate (TBAB) and tert-butyl hydroperoxide (TBHP) resulted in lower yields of 15% and 25%, respectively (entries 4 and 5). Oxygen, when used as an oxidant, did not yield any product in the iodination reaction (entry 6). Subsequently, we explored hypervalent iodine(III) reagents as oxidants, and were pleased to observe a substantial increase in the yield of the desired product 3aa. In the presence of (bis(trifluoroacetoxy)iodo)benzene (PIFA) and (diacetoxyiodo)benzene (PIDA), the yields escalated to 72% and 87%, respectively (entries 7 and 8). Upon conducting oxidant screening, it became apparent that PIDA exhibited superior performance as the oxidant for the iodination reaction. The transition of the halogen source from KI to NaI and tetrabutylammonium iodide (TBAI) resulted in a diminished yield of the desired product 3aa to 64% and 73% respectively (entries 9 and 10), indicating that KI proved to be a more effective halogen source for the iodination reaction. Utilizing 1.0 equivalent of KI in the reaction, as opposed to 1.5 equivalents, resulted in a decrease in the reaction yield to 70% (entry 11).
Table 1 Optimization of reaction conditionsa
|
| Entry |
Oxidant |
Solvent |
Yieldb |
| Reaction conditions: 1a (0.2 mmol), 2a (0.3 mmol), oxidant (1.0 equiv.), H2O (3.0 mL), rt (25–27 °C), 3 h. Isolated yields. TBAI was used instead of KI. NaI was used instead of KI. 1.0 equiv. of KI was used. 0.75 equiv. oxidant was used. 0.5 equiv. oxidant was used. No oxidant. |
| 1 |
K2S2O8 |
H2O |
30% |
| 2 |
Na2S2O8 |
H2O |
35% |
| 3 |
Oxone |
H2O |
38% |
| 4 |
TBPB |
H2O |
15% |
| 5 |
TBHP |
H2O |
25% |
| 6 |
O2 |
H2O |
NR |
| 7 |
PIFA |
H2O |
72% |
| 8 |
PIDA |
H2O |
87% |
| 9c |
PIDA |
H2O |
64% |
| 10d |
PIDA |
H2O |
73% |
| 11e |
PIDA |
H2O |
70% |
| 12f |
PIDA |
H2O |
65% |
| 13g |
PIDA |
H2O |
52% |
| 14h |
— |
H2O |
NR |
Upon reducing the amount of PIDA to 0.75 and 0.5 equivalents, lowering the yields to 65% and 52%, respectively (entries 12 and 13) was observed. When the oxidant was omitted, the reaction did not take place, emphasizing the crucial role of the oxidant in the reaction (entry 14). The optimal yield of 3aa was achieved by employing 1.0 equivalent of 1a, 1.5 equivalents of KI, 1.0 equivalent of PIDA, utilizing water as the solvent, and conducting the reaction for duration of 3 hours. After obtaining optimized reaction conditions, we decided to investigate the substrate scope for halogenation (I/Br/Cl) of pyrazolo[1,5-a]pyrimidines (Scheme 2). Under optimal conditions, iodination of 2-methyl-7-phenylpyrazolo[1,5-a]pyrimidine resulted in product 3aa with an 87% yield. Various derivatives of 2-methyl-7-phenylpyrazolo[1,5-a]pyrimidine, bearing electron-donating (–OMe, –Me) and electron-withdrawing (–Cl, –Br) groups on the phenyl ring, smoothly underwent iodination reactions, producing the respective C3 iodinated products (3ab–3ae) with yields ranging from 83% to 95%. A variety of tri-substituted pyrazolo[1,5-a]pyrimidine derivatives efficiently underwent an iodination reaction under optimised conditions, leading to the formation of products 3af–3ah with excellent yields ranging from 86% to 91%. 