
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStable cycling and low-temperature operation utilizing amorphous carbon-coated graphite anodes for lithium-ion batteries†
Pengfei Xiaoa,
Zhongming Wanga,
Kecheng Long a,
Jixu Yanga,
Xinsheng Liua,
Canhui Linga,
Libao Chenab and
Lin Mei*a
a,
Jixu Yanga,
Xinsheng Liua,
Canhui Linga,
Libao Chenab and
Lin Mei*a
aState Key Laboratory of Powder Metallurgy, Central South University, Changsha, 410083, P. R. China. E-mail: meilin@csu.edu.cn
bNational Energy Metal Resources and New Materials Key Laboratory, Central South University, Changsha, 410083, P. R. China
First published on 24th April 2024
Abstract
With the continuous expansion of the lithium-ion battery market, addressing the critical issues of stable cycling and low-temperature operation of lithium-ion batteries (LIBs) has become an urgent necessity. The high anisotropy and poor kinetics of pristine graphite in LIBs contribute to the formation of precipitated lithium dendrites, especially during rapid charging or low-temperature operation. In this study, we design a graphite coated with amorphous carbon (GC) through the Chemical Vapor Deposition (CVD) method. The coated carbon layer at the graphite interface exhibits enhanced reaction kinetics and expanded lithium-ion diffusion pathways, thereby reduction in polarization effectively alleviates the risk of lithium precipitation during rapid charging and low-temperature operation. The pouch cell incorporating GC‖LiCoO2 exhibits exceptional durability, retaining 87% of its capacity even after 1200 cycles at a high charge/discharge rate of 5C/5C. Remarkably, at −20 °C, the GC-2 maintains a specific capacity of 163 mA h g−1 at 0.5C, higher than that of pristine graphite (65 mA h g−1). Even at −40 °C, the GC-2‖LiCoO2 pouch cell still shows excellent capacity retention. This design realizes the practical application of graphite anode in extreme environments, and have a promising prospect of application.
1. Introduction
In recent years, lithium-ion batteries have been widely used in electric vehicles and portable devices.1–5 This shift in focus has also placed higher demands on lithium-ion batteries.6 The depletion of non-renewable resources has led to an increase in the price of oil and the cost of cars, which has led to a shift in attention from gasoline-powered vehicles to lower-cost electric vehicles.7,8 Currently, individuals must consider range and charging speed as the two primary factors when choosing an electric vehicle.9 Recent advances in high specific energy electrode materials have largely alleviated the challenge of range anxiety, but the focus is now on solving the problem of charging speed and environmental adaptability, such as temperature fluctuation.10,11Graphite is the most commonly used anode material for commercial lithium-ion batteries due to its high theoretical specific capacity, low working potential, low cost and high stability.12–14 However, the poor kinetic properties of graphite make it susceptible to polarization during fast charging and low-temperature charging, which can lead to the precipitation of lithium metal on the surface of anodes, posing significant safety risks. Therefore, there is an immediate need for modifications to graphite materials.
Presently, two main strategies are employed for graphite modification, with one involving alterations to the structure characteristics of graphite, such as reducing the particle size,11 enlarging the layer spacing,15 porous structure,16 doping,17 introducing defects,18 etc., which can shorten the diffusion path of lithium-ion and accelerate the diffusion of lithium-ion. However, this usually destroys the original structure of graphite, increases the specific surface area, and causes instability in the cycling process and reversible capacity loss. The other improving approach involves surface modification of graphite, such as carbon layers,19–21 metal oxides,22 polymers,23 and so on. The coating layers on graphite can isolate it from the electrolyte, thereby reducing the excessive decomposition and co-embedding of the electrolyte. Zhang et al. used turbulent layer carbon for graphite coating and the improved fast-charging and low-temperature performance were attributed to the isotropy of the carbon layers and larger layer spacing, which can provide more channels for lithium-ion and accelerate lithium-ion into the graphite interlayer.24 Lin et al. fabricated an amorphous carbon layer on graphite by carbonization of glucose and achieved stable cycle life and superior multiplicity performance.25 Although these studies have made breakthroughs in the cycle stability and low-temperature performance of graphite anodes, solution-based carbon coating approach involve the impurity components. Additionally, it is an unavoidable issue of uneven coating layer leading to uncontrollable coating thickness on graphite.
