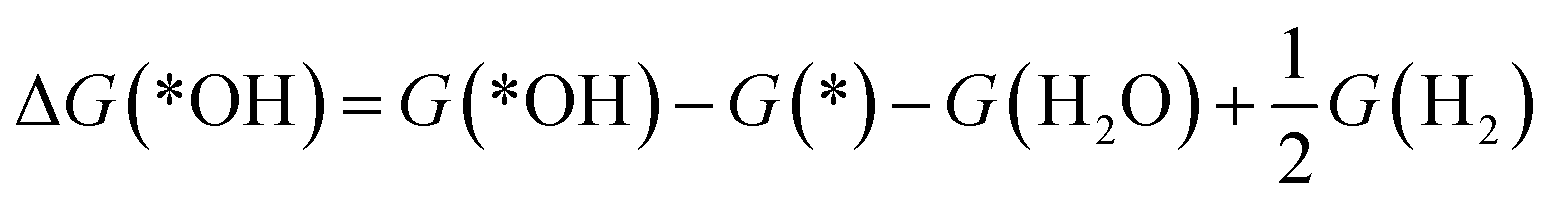DOI:
10.1039/D4RA01754D
(Paper)
RSC Adv., 2024,
14, 16379-16388
Axial heteroatom (P, S and Cl)-decorated Fe single-atom catalyst for the oxygen reduction reaction: a DFT study†
Received
6th March 2024
, Accepted 2nd May 2024
First published on 21st May 2024
Abstract
An FeN4 single-atom catalyst (SAC) embedded in a graphene matrix is considered an oxygen reduction reaction (ORR) catalyst for its good activity and durability, and decoration on the Fe active site can further modulate the performance of the FeN4 SAC. In this work, the axial heteroatom (L = P, S and Cl)-decorated FeN4 SAC (FeN4L) and pure FeN4 were comparatively studied using density functional theory (DFT) calculations. It was found that the rate-determining step (RDS) in the ORR on pure FeN4 is the reduction of OH to H2O in the last step with an overpotential of 0.58 V. However, the RDS of the ORR for the axial heteroatom-decorated FeN4L is the reduction of O2 to OOH in the first step. The axial P and S heteroatom-decorated FeN4P and FeN4S exhibit lower activity than pure FeN4 since the overpotentials of the ORR on FeN4P and FeN4S are 1.02 V and 1.09 V, respectively. Meanwhile, FeN4Cl exhibits the best activity towards the ORR since it possesses the lowest overpotential (0.51 V). The main reason is that the axial heteroatom decoration alleviates the adsorption of all the species in the whole ORR, thus modulating the free energy in every elementary reaction step. A volcano relationship between the d band center and the ORR activity can be determined among the axial heteroatom-decorated FeN4L SACs. The d band center of the Fe atom in various FeN4L SACs follows the order of FeN4 > FeN4Cl > FeN4S > FeN4P, whereas the overpotential of the ORR on various catalysts follows the order of FeN4Cl > FeN4 > FeN4S ≈ FeN4P. ΔG(*OH) is a simple descriptor for the prediction of the ORR activity of various axial heteroatom-decorated FeN4L, although the RDS in the ORR is either the first step or the last step. This paper provides a guide to the design and selection of the ORR over SACs with different axial heteroatom decorations, contributing to the rational design of more powerful ORR electrocatalysts and achieving advances in electrochemical conversion and storage devices.
Introduction
A typical example of a clean energy conversion technology is the proton exchange membrane fuel cell (PEMFC). To ensure energy conversion, the sluggish oxygen reduction reaction (ORR) at the cathode needs a powerful electrocatalyst.1 Currently, the most efficient catalyst for the ORR is platinum group metals (PGMs). However, their major drawbacks (such as huge overpotential, scarcity, and high cost) limit their commercialization. One effective way to address this issue is to explore alternatives to non-precious metal catalysts. Carbon materials doped with metals and heteroatoms have drawn much interest from researchers as promising and exceptional electrocatalysts to efficiently reduce the overpotential of the ORR. Overall, heteroatom dopants (such as N, B, P, and S) can produce charged active sites and cause the polarization of the carbon skeleton, which can effectively reduce the overpotential of the ORR and improve the slow kinetics of the ORR.2–4 As opposed to PGMs and carbon-based metal-free catalysts, single-atom catalysts (SACs) have emerged as a frontier in the field of ORR electrocatalysts owing to their large specific surface area, maximum atomic efficiency, high selectivity, and long-term operational endurance. DFT calculations have been widely and significantly successful in research studies on the ORR mechanism5 since it can greatly save experimental costs and accurately predict the components with high activity. Graphene with defects is one of the numerous substrates that can make an excellent SAC substrate. The electrocatalytic activity of the ORR can be increased by monitoring the d-orbital electrons of the central transition metal (TM) through the coordination of four N atoms,6 and the atomically dispersed TMN4 site has been considered as the active sites for ORR.7–9 Both experimental studies and DFT calculations have shown that the SACs based on non-precious TM (TM = Fe, Co, Mn, Ni, Zn, etc.2,10–12) on nitrogen-doped carbon support can significantly enhance the ORR electrocatalytic activity. Among the aforementioned SACs, the single-atom Fe–N–C catalyst exhibits considerable activity and remarkable selectivity for the ORR,9,13,14 which is even comparable to the commercial platinum catalysts. Furthermore, our previous experiments have proved that the activity and stability of FeN4 is as good as the platinum catalyst for ORR.15,16 The FeN4 catalyst has been reported as a possible replacement for Pt-based catalysts because of its good activity and durability, and the decoration on the Fe active site can further improve the performance of SACs through modulating the electronic and geometric structure.17,18 Thus, the rational design of carbon-based ORR catalysts with low cost, as well as high activity, selectivity, and durability, is of great significance. Lin et al.19 synthesized a new SAC, and each Fe atom is coordinated with five N atoms rather than four N, resulting in a 3D Fe–N5 coordination site. By using DFT calculations, they discovered that the axially coordinated pyridine could positively regulate the binding strength of oxygen on the 2D Fe–N4 site to a higher level. Other scholars came to similar results that were well-researched.20,21 In addition, SACs exhibited good activity for electrochemical N2 reduction,22 CO2 reduction,23–25 lithium–sulfur batteries,26 NO electroreduction27,28 and CO detection.29 Thus, the study on axial heteroatom-decorated Fe SAC for ORR is important, and this paper can shed light on the rational design of SAC catalysts with good performance.
In this work, DFT calculations were performed to investigate the effect of different axial heteroatom decorations on the activity of FeN4L towards ORR. The binding energy and cohesion energy, which describe the stability of the heteroatom-decorated FeN4L, were first studied. Then, the overpotentials of ORR catalyzed by pure FeN4 and various FeN4L were evaluated. Furthermore, the relationship between the overpotential and the OH adsorption free energy (ΔG(*OH)) were studied, since ΔG(*OH) is a good descriptor for ORR activity.30–32 An accurate quantitative description of the ORR activity trends was elucidated with the help of the adsorption free energy of the reaction intermediate, d-band center, partial density of states, differential charge density and Bader charge of the heteroatom-decorated Fe atom.
Computational methods
The spin-polarized DFT calculations were performed by using the Vienna Ab initio Simulation Package (VASP).33,34 The Kohn–Sham wave functions were expanded in a plane wave basis set with a cutoff energy of 500 eV. The projector-augmented wave (PAW) method and PBE potential for the exchange-correlation function were used.35 The 3 × 3 × 1 Monkhorst–Pack k-point mesh was used to sample the Brillouin zone.36 All atoms were allowed to relax until the forces fell below 0.02 eV Å−1. A vacuum region of 15 Å was created to ensure negligible interaction between mirror images.