7-Phenylpyrazolo[1,5-a]pyrimidine and pyrazolo[1,5-a]pyrimidine exhibited excellent reactivity, yielding the corresponding C3 iodinated products 3ai and 3aj with impressive yields of 89% and 79%, respectively. This underscores the excellent regioselectivity of the present methodology. Under optimized conditions, both the pyrazolo[1,5-a]pyrimidine derivative bearing a fluorine substituent on the phenyl ring and the derivative with a pyridine ring attached demonstrated successful reactions, yielding iodination products 3ak and 3al in 84% and 92% yield respectively. Other N-heterocycles like 2-phenylimidazo[1,2-a]pyridine, 2-methyl-4H-pyrido[1,2-a]pyrimidin-4-one and 2-methyl-1H-indole reacted well giving corresponding iodinated products in 86%, 72% and 78% yield respectively. Subsequently, we chose to expand the present approach to conduct the regioselective bromination of pyrazolo[1,5-a]pyrimidine derivatives. To begin with, 2-methyl-7-phenylpyrazolo[1,5-a]pyrimidine was reacted with 1.5 equivalent of potassium bromide in presence of PIDA (1.0 equiv.) using H2O as a solvent for 3 hours. Gratifyingly, the anticipated C3 brominated product 3ba was obtained with a high yield of 88%. The current reaction conditions demonstrated excellent compatibility with both electron-withdrawing (–Cl, –Br) and electron-donating groups (–OMe, –Me) yielding the respective C3 brominated products (3bb–3be) in outstanding yields ranging from 86% to 92%. The reaction exhibited good reactivity with different trisubstituted pyrazolo[1,5-a]pyrimidine derivatives, yielding the corresponding brominated products 3bf–3bh in remarkable yields ranging from 88% to 90%. 7-Phenylpyrazolo[1,5-a]pyrimidine and pyrazolo[1,5-a]pyrimidine displayed excellent reactivity and regioselectivity under optimized conditions, yielding C3 brominated products 3bi (88%) and 3bj (81%). Notably, exclusive bromination was observed solely at the C3 position, with no occurrence of bromination at any other site. Following the successful iodination and bromination, the same strategy was implemented for the site-selective chlorination of the pyrazolo[1,5-a]pyrimidine scaffold. A reaction was conducted by treating 2-methyl-7-phenylpyrazolo[1,5-a]pyrimidine with potassium chloride (1.5 equiv.) utilizing water as a solvent over a period of 3 hours; the chlorinated product 3ca was isolated with a slight lower yield of 67% compared to iodination and bromination. Interestingly, when the same reaction was conducted in MeOH with all other parameters held constant, the yield of the product 3ca significantly increased to 86%. Consequently, based on these findings, we opted to conduct chlorination reactions in water as well as in methanol as the solvent. 2-Methyl-7-phenylpyrazolo[1,5-a]pyrimidines bearing electron donating substituents (–OMe, –Me) as well as electron withdrawing halogen substituents (–Br, –Cl) gave corresponding C3 chlorinated products 3cb–3ce in good yields ranging from 69–73% respectively. Trisubstituted derivatives of pyrazolo[1,5-a]pyrimidine exhibited excellent reactivity in the chlorination protocol, yielding the corresponding mono-chlorinated products 3cf–3ch with commendable yields (68–70%). In line with the regioselectivity observed in iodination and bromination reactions, chlorination exhibited a similar trend.