In this work, we employ CVD to introduce an amorphous carbon layer on the surface of graphite. The amorphous carbon layer on graphite serves as a diffusion channel for lithium-ion, accelerates the embedding of lithium-ion, and reduces the polarization during high power density and low-temperature processes, which has great application prospects for improving performance of lithium-ion batteries. The obtained amorphous carbon layer is uniform and dense compared to the conventional coating method on graphite.26 Furthermore, nanoscale tuning of the thickness of amorphous carbon layer can be realized by precisely controlling the CVD deposition time. The amorphous carbon layer on graphite improves the anisotropy of graphite, provide more lithium-ion diffusion channels, and increase the diffusion rate of lithium-ion in graphite. The introduced carbon layer shows pseudocapacitive behaviors, which accelerate charge transfer, effectively mitigating polarization. As a result, the obtained graphite anode with amorphous carbon layer (GC) exhibits excellent stable cycling performance and low-temperature environment adaptability which solved the phenomenon of lithium precipitation of anode material during fast charging and low temperature charging. When assembled with a LiCoO2 cathode to construct pouch cells, the GC anode illustrates a high specific capacity retention of 87% after 1200 cycles at a high rate of 5C. Additionally, when coupled with a low-temperature electrolyte, the GC anode exhibits a higher capacity (163 mA h g−1) than pristine graphite anode (65 mA h g−1) at 0.5C under −20 °C. And the GC‖LiCoO2 pouch cell maintains excellent charge ability under −40 °C. The graphite with amorphous carbon layer, as prepared in this study, demonstrates significant potential for application in extreme conditions.
2. Experimental section
2.1. Material preparation
The carbon-coated graphite powders were obtained with a tumbling CVD process. During the deposition process, the graphite particles were tumbled in the furnace to obtain a uniform coating. The source gas was liquefied natural gas (LNG) at a flow rate of 0.1 L min−1, and the experiments were carried out at 900 °C for 1, 2 and 3 hours, respectively. Three samples with different coating times were obtained and labeled as GC-1, GC-2, and GC-3, respectively.2.2. Characterization
The surface morphology and microstructure were observed by scanning electron microscopy (SEM, ZEISS Sigma 300) and transmission electron microscopy (TEM, JEOL JEM-F200). The phase composition of the samples was characterized by X-ray diffraction (XRD, Panalytical Empyrean, Cu Kα radiation). The Raman spectra were obtained by a Thermo Scientific DXR3 Raman Microscope with 532 nm laser light. N2 adsorption/desorption at 77.3 K (MicromeriticsTristar 3000) was employed to determine the specific surface area of the samples. The particle size distribution of the samples was determined by the dry method using a laser particle size analyser (Malvern Mastersizer 2000). The conductivity of the samples was measured by the four-point probe method.2.3. Electrochemical measurements
The working electrodes were prepared by coating a mixture of the active material with Super P conductive agent and binder (LA133) in a weight ratio of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 dissolved in deionized water on a copper foil and then drying at 80 °C under vacuum overnight. The dried electrode sheets were then cut into small discs with a diameter of 12 mm and the mass loading of the active material was about 1.7 mg cm−2. CR2016 coin-type cells were assembled in an argon-filled glovebox with less than 0.1 ppm O2 and 0.1 ppm H2O, while a lithium-metal disk as the counter electrode and a Celgard 2400 membrane as the separator. Meanwhile, LiPF6 (1.0 M) in EC/EMC/FEC (3:7:1 vol%) was used as the electrolyte at 30 °C test, and LiTFSI (1 M) in DOL at −20 °C test. The charge/discharge performance of the half-cells was tested in the voltage window of 0.01–2 V at 30 °C. The electrochemical impedance spectroscopy (EIS) analyses were carried out from 100 kHz to 0.1 Hz at a voltage amplitude of 10 mV. The cyclic voltammetry (CV) profiles were tested at a scan rate of 0.1–2 mV s−1. For galvanostatic intermittent titration technique (GITT) measurement, the pulsed current was 0.1C. The cell was discharged for 0.5 hour followed by 2 hours rest to reach the equilibrium state.
