
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe conjugates of 5′-deoxy-5-fluorocytidine and hydroxycinnamic acids – synthesis, anti-pancreatic cancer activity and molecular docking studies†
Marcin Cybulski *a,
Magdalena Zaremba-Czogallab,
Bartosz Trzaskowskic,
Marek Kubiszewskid,
Joanna Tobiasza,
Anna Jarominb,
Piotr Krzeczyńskia,
Jerzy Gubernatorb and
Olga Michalak*a
*a,
Magdalena Zaremba-Czogallab,
Bartosz Trzaskowskic,
Marek Kubiszewskid,
Joanna Tobiasza,
Anna Jarominb,
Piotr Krzeczyńskia,
Jerzy Gubernatorb and
Olga Michalak*a
aPharmacy, Cosmetic Chemistry and Biotechnology Research Group, Łukasiewicz Research Network-Industrial Chemistry Institute, Rydygiera 8, 01-793 Warsaw, Poland. E-mail: olga.michalak@ichp.lukasiewicz.gov.pl; marcin.cybulski@ichp.lukasiewicz.gov.pl; Tel: +48 453 056 175 Tel: +48 453 056 177
bDepartment of Lipids and Liposomes, Faculty of Biotechnology, University of Wroclaw, Fryderyka Joliot-Curie 14a, 50-383 Wroclaw, Poland
cChemical and Biological Systems Simulation Laboratory, Center of New Technologies, University of Warsaw, Banacha 2c, 02-097 Warsaw, Poland
dPharmaceutical Analysis Laboratory, Łukasiewicz Research Network-Industrial Chemistry Institute, Rydygiera 8, 01-793 Warsaw, Poland
First published on 23rd April 2024
Abstract
New amide conjugates 1–6 of hydroxycinnamic acids (HCA) and 5′-deoxy-5-fluorocytidine (5-dFCR), the prodrug of 5-fluorouracil (5-FU), were synthesized and tested in vitro against pancreatic cancer lines (PDAC). The compounds showed slightly higher efficacy against primary BxPC-3 cells (IC50 values of 14–45 μM) than against metastatic AsPC-1 (IC50 values of 37–133 μM), and similar to that of 5-FU for both PDAC lines. Compound 1, which has a para-(acetyloxy)coumaroyl substituent, was found to be the most potent (IC50 = 14 μM) with a selectivity index of approximately 7 to normal dermal fibroblasts (IC50 = 96 μM). The potential pharmacological profiles were discussed on the basis of the ADME data. Docking to the carboxylesterase CES2 showed that the synthesized compounds have the ability to bind via hydrogen bonding between a specific acetate group of the sugar moiety and Ser228, which belongs to the catalytic triad that causes hydrolysis. Docking to albumin, a major transport protein in the circulatory system, revealed a strong interaction of the conjugates at the binding site which is native to warfarin and responsible for its transport in the body.
Introduction
Combination therapy with drugs targeting a different or similar pathway is the gold standard in oncological treatments.1–5 In addition to the efficient therapies with anticancer cocktails,6 the effective combination therapy of natural plant medicines and known anticancer drugs has been reported in a limited number of studies. Lee et al.7 postulated the benefits in postoperative gastric cancer patients in terms of overall and disease-free survival after chemotherapy supported by traditional herbal medicines. Kamran et al.8 evidenced a synergistic effect in HCT116 colon cancer cells after co-administration of 5-fluorouracil and diosmetin. However, for some types of cancer, there are still limitations that need to be overcome to achieve a better survival rate. Besides, in some clinical analyses, the combination therapies afford predictable and clinically unsignificant benefits.9 The obstacles of multidrug therapies require work on more effective anti-cancer drugs. One of the directions is to design and study single molecules that combine structures of more than one anti-cancer drug or contain their haptophore fragments.10–12 The other approach is based on the conjugation of a known anticancer agent with a molecule that allows targeted delivery to tumour cells. As a result of research, eleven antibody-drug conjugates for cancer therapy have already been approved by FDA.13,14 Furthermore, glufosfamide, the first sugar conjugate of DOX, is still under investigation to determine whether it provides additional survival benefit in patients with metastatic pancreatic cancer after gemcitabine-based first-line regimen.15Since the 1960s, 5-fluorouracil (5-FU) has been used as a time-dependent antimetabolite in the treatment of diverse types of malignancies.16 The effectiveness of oral 5-FU is limited due to its low bioavailability related to poor membrane permeability and GIT absorption.17 Therefore, continuous intravenous infusion of 5-FU is the most effective chemotherapeutic regimen.18 In the last four decades of the 20th century, various clinical trials showed that oral 5-FU prodrugs could be similarly effective to continuous infusion of 5-FU. Two orally administered prodrugs, tegafur and capecitabine, have already been registered. Both act through the dihydropyrimidine dehydrogenase (DPD) inhibition mechanism. Capecitabine (brand name Xeloda) has been shown to be effective in treating colon, colorectal, gastric, and breast cancer in monotherapy. It may also be used with other cancer drugs, such as cisplatin, docetaxel, oxaliplatin, irinotecan or bevacizumab against gastric cancer, metastatic breast cancer19 or metastatic colorectal cancer.20 Various capecitabine derivatives have been designed and synthesized to date.21,22 Recently, the PCC0208037 conjugate containing the haptophoric groups of irinotecan and capecitabine has been synthesized and evaluated in vitro.23 Biological studies confirmed its anti-tumour activity and explored the potential molecular mechanism of action: cell cycle arrest and DNA damage. In vivo studies showed comparable anti-tumour efficacy of PCC0208037 in the xenograft colorectal cancer model and more favourable drug-like properties, as compared to irinotecan. Another ZRX1 conjugate acting as a prodrug of both the EGFR and the 5-FU inhibitor was also synthesized and biologically evaluated.24 ZRX1 was designed to release 5-dFCR from its ester and the EGFR inhibitor in the first step of the metabolic pathway via hydrolysis by carboxylesterases (CES). The conjugate had the potential to induce a stronger growth inhibitory effect against human breast carcinoma cells than capecitabine or a single EGFR inhibitor and equivalent potency when compared with their combination.
Capecitabine (oral) and 5-FU (injection) are important drugs approved for the treatment of pancreatic cancer, either alone or in combination with FOLFIRINOX (folinic acid, fluorouracil, irinotecan, oxaliplatin) or OFF (oxaliplatin, fluorouracil, folinic acid) chemotherapy regimens.25 Other mono- and combination therapies, such as paclitaxel nanoparticles, everolimus, erlotinib, gemcitabine, irinotecan liposomal, olaparib, mitomycin, and sunitinib, as well as gemcitabine with platinum compounds (cisplatin, oxaliplatin), have also not demonstrated significant benefits for pancreatic cancer treatment.26–28
Hydroxycinnamic acids have shown promising anticancer effects by inducing apoptosis through the modulation of intracellular reactive oxygen species (ROS).29–32 Recently, intensive research on hydroxycinnamic acid derivatives has been focused in order to find new hybrid drugs.33,34 Our previous results demonstrated that conjugates of indolo[2,3-b]quinoline antineoplastic with hydroxycinnamic acid analogues exhibit cytotoxic activity against primary AsPC-1 and metastatic BxPC-3 pancreatic cancer cells.35 Furthermore, simple hydroxycinnamic acid analogues, when combined with curcumin and/or carnosic acid, exhibit synergistic activity against leukaemia cells.36 One of the compounds in the metabolic pathway of capecitabine, to release active 5-FU, is 5′-deoxy-5-fluorocytidine 5-dFCR (Fig. 1). This compound is endogenously synthesized by hydrolysis of the amide group in capecitabine. Due to the presence of a free amino group in the chemical structure of 5-dFCR it was selected as a scaffold to design new group of its conjugates with hydroxycinnamic acids (HCA) by connection through amide-type bond.
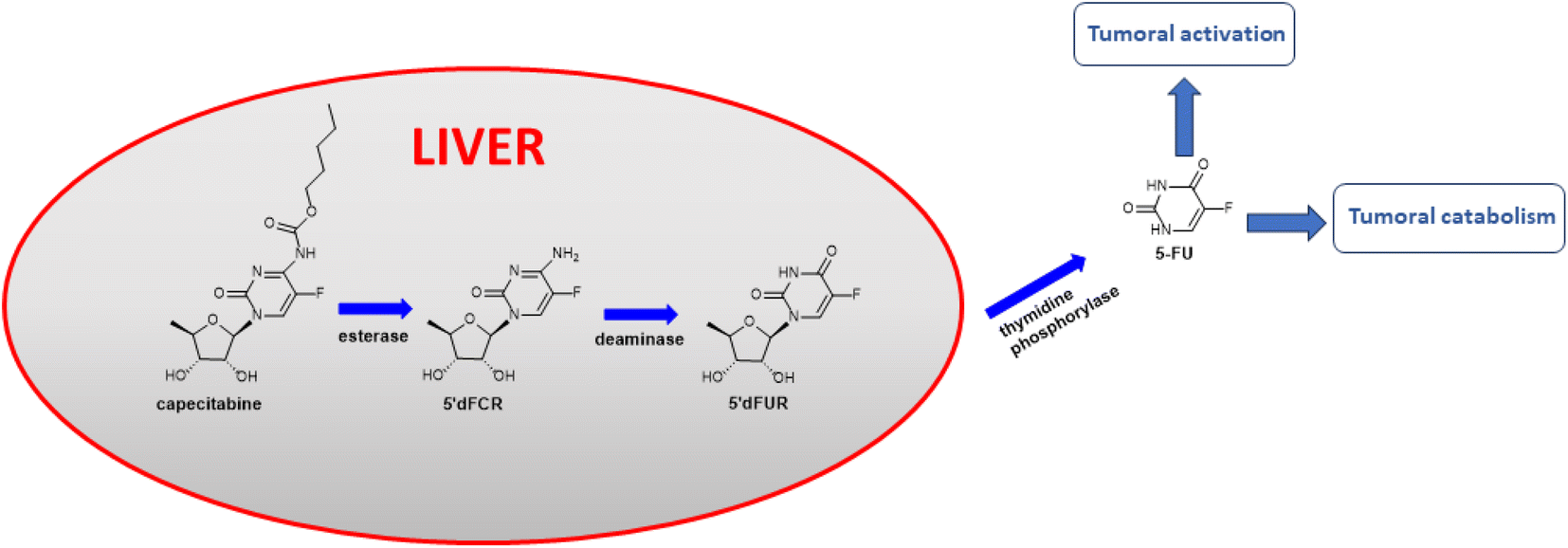 | ||
| Fig. 1 Capecitabine metabolic pathway.37 | ||
The two predominant carboxylesterases CES1 and CES2 play a crucial role in the human body in the metabolism of a wide variety of esters.38 CES2 prefers the hydrolysis of esters with a relatively large alcohol group and a small acyl group.39 This characteristic was used in the successful design of prodrug compounds, in which biotransformation by CES2 is the first metabolic step to give an active drug, such as prasugrel, capecitabine, flutamide, and fluorescein diacetate. High CES2 activity in PDAC pancreatic ductal adenocarcinoma (e.g. BxPC-3, AsPC-1 cell lines) may be associated with the patient's increased sensitivity to irinotecan.40,41 Moreover, elevated tumoral CES2 was a statistically significant predictor of poor overall survival in an orthotopic mouse model of PDAC.42
The systematic increase in cancer incidence combined with low survival rates in some types, including pancreatic cancer,43 encourages the search for new effective molecules. Studies of conjugates of known chemotherapeutic drugs/or their prodrugs are one of the promising approaches to find compounds with an enhanced profile of action or better targeting known cancer pathways. As drug resistance limits the clinical utility of 5-FU, ROS modulation by phenolic compounds may be one method to overcome 5-FU resistance in cancer cells due to the different balance of ROS regulation in normal and cancer cells that affect their apoptosis mechanism.44
This work presents the synthesis, computational studies, and biological evaluation of new potential prodrugs 1–6 containing 5-dFCR and selected hydroxycinnamic acids in their structure (Fig. 2). The compounds were evaluated in vitro for their antiproliferative profile against pancreatic cancer cell lines. Cytotoxicity profiles were determined using the MTT assay. For the model compounds, 6 and its peracetylated precursor 5, the difference in their potency on two PDAC cell lines related to the number of acetylated hydroxyl groups in their structures was discussed.