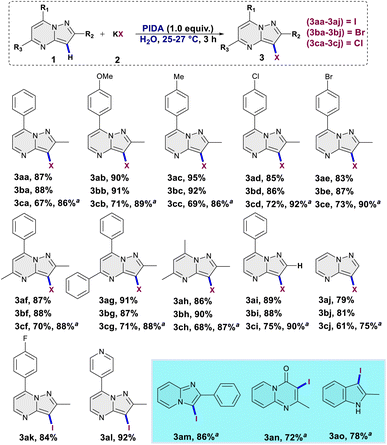 |
| | Scheme 2 Substrate scope for the regioselective halogenation of pyrazolo[1,5-a]pyrimidines and other N-heterocycles. Reaction conditions: 1 (0.2 mmol), KX (0.3 mmol), PIDA (1.0 equiv.), H2O (3.0 mL), rt (25–27 °C), 3 h. Yields are isolated yields. aReactions were carried out in MeOH. | |
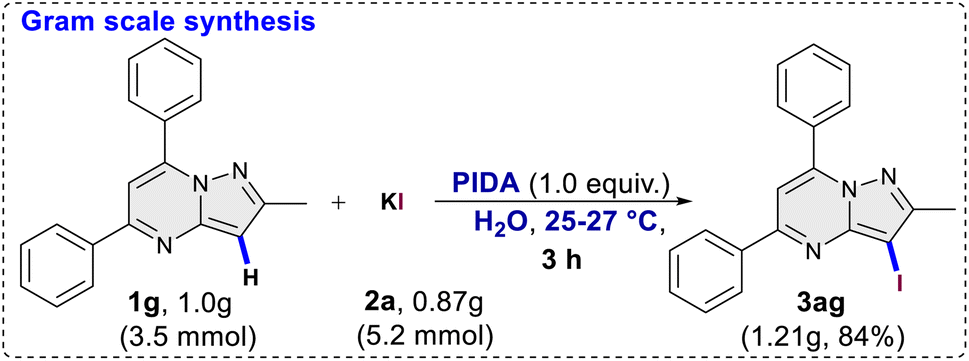
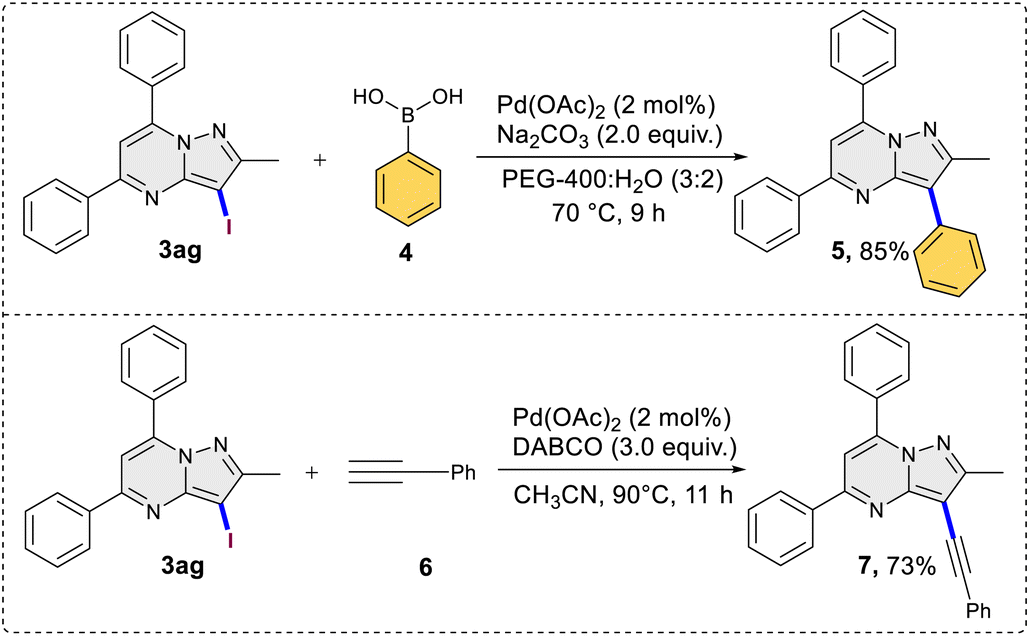
This was evident in the selective formation of products 3ci and 3cj, providing yields of 75% and 61%, respectively. To ascertain the reliability and efficacy of the devised methodology, we decided to carry out iodination of the substrate on gram scale. A 3.5 mmol (1.0 g) quantity of 2-methyl-5,7-diphenylpyrazolo[1,5-a]pyrimidine (1g) was subjected to a reaction with 5.2 mmol (0.87 g) of KI, in water as a solvent. The reaction proceeded for 3 hours at room temperature, yielding the desired C3 iodinated product 3ag with a 84% yield (1.21 g) (Scheme 3). Emphasizing the utility of the synthesized halogenated compounds, their potential to undergo transformation into substituted pyrazolo[1,5-a]pyrimidine derivatives was investigated. Specifically, a targeted approach involved subjecting 3-iodo-2-methyl-5,7-diphenylpyrazolo[1,5-a]pyrimidine to well-established cross-coupling reactions, namely Suzuki–Miyaura coupling and Sonogashira coupling (Scheme 4).12 Under the conditions outlined in Scheme 4, compound 3ag underwent reactions with phenyl boronic acid and phenyl acetylene, yielding products 5 and 7 with good yields of 85% and 73%, respectively. Following this, a series of controlled experiments were meticulously carried out to clarify the mechanistic foundations governing the halogenation reaction of pyrazolo[1,5-a]pyrimidine. These reactions were systematically conducted under optimized conditions, adding radical scavengers, namely (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) and 2,6-di-tert-butyl-4-methylphenol (BHT), to assess the potential participation of radical intermediates (Scheme 5). Despite the addition of TEMPO and BHT in the halogenation reactions, these radical scavengers proved ineffective in completely suppressing product formation. Only a modest reduction in product yield in case of iodination and bromination was observed, with the iodinated product 3af being obtained in 77% and 69% yields in the presence of TEMPO and BHT, respectively. Similarly, the bromination product 3bf was isolated in 72% and 71% yields in the presence of TEMPO and BHT, demonstrating a marginal decrease in overall efficiency. In