The ORR occurs via the following steps:
| | |
O2(g) + * + H+ + e− → *OOH
| (1) |
| |
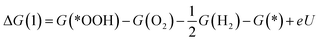 | (2) |
| | |
*OOH + H+ + e− → *O + H2O
| (3) |
| |
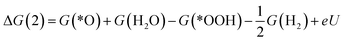 | (4) |
| |
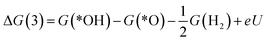 | (6) |
| | |
*OH + H+ + e− → * + H2O
| (7) |
| |
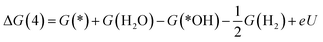 | (8) |
where * represents the preferable adsorption site for intermediates, and
G(*),
G(*OOH),
G(*O) and
G(*OH) are the Gibbs free energies of the clean catalyst, and the catalyst adsorbed with *OOH, *O and *OH, respectively.
G(H
2O) and
G(H
2) are the Gibbs free energies of H
2O and H
2, respectively.
The Gibbs free energy (ΔG) diagram of the ORR was calculated using the computational hydrogen electrode (CHE) model proposed by Nørskov et al.,37 where the free energy of (H+ + e−) under standard conditions is equal to the value of 1/2H2. The free energy change (ΔG) of the primitive step of the ORR is calculated according to the following equation.
| | |
ΔG = ΔE + ΔZPE − TΔS + Δ∫CPdT + ΔGU + ΔGpH
| (9) |
where Δ
E is the total energy difference between the reactants and products of the reactions, ΔZPE is the zero-point energy correction, Δ
S is the vibrational entropy change and
T is temperature with the value of 298.15 K. Δ∫
CPd
T is the difference in enthalpic correction, Δ
GU = −
eU, where
e is the elementary charge,
U is the electrode potential, and Δ
GpH is the correction of the H
+ free energy.
The overpotential for ORR (ηORR) is calculated according to the following equation:
| | |
ηSHE = 1.23 V + ΔGmax/e
| (10) |
where 1.23 V is defined as the equilibrium potential of the overall 4-electron ORR at the standard state, and Δ
Gmax represents the maximum Δ
G associated with the proton–electron-transfer steps.
The binding energies (EBinding) of the axial heteroatom with FeN4 and the adsorption energy (Eads) of the intermediates in ORR have been calculated according to formulas (11) and (12):
| | |
EBinding = EFeN4X − EFeN4 − EX
| (11) |
| | |
Eads = Esystem − EC − Especies
| (12) |
where
EFeN4X,
EFeN4 and
EX are the total electronic energies (non-ZPE corrected) of the system, FeN
4 and heteroatoms, respectively.
Esystem,
EC and
Especies denote the total electronic energy (non-ZPE corrected) of the adsorption system, the individual surface and adsorbate, respectively.
Result and discussion
A 6 × 6 graphene supercell containing 72 carbon atoms was used as the basis for the construction of the axial heteroatom-decorated FeN4 SAC. A double carbon atom vacancy was constructed at the center of graphene. Then, an Fe atom was embedded in the center of the double vacancy, and the four closest C atoms surrounding the vacancy were replaced by four N atoms, resulting in a structural model of the FeN4 catalyst.
Fig. 1 shows the top and side views of FeN4, FeN4P, FeN4S, and FeN4Cl. The metal and the four coordinated N atoms almost remain in place in the pure FeN4, while a slight deviation from the original plane can be determined after the axial heteroatom decoration. The Bader charge in Fig. S1† shows that the central Fe atom in FeN4, FeN4P, FeN4S, and FeN4Cl loses electrons (−1.03e, −0.86e, −1.06e, and −1.15e; the negative value represents the deficiency of electrons), and electrons are transferred to the surrounding coordinated nonmetal atoms, where the P, S, and Cl atoms get 0.04e, 0.44e, and 0.54e, respectively.
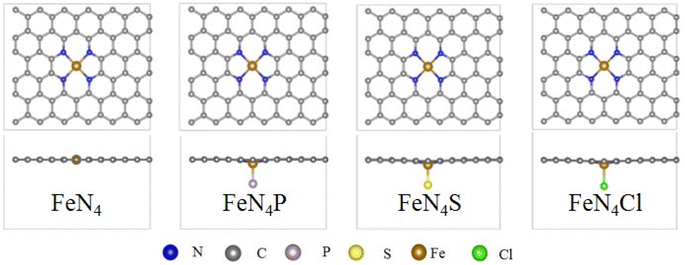 |
| | Fig. 1 Top and side views of the optimized FeN4, FeN4P, FeN4S, and FeN4Cl, respectively. | |
As shown in Table 1, the binding energy between the heteroatom and Fe are all negative with values of −1.79 eV, −3.30 eV, and −2.97 eV, respectively, indicating that these structures can be stabilized. Thus, the subsequent adsorption of the ORR intermediates can be carried out on these structures.
Table 1 Binding energy (EBinding) between the axial heteroatom and Fe in different FeN4L
| System |
FeN4P |
FeN4S |
FeN4Cl |
| EBinding (eV) |
−1.79 |
−3.03 |
−2.97 |
The optimized structures of the *OOH, *O, and *OH intermediates adsorbed on FeN4, FeN4P, FeN4S, and FeN4Cl are depicted in Fig. S2.† The Fe atom is located at the center of the double vacant graphene in the FeN4, FeN4P, FeN4S, and FeN4Cl systems. The bond lengths between the central Fe and 4 coordinated N atoms are almost similar, and the deviation of the central Fe atom from the graphene plane became smaller compared with the pure FeN4. Table 2 lists the bond lengths between the central Fe and various O atoms from the *OOH, *O, *OH intermediates and the inter-bond lengths of various ORR intermediates as well. For the adsorbed *OOH intermediates on various axial heteroatom FeN4L, the bond lengths of Fe–O follow the order of FeN4S > FeN4P > FeN4Cl > FeN4, and the values are 2.02 Å, 1.95 Å, 1.86 Å, and 1.80 Å, respectively. This is consistent with the fact that the relatively large binding energy usually corresponds to a short distance. For the *O intermediates, a similar phenomenon can also be observed. The bond lengths of Fe–O follow the order of FeN4S > FeN4P = FeN4Cl > FeN4, and the values are 1.69 Å, 1.68 Å, 1.68 Å, and 1.65 Å, respectively. Similarly, for the *OH intermediate, the bond lengths of Fe–O follow the order of FeN4S > FeN4P > FeN4Cl > FeN4, and the values are 1.94 Å, 1.92 Å, 1.87 Å, and 1.83 Å, respectively. Thus, it can be concluded that the axial heteroatom decoration of FeN4L leads to a decreased binding between various ORR intermediates and axial heteroatom-decorated Fe SACs.