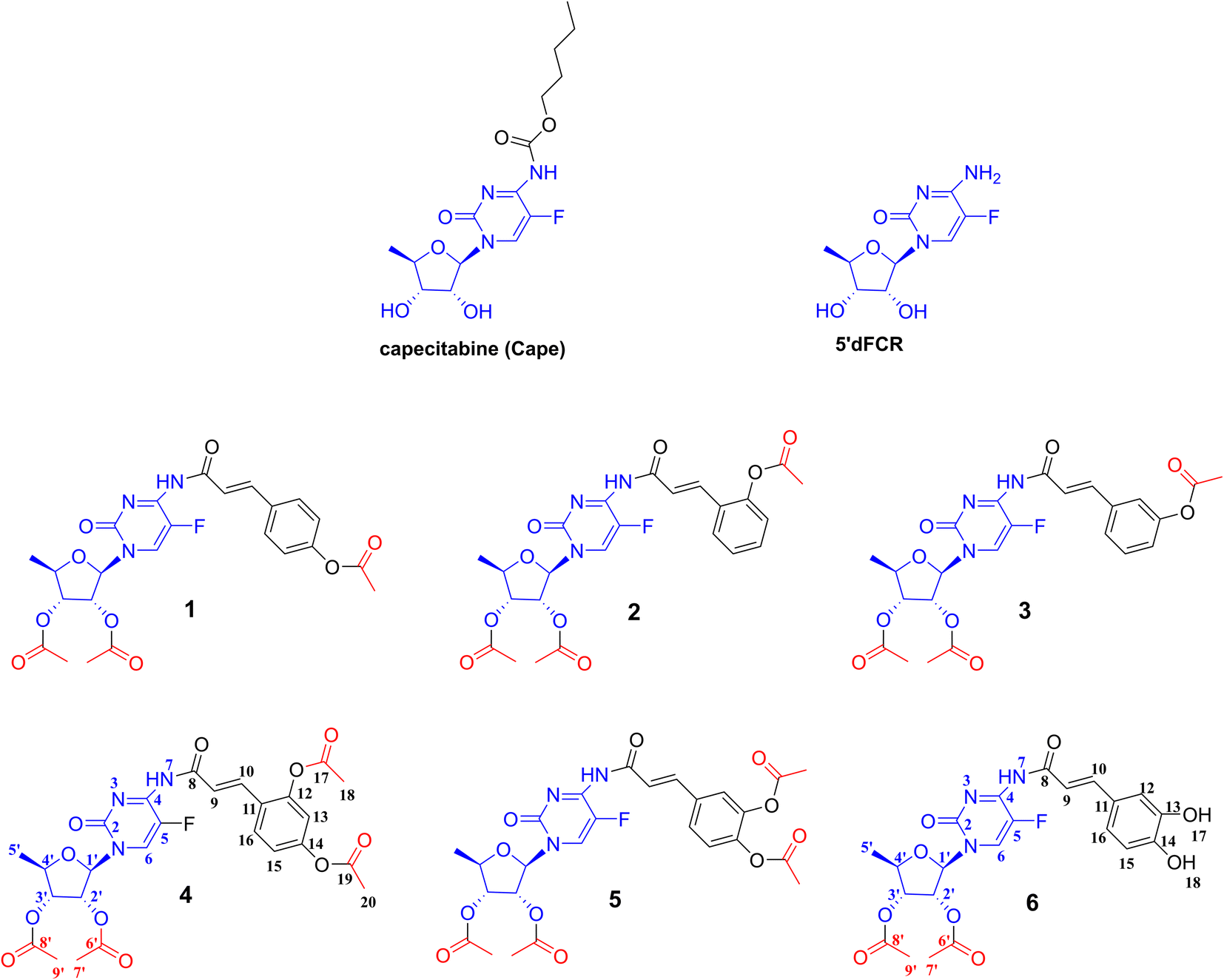 | ||
| Fig. 2 Chemical structures of synthetised 1–6 prodrug conjugates of 5-dFCR and HCA. | ||
Results and discussion
Chemistry
Caffeic acid was selected as a typical substrate from the hydroxycinnamic acids group. The aim of the first set of experiments was to investigate the feasibility of synthesizing a model conjugate from 5-dFCR and caffeic acid. It was intended to obtain the dual molecule with all-free hydroxyl groups and connected by an amide bond. Due to the presence of phenolic-type hydroxyl groups, which are unstable at high pH45 and also prone to oxidation, synthesis with unprotected phenolic acids as substrates is unfavourable. The HCA protection strategy may affect the effectiveness of the next coupling step. Therefore, it was decided to introduce various protecting groups to the hydroxyls of caffeic acid, to obtain stable coupling HCA substrates. Based on literature data,46 three different protecting groups, which tolerate different deprotection conditions and stability, were selected for preliminary studies, that is, silyl,47 acetyl48 and allyl.49 Similarly, the 5-dFCR substrate with isopropylidene or acetyl at the hydroxyl groups of the ribose ring was used for the syntheses. At first, di-O-protected silyl, acetyl, and allyl caffeic acid were synthesised according to known procedures.50–52 Many methods of converting hydroxycinnamic acids to their corresponding amides by using classical reagents have been described in the literature. The classic HBTU/HOBt coupling of caffeic acid derivatives with L-serine methyl ester was described by Monteiro et al.53 with the yields of 57–70%. Spasova et al.54 used the EDCI/HOBt system to synthetise a group of hydroxycinnamic acid derivatives in order to study their antioxidant properties. A similar strategy was used by Takao55 who obtained the suitable amides for antioxidant SAR studies. Rajan et al.56 described the synthesis of amide hydroxycinnamic acids derivatives in the presence of BOP with a yield of 65–85%.Thus, for our test reaction between caffeic acid and 5′-deoxy-5-fluoro-2′,3′-O-isopropylidenocytidine (27) PyBOP reagent was selected. Unfortunately, the TLC monitoring showed a complicated reaction mixture. After workup, the unreacted 27 was recovered with a yield of 70%. When EDCI/DMF reagent system was used, no cytidine derivative consumption was observed on TLC. Then, silyl-protected 3,4-di-O-tert-butyldimethylsilyl caffeic acid was reacted with 5′-deoxy-5-fluoro-2′,3′-O-isopropylidenocytidine (27) using various coupling agents: DCC, EDCI, TBTU, COMU. Due to ambiguous TLC results, the post-reaction mixtures were purified using column chromatography to 27 as the main product.
Then, the relatively labile silyl groups were replaced with a stable allyl ether protection. Reaction with 3,4-di-O-allyl caffeic acid in the presence of COMU led to the activation of the carboxylic group. However, no further progress in amide formations was observed. In contrast, the reaction with PyBOP resulted in a complex mixture of TLC spots from various compounds, including a small spot from the anticipated amide product. Therefore, we decided to test the efficiency of carboxyl group activation through acid chlorides due to the small size of the leaving group.
At first, the protocol described by Quéléver et al.,57 where acid chloride is generated in situ by POCl3, was applied to give two reaction products: mono-28 and disubstituted 26 derivatives of 5′-deoxy-5-fluoro-2′,3′-O-isopropylidenocytidine (Fig. 3). Despite the use of various molar excesses of the amine substrate, the reaction with POCl3 gave the disubstituted derivative 26 as the main product. Since this observation, the allyl-protected acid 18 was transformed into the respective chloride 24 and conjugated with cytidine derivative 27 in the series of test reactions. The best reaction condition, that was, methylene chloride and 50% NaOH aqueous solvent, allowed to obtain the compound 28 in 91% yield. Experiments to remove allyl ethers from the completely protected derivative 28 were next carried out in the presence of tetrakis(triphenylphosphine)palladium49 (Pd(PPh3)4), or the DMSO-I2 and DMSO-NaI reagent systems.58–60 The attempts with Pd(0) catalyst led to a multispots reaction mixture on TLC plates. Reactions with iodine/iodide at room temperature showed no progress. However, when heated to 90 °C the cleavage of conjugates to the O-allyl-hydroxycinnamic acid was observed. For the reaction with tetrakis(triphenylphosphine)-palladium, the various solvents, including methanol or DMF, as well as different molar excesses of the catalyst were also verified. The results showed that the decomposition products, without a predominant compound, were detected on TLC. Reactions with palladium(II) chloride led to black suspensions containing the unreacted substrate with a lot of small impurities and a distinct tarry starting spot on TLC. Due to the unfavourable results of the removal of allyl from the compound with an isopropylidene protected sugar moiety, it was decided to change the sequence of deprotection. The isopropylidene removal was achieved in the single experiment by adding 5 M HCl in methanol to the crude post-reaction mixture of 28. After 1 hour at ambient temperature, followed by extraction and chromatographic purification on silica gel compound 29 was isolated with a low yield of 23%.