:1:1 dissolved in deionized water on a copper foil and then drying at 80 °C under vacuum overnight. The dried electrode sheets were then cut into small discs with a diameter of 12 mm and the mass loading of the active material was about 1.7 mg cm−2. CR2016 coin-type cells were assembled in an argon-filled glovebox with less than 0.1 ppm O2 and 0.1 ppm H2O, while a lithium-metal disk as the counter electrode and a Celgard 2400 membrane as the separator. Meanwhile, LiPF6 (1.0 M) in EC/EMC/FEC (3:7:1 vol%) was used as the electrolyte at 30 °C test, and LiTFSI (1 M) in DOL at −20 °C test. The charge/discharge performance of the half-cells was tested in the voltage window of 0.01–2 V at 30 °C. The electrochemical impedance spectroscopy (EIS) analyses were carried out from 100 kHz to 0.1 Hz at a voltage amplitude of 10 mV. The cyclic voltammetry (CV) profiles were tested at a scan rate of 0.1–2 mV s−1. For galvanostatic intermittent titration technique (GITT) measurement, the pulsed current was 0.1C. The cell was discharged for 0.5 hour followed by 2 hours rest to reach the equilibrium state.
The GC-2 electrode for pouch cell consisted of 94 wt% active materials, 3 wt% LA133 binder, and 3 wt% acetylene black conductive additive. The mass loading of the electrode reached ∼11 mg cm−2 (double-side) and the compaction density was 1.2 g cm−3. The LiCoO2 electrode for pouch cell consisted of 95 wt% active material, 1.8 wt% PVDF binder, and 3.2 wt% acetylene black. It possessed a mass loading of ∼18 mg cm−2 (double-side) and a compaction density of 3.0 g cm−3. The pouch cell comprised stacked layers of cathode, separator, and anode, all of which were packaged by aluminum plastic film. The pouch cells were assembled in a drying room. Using a turn stacking process, the 1 A h GC-2‖LiCoO2 pouch cells were assembled by 8 pieces of cathode electrodes with a size of 5.5 cm × 7.9 cm and 9 pieces of anode electrodes (5.7 cm × 8.2 cm). The electrolyte was LB-091 (buy from CAPCHEM Co. Ltd). After the electrolyte filling, the pouch cells were vacuumed before electrochemical test. The cut-off voltage ranges for pouch cells were 3.0–4.2 V (30 °C) and 2.4–4.2 V (−40 °C). During the charging process, the pouch cells were constant-current charge to 4.2 V and then constant-voltage charge to cut-off current (20 mA).