Table 2 Bond length (Å) between the ORR intermediates OOH, O, and OH and the central active Fe site
| System |
Bond length/Å |
| *OOH |
*O |
*OH |
| dFe–O |
dO–O |
dO–H |
dFe–O |
dFe–O |
dO–H |
| FeN4 |
1.80 |
1.47 |
0.98 |
1.65 |
1.83 |
0.98 |
| FeN4P |
1.95 |
1.45 |
0.98 |
1.68 |
1.92 |
0.98 |
| FeN4S |
2.02 |
1.44 |
0.98 |
1.69 |
1.94 |
0.97 |
| FeN4Cl |
1.86 |
1.47 |
0.98 |
1.68 |
1.87 |
0.98 |
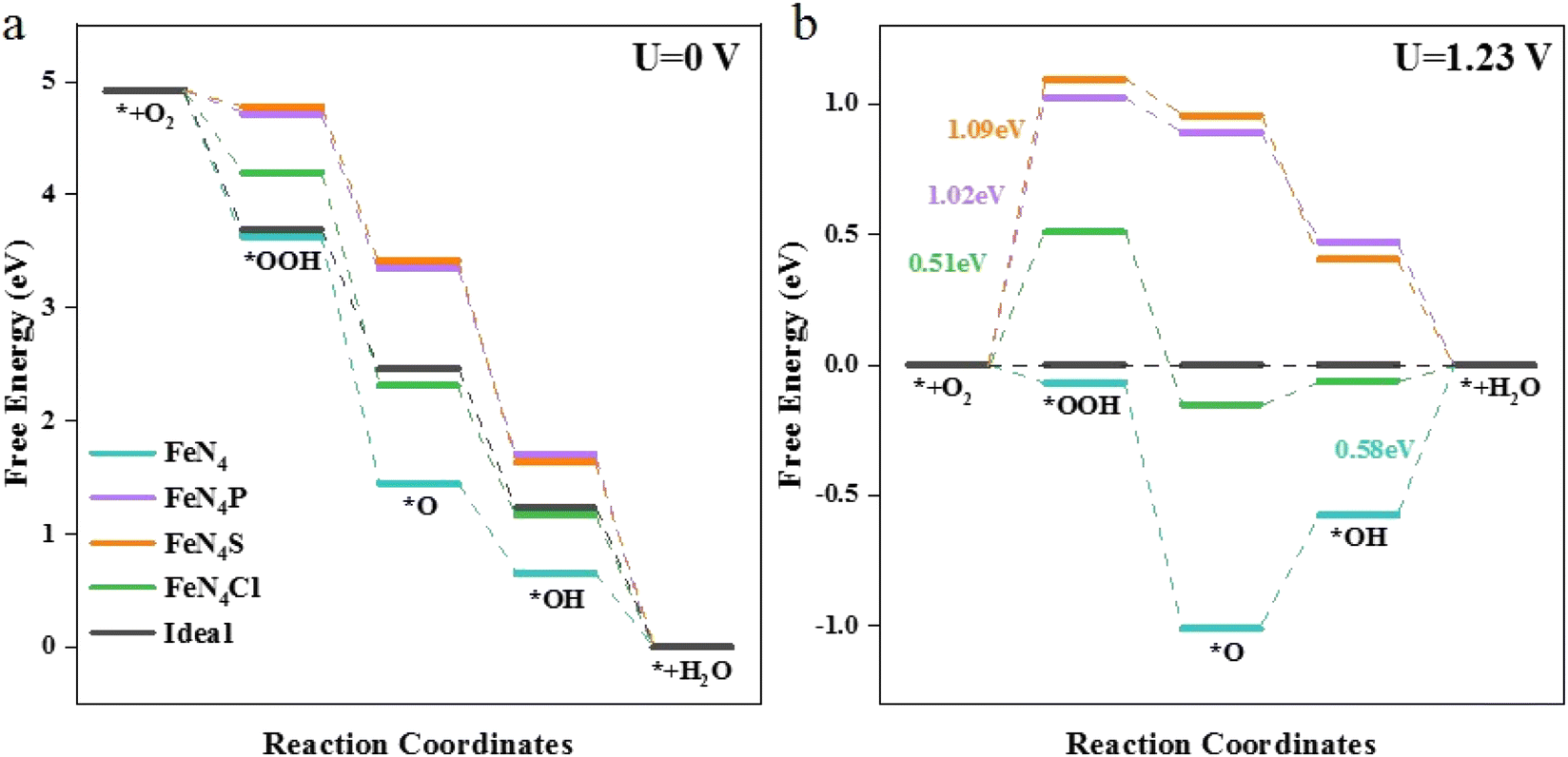
The free energy changes (ΔG) on the different catalysts are shown in Fig. 2. The ΔG for each elementary step at the active sites of various FeN4L are based on the following four reactions: (1) O2 adsorbs at the active site and hydrogenates to form *OOH; (2) *OOH continues to hydrogenate to form *O; (3) *O continues to combine with H+ and forms *OH; (4) H2O is produced through hydrogenation of *OH and eventually desorbs.
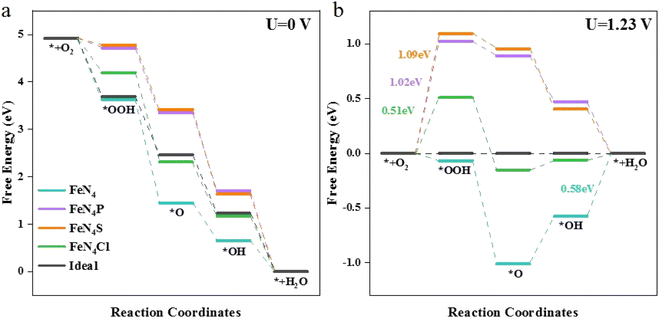 |
| | Fig. 2 Free energy diagrams for the ORR on FeN4, FeN4P, FeN4S, FeN4Cl, and the ideal catalyst at (a) U = 0 V and (b) U = 1.23 V. | |
At the ORR equilibrium potential, all four ΔG values are negative, indicating that each reaction of the ORR is thermodynamically favoured to occur. For a perfect ORR catalyst, ΔG should be −1.23 eV for each elementary reaction step involving electron transfer, causing zero overpotential (Fig. 2a). The rate-determining step (RDS) of ORR on FeN4 differs from that on the axial heteroatom-decorated FeN4L, as illustrated in Fig. 2. The RDS is defined by eqn (10). For FeN4, the reaction is determined by the last step of ORR, i.e., the formation of H2O from *OH (ΔG(4)). However, for the axial heteroatom-decorated FeN4P, FeN4S, and FeN4Cl, the reaction is controlled by the first proton–electron transfer step (ΔG(1)), i.e., the protonation of oxygen to *OOH. At 1.23 V, the two steps of hydrogenation of *O and the formation of H2O from *OH are free energy increasing processes at the FeN4 active site, with the largest free energy increase for *OH. This suggests that the *OH removal step is the RDS with an overpotential of 0.58 V. Whereas the energy of the first step for FeN4P, FeN4S, and FeN4Cl is 1.02 eV, 1.09 eV, and 0.51 eV, respectively. The FeN4Cl has the lowest overpotential, indicating that the axial Cl decoration contributed the most to the ORR activity of FeN4L compared to the pure FeN4 structure, the P and S heteroatom-decorated FeN4L.