case of chlorination reaction product 3cf was obtained in 45% and 48% yields in presence of TEMPO and BHT respectively. Based on the results of control experiments and literature study13 a possible mechanism has been postulated for the halogenation of pyrazolo[1,5-a]pyrimidines (Scheme 6). The process commences with PIDA initiating a ligand exchange with a halide salt, resulting in the formation of intermediate A. This intermediate then undergoes a transformation into hypohalite salt B, serving as a source for an electrophilic halogen species. Following this, substrate 1a engages with the electrophilic halogen species B, leading to the formation of intermediate C. In the final step, intermediate C undergoes deprotonation, ultimately yielding the halogenated product 3.
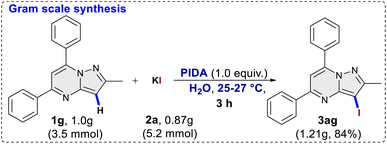 |
| | Scheme 3 Gram scale synthesis. | |
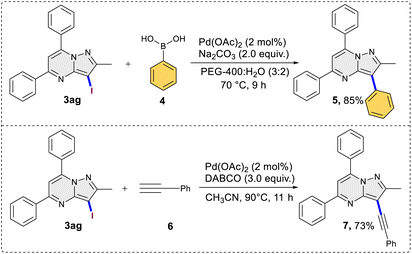 |
| | Scheme 4 Synthetic application in cross coupling reactions. | |
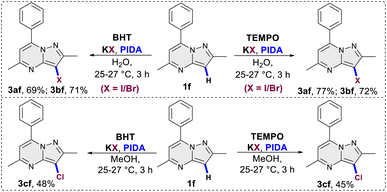 |
| | Scheme 5 Control experiments. | |
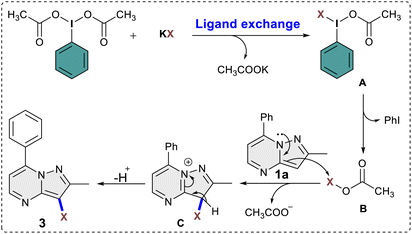 |
| | Scheme 6 Plausible reaction mechanism. | |
Conclusions
In conclusion, we have developed an environmentally friendly and efficient method for the regio-selective C3 halogenation of pyrazolo[1,5-a]pyrimidines, using a hypervalent iodine(III) reagent, readily and cheaply available potassium halide salts, and water as a green solvent at ambient temperature. The method facilitates clean and effective halogenation, resulting in the synthesis of diverse C3 halogenated pyrazolo [1,5-a]pyrimidine derivatives with consistently good to excellent yields. Our preliminary mechanistic studies suggest the involvement of an electrophilic substitution mechanism in this process. Beyond its environmental merits, this method demonstrates characteristics of rapidity, operational simplicity, scalability, and an expansive substrate scope, thereby enhancing its potential as a versatile and valuable tool in organic synthesis.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We express gratitude to the CSIR research grant (02/0486/23/EMR-II) from the Government of India for providing generous financial assistance, sophisticated Instrumentation Centre at IIT Indore for their invaluable assistance. Additionally, our deep appreciation goes to the 500 MHz NMR facility at department of chemistry, supported by DST-FIST, Govt. of India. A. S. C. acknowledges the fellowship granted by CSIR-New Delhi, and R. T. B. expresses gratitude for the fellowship received from IIT Indore.