3. Results and discussion
The surface morphology of graphite and carbon-coated graphite was analyzed by SEM. As shown in Fig. 1a, the surface of pristine graphite is rough, and scale-like graphite lamellae structure is visible. The exposed graphite sheets directly contact with the electrolyte, and during the repeated lithiation/de-lithiation process, the graphite sheets flake off due to the co-insertion of solvent molecules, ultimately leading to capacity loss and battery failure. Though carbon coating, the surface of the sample becomes smooth and unstable structures are covered. To investigate the effect of coating thickness of amorphous carbon layer, we set up a gradient experiment with different deposition times to control the thickness of the samples. When deposited for 1 hour, it is apparent that the surface of samples is not sufficiently encapsulated and the unstable structure still exists (Fig. S1a†). As the deposition time is increased to 2 hours, the unstable structure was completely covered (Fig. 1b). The cross-section SEM (Fig. S1c and d† and 1c) demonstrated that the thickness of coating layer on graphite anode increased as prolong CVD deposition time.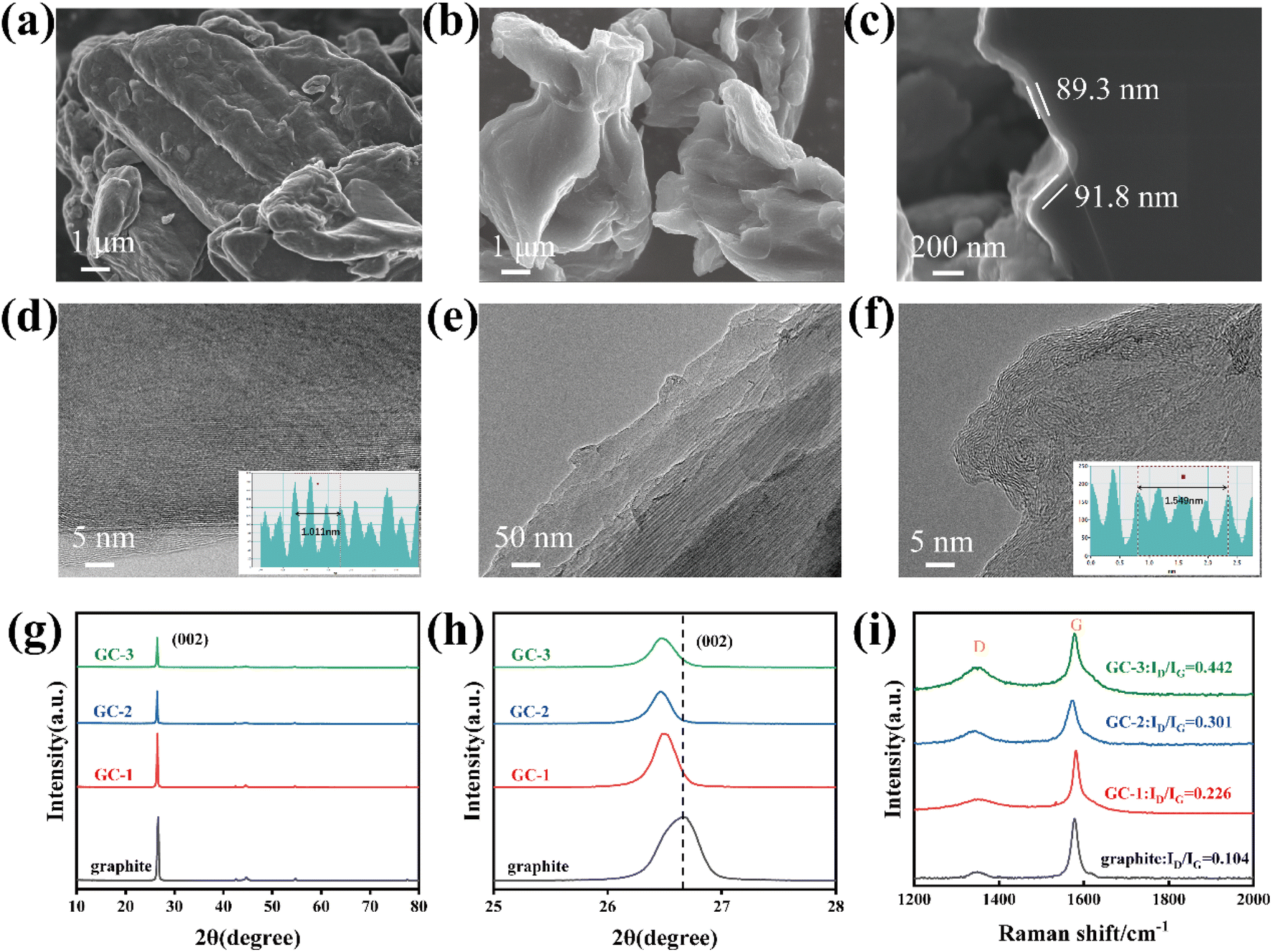 | ||
| Fig. 1 Morphological and spectral characterizations of samples: SEM images of pristine graphite (a) and GC-2 (b); cross-section SEM of GC-2 (c); TEM images of pristine graphite (d) and GC-2 (e and f); XRD patterns (g and h); Raman spectra (i). | ||
In order to reveal more information about the surface microstructure of the coatings, TEM was used for further observation of the composites. From the TEM images, it demonstrates that the edge region of pristine graphite has clear lattice stripes with an average layer spacing of 0.337 nm (Fig. 1d), while the carbon layer on the GC-2 surface has a chaotic layer structure (Fig. 1e and f), which is consistent with the typical characteristics of amorphous carbon.27 The average layer spacing of the carbon layer is 0.387 nm, which is larger than that of the pristine graphite and is favorable for the embedding of lithium-ion.