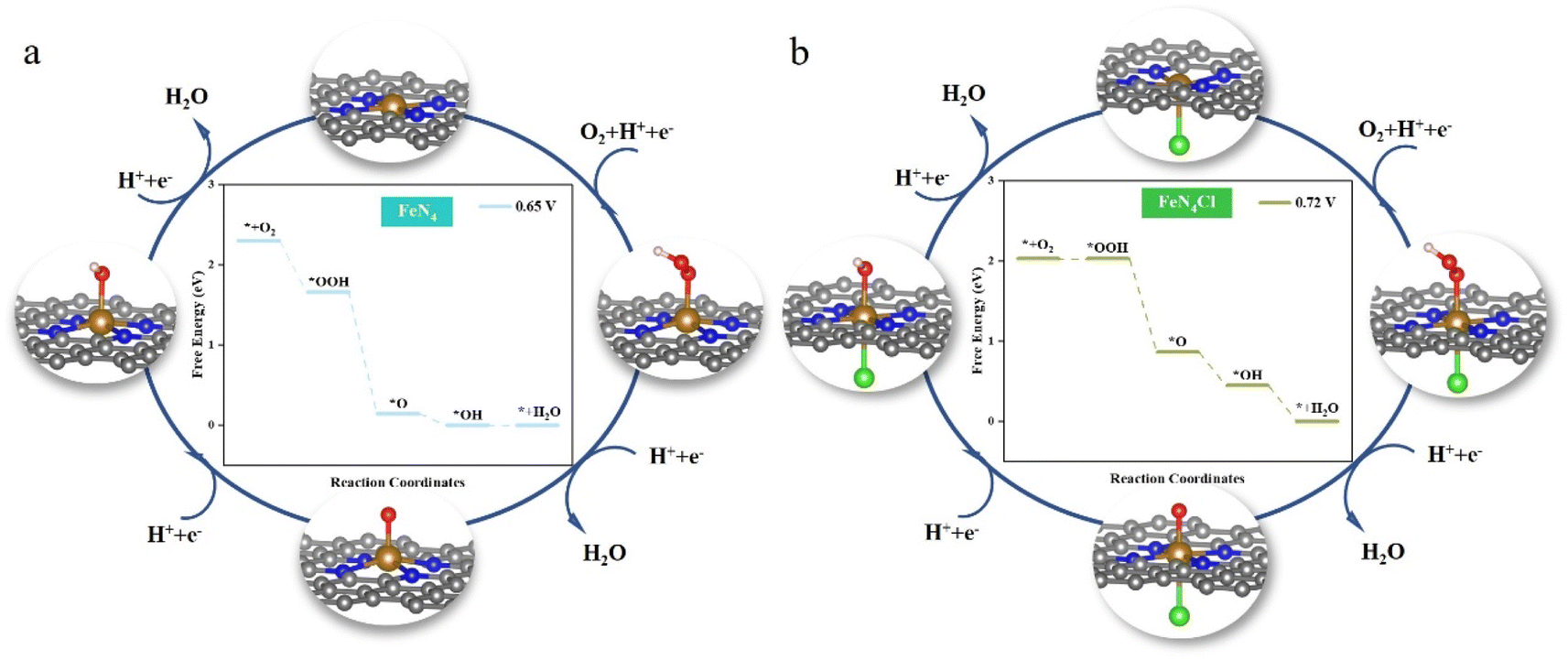
Fig. 3 delves deeper into the ORR catalytic cycle and the optimized conformation of the ORR intermediates on FeN4 and FeN4Cl to reveal the origin of their high ORR activity (FeN4P and FeN4S in Fig. S3†). As a transition metal element, the central Fe atom can transfer partial electrons throughout the ORR process, resulting in stronger binding to diverse intermediates. Thus, the obtained conclusion is consistent with the above calculation results, i.e., at the central iron atom, some electron transfer occurs, followed by hydroxide ion desorption. On the contrary, after the central iron atom has been axially decorated with heteroatoms, a portion of the electrons are transferred to the heteroatoms. The electrons provided by the iron atom to the oxygen reduction intermediates are reduced, and the binding becomes weaker, making desorption of the hydroxide ion easier on FeN4X. Conversely, the adsorption of O2 to generate OOH is the most difficult step in the entire ORR process.
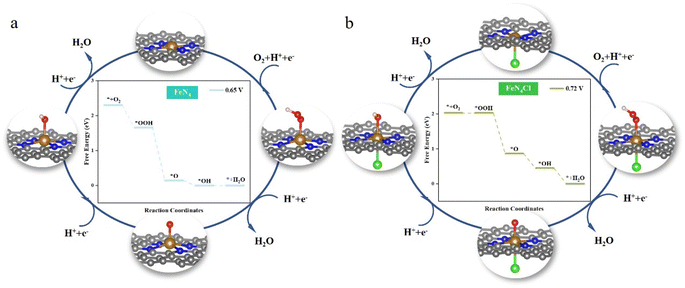 |
| | Fig. 3 Schematic diagram of the cycle of the ORR. The middle diagram shows ΔG of the ORR at different potentials on (a) FeN4 and (b) FeN4Cl. For U < 0.65 V and U < 0.72 V, all steps on FeN4 and FeN4Cl are thermodynamically accessible. | |
Table 3 demonstrates that the adsorption of various intermediates of the ORR on FeN4, FeN4P, FeN4S, and FeN4Cl is an exothermic process. The adsorption of OOH was strongest on the FeN4 active site with an adsorption energy of −1.68 eV, moderate adsorption energy of −1.12 eV on FeN4Cl, and the smallest adsorption energy on the FeN4S surface with a value of −0.53 eV. Meanwhile, it is −0.60 eV on FeN4P. Correspondingly, O also exhibited the strongest adsorption on the FeN4 active site with −4.36 eV, followed by FeN4Cl at −3.52 eV. The adsorption energies on the FeN4P and FeN4S surfaces are also relatively strong at −2.46 eV and −2.41 eV, respectively. Similarly, the exothermic processes that occur during the OH intermediate adsorption on the surfaces of FeN4, FeN4P, FeN4S, and FeN4Cl are all thermodynamically advantageous. *OH has the highest adsorption energy (−2.73 eV) on FeN4, the lowest (−1.68 eV) on FeN4P, and −1.74 eV and −2.23 eV on FeN4S and FeN4Cl, respectively. In comparison, OOH, O, and OH on FeN4 have the strongest adsorption. Meanwhile, FeN4P, FeN4S, and FeN4Cl, which were decorated by axial heteroatoms, decreased the adsorption of the oxygen-containing ORR intermediates to varying degrees. Furthermore, the adsorption energy of the *O intermediate was higher than both *OOH and *OH on the same catalyst. This indicated that the dissociation of OOH in the first step is relatively simple, and that the hydrogenation reaction of *O occurring in the latter step is also favored by the strong adsorption of the intermediate O.32
Table 3 The adsorption energy (Eads) of OOH, O, and OH on FeN4, FeN4P, FeN4S, and FeN4Cl
| System |
Adsorption energy/eV |
| *OOH |
*O |
*OH |
| FeN4 |
−1.68 |
−4.36 |
−2.73 |
| FeN4P |
−0.60 |
−2.46 |
−1.68 |
| FeN4S |
−0.53 |
−2.41 |
−1.74 |
| FeN4Cl |
−1.12 |
−3.52 |
−2.23 |
The reaction free energy of the elementary reaction in ORR is determined by the adsorption free energy (ΔGads) of the intermediate species, *OH, *O and *OOH (abbreviated as ΔG(*OH), ΔG(*O), and ΔG(*OOH)).38 The ΔGads of *OOH, *O, and *OH can be calculated according to the following equations:
| |
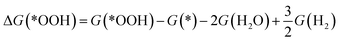 | (13) |
| | |
ΔG(*O) = G(*O) − G(*) − G(H2O) + G(H2)
| (14) |
| |
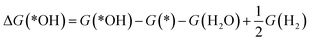 | (15) |
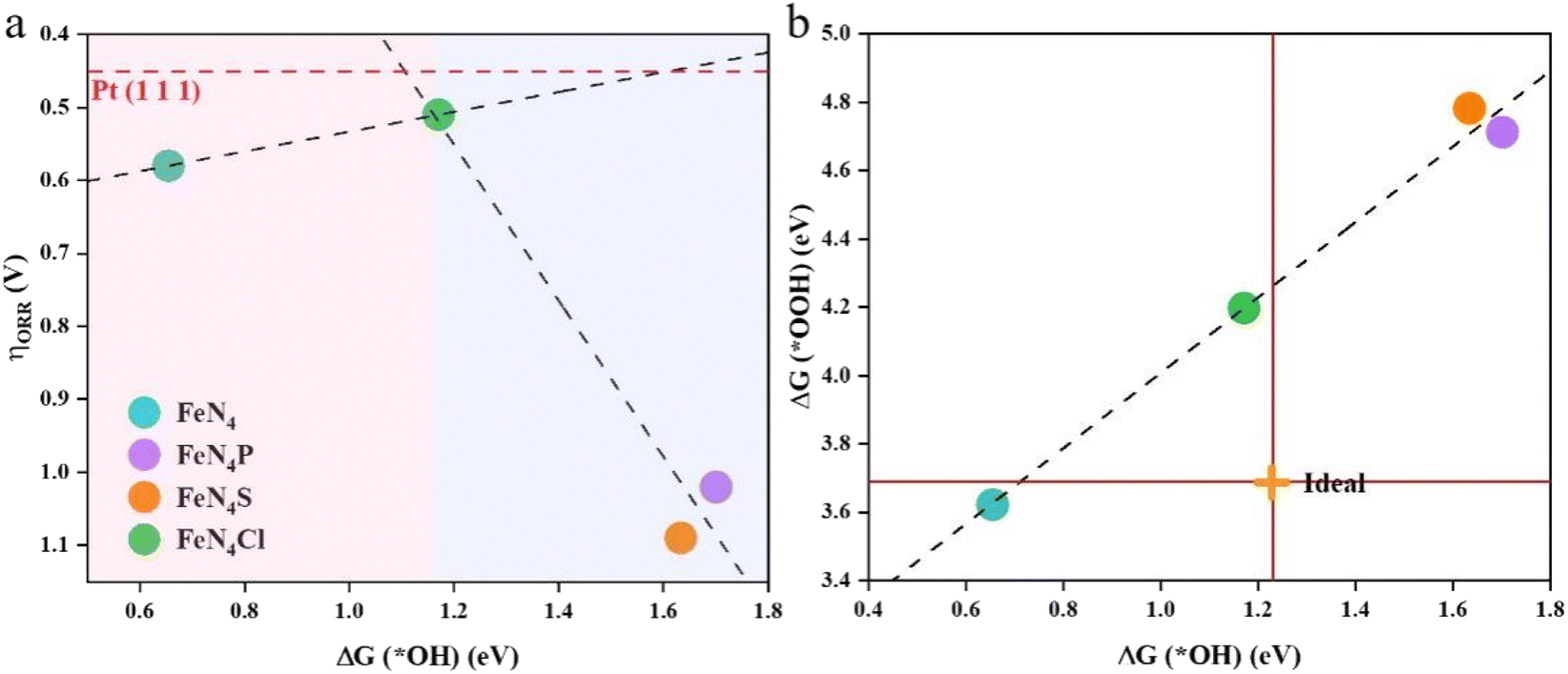
Table S1† lists the calculated ΔG(*OH), ΔG(*O), and ΔG(*OOH) on various catalysts. ΔG(*OH) is the variable used to describe the ORR overpotential because RDS is related to either ΔG(*OH) or ΔG(*OOH). As ΔG(*OH) becomes more negative, the strength of the bond between *OH and the catalyst increases.