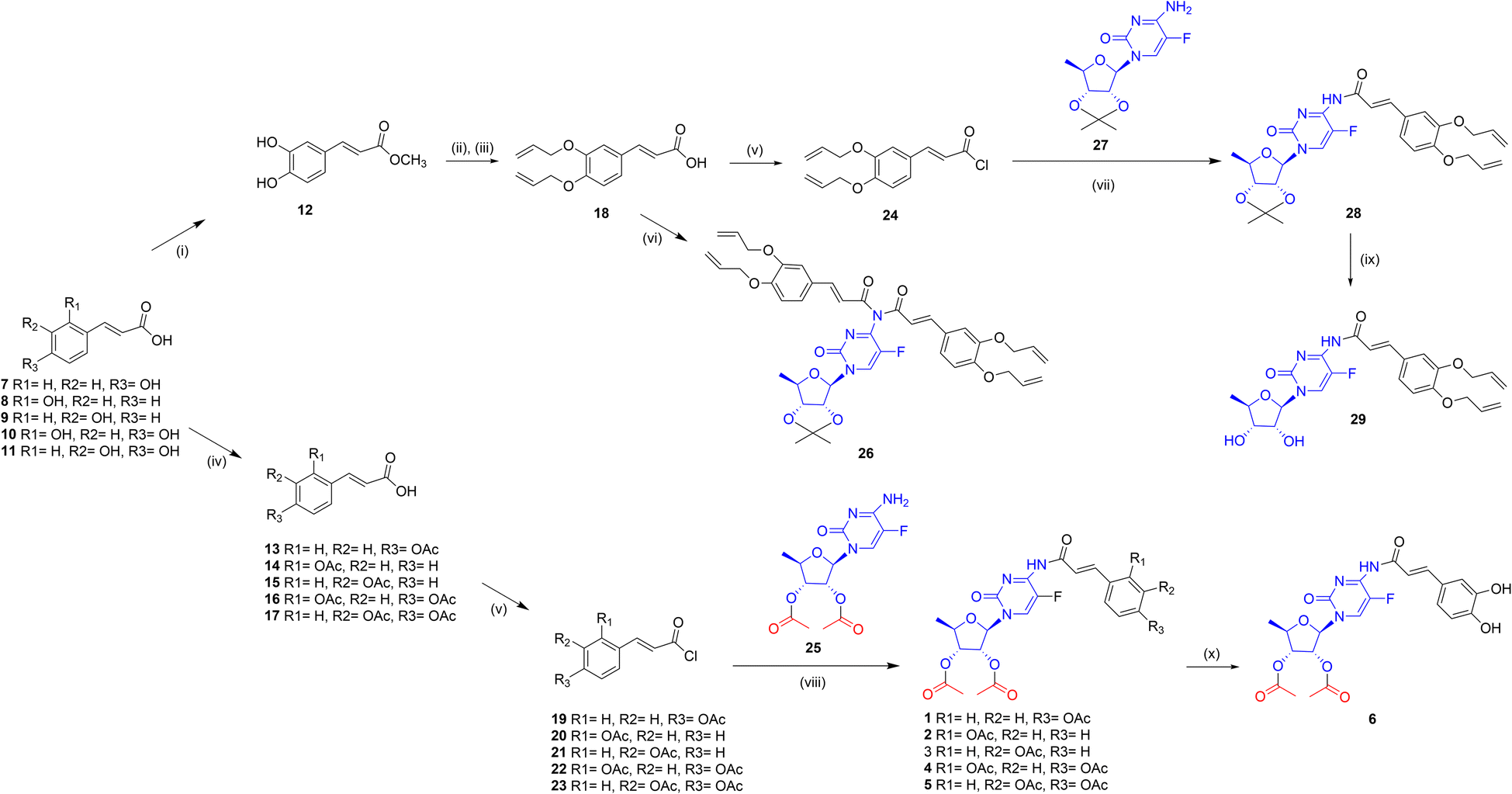 | ||
| Fig. 3 Synthesis of 1–6. Reagents and reaction conditions: (i) CH3OH, CH3COCl, r.t.; (ii) allyl bromide, K2CO3, acetone, r.t.; (iii) NaOH, H2O/CH3OH; (iv) pyridine, DMAP, (CH3CO)2O; (v) SOCl2, DCM; (vi) POCl3, pyridine; (vii) 50% NaOH aq./DCM; (viii) pyridine, DCM, −20 to −5 °C then r.t; (ix) 5 M HCl/MeOH; (x) K2CO3/chloroform. | ||
We also investigated other conditions for removing the isopropylidene group, such as THF/1 M HCl or indium chloride in methanol. Unfortunately, these conditions resulted in substrate degradation without the expected product. Next, we attempted to remove the allyl groups from 29 using either the Pd(PPh3)4/MeOH/K2CO3 or [Pd(P(C6H5)3)4]/DCM system. However, the main product obtained was 5′-deoxy-5-fluorocytidine as a result of amide bond cleavage. Taking into consideration these results, it was decided to obtain the new peracetylated conjugates 1–6 as CES2 prodrugs. For this reason, the following substrates were synthesized: 5′-deoxy-2′,3′-di-O-acetyl-5-fluorocytidine (25) and O-acyl protected hydroxycinnamic acids 13–17, all with yields greater than 90%. The coupling of 25 with 13–17 carried out in methylene chloride/50% NaOH aq., which was effective for 28 formations, did not yield products 1–5 and caused degradation of 13–17. Finally, compounds 1–5 were obtained according to the procedure Shimma et al.,61 by using methylene chloride with pyridine. Compound 5 was chosen as the model substrate for hydrolysis studies to remove acetyl groups. Various hydrolysis conditions were tested, including K2CO3 in methanol or 25% NH3 aq. in methanol. The reaction progressed quickly, and several compounds were observed on TLC with no expected product or its traces. Only the reaction in chloroform and the presence of anhydrous K2CO3 produced the hydrolysis product. After purification on preparative TLC, we obtained conjugate 6 with a total yield of 42%. NMR analysis confirmed the structure of 6 and showed the absence of acetyl groups in the caffeic acid fragment of the conjugate. Compound 6, which possesses two unbound hydroxyl groups, was tested to compare its in vitro activity with that of its peracetylated analogue 5, and to determine the effect of the presence of CES2-sensitive ester groups on anti-PDAC activity.
In vitro anticancer activity against AsPC-1 and BxPC-3 pancreatic cancer lines and toxicity effect on normal human NHDF cells
The cytotoxic activity of conjugates 1–6 and the reference capecitabine (Cape) was tested using the MTT method adapted from Mosmann.62 The compounds were dissolved in DMSO, diluted in culture medium to appropriate concentrations, and applied to metastatic AsPC-1 and primary BxPC-3 pancreatic cancer cells. After 72 hours of incubation, compounds showed similar moderate activity against AsPC-1 cancer cells in the IC50 range of 37–133 μM (Table 1, Fig. 4). Compound 6, with unprotected hydroxyl groups in the caffeic acid fragment, showed the lowest activity towards AsPC-1 cells. All tested conjugates 1–6 exhibited significantly better activity against primary BxPC-3 pancreatic cancer cells compared to the metastatic line, with IC50 values ranging from 14–45 μM. Compound 4, with a (2,4-diacetyloxy)cinnamoyl substituent, showed the lowest activity toward BxPC-3 cells, while compound 1, with a para-(acetyloxy)coumaroyl substituent, showed the highest activity. It is worth emphasizing that peracetylated conjugate 5 and its simple analogue 6, with unprotected hydroxyl groups in the caffeic acid part, exhibited similar activity against BxPC-3 cells (IC50 = 31.6 μM and 23.1 μM, respectively) and AsPC-1 cells (IC50 = 90.1 μM and 132.6 μM, respectively). However, peracetylated 5 was slightly more potent against AsPC-1 than semiacetylated conjugate 6. The initial phase of in vitro research involves testing the compound on normal cell lines to confirm lower toxicity to healthy cells than to cancer cells, which is essential for identifying a potential clinical therapeutic window.63 Therefore, we used normal human NHDF fibroblasts as model cells to evaluate the in vitro toxicity of conjugates 1–6. All tested conjugates exhibited low selectivity towards the metastatic cell line, with a selectivity index (SI) between 1–2. However, the compounds acted more selectively in the case of the primary cell line BxPC-3. The caffeic acid derivative 6 and the para-coumaric acid derivative 1 showed the highest selectivity with an SI of approximately 7. The tests conducted on the PDAC lines did not reveal any significant differences in the activity of compounds 5 and 6. This suggests that both compounds may undergo enzymatic hydrolysis to form the same active substance.| Compound | IC50 (μM) | IC50 (μM) | SI | ||
|---|---|---|---|---|---|
| AsPC-1 | BxPC-3 | NHDF | AsPC-1 | BxPC-3 | |
| 1 | 57.15 ± 2.06 | 13.60 ± 2.48 | 96.49 ± 5.79 | 1.69 | 7.10 |
| 2 | 69.26 ± 4.00 | 26.93 ± 1.64 | 98.54 ± 10.46 | 1.42 | 3.66 |
| 3 | 37.00 ± 3.20 | 20.18 ± 2.43 | 60.04 ± 3.17 | 1.62 | 2.98 |
| 4 | 81.47 ± 3.60 | 45.09 ± 4.20 | 116.17 ± 2.71 | 1.43 | 2.58 |
| 5 | 90.07 ± 4.53 | 31.59 ± 0.82 | 121.30 ± 4.32 | 1.35 | 3.83 |
| 6 | 132.60 ± 5.67 | 23.16 ± 1.25 | 163.40 ± 4.33 | 1.23 | 7.06 |
| Capecitabine (Cape) | >200 | >200 | >200 | — | — |
| 5-Fluorouracil (5-FU) | 52.39 ± 11.56 | 12.74 ± 2.73 | >200 | — | — |
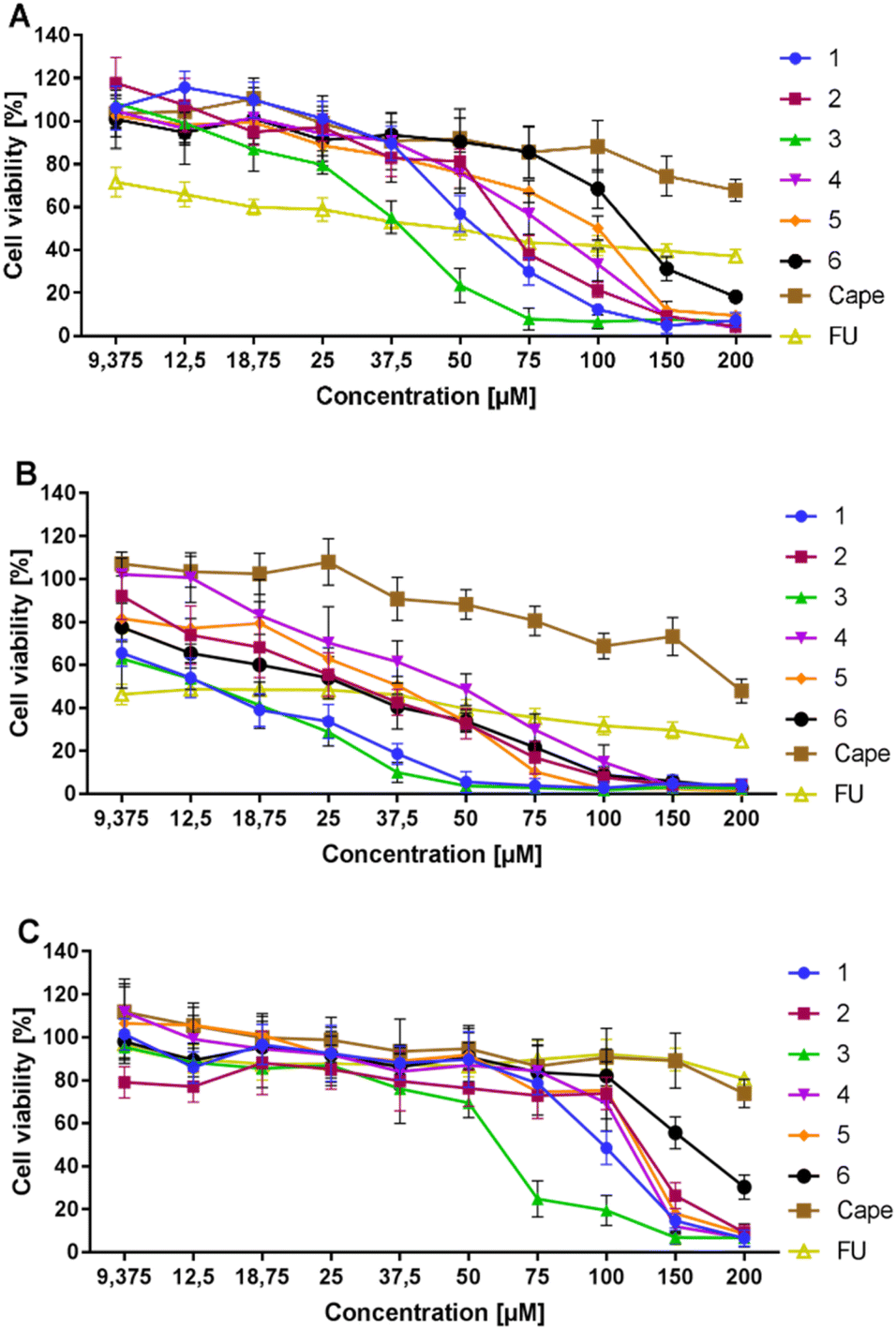 | ||
| Fig. 4 Cytotoxic effects of 1–6 and capecitabine on AsPC-1 (A), BxPC-3 cells (B), and NHDF (C) expressed as % of viability compared to the untreated control. | ||
Computational studies
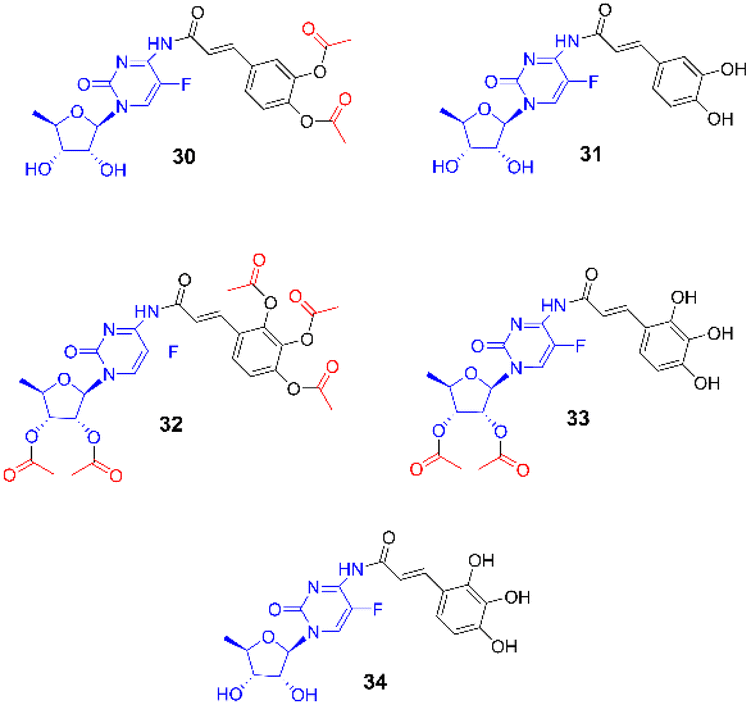 | ||
| Fig. 5 Hypothetical structures of conjugates 30–34 used in computational studies. | ||
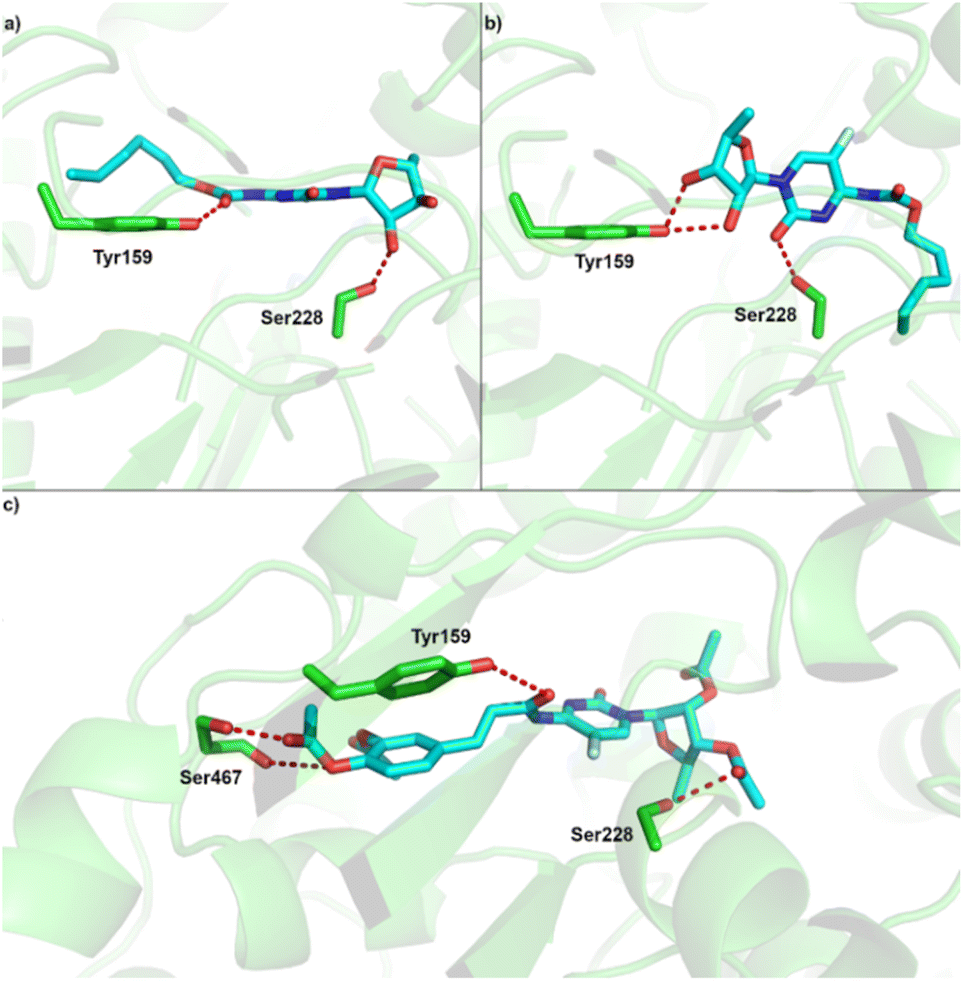 | ||
| Fig. 6 Predicted binding site of CES2 and binding pose of (a) capecitabine (pose 1) (b) capecitabine (pose 2); (c) compound 5. | ||
| Compound | Site 1 | |
|---|---|---|
| ΔGa (kcal mol−1) | Kib (μm) | |
| a ΔG – Gibbs free energy of binding (in kcal mol−1).b Ki – inhibition constant at 298 K (μm). | ||
| 1 | −12.1 | 0.001 |
| 2 | −8.8 | 0.334 |
| 3 | −12.1 | 0.001 |
| 4 | −9.6 | 0.100 |
| 5 | −11.7 | 0.002 |
| 6 | −10.3 | 0.030 |
| 30 | −10.8 | 0.013 |
| 31 | −7.4 | 4.1 |
| 32 | −5.2 | 143.1 |
| 33 | −9.4 | 0.135 |
| 34 | −6.7 | 13.1 |
| 5-dFCR | −7.5 | 5.0 |
| Capecitabine | −7.6 | 4.0 |