The particle size distribution of the samples before and after coating was then examined by dry powder laser particle sizer, and it can be seen that the D50 diameter of the particles gradually increases with the increase of deposition time (Fig. S2a†). The specific surface area of the samples was tested by BTE, and the specific surface area of GCs samples with coating were decreased, which was due to the covering of defective sites on the surface of the pristine graphite (Fig. S2b†). The conductivity was tested by the four-point probe method at 30 MPa, the electrical conductivity of graphite is 144.82 S cm−1 and the GC-2 is 148.8 S cm−1. This contrasts to the previous literature reporting24 that the conductivity of graphite decreases after amorphous carbon coating.
The XRD patterns of the samples before and after coating were analyzed in Fig. 1g. The positions of the diffraction peaks of all the carbon-coated materials are consistent with those of the pristine graphite, indicating that the crystal structure of the materials was not damaged after coating. The (002) diffraction peak intensities of the carbon-coated materials are lower than those of the pristine graphite, which is due to the low crystallinity of the carbon layer on the surface of graphite. The enlarged view of the XRD patterns shows that the (002) diffraction peak of the GCs shifted to a lower diffraction angle, indicating a larger layer spacing of the surface carbon layer28 (Fig. 1h). It facilitates the promotion of the rapid embedding/de-embedding of lithium-ion and alleviates the charge transport resistance during lithiation/de-lithiation, thus contributing to the improvement of cycling stability and rate performance. Notably, due to the anisotropy of graphite, lithium-ion can only enter the graphite interlayers from the end faces of graphite.18 The isotropy is improved by the coating modification, which is manifested by the change of OI value (the ratio of peak intensities of the (004) and (110) crystal faces). As shown in Fig. S3,† the modified graphite has a smaller OI value, which corresponds to a larger (110) area, to increase the lithium-ion embedded active sites.
According to the Raman spectroscopy results (Fig. 1i), the peaks located at 1352 cm−1 (D-band) and 1583 cm−1 (G-band) are attributed to sp3-type disordered and sp2-type graphitized carbon.29 The peak intensity ratio of the D and G bands (ID/IG) represents the degree of disordering of the carbon material.30 The degree of disorder of the coated graphite significantly increases, indicating more defects and electrochemically active sites. In addition, the GCs possess the same XPS survey spectra as graphite, indicating that no new elements have been introduced into the amorphous carbon (Fig. S4†).