The adsorption free energy is not equivalent to the adsorption energy, but reflects the trend of the binding strength of the same substance on different substrates. An appropriate catalyst should have a reasonable ΔGads to make sure that the adsorption of intermediates is neither too high nor too low. The correlation between the ORR overpotential and the Gibbs free energy of the *OH intermediates can be seen in Fig. 4a. The overpotential and ΔG(*OH) have a perfect volcanic relationship, and the catalytic process will be hindered by too strong or too weak interactions between the catalysts and intermediates. The inflection point of the volcano diagram is the turning point for both strong and weak adsorption of OH. The ORR process is constrained in the strong binding region (the left pink zone of the volcano diagram) by the reduction of *OH to H2O in the last step. The limit of the catalytic process in the weak binding zone (the right blue zone of the volcano diagram) is the first step of O2 reduction to *OOH. Among all the studied catalysts here, FeN4Cl is nearly at the top of the volcano diagram, and is closest to the theoretical overpotential of Pt(111).37 This indicates that FeN4Cl possesses a comparable catalytic activity with Pt(111). For the ideal ORR electrocatalyst, ΔG(*OH) should be 1.23 eV and ΔG(*OOH) should be 3.69 eV. As shown in Fig. 4b, a proportional relationship ΔG(*OOH) = 1.10ΔG(*OH) + 3.41 with a coefficient of determination R2 = 0.98 can be determined, indicating a strong linear relationship between the adsorption free energy of *OH and *OOH. The axial heteroatoms decoration of FeN4 with Cl, P, and S elements can effectively tune the binding energy of *OH and *OOH, with FeN4Cl being closer to the ideal electrocatalyst value than FeN4P and FeN4S, indicating that the Cl heteroatom decoration can enhance the electrocatalytic activity of FeN4 more effectively than both P and S heteroatoms. Thus, the evaluation of ORR activity via ΔG(*OH) is applicable for the axial heteroatom-decorated FeN4L, although the RDS is either the last step for pure FeN4 or the first step for various FeN4L.