For albumin, we performed molecular docking of all studied compounds to the two potential binding sites, as predicted in previous work.68 In all cases except 5-dFCR this computational approach predicts more favourable binding to site 1, which is also the native binding site of warfarin in the crystal structure of the warfarin-albumin complex.69 Also, our computational protocol predicts that all compounds are bound substantially stronger to albumin than warfarin. The lowest inhibition constant value among all studied systems was predicted for compound 3 with low nanomolar value (Fig. 7, Table 3).
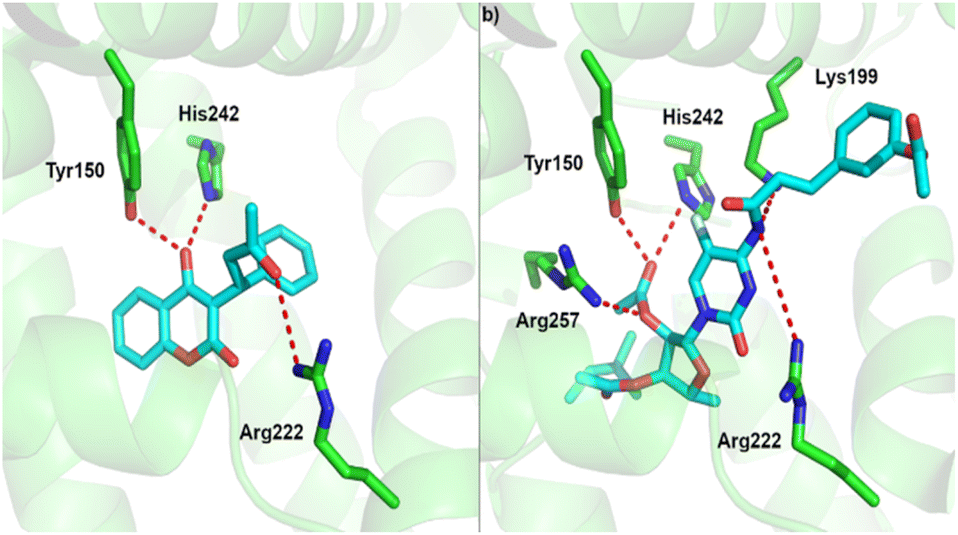 | ||
| Fig. 7 Binding site of albumin and (a) experimental binding pose of warfarin and, (b) predicted binding pose of compound 3. | ||
| Compound | Site 1 | Site 2 | ||
|---|---|---|---|---|
| ΔGa (kcal mol−1) | Kib (μm) | ΔG (kcal mol−1) | Kib (μm) | |
| a ΔG – Gibbs free energy of binding (in kcal mol−1).b Ki – inhibition constant at 298 K (μm). | ||||
| 1 | −10.6 | 0.017 | −9.5 | 0.107 |
| 2 | −10.5 | 0.020 | −7.9 | 1.601 |
| 3 | −11.1 | 0.007 | −7.6 | 2.658 |
| 4 | −10.3 | 0.028 | −8.9 | 0.296 |
| 5 | −9.8 | 0.065 | −9.3 | 0.150 |
| 6 | −10.3 | 0.028 | −8.0 | 1.353 |
| 30 | −10.3 | 0.028 | −8.0 | 1.353 |
| 31 | −9.6 | 0.091 | −7.8 | 1.896 |
| 32 | −10.1 | 0.039 | −7.6 | 2.658 |
| 33 | −9.7 | 0.077 | −8.5 | 0.581 |
| 34 | −9.6 | 0.091 | −7.7 | 2.245 |
| 5-dFCR | −7.2 | 5.224 | −8.0 | 1.353 |
| Capecitabine | −8.5 | 0.581 | −8.0 | 1.353 |
| Warfarin | −7.8 | 1.995 | — | — |
Conclusion
The new amide conjugates 1–6 of natural hydroxycinnamic acid (ortho-, meta-, para-coumaric, 2,4-dihydroxycinnamic, caffeic) and 5′-deoxy-5-fluorocytidine, the intermediate metabolite of oral capecitabine and prodrug of 5-fluorouracyl (5-FU), were obtained by evaluating different synthetic strategies. The most efficient method leading to peracetylated conjugates was the simple direct N-acylation of acetyl-protected 5′-deoxy-5-fluorocytidine with acetylated HCA acid chlorides. In in vitro studies, compounds 1–6 showed slightly higher efficacy against primary BxPC-3 than metastatic AsPC-1 pancreatic cancer cells with IC50 values ranging from 14–45 μM and 37–133 μM, respectively. They were also more selective in the case of the primary line, with the highest selectivity against normal dermal fibroblasts calculated to be about 7 for the caffeic acid derivative 6 and the para-coumaric acid derivative 1. Considering that the compounds can act as substrates of CES2 carboxylesterase, we compared the in vitro activity of two compounds differing in the number of acetyl groups in the molecule, i.e. 5 and 6. As a result, both showed similar activity against BxPC-3 cells with IC50 of 32 μM and 23 μM, and against AsPC-1 cells with IC50 of 90 μM and 133 μM, which may indicate the enzymatic hydrolysis of both compounds to the same active compound. Although the conjugates violated two of Lipinski's rules, related to molecular mass and number of hydrogen bonds, there are many examples of drugs or drug candidates that violate these rules to a similar extent. The toxicity predictions using ProTox-II showed high estimated LD50 values and no potential toxicity issues, similarly to capecitabine. Molecular docking to CES2 shows two potential binding modes with the critical Ser228. Compounds 1–6 were predicted to bind rather similarly to the binding mode of capecitabine and close to Ser228 from the catalytic triad, that catalysed the hydrolysis of ester groups. The strongest binding based on hydrogen bonding with acetyl group of sugar moieties was predicted for compounds 1, 3 with Ki values of 0.001 μm and 5 with Ki value of 0.002 μm. Our computational protocol also estimated that all compounds bound significantly stronger to human serum albumin than warfarin, the most widely used oral anticoagulant. In summary, based on in vitro and in silico studies, compounds 1–6 could be viewed as potential lead structures for further modifications aimed at enhancing 5-FU activity against pancreatic cancer.Experimental
For monitoring of reactions Merck DC-Alufolien Kieselgel 60 F25 TLC plates were used. Column chromatography was performed on Merck silica gel 60 (230–400 mesh). Melting points were measured on a Mettler Toledo MP90 apparatus and were uncorrected. NMR spectra were recorded on a Bruker AVANCE III HD 500 MHz spectrometer (at 298 K) in CDCl3 and/or DMSO-d6 using TMS as an internal standard. High-resolution mass spectrometry (HRMS) measurements were performed using Synapt G2-Si mass spectrometer (Waters) equipped with an ESI source and quadrupole-Time-of-Flight mass analyzer. The mass spectrometer operated in positive or negative ion detection modes. The measurement was performed with a capillary voltage set to 2.7 kV and a sampling cone set to 20 V. The source temperature was 110 °C. To ensure accurate mass measurements, the data was collected in centroid mode, and the mass was corrected during acquisition using leucine encephalin solution as an external reference, Lock-SprayTM, (Waters Corp., Milford, MA, USA), which generated a reference ion at m/z 554.2615 ([M − H]−) in negative ESI mode and at m/z 556.2771 Da ([M + H]+) in positive ESI mode. The results of the measurements were processed using the MassLynx 4.1 software (Waters).MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) were purchased from Sigma-Aldrich (Poznan, Poland) and DMSO from Archem (Kamieniec Wroclawski, Poland). Cell culture media (RPMI-1640, alpha-MEM and DMEM), stable glutamine 100×, trypsin–EDTA, heat inactivated fetal bovine serum premium (FBS) and antibiotic–antimycotic 100× were purchased from BioWest BioWest by CytoGen (Zgierz, Poland). The normal human dermal fibroblast cell line (NHDF) was purchased from Lonza (Lonza, Warsaw, Poland), and the BxPC-3 cell line was purchased from the American Tissue Culture Collection (ATCC, Manassas, VA, USA). The AsPC lines was kindly provided by the Institute of Immunology and Experimental Therapy (Wroclaw, Poland). The trans-hydroxycinnamic acids (HCA), 5′-deoxy-5-fluoro-2′,3′-O-isopropylidenocytidine (27, CAS 66335-37-3), 5′-deoxy-5-fluoro-2′,3′-di-O-acetylcytidine (25, CAS 161599-46-8), capecitabine (154361-50-9), chemical reagents and solvents were purchased from chemical suppliers. Acetyl protected HCA and methyl HCA esters were synthesized according to the common respective procedures.48,70 Acetyl protected HCA were converted to the corresponding acid chlorides with an excess of thionyl chloride.