GCs-based half cells were assembled to analyze the electrochemical performances of anodes. According to the first charge/discharge curves, the GCs show similar reversible specific capacity as pristine graphite and the initial coulombic efficiency is not reduced by introducing the carbon layer (Fig. 2a). The rate performance of GCs anode was tested with different current densities (Fig. 2b), the GCs samples show higher discharge specific capacity than pristine graphite. Fig. 2c shows the charge/discharge curves of pristine graphite and GCs at 0.5C. GC-2 exhibits the highest discharge specific capacity of 338.03 mA h g−1, higher than that of pristine graphite (300.08 mA h g−1). Especially, the GC-2 anode exhibits a superior coulombic efficiency at various current densities, exceedingly even in presently reported results of modifications of graphite anodes,23–25,31 as shown in Fig. S5.†
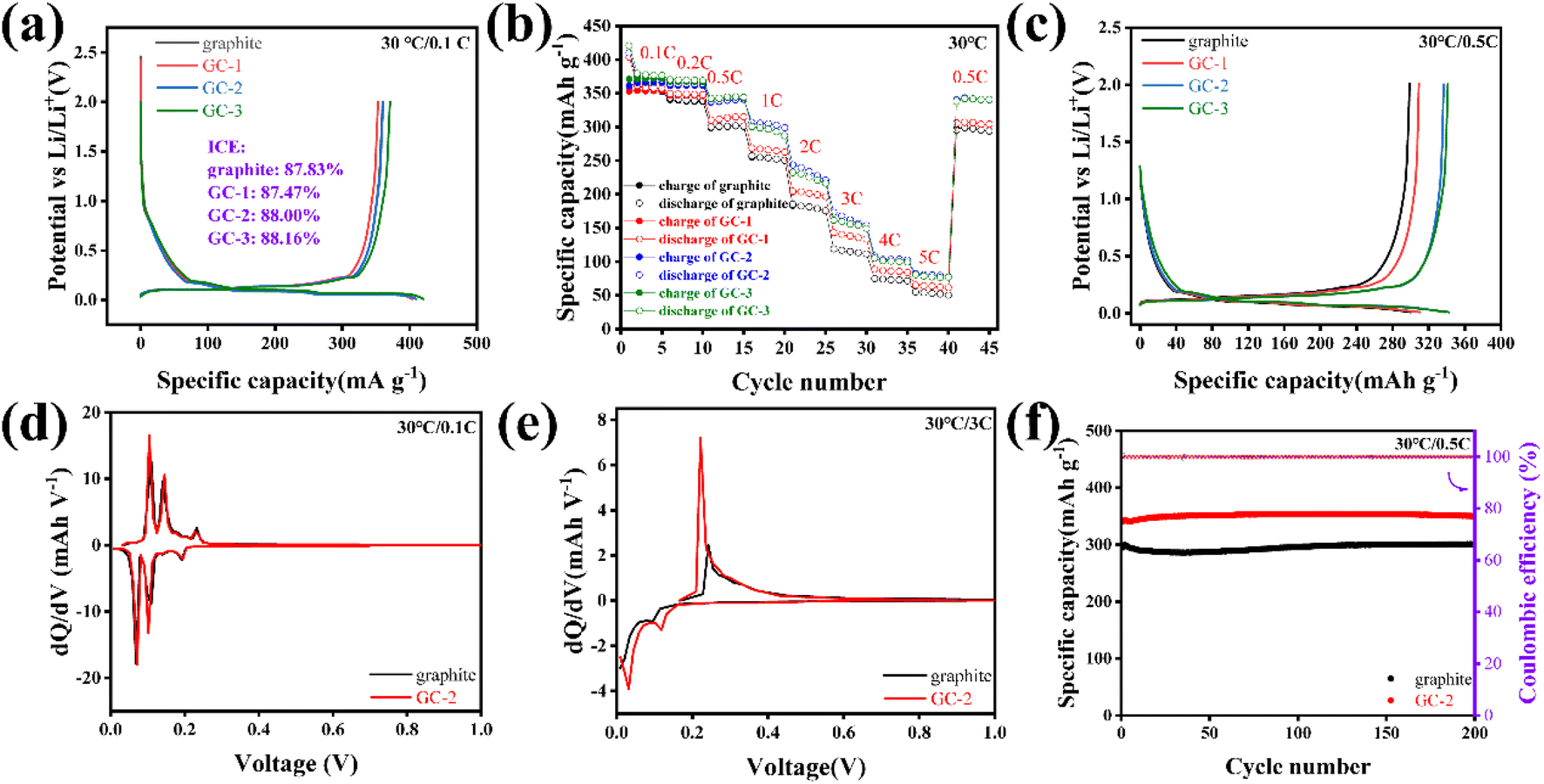 | ||
| Fig. 2 Electrochemical performance of pristine graphite and GCs: (a) the first charge/discharge curves and initial coulombic efficiency at 0.1C; (b) rate performance from 0.1 to 5C (1C = 370 mA h g−1); (c) charge/discharge curves at 0.5C; dQ/dV curves of pristine graphite and GC-2 at 0.1C (d) and 3C (e); (f) cycle performance of pristine graphite and GC-2 at 0.5C. | ||
The DQ/DV curves of pristine graphite and GC-2 were then analyzed. The distance between the oxidation and reduction peaks of pristine graphite gradually increases with increasing current density, indicating greater polarization. Even at a rate of 3C, GC-2 still has obvious redox peaks, while those of pristine graphite almost disappear, indicating that the surface coating strategy can greatly reduce the polarization (Fig. 2d and e and S6†). The half cells of pristine graphite and GC-2 after conducting rate tests are selected to proceed with the 0.5C cycling test (Fig. 2f). After 200 cycling tests at 0.5C, GC-2 anode exhibits the higher discharge specific capacity of 350.2 mA h g−1 compared to pristine graphite, with no clearly capacity drop observed. For GC-2 anodes, the graphite substrate is effectively protected by amorphous carbon coating, facilitating increased Li-ion reversible intercalation and stable cycling process.