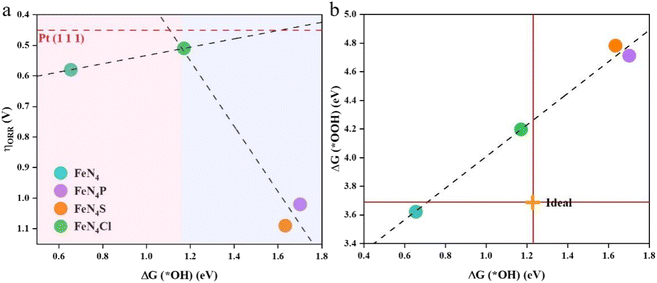 |
| | Fig. 4 (a) Volcano plot between the theoretical overpotential (ηORR) and the adsorption free energy of *OH for the ORR on various catalysts. The red dashed line represents the ORR overpotential on Pt(111). (b) The relationship between the adsorption free energy of *OOH and *OH. The ideal intersection point is marked with an orange cross. | |
The electronic properties of a substance have a significant impact on its catalytic activity. The study of the electronic properties of catalysts can provide a greater understanding of the origin of the catalytic activity. Therefore, various electronic properties were calculated and analysed to provide insight into the ORR electrocatalytic performances of FeN4, FeN4P, FeN4S, and FeN4Cl. The projected density of states (PDOS) was used to evaluate the d-band center of the materials, and the Bader charge and differential charge density of several intermediates were calculated as well.
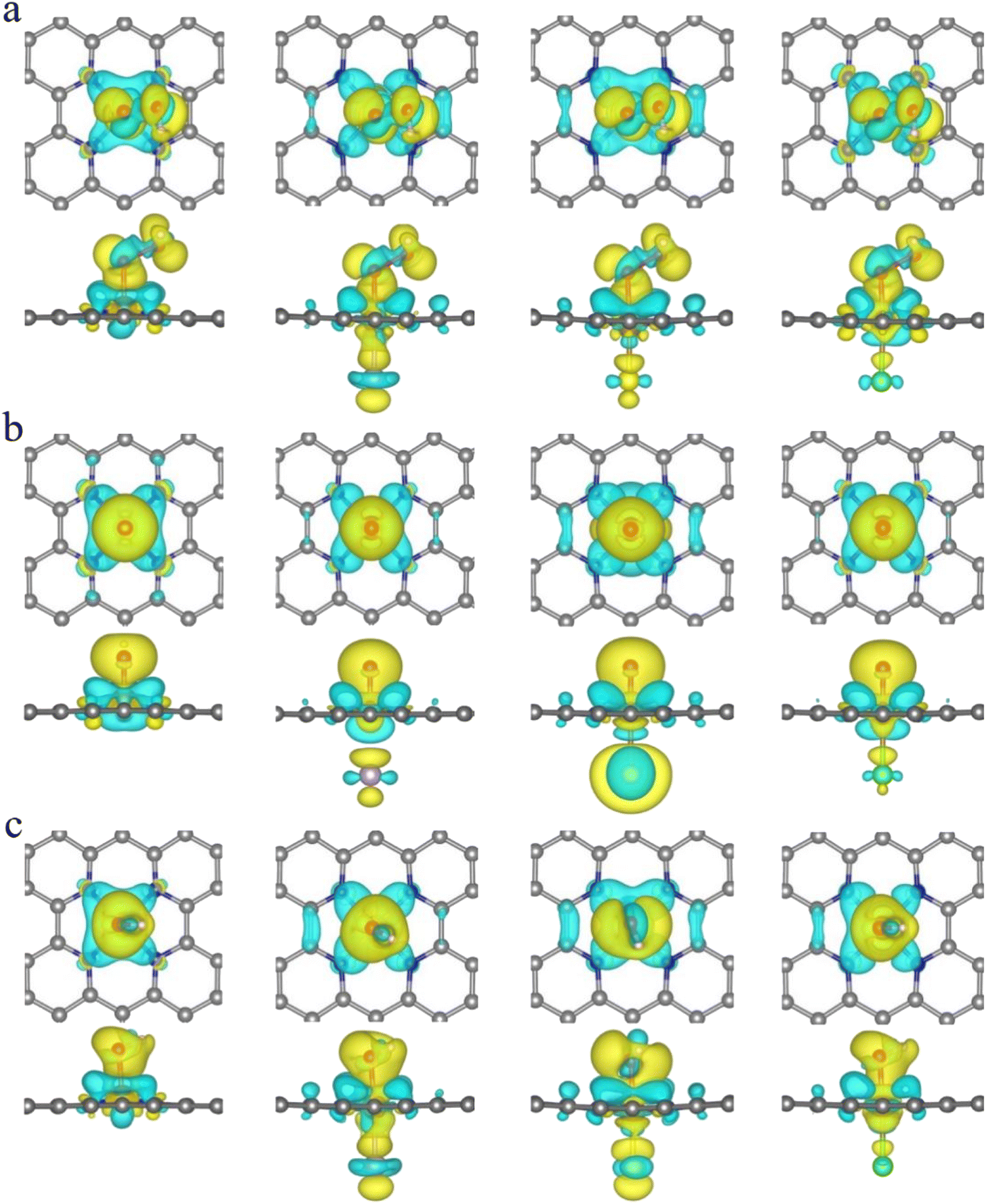
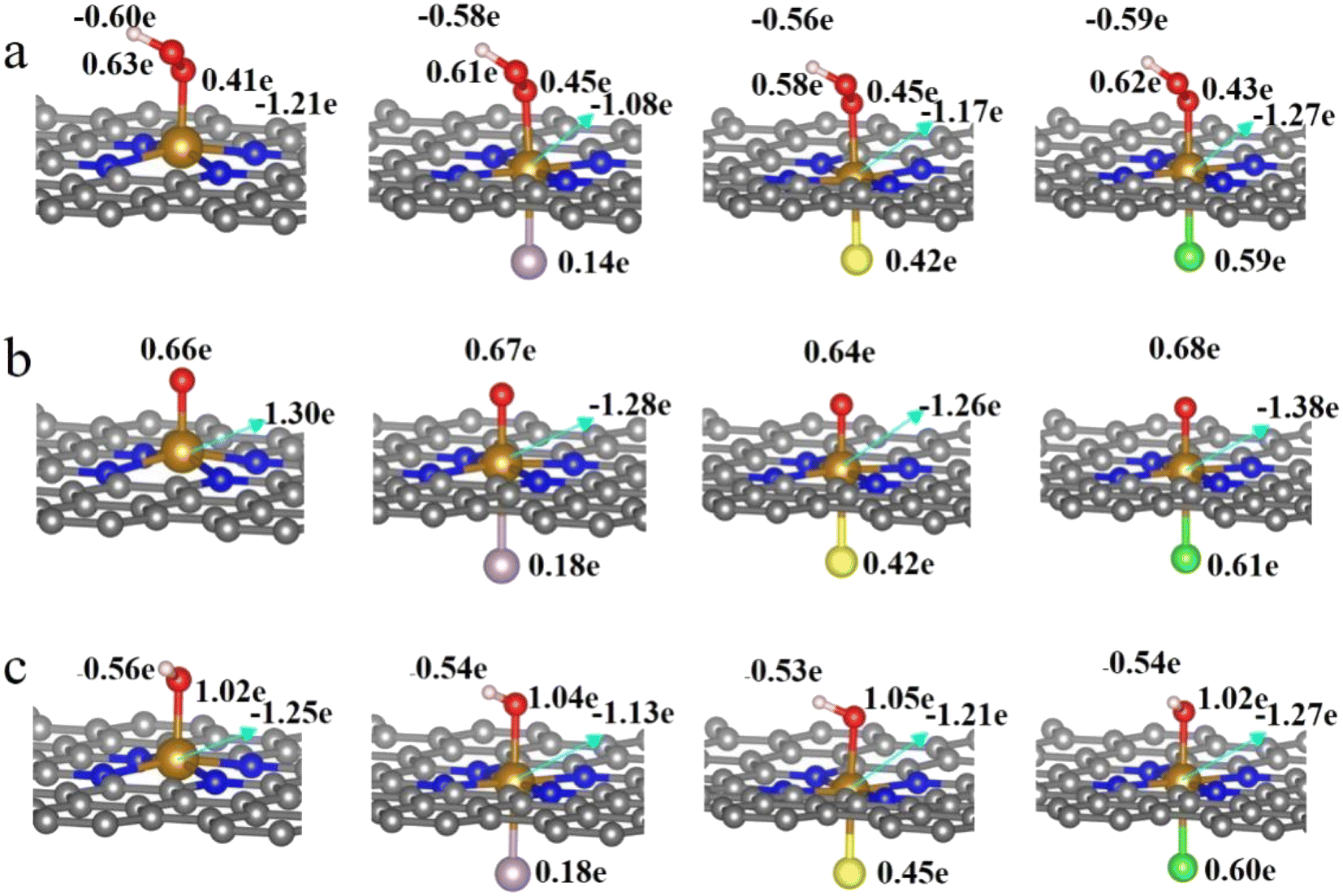
It is clear from the results of the differential charge density (Fig. 5) that charge transfer and redistribution occur between the ORR intermediates (*OOH, *O, *OH) and the FeN4, FeN4P, FeN4S, and FeN4Cl catalysts. The O atom shows the strongest electronegativity compared to the H, C, N and Fe atoms. Thus, as shown in Fig. 6, in the FeN4 system, the O atom receives electrons from the directly attached Fe and/or H atoms, with OH*, O* and OOH* receiving 0.41e, 0.66e and 1.02e, respectively. When FeN4 is axially decorated with P, S, and Cl, the electrons obtained by the O atom from the directly attached Fe and/or H atoms are very close to those of FeN4, differing only by ±0.01–0.04e.