References
-
(a) D. G. Fujimori and C. T. Walsh, Curr. Opin. Chem. Biol., 2007, 11, 553–560 CrossRef CAS PubMed;
(b) C. C. Hughes, J. B. MacMillan, S. P. Gaudêncio, P. R. Jensen and W. Fenical, Angew. Chem., Int. Ed., 2009, 48, 725–727 CrossRef CAS PubMed;
(c) K. Kaur, M. Jain, R. P. Reddy and R. Jain, Eur. J. Med. Chem., 2010, 45, 3245–3264 CrossRef CAS PubMed;
(d) S. Vandekerckhove, H. G. Tran, T. Desmet and M. D’hooghe, Bioorg. Med. Chem., 2013, 23, 4641–4643 CrossRef CAS PubMed.
-
(a) B. Gál, C. Bucher and N. Z. Burns, Mar. Drugs, 2016, 14, 206 CrossRef PubMed;
(b) M. Quémener, S. Kikionis, M. Fauchon, Y. Toueix, F. Aulanier, A. M. Makris, V. Roussis, E. Ioannou and C. Hellio, Mar. Drugs, 2022, 20, 32 CrossRef PubMed;
(c) C. Wang, W. Du, H. Lu, J. Lan, K. Liang and S. Cao, Molecules, 2021, 26, 2754 CrossRef CAS PubMed;
(d) N. Winterton, Green Chem., 2000, 2, 173–225 RSC.
-
(a) I. S. Nawghare, A. K. Singh, A. Maibam, S. S. Deshmukh, S. Krishnamurty, K. Krishnamoorthy and J. Nithyanandhan, Mater. Adv., 2023, 4, 3270–3284 RSC;
(b) M. Tuikka, P. Hirva, K. Rissanen, J. Korppi-Tommola and M. Haukka, ChemComm, 2011, 47, 4499–4501 RSC.
-
(a) I. J. S. Fairlamb, Chem. Soc. Rev., 2007, 36, 1036–1045 RSC;
(b) S. E. Hooshmand, B. Heidari, R. Sedghi and R. S. Varma, Green Chem., 2019, 21, 381–405 RSC;
(c) N. Kambe, T. Iwasaki and J. Terao, Chem. Soc. Rev., 2011, 40, 4937–4947 RSC;
(d) P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS PubMed;
(e) C. L. Sun, H. Li, D. G. Yu, M. Yu, X. Zhou, X. Y. Lu, K. Huang, S. F. Zheng, B. J. Li and Z. J. Shi, Nat. Chem., 2010, 2, 1044–1049 CrossRef CAS PubMed;
(f) C. L. Sun and Z. J. Shi, Chem. Rev., 2014, 114, 9219–9280 CrossRef CAS PubMed.