General procedure of synthesis of compounds 1–5
5′-Deoxy-5-fluoro-2′,3′-di-O-acetylcytidine (1 eq.) was dissolved in methylene chloride (4 mL) and dry pyridine (2 eq.) and cooled to −20 °C. To the stirred solution the respective hydroxycinnamoyl chloride (19–23) was added portionwise over a period of 15 min. with the temperature kept from −10 °C to −20 °C. The reaction was stirred 1 hour at −20 °C, then it was allowed to reach ambient temperature in 1 hour. After extraction in water/methylene chloride system, the separated organic layer was dried over anhydrous MgSO4, and the solvent was distilled off under reduced pressure. Then crude product was purified using flash chromatography on silica gel.2′,3′-Di-O-acetyl-5′-deoxy-5-fluoro-N4-4-acetyloxycinnamoyl-cytidine (1)
Compound 1 was obtained as a white solid from 4-acetyloxycinnamoyl chloride 19 (0.17 g, 0.76 mM) and 5′-deoxy-5-fluoro-2′,3′-di-O-acetylcytidine (25) (0.25 g, 0.76 mM). The crude product (0.33 g) was purified by chromatography on a silica gel column with chloroform/methanol 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, followed by hexanes:ethyl acetate 1:2. Yield 0.11 g (28%); m.p. 79 °C (dec.).
:1, followed by hexanes:ethyl acetate 1:2. Yield 0.11 g (28%); m.p. 79 °C (dec.).
1H NMR (500 MHz, DMSO-d6) δ 11.96 (d, 1H, J = 4.9 Hz, H-7, NH), 8.17 (d, 1H, JHF = 6.9 Hz, H-6, double bond in flucytosine ring), 7.59 (d, 2H, J = 8.6 Hz, H-12, H-16, aromatic CA), 7.41 (d, 1H, J = 15.9 Hz, H-10, double bond CA), 7.16 (d, 2H, J = 8.6 Hz, H-13, H-15, aromatic CA), 6.57 (d, 1H, J = 15.9 Hz, H-9, double bond CA), 5.80 (d, 1H, J = 4.9 Hz, H-1′, CHN deoxyribose), 5.44 (dd, 1H, J1 = 5.4 Hz, J2 = 6.3 Hz, H-2′, deoxyribose), 5.08 (dd, 1H, J1 = J2 = 6.1 Hz, H-3′, deoxyribose), 4.07 (dq, 1H, J1 = J2 = 6.1 Hz, H-4′, deoxyribose), 2.27 (s, 3H, H-18, CH3CO CA), 2.06 (s, 3H, H-9′, deoxyribose COCH3), 2.04 (s, 3H, H-7′, CH3CO deoxyribose), 1.34 (d, 3H, J = 6.3 Hz, H-5′, deoxyribose CH3); 13C NMR (125 MHz, DMSO-d6) δ 169.5 (C-8′, deoxyribose C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 169.4 (C-6′, deoxyribose CO), 169.1 (C-17, CO CA), 166.6 (C-8, CO CA), 157.0 (d, J2CF = 26.2 Hz, C-4, flucytosine ring), 151.2 (C-14, aromatic CA), 149.0 (C-2, flucytosine ring CO), 140.2 (d, JCF = 231.6 Hz, C-5, flucytosine ring), 138.2 (C-10, double bond CA), 132.6 (C-11, aromatic CA), 128.7 (C-12, aromatic CA), 126.0 (d, J = 34.5 Hz, C-6, double bond), 122.4 (C-9, double bond CA), 122.4 (C-15, aromatic CA), 87.6 (C-1′, deoxyribose), 77.2 (C-4′, deoxyribose), 73.2 (C-3′, deoxyribose), 71.9 (C-2′, deoxyribose), 20.9 (C-18, CH3CO CA), 20.3 (C-9′, deoxyribose COCH3), 20.2 (C-7′, deoxyribose COCH3), 17.9 (C-5′, deoxyribose CH3); HRMS (ESI, m/z): calculated for C24H25N3O9F [M + H]+ 518.1575; found 518.1577, calculated for C24H24N3O9FNa [M + Na]+ 540.1394; found 540.1400.
O), 169.4 (C-6′, deoxyribose CO), 169.1 (C-17, CO CA), 166.6 (C-8, CO CA), 157.0 (d, J2CF = 26.2 Hz, C-4, flucytosine ring), 151.2 (C-14, aromatic CA), 149.0 (C-2, flucytosine ring CO), 140.2 (d, JCF = 231.6 Hz, C-5, flucytosine ring), 138.2 (C-10, double bond CA), 132.6 (C-11, aromatic CA), 128.7 (C-12, aromatic CA), 126.0 (d, J = 34.5 Hz, C-6, double bond), 122.4 (C-9, double bond CA), 122.4 (C-15, aromatic CA), 87.6 (C-1′, deoxyribose), 77.2 (C-4′, deoxyribose), 73.2 (C-3′, deoxyribose), 71.9 (C-2′, deoxyribose), 20.9 (C-18, CH3CO CA), 20.3 (C-9′, deoxyribose COCH3), 20.2 (C-7′, deoxyribose COCH3), 17.9 (C-5′, deoxyribose CH3); HRMS (ESI, m/z): calculated for C24H25N3O9F [M + H]+ 518.1575; found 518.1577, calculated for C24H24N3O9FNa [M + Na]+ 540.1394; found 540.1400.
2′,3′-Di-O-acetyl-5′-deoxy-5-fluoro-N4-2-acetyloxycinnamoyl-cytidine (2)
Compound 2 was obtained as a white solid from 2-acetyloxycinnamoyl chloride 20 (0.17 g, 0.76 mM) and 5′-deoxy-5-fluoro-2′,3′-di-O-acetylcytidine (25) (0.25 g, 0.76 mM). The crude product (0.43 g) was purified by chromatography on a silica gel column with hexanes:ethyl acetate 1:2. Yield 0.17 g (42%); m.p. 74 °C (dec.).
1H NMR (500 MHz, DMSO-d6) δ 11.01 (bs, 1H, H-7, NH), 8.40 (bs, 1H, H-6, double bond in flucytosine ring), 7.75 (d, 1H, J = 6.9 Hz, H-16, aromatic CA), 7.61 (d, 1H, J = 15.9 Hz, H-10, double bond), 7.49 (m, 1H, H-14, aromatic CA), 7.36 (dd, 1H, J1 = J2 = 7.5 Hz, H-15, aromatic CA), 7.24 (d, 1H, J = 8.0 Hz, H-13, aromatic CA), 7.14 (d, 1H, J = 15.4 Hz, H-9, double bond), 5.83 (d, 1H, J = 4,1 Hz, H-1′, CHN deoxyribose), 5.49 (dd, 1H, J1 = 4.2 Hz, J2 = 6.2 Hz, H-2′, deoxyribose), 5.13 (dd, 1H, J1 = J2 = 6.4 Hz, H-3′, deoxyribose), 4.14 (dq, 1H, J1 = J2 = 6.4 Hz, H-4′, deoxyribose), 2.37 (s, 3H, H-18, CH3CO CA), 2.06 (s, 3H, H-9′, deoxyribose COCH3), 2.06 (s, 3H, H-7′, deoxyribose COCH3), 1.37 (d, 3H, J = 6,3 Hz, H-5′, deoxyribose CH3); 13C NMR (125 MHz, DMSO-d6) δ 169.5 (C-8′, deoxyribose CO), 169.3 (C-6′, deoxyribose CO), 169.2 (C-17, CO CA), 163.8 (C-8, CO CA), 154.5 (C-4, flucytosine ring), 152.3 (C-2, flucytosine ring CO), 149.2 (C-12, aromatic CA), 137.5 (d, C-5, flucytosine ring), 136.4 (C-10, double bond CA), 131.6 (C-6, flucytosine ring), 131.4 (C-14, aromatic CA), 128.2 (C-16, aromatic CA), 126.8 (C-11, aromatic CA), 126.6 (C-15, aromatic CA), 123.6 (C-13, aromatic CA), 122.9 (C-9, double bond CA), 90.00 (C-1′, deoxyribose), 77.4 (C-4′, deoxyribose), 73.4 (C-3′, deoxyribose), 72.7 (C-2′, deoxyribose), 20.7 (C-18, CH3CO CA), 20.3 (C-9′, deoxyribose COCH3), 20.3 (C-7′, deoxyribose COCH3), 17.7 (C-5′, deoxyribose CH3); HRMS (ESI, m/z): calculated for C24H25N3O9F [M + H]+ 518.1575; found 518.1580, calculated for C24H24N3O9FNa [M + Na]+ 540.1394; found 540.1398.
2′,3′-Di-O-acetyl-5′-deoxy-5-fluoro-N4-3-acetyloxycinnamoyl-cytidine (3)
Compound 3 was obtained as a white solid from 3-acetyloxycinnamoyl chloride 21 (0.17 g, 0.76 mM) and 5′-deoxy-5-fluoro-2′,3′-di-O-acetylcytidine (25) (0.25 g, 0.76 mM). The crude product (0.33 g) was purified by chromatography on a silica gel column with chloroform/methanol 50:1, followed by hexanes:ethyl acetate 1:2. Yield 0.16 g (40%); m.p. 75 °C (dec.).