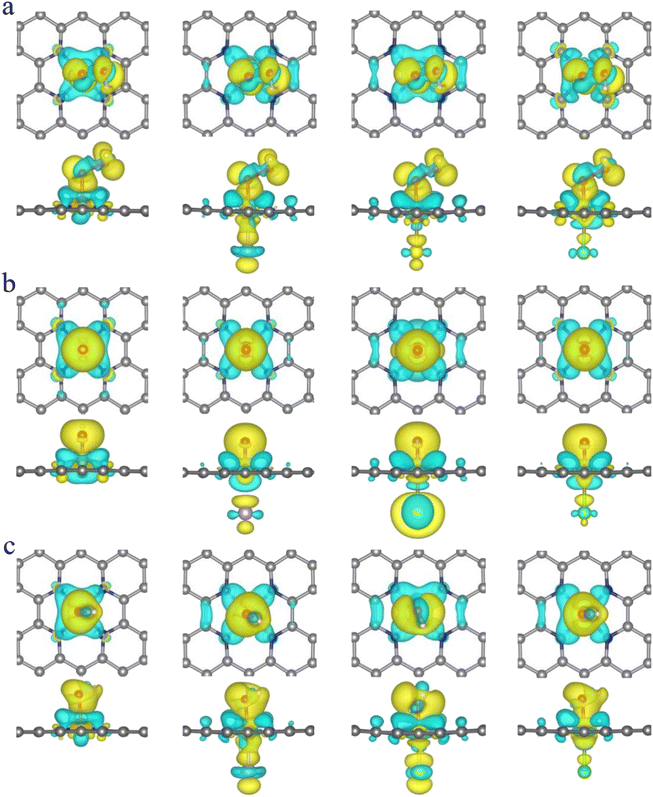 |
| | Fig. 5 The differential charge density of FeN4, FeN4P, FeN4S and FeN4Cl for (a) *OOH, (b) *O and (c) *OH, respectively. The isosurface is 0.002 e Å−3. | |
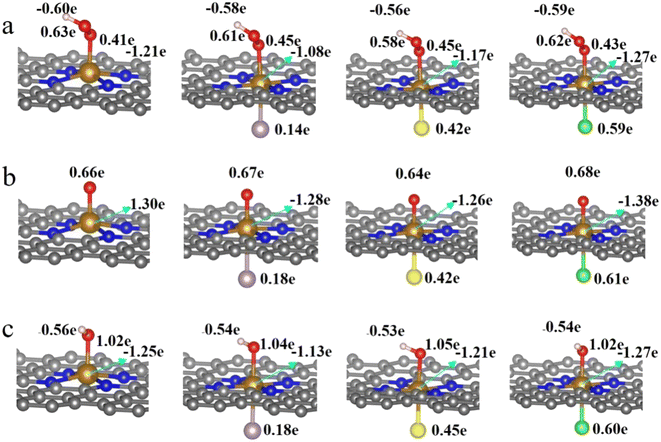 |
| | Fig. 6 Bader charge analysis of FeN4, FeN4P, FeN4S and FeN4Cl for (a) *OOH, (b) *O and (c) *OH, respectively. | |
The d-bands of the Fe atom in FeN4, FeN4P, FeN4S, and FeN4Cl are depicted in Fig. 7a. There are some electrons near the Fermi level; thus, it can be deemed that all of the FeN4L SACs possess good conductivity. Furthermore, the d-band center of Fe in FeN4 is −1.39 eV, which is closest to the Fermi energy level (EF) compared with the axial heteroatom-decorated FeN4L. The d-band centers of Fe in FeN4P, FeN4S, and FeN4Cl are all farther away from EF compared to that in FeN4, with the values of −2.41 eV, −2.13 eV and −1.89 eV, respectively. The d-band center theory has successfully described the chemisorption capacity and electrocatalytic properties of transition metals.39,40 Generally, a more negative d-band center indicates a weaker adsorption. The d-band center of FeN4Cl is closest to EF among all the axial heteroatom-decorated FeN4L, indicating that FeN4Cl is the best catalyst for the ORR intermediate adsorption. Fig. 7b shows the scaling relationship between the d-band center of Fe and ΔG(*OH). A negative correlation between the d-band center and ΔG(*OH) can be observed, indicating that the intensity of adsorption of OH on Fe atoms increases with the upward shift of the d-band center of the Fe atom, which is consistent with the classical d-band theory.39,40
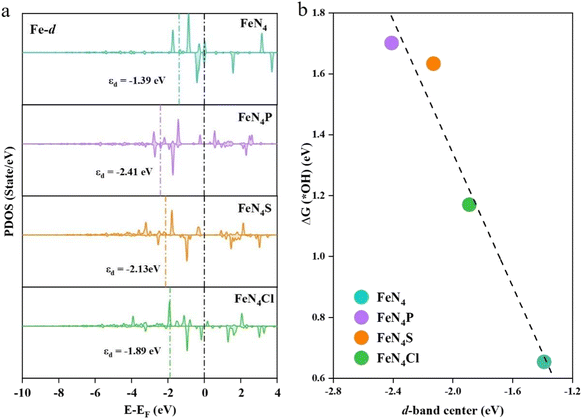 |
| | Fig. 7 (a) Projected density of states of the Fe atom on FeN4, FeN4P, FeN4S and FeN4Cl, and their corresponding d-band centers. (b) Relationship between the d-band center of the Fe atom in FeN4, FeN4P, FeN4S and FeN4Cl and ΔG(*OH). | |
Conclusions
We performed a detailed calculation and analysis of the structural stability, electronic property, and catalytic activity of both FeN4 and axial heteroatom-decorated FeN4L (L = P, S and Cl) using DFT calculations. Our results show that all of the axial heteroatom-decorated FeN4L SACs have good structural stability. The strong adsorption of the reaction intermediates on FeN4 resulted in suboptimal catalytic activity for ORR, and the RDS of ORR on FeN4 is the reduction of OH to H2O with an overpotential of 0.58 eV. For the axial heteroatom-decorated FeN4L, all of the heteroatoms can bind tightly with the central Fe atom in FeN4. Meanwhile, the RDS for FeN4P, FeN4S and FeN4Cl is the hydrogenation of O2 to OOH with the overpotentials of 1.02 V, 1.09 V and 0.51 V, respectively. FeN4Cl is nearly at the top of the volcano diagram, and the overpotential of ORR is closest to Pt(111). The axial Cl heteroatom decoration improves the ORR catalytic activity compared with pure FeN4, while the axial P and S heteroatoms have a negative effect on the ORR. The evaluation of ORR activity via ΔG(*OH) is applicable for the axial heteroatom-decorated FeN4L, although the RDS in the ORR is either the first step or the last step.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was financially supported by the Natural Science Foundation of Chongqing (No. 2023NSCQ-MSX1669).
References
- X. Xie, C. He, B. Li, Y. He, D. A. Cullen, E. C. Wegener, A. J. Kropf, U. Martinez, Y. Cheng and M. H. Engelhard, et al., Performance enhancement and degradation mechanism identification of a single-atom Co–N–C catalyst for proton exchange membrane fuel cells, Nat. Catal., 2020, 3, 1044–1054 CrossRef CAS.
- J. Dong, Z. Gao, W. Yang, A. Li and X. Ding, Adsorption characteristics of Co-anchored different graphene substrates toward O2 and NO molecules, Appl. Surf. Sci., 2019, 480, 779–791 CrossRef CAS.
- C. Han and Z. Chen, Adsorption properties of O2 on the unequal amounts of binary co-doped graphene by B/N and P/N: a density functional theory study, Appl. Surf. Sci., 2019, 471, 445–454 CrossRef CAS.
- C. Li, Z. Yu, H. Liu and M. Xiong, Synergetic contribution of Fe/Co and N/B dopants in mesoporous carbon nanosheets as remarkable electrocatalysts for zinc-air batteries, Chem. Eng. J., 2019, 371, 433–442 CrossRef CAS.
- Y. Tang, W. Chen, H. Zhang, Z. Shen, G. Zhao and X. Dai, Modulating the electronic structures and sensing properties of metal and non-metal atoms modified graphene sheets, Phys. E, 2019, 111, 206–217 CrossRef CAS.
- H. Niu, X. Wang, C. Shao, Y. Liu, Z. Zhang and Y. Guo, Revealing the oxygen reduction reaction activity origin of single atoms supported on g-C3N4 monolayers: a first-principles study, J. Mater. Chem. A, 2020, 8, 6555–6563 RSC.
- Z. Yang, Y. Wang, M. Zhu, Z. Li, W. Chen, W. Wei, T. Yuan, Y. Qu, Q. Xu and C. Zhao, et al., Boosting Oxygen Reduction Catalysis with Fe–N4 Sites Decorated Porous Carbons toward Fuel Cells, ACS Catal., 2019, 9, 2158–2163 CrossRef CAS.
- H. Zhang, H. T. Chung, D. A. Cullen, S. Wagner, U. I. Kramm, K. L. More, P. Zelenay and G. Wu, High-performance fuel cell cathodes exclusively containing atomically dispersed iron active sites, Energy Environ. Sci., 2019, 12, 2548–2558 RSC.
- X. Wan, X. Liu, Y. Li, R. Yu, L. Zheng, W. Yan, H. Wang, M. Xu and J. Shui, Fe–N–C electrocatalyst with dense active sites and efficient mass transport for high-performance proton exchange membrane fuel cells, Nat. Catal., 2019, 2, 259–268 CrossRef CAS.