-
(a) A. Dorababu, Arch. Pharm., 2022, 355, 2200154 CrossRef CAS PubMed;
(b) N. G. Johansson, L. Dreano, K. Vidilaseris, A. Khattab, J. Liu, A. Lasbleiz, O. Ribeiro, A. Kiriazis, G. Boije af Gennäs, S. Meri, A. Goldman, J. Yli-Kauhaluoma and H. Xhaard, ChemMedChem, 2021, 16, 3360–3367 CrossRef CAS PubMed;
(c) A. Kiessling, R. Wiesinger, B. Sperl and T. Berg, ChemMedChem, 2007, 2, 627–630 CrossRef CAS PubMed;
(d) P. McGillan, N. G. Berry, G. L. Nixon, S. C. Leung, P. J. H. Webborn, M. C. Wenlock, S. Kavanagh, A. Cassidy, R. H. Clare, D. A. Cook, K. L. Johnston, L. Ford, S. A. Ward, M. J. Taylor, W. D. Hong and P. M. O'Neill, ACS Med. Chem. Lett., 2021, 12, 1421–1426 CrossRef CAS PubMed.
-
(a) C. Almansa, A. F. de Arriba, F. L. Cavalcanti, L. A. Gómez, A. Miralles, M. Merlos, J. García-Rafanell and J. Forn, J. Med. Chem., 2001, 44, 350–361 CrossRef CAS PubMed;
(b) A. Arias-Gómez, A. Godoy and J. Portilla, Molecules, 2021, 26, 2708 CrossRef PubMed;
(c) L. F. S. P. Azeredo, J. P. Coutinho, V. A. P. Jabor, P. R. Feliciano, M. C. Nonato, C. R. Kaiser, C. M. S. Menezes, A. S. O. Hammes, E. R. Caffarena, L. V. B. Hoelz, N. B. de Souza, G. A. N. Pereira, I. P. Cerávolo, A. U. Krettli and N. Boechat, Eur. J. Med. Chem., 2017, 126, 72–83 CrossRef CAS PubMed;
(d) S. Cherukupalli, R. Karpoormath, B. Chandrasekaran, G. A. Hampannavar, N. Thapliyal and V. N. Palakollu, Eur. J. Med. Chem., 2017, 126, 298–352 CrossRef CAS PubMed;
(e) J. Y. Hwang, M. P. Windisch, S. Jo, K. Kim, S. Kong, H. C. Kim, S. Kim, H. Kim, M. E. Lee, Y. Kim, J. Choi, D.-S. Park, E. Park, J. Kwon, J. Nam, S. Ahn, J. Cechetto, J. Kim, M. Liuzzi, Z. No and J. Lee, Bioorg. Med. Chem., 2012, 22, 7297–7301 CrossRef CAS PubMed;
(f) Y. Liu, R. Laufer, N. K. Patel, G. Ng, P. B. Sampson, S.-W. Li, Y. Lang, M. Feher, R. Brokx, I. Beletskaya, R. Hodgson, O. Plotnikova, D. E. Awrey, W. Qiu, N. Y. Chirgadze, J. M. Mason, X. Wei, D. C.-C. Lin, Y. Che, R. Kiarash, G. C. Fletcher, T. W. Mak, M. R. Bray and H. W. Pauls, ACS Med. Chem. Lett., 2016, 7, 671–675 CrossRef CAS PubMed;
(g) G. Sabita, R. Savitha, K. Divya and K. Bhaskar, Chem. Data Collect., 2022, 38, 100822 CrossRef CAS;
(h) C. Soares de Melo, T.-S. Feng, R. van der Westhuyzen, R. K. Gessner, L. J. Street, G. L. Morgans, D. F. Warner, A. Moosa, K. Naran, N. Lawrence, H. I. M. Boshoff, C. E. Barry, C. J. Harris, R. Gordon and K. Chibale, Bioorg. Med. Chem., 2015, 23, 7240–7250 CrossRef CAS PubMed.