1H NMR (500 MHz, DMSO-d6) δ 11.96 (d, 1H, J = 4,9 Hz, H-7, NH), 8.17 (d, 1H, J = 6.9 Hz, H-6, double bond in flucytosine ring), 7.44 (m, 1H, H-15, aromatic CA), 7.44 (m, 1H, H-12, aromatic CA), 7.39 (d, 1H, J = 15.9 Hz, H-10, double bond CA), 7.32 (m, 1H, H-16, aromatic CA), 7.13 (m, 1H, H-14, aromatic CA), 6.61 (d, 1H, J = 15.9 Hz, H-9, double bond CA), 5.80 (d, 1H, J = 5.1 Hz, H-1′, CHN deoxyribose), 5.44 (dd, 1H, J1 = 5.6 Hz, J2 = 6.1 Hz, H-2′, deoxyribose), 5.08 (dd, 1H, J1 = J2 = 6.1 Hz, H-3′, deoxyribose), 4.07 (dq, 1H, J1 = J2 = 6.1 Hz, H-4′, deoxyribose), 2.27 (s, 3H, H-18, CH3CO CA), 2.06 (s, 3H, H-9′, deoxyribose COCH3), 2.04 (s, 3H, H-7′, deoxyribose COCH3), 1.35 (d, 1H, J = 6.3 Hz, H-5′, deoxyribose CH3); 13C NMR (125 MHz, DMSO-d6) δ 169.5 (C-8′, deoxyribose CO), 169.4 (C-6′, deoxyribose CO), 169.2 (C-17, CO CA), 166.4 (C-8, CO CA), 157.0 (d, J = 26.0 Hz, C-4, flucytosine ring), 150.9 (C-13, aromatic CA), 149.0 (C-2, flucytosine ring CO), 140.2 (d, J = 231.6 Hz, C-5, flucytosine ring), 138.1 (C-10, double bond CA), 136.5 (C-11, aromatic CA), 130.0 (C-15, aromatic CA), 126.0 (d, J = 34.4 Hz, C-6, flucytosine ring), 125.1 (C-12, aromatic CA), 123.4 (C-9, double bond CA), 122.8 (C-14, aromatic CA), 120.5 (C-16, aromatic CA), 87.6 (C-1′, deoxyribose), 77.2 (C-4′, deoxyribose), 73.2 (C-3′, deoxyribose), 71.9 (C-2′, deoxyribose), 20.8 (C-18, CH3CO CA), 20.3 (C-9′, deoxyribose COCH3), 20.2 (C-7′, deoxyribose COCH3), 17.9 (C-5′, deoxyribose CH3); HRMS (ESI, m/z): calculated for C24H25N3O9F [M + H]+ 518.1575; found 518.1579, calculated for C24H24N3O9FNa [M + Na]+ 540.1394; found 540.1398.
2′,3′-Di-O-acetyl-5′-deoxy-5-fluoro-N4-2,4-acetyloxycinnamoyl-cytidine (4)
Compound 4 was obtained as a white solid from 2,4-diacetyloxycinnamoyl chloride 22 (0.13 g, 0.46 mM) and 5′-deoxy-5-fluoro-2′,3′-di-O-acetylcytidine (25) (0.15 g, 0.46 mM). The crude product (0.21 g) was purified by chromatography on a silica gel column with hexanes:ethyl acetate:methanol 5:5:1. Yield 0.1 g (38%); m.p. 96 °C (dec.).
1H NMR (500 MHz, DMSO-d6) δ 11.04 (bs, 1H, H-7, NH), 8.39 (bs, 1H, H-6, double bond in flucytosine ring), 7.79 (d, 1H, J = 7.6 Hz, H-16, aromatic CA), 7.59 (d, 1H, J = 15.9 Hz, H-10, double bond CA), 7.17 (m, 1H, H-15, aromatic CA), 7.14 (m, 1H, H-13, aromatic CA), 7.11 (m, 1H, H-9, double bond CA), 5.83 (d, 1H, J = 4.0 Hz, H-1′, CHN deoxyribose), 5.49 (dd, 1H, J1 = 4.5 Hz, J2 = 6.1 Hz, H-2′, deoxyribose), 5.13 (dd, 1H, J1 = J2 = 6.4 Hz, H-3′, deoxyribose), 4.13 (dq, 1H, J1 = J2 = 6.3 Hz, H-4′, deoxyribose), 2.37 (s, 3H, H-18, CH3CO CA), 2.28 (s, 3H, H-20, CH3CO CA), 2.06 (s, 3H, H-9′, deoxyribose COCH3), 2.06 (s, 3H, H-7′, deoxyribose COCH3), 1.37 (d, 1H, J = 6.3 Hz, H-5′, deoxyribose CH3); 13C NMR (125 MHz, DMSO-d6) δ 169.5 (C-8′, deoxyribose CO), 169.3 (C-6′, deoxyribose CO), 168.9 (C-17, CO CA), 168.8 (C-19, CO CA), 163.9 (C-8, CO CA), 154.5 (C-4, flucytosine ring), 152.2 (C-2, flucytosine ring CO), 152.1 (C-14, aromatic CA), 149.6 (C-12, aromatic CA), 137.5 (d, C-5, flucytosine ring), 135.7 (C-10, double bond CA), 131.4 (C-6, flucytosine ring), 129.00 (C-16, aromatic CA), 124.5 (C-11, aromatic CA), 123.0 (C-9, double bond CA), 120.3 (C-15, aromatic CA), 117.4 (C-13, aromatic CA), 89.9 (C-1′, deoxyribose), 77.4 (C-4′, deoxyribose), 73.4 (C-3′, deoxyribose), 72.7 (C-2′, deoxyribose), 20.8 (C-20, CH3CO CA), 20.7 (C-18, CH3CO CA), 20.3 (C-9′, deoxyribose COCH3), 20.3 (C-7′, deoxyribose COCH3), 17.7 (C-5′, deoxyribose CH3); HRMS (ESI, m/z): calculated for C26H27N3O11F [M + H]+ 576.1630; found 576.1631, calculated for C26H26N3O11FNa [M + Na]+ 598.1449; found 598.1451.
2′,3′-Di-O-acetyl-5′-deoxy-5-fluoro-N4-3,4-diacetyloxycinnamoyl-cytidine (5)
Compound 5 was obtained as a white solid from 3,4-diacetyloxycinnamoyl chloride 23 (0.86 g, 3.04 mM) and 5′-deoxy-5-fluoro-2′,3′-di-O-acetylcytidine (25) (1.00 g, 3.04 mM). The crude product (1.69 g) was purified by chromatography on a silica gel column with chloroform/methanol 50:1, followed by hexanes:ethyl acetate:methanol 5:5:1. Yield 0.85 g (49%); m.p. 88 °C (dec.).
1H NMR (500 MHz, CDCl3) δ 7.84 (d, 1H, J = 15.7 Hz, H-10, double bond), 7.53 (s, 1H, H-7, NH), 7.49 (d, 1H, J = 8.5 Hz, H-16, aromatic CA), 7.42 (d, 1H, J = 1.8 Hz, H-12, aromatic CA), 7.23 (d, 1H, J = 8.5 Hz, H-15, aromatic CA), 5.98 (d, 1H, J = 4.6 Hz, H-1′, CHN deoxyribose), 5.32 (dd, J1 = J2 = 5.1 Hz, H-2′, deoxyribose), 5.01 (dd, J1 = J2 = 5.7 Hz, H-3′, deoxyribose), 4.27 (dq, 1H, J1 = J2 = 6.1 Hz, H-4′, deoxyribose), 2.30 (s, 3H, H-20, CH3CO CA), 2.29 (s, 3H, H-18, CH3CO CA), 2.11 (s, 3H, H-7′, deoxyribose COCH3), 2.10 (s, 3H, H-9′, deoxyribose COCH3), 1.48 (d, 3H, J = 6.4 Hz, H-5′, deoxyribose CH3); 13C NMR (125 MHz, CDCl3) δ 169.7 (C-8′, CO deoxyribose), 169.6 (C-6′, CO deoxyribose), 168.0 (C-17, CO CA), 167.9 (C-19, CO deoxyribose), 152.4 (C-2, CO flucytosine ring), 144.0 (C-10, double bond), 143.6 (C-14, aromatic CA), 142.4 (C-13, aromatic CA), 133.5 (C-11, aromatic CA), 126.8 (C-16, aromatic CA), 123.9 (C-15, aromatic CA), 123.3 (C-12, aromatic CA), 88.4 (C-1′, deoxyribose CH), 78.2 (C-4′, deoxyribose), 73.9 (C-3′, deoxyribose), 73.3 (C-2′, deoxyribose), 20.6 (C-18, CH3CO CA), 20.6 (C-20, CH3CO CA), 20.5 (C-9′, deoxyribose COCH3), 20.4 (C-7′, deoxyribose COCH3), 18.7 (C-5′, deoxyribose CH3).
1H NMR (500 MHz, DMSO-d6) δ 11.96 (d, 1H, J = 5.0 Hz, H-7, NH), 8.17 (d, 1H, J = 6.9 Hz, H-6, double bond in flucytosine ring), 7.49 (m, 1H, H-16, aromatic CA), 7.47 (m, 1H, H-12, aromatic CA), 7.39 (d, 1H, J = 15.9 Hz, H-10, double bond CA), 7.30 (d, 1H, J = 8.3 Hz, H-15, aromatic CA), 6.68 (d, 1H, J = 15.9 Hz, H-9, double bond CA), 5.80 (d, 1H, J = 5,2 Hz, H-1′, CHN deoxyribose), 5.43 (dd, 1H, J1 = 5.3 Hz, J2 = 6.3 Hz, H-2′, deoxyribose), 5.08 (dd, 1H, J1 = J2 = 6.1 Hz, H-3′, deoxyribose), 4.07 (dq, 1H, J1 = J2 = 6.1 Hz, H-4′, deoxyribose), 2.28 (s, 6H, H-18, H-20, CH3CO CA), 2.06 (s, 3H, H-9′, deoxyribose COCH3), 2.04 (s, 3H, H-7′, deoxyribose COCH3), 1.34 (d, 3H, J = 6.3 Hz, 3H, H-5′, deoxyribose CH3); 13C NMR (125 MHz, DMSO-d6) δ 169.5 (C-8′, deoxyribose CO), 169.4 (C-6′, deoxyribose CO), 168.2 (C-19, CO CA), 168.2 (C-17, CO CA), 166.35 (C-8, CO CA), 157.0 (d, J = 26.1 Hz, C-4, flucytosine ring), 149.0 (C-2, flucytosine ring CO), 142.7 (C-14, aromatic CA), 142.3 (C-13, aromatic CA), 140.2 (d, JCF = 231.6 Hz, C-5, flucytosine ring), 137.5 (C-10, double bond CA), 133.8 (C-11, aromatic CA), 126.0 (d, J = 33.7 Hz, C-6, flucytosine ring), 124.1 (C-15, aromatic CA), 123.4 (C-9, double bond CA), 122.3 (C-12, aromatic CA), 87.6 (C-1′, deoxyribose), 77.2 (C-4′, deoxyribose), 73.2 (C-3′, deoxyribose), 71.9 (C-2′, deoxyribose) 20.3 (C-18, CH3CO CA), 20.3 (C-20, CH3CO CA), 20.3 (C-9′, deoxyribose COCH3), 20.2 (C-7′, deoxyribose COCH3), 17.9 (C-5′, deoxyribose CH3); HRMS (ESI, m/z): calculated for C26H27N3O11F [M + H]+ 576.1630; found 576.1633, calculated for C26H26N3O11FNa [M + Na]+ 598.1449; found 598.1448.