In order to better clarify the enhancement mechanism of the carbon-coated graphites, various electrochemical characterization techniques were used to analyze the surface kinetic behavior of the samples. The EIS of GCs and pristine graphite were performed in Fig. 3a and S7.† After fitting, the charge transfer impedance (Rct) of GC-2 is significantly smaller than that of pristine graphite, indicating that GC-2 possesses better interface dynamics. To better understand the reasons for the performance improvements in GC-2, the diffusion coefficients of lithium-ion in the bulk phase are calculated by fitting the low frequency region of the electrochemical impedance spectra using the following equation:32
| DLi+ = R2T2/2A2n4F4CLi+2δ2 |
| Z′ = Rs + Rct + δω−1/2 |
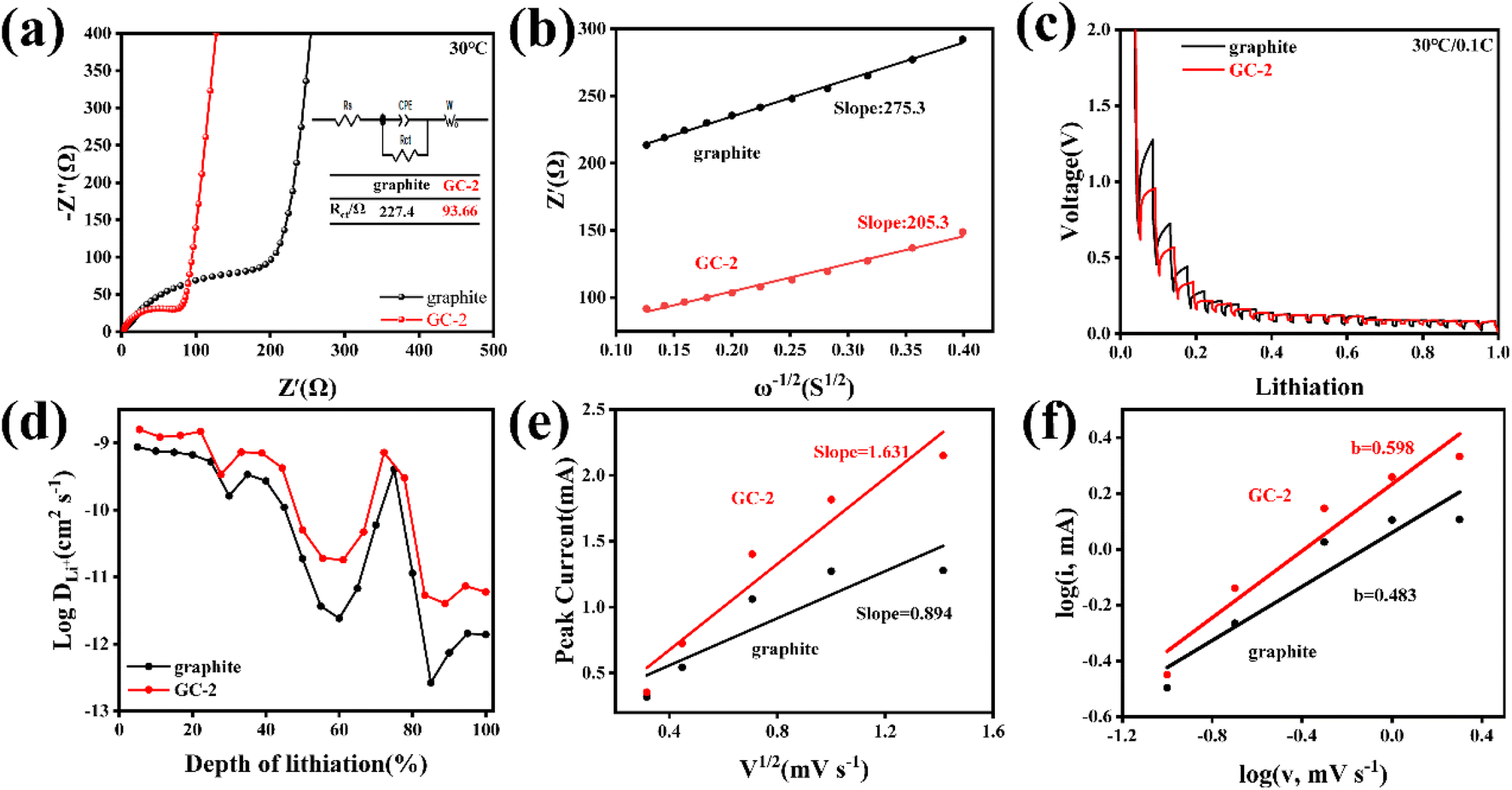 | ||
| Fig. 3 Kinetic analysis of the electrochemical behavior for pristine graphite and GC-2 electrodes: (a) pristine EIS Nyquist curves; (b) the relationship curves between Z′ and ω−1/2 in the low-frequency region. (c) GITT profiles (current pulse at 0.1C for 30 min followed by 2 h relaxation). (d) The lithium-ion diffusion coefficient obtained via the GITT technique. (e) Relationship between the square root of the scan rate and the peak current (the slope is related to the relative electrochemically active surface area). (f) The b value using the relationship between the peak current and the sweep rate. | ||
As shown in Fig. 3b, the δ value of the graphite and GC-2 electrodes are 275.3 Ω S−1/2 and 205.3 Ω S−1/2, respectively. The average lithium-ion diffusion coefficient of GC-2 is calculated to be 6.59 × 10−13 cm2 s−1, which is higher than 3.66 × 10−13 cm2 s−1 of the pristine graphite. The results show that the introduction of the carbon coating on the graphite surface achieves faster diffusion kinetics of lithium-ion and thus improves the rate performance.
In addition, the galvanostatic intermittent titration technique (GITT) was used to more intuitively observe the diffusion kinetics during lithium insertion. As shown in Fig. 3c, GC-2 shows less voltage hysteresis. And GC-2 was calculated to possess higher lithium-ion diffusion coefficients in all lithiation states, indicating that the carbon coating can improve diffusion kinetics (Fig. 3d). The diffusion coefficients can be calculated according to the following equation:34