- H. Kumar, R. Das, A. Choithramani, A. Gupta, D. Khude, G. Bothra and A. Shard, ChemistrySelect, 2021, 6, 5807–5837 CrossRef CAS.
-
(a) A. G. Al-Sehemi, A. Irfan and A. M. Fouda, Spectrochim. Acta, Part A, 2013, 111, 223–229 CrossRef CAS PubMed;
(b) M. A. El-Gahami, A. E. M. Mekky, T. S. Saleh and A. S. Al-Bogami, Spectrochim. Acta, Part A, 2014, 129, 209–218 CrossRef CAS PubMed;
(c) H. Kim, A. Jo, J. Ha, Y. Lee, Y. S. Hwang and S. B. Park, ChemComm, 2016, 52, 7822–7825 RSC;
(d) A. Z. Sayed, M. S. Aboul-Fetouh and H. S. Nassar, J. Mol. Struct., 2012, 1010, 146–151 CrossRef CAS;
(e) M. Singsardar, R. Sarkar, K. Majhi, S. Sinha and A. Hajra, ChemistrySelect, 2018, 3, 1404–1410 CrossRef CAS;
(f) A. Tigreros, J. Zapata-Rivera and J. Portilla, ACS Sustain. Chem. Eng., 2021, 9, 12058–12069 CrossRef CAS.
- W. E. Kirkpatrick, T. Okabe, I. W. Hillyard, R. K. Robins, A. T. Dren and T. Novinson, J. Med. Chem., 1977, 20, 386–393 CrossRef CAS PubMed.
- L. Yin and J. Liebscher, Synthesis, 2004, 2004, 2329–2334 CrossRef.
-
(a) J.-C. Castillo, H.-A. Rosero and J. Portilla, RSC Adv., 2017, 7, 28483–28488 RSC;
(b) M. A. P. Martins, E. Scapin, C. P. Frizzo, F. A. Rosa, H. G. Bonacorso and N. Zanatta, J. Braz. Chem. Soc., 2009, 20, 205–213 CrossRef CAS.
-
(a) J.-H. Li, Y. Liang and Y.-X. Xie, J. Org. Chem., 2005, 70, 4393–4396 CrossRef CAS PubMed;
(b) L. Liu, Y. Zhang and Y. Wang, J. Org. Chem., 2005, 70, 6122–6125 CrossRef CAS PubMed;
(c) P. Sikdar, T. Choudhuri, S. Paul, S. Das and A. K. Bagdi, ACS Omega, 2023, 8, 23851–23859 CrossRef CAS PubMed.
-
(a) N. Mali, J. G. Ibarra-Gutiérrez, L. I. Lugo Fuentes, R. Ortíz-Alvarado, L. Chacón-García, P. Navarro-Santos, J. O. C. Jiménez-Halla and C. R. Solorio-Alvarado, Eur. J. Org Chem., 2022, 2022, e202201067 CrossRef CAS;
(b) F. Yang, X. Wang, W. Zhao, F. Yu and Z. Yu, ACS Omega, 2021, 6, 34044–34055 CrossRef CAS PubMed;
(c) L. Gu, T. Lu, M. Zhang, L. Tou and Y. Zhang, Adv. Synth. Catal., 2013, 355, 1077–1082 CrossRef CAS;
(d) P. Katrun and C. Kuhakarn, Tetrahedron Lett., 2019, 60, 989–993 CrossRef CAS;
(e) L. A. Segura-Quezada, K. R. Torres-Carbajal, K. A. Juárez-Ornelas, A. J. Alonso-Castro, R. Ortiz-Alvarado, T. Dohi and C. R. Solorio-Alvarado, Org. Biomol. Chem., 2022, 20, 5009–5034 RSC;
(f) Y. Wang, Y. Wang, K. Jiang, Q. Zhang and D. Li, Org. Biomol. Chem., 2016, 14, 10180–10184 RSC.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Umesh A. Kshirsagar
and
Umesh A. Kshirsagar