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Single-Atom Catalysts: Synthetic Strategies and Electrochemical Applications, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- X. Li, W. Bi, M. Chen, Y. Sun, H. Ju, W. Yan, J. Zhu, X. Wu, W. Chu and C. Wu, et al., Exclusive Ni–N4 Sites Realize Near-Unity CO Selectivity for Electrochemical CO2 Reduction, J. Am. Chem. Soc., 2017, 139, 14889–14892 CrossRef CAS PubMed.
- B. Wang, X. Wang, J. Zou, Y. Yan, S. Xie, G. Hu, Y. Li and A. Dong, Simple-Cubic Carbon Frameworks with Atomically Dispersed Iron Dopants toward High-Efficiency Oxygen Reduction, Nano Lett., 2017, 17, 2003–2009 CrossRef CAS PubMed.
- A. Zitolo, V. Goellner, V. Armel, M.-T. Sougrati, T. Mineva, L. Stievano, E. Fonda and F. Jaouen, Identification of catalytic sites for oxygen reduction in iron- and nitrogen-doped graphene materials, Nat. Mater., 2015, 14, 937–942 CrossRef CAS PubMed.
- H. Zhang, S. Hwang, M. Wang, Z. Feng, S. Karakalos, L. Luo, Z. Qiao, X. Xie, C. Wang and D. Su, et al., Single Atomic Iron Catalysts for Oxygen Reduction in Acidic Media: Particle Size Control and Thermal Activation, J. Am. Chem. Soc., 2017, 139, 14143–14149 CrossRef CAS PubMed.
- P. Li, X. Qi, L. Zhao, J. Wang, M. Wang, M. Shao, J. S. Chen, R. Wu and Z. Wei, Hierarchical 3D Porous Carbon with Facilely Accessible Fe-N4 Single-Atom Sites for Zn-Air Batteries, J. Mater. Chem. A, 2022, 10, 5925–5929 RSC.
- C. Fu, X. Qi, L. Zhao, T. Yang, Q. Xue, Z. Zhu, P. Xiong, J. Jiang, X. An and H. Chen, et al., Synergistic cooperation between atomically dispersed Zn and Fe on porous nitrogen-doped carbon for boosting oxygen reduction reaction, Appl. Catal., B, 2023, 335, 122875 CrossRef CAS.
- J. Zhang, Z. Zhao, Z. Xia and L. Dai, A metal-free bifunctional electrocatalyst for oxygen reduction and oxygen evolution reactions, Nat. Nanotechnol., 2015, 10, 444–452 CrossRef CAS PubMed.
- R. Li, Z. Wei and X. Gou, Nitrogen and Phosphorus Dual-Doped Graphene/Carbon Nanosheets as Bifunctional Electrocatalysts for Oxygen Reduction and Evolution, ACS Catal., 2015, 5, 4133–4142 CrossRef CAS.
- Y. Lin, P. Liu, E. Velasco, G. Yao, Z. Tian, L. Zhang and L. Chen, Fabricating Single-Atom Catalysts from Chelating Metal in Open Frameworks, Adv. Mater., 2019, 31, e1808193 CrossRef PubMed.
- K. Liu, Z. Qiao, S. Hwang, Z. Liu, H. Zhang, D. Su, H. Xu, G. Wu and G. Wang, Mn- and N- doped carbon as promising catalysts for oxygen reduction reaction: theoretical prediction and experimental validation, Appl. Catal., B, 2019, 243, 195–203 CrossRef CAS.
- R. Venegas, F. J. Recio, C. Zuniga, M. Viera, M. P. Oyarzun, N. Silva, K. Neira, J. F. Marco, J. H. Zagal and F. Tasca, Comparison of the catalytic activity for O(2) reduction of Fe and Co MN4 adsorbed on graphite electrodes and on carbon nanotubes, Phys. Chem. Chem. Phys., 2017, 19, 20441–20450 RSC.
- A. Rasool, I. Anis, S. A. Bhat and M. A. Dar, Optimizing the NRR activity of single and double boron atom catalysts using a suitable support: a first principles investigation, Phys. Chem. Chem. Phys., 2023, 25, 22275–22285 RSC.
- D. Wu, J. Wu, P. Lv, H. Li, K. Chu and D. Ma, Atomically Dispersed Alkaline-Earth Metals as Active Centers for CO2 Electroreduction to Exclusively Produce Formate, Small Struct., 2023, 4, 2200358 CrossRef CAS.
- A. Hassan and M. A. Dar, Understanding the activity of single atom catalysts for CO2 reduction to C2 products: a high throughput computational screening, New J. Chem., 2023, 47, 7225–7231 RSC.
- R. Khanam, A. Hassan, Z. Nazir and M. A. Dar, Nickel single atom catalyst supported on the gallium nitride monolayer: first principles investigations on the decisive role of support in the electrocatalytic reduction of CO2, Sustainable Energy Fuels, 2023, 7, 5046–5056 RSC.
- Q. Zhou, X. Qi, Y. Zhou, J. Li, J. Jiang, H. Li, X. Niu, R. Wu and J. S. Chen, Zinc-cobalt-bimetallic catalyst on three-dimensional ordered nitrogen-doped porous carbon for high-performance lithium-selenium batteries, J. Alloys Compd., 2023, 942, 168944 CrossRef CAS.
- K. Chen, G. Wang, Y. Guo, D. Ma and K. Chu, Iridium single-atom catalyst for highly efficient NO electroreduction to NH3, Nano Res., 2023, 16, 8737–8742 CrossRef CAS.
- K. Chen, J. Wang, H. Zhang, D. Ma and K. Chu, Self-Tandem Electrocatalytic NO Reduction to NH3 on a
W Single-Atom Catalyst, Nano Lett., 2023, 23, 1735–1742 CrossRef CAS PubMed.
- S. Impeng, A. Junkaew, P. Maitarad, N. Kungwan, D. Zhang, L. Shi and S. Namuangruk, A MnN4 moiety embedded graphene as a magnetic gas sensor for CO detection: a first principle study, Appl. Surf. Sci., 2019, 473, 820–827 CrossRef CAS.
- Z. Liang, M. Luo, M. Chen, X. Qi, J. Liu, C. Liu, S. G. Peera and T. Liang, Exploring the oxygen electrode bi-functional activity of Ni–N–C-doped graphene systems with N, C co-ordination and OH ligand effects, J. Mater. Chem. A, 2020, 8, 20453–20462 RSC.
- D. Kan, R. Lian, D. Wang, X. Zhang, J. Xu, X. Gao, Y. Yu, G. Chen and Y. Wei, Screening effective single-atom ORR and OER electrocatalysts from Pt decorated MXenes by first-principles calculations, J. Mater. Chem. A, 2020, 8, 17065–17077 RSC.
- Z. Xue, X. Zhang, J. Qin and R. Liu, TMN4 complex embedded graphene as bifunctional electrocatalysts for high efficiency OER/ORR, J. Energy Chem., 2021, 55, 437–443 CrossRef CAS.
- G. Kresse and J. Hafner, Ab initio molecular dynamics for liquid metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- P. E. Blochl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- A. Kulkarni, S. Siahrostami, A. Patel and J. K. Norskov, Understanding Catalytic Activity Trends in the Oxygen Reduction Reaction, Chem. Rev., 2018, 118, 2302–2312 CrossRef CAS PubMed.
- X. Liu, Y. Jiao, Y. Zheng, K. Davey and S.-Z. Qiao, A computational study on Pt and Ru dimers supported on graphene for the hydrogen evolution reaction: new insight into the alkaline mechanism, J. Mater. Chem. A, 2019, 7, 3648–3654 RSC.
- Y. Wu, C. Li, W. Liu, H. Li, Y. Gong, L. Niu, X. Liu, C. Sun and S. Xu, Unexpected monoatomic catalytic-host synergetic OER/ORR by graphitic carbon nitride: density functional theory, Nanoscale, 2019, 11, 5064–5071 RSC.
|
| This journal is © The Royal Society of Chemistry 2024 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab,
Li Li*c and
Andreu Cabot
*ab,
Li Li*c and
Andreu Cabot