2′,3′-Di-O-acetyl-5′-deoxy-5-fluoro-N4-3,4-dihydroxycinnamoyl-cytidine (6)
To 2′,3′-di-O-acetyl-5′-deoxy-5-fluoro-N4-3,4-diacetyloxycinnamoyl-cytidine 5 (0.15 g; 0.261 mmol) in methylene chloride (6.5 mL), anhydrous potassium carbonate (0.269 g; 0.339 mmol) was added. The reaction mixture was stirred overnight and then filtered. The precipitate was washed with methylene chloride, then ethyl acetate. Next it was dissolved in water, acidified to pH = 5 using 0.5 M HCl aq. and extracted with ethyl acetate (3 × 20 mL). The combined organic fractions were dried over anhydrous MgSO4, and the solvent was distilled off under reduced pressure The crude product (0.113 mg) was purified using flash column chromatography on a silica gel with chloroform: methanol 95:5 to 9:1, then on a preparative TLC plate/SiO2/1 mm with chloroform:methanol 95:5. After evaporation to oil, the compound 6 was precipitated off using ethyl acetate/n-hexane mixture to give 6 as a yellow solid. Yield 0.054 g (42%); m.p. 116 °C (dec.); 1H NMR (500 MHz, DMSO-d6) δ 10.73 (s, 1H, H-7, NH), 9.59 (s, 1H, H-17, phenolic OH), 9.25 (s, 1H, H-18, phenolic OH), 8.37 (d, 1H, J = 4.7 Hz, H-6, double bound in flucytosine ring), 7.50 (d, 1H, J = 15.6 Hz, H-10, double bound), 7.03 (d, 1H, J = 1.9 Hz, H-12, aromatic CA), 6.95 (dd, 1H, J1 = 8.2 Hz, J2 = 1.7 Hz, H-16, aromatic CA), 6.84 (d, 1H, J = 15.4 Hz, H-9, double bond), 6.78 (d, 1H, J = 8.2 Hz, H-15, aromatic CA), 5.82 (d, 1H, J = 4.2 Hz, H-1′, CHN deoxyribose), 5.49 (dd, 1H, J1 = 6.2 Hz, J2 = 4.3 Hz, H-2′, deoxyribose), 5.13 (dd, 1H, J1 = J2 = 6.4 Hz, H-3′, deoxyribose), 4.13 (dt, J1 = J2 = 6.3 Hz, H-4′, deoxyribose) 2.06 (s, 6H, H-7′, H-9′, deoxyribose C(O)CH3), 1.37 (d, 3H, J = 6.3 Hz, H-5′, deoxyribose CH3), 13C NMR (125 MHz, DMSO-d6) δ 169.5 (C-8′, deoxyribose CO), 169.3 (C-6′, deoxyribose CO), 164.3 (C-8, CO CA), 154.6 (C-4, flucytosine ring), 152.3 (C-2, flucytosine ring CO), 148.6 (C-14, aromatic CA), 145.6 (C-13, aromatic CA), 144.2 (C-10, double bond), 137.3 (d, JCF = 247.1 Hz, C-5, flucytosine ring), 131.3 (d, JCF = 36.0 Hz, C-6, flucytosine ring), 125.8 (C-11, aromatic CA), 121.8 (C-16, aromatic CA), 116.6 (C-9, double bond), 115.8 (C-15, aromatic CA), 114.2 (C-12, aromatic CA), 89.9 (C-1′, deoxyribose), 77.3 (C-4′, deoxyribose), 73.4 (C-3′, deoxyribose), 72.7 (C-2′, deoxyribose), 20.3 (C-7′, deoxyribose COCH3), 20.3 (C-9′, deoxyribose COCH3), 17.7 (C-5′, deoxyribose CH3); HRMS (ESI, m/z): calculated for C22H23N3O9F [M + H]+ 492.1418; found 492.1418, calculated for C22H22N3O9FNa [M + Na]+ 514.1238; found 514.1234.
Cell culture
In vitro cell culture procedures were performed under aseptic conditions and cells were propagated in humidified Innova CO-180 incubator (New Brunswick Scientific, Edison, NJ, USA) supplied with 5% CO2 and maintained at 37 °C. Subculturing was performed twice per week (at approximately 72 h intervals) and cell growth was monitored with a Nikon Eclipse microscope. Pancreatic cancer cell lines: BxPC-3 (from primary tumour) and AsPC-1 (from ascites) were cultured in RPMI-1640 medium supplemented with 10% heat inactivated fetal bovine serum (FBS), glutamine and antibiotic–antimycotic mixture. NHDF grew in alpha-MEM supplemented with 10% FBS, 2 mM glutamine, and antibiotic–antimycotic.Cell viability assay (MTT)
The cytotoxicity impact of the ligands on cancer cell and healthy control cell lines was evaluated as described earlier71–73 by performing the MTT assay.62 Cells were seeded in 96-well plates (at a density of 5 × 103 cells per well) in appropriate culture medium and plates were incubated for 24 h. Afterwards, the media were replaced with fresh media supplemented with varying concentration (in the range of 125–4000 nM) of synthesized complexes (compounds were dissolved in DMSO, heated and sonicated at 55 °C for 15 min before dilution in medium) or an equivalent volume of DMSO, considered as solvent control and incubation proceeded for another 72 h. Subsequently, the medium containing the chemicals tested was removed and the MTT solution (50 μL per well of 10 times diluted in medium from the solution stock 0.5 mg mL−1) was added. After 3 h of incubation, the MTT solution was replaced with DMSO (50 μL per well) to dissolve the purple formazan crystals and developed color. Absorbance was measured at 560 nm with a reference wavelength of 670 nm on an Asys UVM 340 Microplate Reader (Cambridge, UK). The results were expressed as a percentage of viable cells in comparison to the control (the untreated cells, taken as 100%) implementing the formula:| Cell viability (%) = (AT/AC) × 100 |
Computational methods
In the computational part of this work, we used a similar approach to our previous studies.74–76 Models of all studied ligands were built starting from the structure of capecitabine (Pubchem CID: 60953).77 LigProp 3.3 software (Schrodinger Inc.) was used to prepare all-atom 3D structures. Subsequently, we evaluated all ADME properties using QikProp 4.6 software (Schrodinger Inc.) with default options. For the toxicity prediction, we used ProTox-II webserver.67 In the molecular docking part, we used Autodock Vina ver. 1.1.2 (ref. 78) with default options and exhaustiveness level set at 18. For molecular docking to CES2, we used the AlphaFold model of this protein.79 with the following residues set as flexible: Trp145, Tyr159, Glu227, Ser228, Thr232, Ser233, Ser235, Leu259, Phe416, His457, Glu460, Leu461. After the docking we selected the lowest-energy pose of ligand 5 and used it as a template to build all other ligand models, which were redocked to the protein using Autodock ver. 4.2 (ref. 80) and the local search algorithm with 1000 repetitions. For albumin docking we used the available crystal structure of this protein (PDB: 2BXD)69 and two potential binding sites, as predicted in previous studies.68 For site 1 the following residues were treated as flexible: Tyr150, Lys199, Phe211, Arg222, Leu238, His242, Arg257, Leu260, while for site 2 the flexible residues were: Leu387, Asn391, Arg410, Tyr411, Lys414, Arg485, Ser489. In both cases we used a 25 × 25 × 25 Å box centered on the corresponding binding sites.Author contributions
Conceptualization, M. C. and O. M.; methodology, M. C., M. Z.-C., A. J., P. K., and B. T.; software, B. T.; formal analysis, B. T. and M. Z.-C.; investigation, M. C., J. T., M. Z.-C., P. K., B. T. and M. K.; resources, M. C., M. Z.-C., B.·K., A. J. and O. M.; data curation, B. T., M. K., M. Z.-C.; writing—original draft preparation, M. C. and B. T.; writing—review and editing, M. C., M. Z.-C., O. M. and B. T.; visualization, M. C., M. Z.-C. and B. T.; supervision, M. C., P. K. M. K., A. J. and O. M.; project administration, M. C. and O. M.; funding acquisition, M. C. and O. M. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by Łukasiewicz Center, grant agreement no. 4/Ł-ICHP/CŁ/2021. Authors wish to acknowledge our colleagues: Katarzyna Sidoryk (Pikralida sp. z o.o, Poland) and Sylwia Żelazowska (Łukasiewicz Research Network–Industrial Chemistry Institute, Warsaw, Poland) for scientific comments and NMR data entry.References
- R. Mokhtari, T. Homayouni, N. Baluch, E. Morgatskaya, S. Kumar, B. Das and H. Yeger, Oncotarget, 2017, 8(23), 38022 CrossRef PubMed
.
- M. Kudo, World J. Gastroenterol., 2019, 25(7), 789 CrossRef CAS PubMed
- A. Eras, D. Castillo, M. Suárez, N. Vispo, F. Albericio and H. Rodriguez, Front. Chem., 2022, 10, 889083 CrossRef CAS PubMed
- M. Szumilak, A. Wiktorowska-Owczarek and A. Stanczak, Molecules, 2021, 26(9), 2601 CrossRef CAS PubMed
- K. Tkaczuk, Clin. Ther., 2009, 31(2), 2273 CrossRef PubMed
- A. VanHook, Sci. Signaling, 2014, 7(347), 284 Search PubMed
- Y. Lee, K. Bae, H. Yoo and S. Cho, Integr. Cancer Ther., 2018, 17, 619 CrossRef PubMed
- S. Kamran, A. Sinniah, Z. Chik and M. Alshawsh, Biomedicines, 2022, 10(3), 531 CrossRef CAS PubMed
- A. Palmer, B. Izar, H. Hwangbo and P. Sorger, Clin. Cancer Res., 2022, 28(2), 368 CrossRef PubMed
- B. Fox, X. Xiao, S. Antony, G. Kohlhagen, Y. Pommier, B. Staker, L. Stewart and M. Cushman, J. Med. Chem., 2003, 46(15), 3275 CrossRef CAS PubMed
- K. Sashidhara, A. Kumar, M. Kumar, J. Sarkar and S. Sinha, Bioorg. Med. Chem. Lett., 2010, 20(24), 7205 CrossRef CAS PubMed
- L. Zhang and Z. Xu, Eur. J. Med. Chem., 2019, 181, 111587 CrossRef CAS PubMed
- C. do Pazo, K. Nawaz and M. Webster, Nat. Rev. Drug Discovery, 2021, 20(8), 583 CrossRef CAS PubMed
- J. Tong, P. Harris, M. Brimble and I. Kavianinia, Molecules, 2021, 26(19), 5847 CrossRef CAS PubMed
- U.S. National Library of Medicine, https://clinicaltrials.gov/, https://beta.clinicaltrials.gov/study/NCT01954992, accessed February 2024.
- K. Miura, M. Kinouchi, K. Ishida, W. Fujibuchi, T. Naitoh, H. Ogawa, T. Ando, N. Yazaki, K. Watanabe, S. Haneda, C. Shibata and I. Sasaki, Cancers, 2010, 2(3), 1717 CrossRef CAS PubMed
- N. Ahmad, A. Albassam, M. Khan, Z. Ullah, T. Buheazah T, H. AlHomoud and H. Al-Nasif, Saudi J. Biol. Sci., 2022, 29(5), 3704 CrossRef PubMed
- Meta-analysis Group In Cancer, P. Piedbois, P. Rougier, M. Buyse, J. Pignon, L. Ryan, R. Hansen, B. Zee, B. Weinerman, J. Pater, C. Leichman, J. Macdonald, J. Benedetti, J. Lokich, J. Fryer, G. Brufman, R. Isacson, A. Laplanche and E. Levy, J. Clin. Oncol., 1998, 16(1), 301 CrossRef PubMed
- European Medicines Agency, Xeloda (Capecitabine), https://www.ema.europa.eu/en/medicines/human/EPAR/xeloda, accessed February 2024.
- P. García-Alfonso, A. Muñoz Martín, L. Ortega Morán, J. Soto Alsar, G. Torres Pérez-Solero, M. Blanco Codesido, P. Calvo Ferrandiz and S. Grasso Cicala, Ther. Adv. Med. Oncol., 2021, 13, 1 Search PubMed
- X. Jia, X. Liu, J. Wang, M. Wang, H. Guo and M. Liu, Chem. Res. Chin. Univ., 2015, 31, 78 CrossRef CAS
- V. Jhansi Rani, A. Raghavendra, P. Kishore, Y. Nanda Kumar, K. Hema Kumar and K. Jagadeeswarareddy, Eur. J. Med. Chem., 2012, 54, 690 CrossRef CAS PubMed
- M. Li, L. Wang, Y. Wei, W. Wang, Z. Liu, A. Zuo, W. Liu, J. Tian and H. Wang, Pharmaceuticals, 2022, 16(1), 53 CrossRef PubMed
- M. Ait-Tihyaty, Z. Rachid, A. Larroque-Lombard and B. Jean-Claude, Invest. New Drugs, 2013, 31(6), 1409 CrossRef CAS PubMed
- National Cancer Institute at the National Institutes of Health, https://www.cancer.gov/about-cancer/treatment/drugs/pancreatic#2, accessed February 2024 Search PubMed.
- A. Lambert, C. Gavoille and T. Conroy, Ther. Adv. Gastroenterol., 2017, 10(8), 631 CrossRef CAS PubMed
- F. Bray, J. Ferlay, I. Soerjomataram, R. Siegel, L. Torre and A. Jemal, Ca-Cancer J. Clin., 2018, 68(6), 394 CrossRef PubMed
- J. Ferlay, M. Colombet, I. Soerjomataram, T. Dyba, G. Randi, M. Bettio, A. Gavin, O. Visser and F. Bray, Eur. J. Cancer, 2018, 103, 356 CrossRef CAS PubMed
- M. Zaremba-Czogalla, A. Jaromin, K. Sidoryk, A. Zagórska, M. Cybulski and J. Gubernator, Materials, 2020, 13(24), 5813 CrossRef CAS PubMed
- L. Rocha, M. Monteiro and A. Teodoro, Cancer Clin. Oncol., 2012, 1(2), 109 Search PubMed
- M. Yamaguchi, T. Murata, B. El-Rayes and M. Shoji, Oncol. Rep., 2015, 34(6), 3304 CrossRef CAS PubMed
- K. Espíndola, R. Ferreira, L. Narvaez, A. Silva Rosario, A. da Silva, A. Silva, A. Vieira and M. Monteiro, Front. Oncol., 2019, 9, 541 CrossRef PubMed
- X. Zhang, X. He, Q. Chen, J. Lu, S. Rapposelli and R. Pi, Bioorg. Med. Chem., 2018, 26(3), 543 CrossRef CAS PubMed
- S. Khargharia, R. Rohman and R. Kar, ChemistrySelect, 2022, 7(24), e202201440 CrossRef CAS
- M. Cybulski, K. Sidoryk, M. Zaremba-Czogalla, B. Trzaskowski, M. Kubiszewski, J. Tobiasz, A. Jaromin and O. Michalak, Int. J. Mol. Sci., 2024, 25(5), 2573 CrossRef CAS PubMed
- A. Trachtenberg, S. Muduli, K. Sidoryk, M. Cybulski and M. Danilenko, Front. Pharmacol, 2019, 10, 507 CrossRef CAS PubMed
- J. Ciccolini, C. Mercier and G. Milano, Genomics and Pharmacogenomics in Anticancer Drug Development and Clinical Response, Cancer Drug Discovery and Development™, ed. F. Innocenti, Humana Press, 2008, DOI:10.1007/978-1-60327-088-5_14
- D. Wang, L. Zou, Q. Jin, J. Hou, G. Ge and L. Yang, Acta Pharm. Sin. B, 2018, 8(5), 699 CrossRef PubMed
- L. Di, Curr. Drug Metab., 2019, 20(2), 91 CrossRef CAS PubMed
- M. Capello, J. Fahrmann, M. Rios Perez, J. Vykoukal, E. Irajizad, S. Tripathi, D. Roife, L. Bantis, Y. Kang, D. Kundnani, H. Xu, L. Prakash, J. Long, H. Katayama, A. Fleury, S. Ferri-Borgogno, D. Baluya, J. Dennison, C. Aguilar-Bonavides, J. Casabar, M. Celiktas, K. Do, O. Fiehn, A. Maitra, H. Wang, Z. Feng, P. Chiao, M. Katz, J. Fleming and S. Hanash, JCO Precis. Oncol., 2020, 4, 426 CrossRef PubMed
- M. Capello, M. Lee, H. Wang, I. Babel, M. Katz, J. Fleming, A. Maitra, H. Wang, W. Tian, A. Taguchi, S. Hanash and J. Natl, Cancer. Inst., 2015, 107(8), djv132 Search PubMed
- Y. Chen, M. Capello, M. Rios Perez, J. Vykoukal, D. Roife, Y. Kang, L. Prakash, H. Katayama, E. Irajizad, A. Fleury, S. Ferri-Borgogno, D. Baluya, J. Dennison, K. Do, O. Fiehn, A. Maitra, H. Wang, P. Chiao, M. Katz, J. Fleming, S. Hanash and J. Fahrmann, Mol. Metab., 2022, 56, 101426 CrossRef CAS PubMed
- Global Burden of Disease 2019 Cancer Collaboration, JAMA Oncol., 2022, 8(3), 420 CrossRef PubMed
- K. Chun and S. Joo, Biomol. Ther., 2022, 30(6), 479 CrossRef CAS PubMed
- M. Friedman and H. Jürgens, J. Agric. Food Chem., 2000, 48(6), 2101 CrossRef CAS PubMed
- P. G. M. Wuts, Greene's Protective Groups in Organic Synthesis, Wiley, edn 5, 2014 Search PubMed
- R. Crouch, Tetrahedron, 2004, 60, 5833 CrossRef CAS
- D. Rabiej-Kozioł, M. Krzemiński and A. Szydłowska-Czerniak, Materials, 2020, 13, 4536 CrossRef PubMed
- A. Franks, Q. Wang and K. Franz, Bioorg. Med. Chem. Lett., 2015, 25(21), 4843 CrossRef CAS PubMed
- L. Bian, S. Cao, L. Cheng, A. Nakazaki, T. Nishikawa and J. Qi, ChemMedChem, 2018, 13, 1972 CrossRef CAS PubMed
- J. Giessel, A. Loesche, R. Csuk and I. Serbian, Eur. J. Med. Chem., 2019, 177, 259 CrossRef CAS PubMed
- M. D'Ambrosio, Food Chem., 2013, 138(4), 2079 CrossRef PubMed
- L. Monteiro, S. Oliveira, F. Paiva-Martins, P. Ferreira, D. Pereira, P. Andrade and P. Valentão, Tetrahedron, 2017, 73(43), 6199 CrossRef CAS
- M. Spasova, V. Kortenska-Kancheva, I. Totseva, G. Ivanova, L. Georgiev and T. Milkova, J. Pept. Sci., 2006, 12(5), 369 CrossRef CAS PubMed
- K. Takao, K. Toda, T. Saito and Y. Sugita, Chem. Pharm. Bull., 2017, 65(11), 1020 CrossRef CAS PubMed
- P. Rajan, I. Vedernikova, P. Cos, D. Berghe, K. Augustyns and A. Haemers, Bioorg. Med. Chem. Lett., 2001, 11(2), 215 CrossRef CAS PubMed
- G. Quéléver, S. Burlet, C. Garino, N. Pietrancosta, Y. Laras and J. Kraus, J. Comb. Chem., 2004, 6(5), 695 CrossRef PubMed
- S. Gaikwad, M. Gaikwad and P. Lokhande, J. Heterocyclic Chem., 2021, 58, 1408 CrossRef CAS
- P. Lokhande and B. Nawghare, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2012, 51(1), 328 Search PubMed
- M. Nagaraju, A. Krishnaiah and H. Mereyala, Synth. Commun., 2007, 37(15), 2467 CrossRef CAS
- N. Shimma, I. Umeda, M. Arasaki, C. Murasaki, K. Masubuchi, Y. Kohchi, M. Miwa, M. Ura, N. Sawada, H. Tahara, I. Kuruma, I. Horii and H. Ishitsuka, Bioorg. Med. Chem., 2000, 8(7), 1697 CrossRef CAS PubMed
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55 CrossRef CAS PubMed
- B. Liu, L. Ezeogu, L. Zellmer, B. Yu, N. Xu and D. Liao, Cancer Med., 2015, 4(9), 1394 CrossRef PubMed
- C. Lipinski, F. Lombardo, B. Dominy and P. Feeney, Adv. Drug Delivery Rev., 2001, 46(1–3), 3 CrossRef CAS PubMed
- B. Doak, B. Over, F. Giordanetto and J. Kihlberg, Chem. Biol., 2014, 21(9), 1115 CrossRef CAS PubMed
- M. Sanguinetti and M. Tristani-Firouzi, Nature, 2006, 440(7083), 463 CrossRef CAS PubMed
- P. Banerjee, A. Eckert and A. Schrey, Nucleic Acids Res., 2018, 46(W1), W257 CrossRef CAS PubMed
- S. Mousavi and M. Fatemi, Bioorg. Chem., 2019, 90, 103037 CrossRef CAS PubMed
- J. Ghuman, P. Zunszain, I. Petitpas, A. Bhattacharya, M. Otagiri and S. Curry, J. Mol. Biol., 2005, 353(1), 38 CrossRef CAS PubMed
- N. Bukowski, J. Pandey, L. Doyle, T. Richard, C. Anderson and Y. Zhu, Bioconjug. Chem., 2014, 25(12), 2189 CrossRef CAS PubMed
- N. Filipczak, A. Jaromin, A. Piwoni, M. Mahmud, C. Sarisozen, V. Torchilin and J. Gubernator, Cancers, 2019, 11(12), 1982 CrossRef CAS PubMed
- M. Fandzloch, A. Jaromin, M. Zaremba-Czogalla, A. Wojtczak, A. Lewińska, J. Sitkowski, J. Wiśniewska, I. Łakomska and J. Gubernator, Dalton Trans., 2020, 49(4), 1207 RSC
- A. Jaromin, S. Parapini, N. Basilico, M. Zaremba-Czogalla, A. Lewińska, A. Zagórska, M. Walczak, B. Tyliszczak, A. Grzeszczak, M. Łukaszewicz, Ł. Kaczmarek and J. Gubernator, Bioact. Mater., 2020, 6(4), 1163 Search PubMed
- O. Michalak, M. Cybulski, W. Szymanowski, A. Gornowicz, M. Kubiszewski, K. Ostrowska, P. Krzeczyński, K. Bielawski, B. Trzaskowski and A. Bielawska, Int. J. Mol. Sci., 2023, 24(10), 8875 CrossRef CAS PubMed
- P. Krzeczyński, M. Dutkiewicz, O. Zegrocka-Stendel, B. Trzaskowski and K. Koziak, Molecules, 2023, 28(5), 2287 CrossRef PubMed
- K. Ostrowska, A. Leśniak, W. Gryczka, Ł. Dobrzycki, M. Bujalska-Zadrożny and B. Trzaskowski, Int. J. Mol. Sci., 2023, 24(3), 2779 CrossRef CAS PubMed
- S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He, S. He, Q. Li, B. Shoemaker, P. Thiessen, B. Yu, L. Zaslavsky, J. Zhang and E. Bolton, Nucleic Acids Res., 2023, 51(D1), D1373 CrossRef PubMed
- O. Trott and A. Olson, J. Comput. Chem., 2010, 31(2), 455 CrossRef CAS PubMed
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. Kohl, A. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596(7873), 583 CrossRef CAS PubMed
- G. Morris, R. Huey, W. Lindstrom, M. Sanner, R. Belew, D. Goodsell and A. Olson, J. Comput. Chem., 2009, 30(16), 2785 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Tables S1 and S2: ADMET properties of the tested compounds; synthesis of 18, 26, 28 and 29; Fig. S1–S22 NMR spectra of 1–6, 18, 26, 28, 29; Fig. S23–S28 HRMS spectra of 1–6. See DOI: https://doi.org/10.1039/d4ra01683a |
| This journal is © The Royal Society of Chemistry 2024 |