Among them, τ is the constant current discharging time, mB is the mass of the active material, VM is the molar volume, MB is the molar mass, S is the electrode area, ΔEs is the voltage change caused by the pulse; ΔEτ is the voltage change of constant current charging (discharging).
To further reveal the reason for the enhanced interface reaction kinetics of the GC-2, CV curves were measured at different scan rates ranging from 0.1 to 2 mV s−1 (Fig. S8a and b†). At different scanning rates, the redox peak currents of GC-2 are larger than those of pristine graphite, indicating higher electrochemical reactivity. In addition, the electrochemically active area can be calculated by the relationship between scan rate and peak current from the Randles–Sevcik equation:32
| Ip = (2.69 × 105)n3/2AD1/2C0v1/2 |
The CV curves at different sweep speeds can also be used to analyze the lithium-ion storage mechanism of the GC-2 and graphite. The relationship between the peak current (i) and scanning rate (v) is defined as:35
| i = avb |
| log(i) = blog(v) + log(a) |
The value of b can be obtained via the slope of the log(v) versus the log(i) plot. When b = 0.5, the process is diffusion-controlled. When b = 1, the process is surface-controlled.36 As shown in Fig. 3f, for the pristine graphite electrode, the b value is 0.483 which represents typical diffusion-controlled processes. In contrast, the b value of GC-2 is 0.598, which suggests that the lithiation process is in part related to the pseudocapacitive ion storage behavior that causes the fast kinetics.37 It demonstrates that the introduction of the carbon layer improves the lithium storage behavior of graphite and enhances the adsorption capacity of lithium-ion, which induces a faster charge transfer process and greatly improves the kinetics of the graphite surface reaction, resulting in an attractive fast charging capability.
The ex situ XRD was used to reveal the structural stability of anodes during the cycling process. In the first charge/discharge process, we can observe that the (002) peak disappeared when discharged to 0.01 V, indicating the formation of graphite intercalation compound. When returning to 2 V, the (002) peak of pristine graphite is shifted to a smaller angle (Fig. 4a), demonstrating poor structural reversibility of the pristine graphite. Whereas GC-2 has good reversibility (Fig. 4b), which enhance its electrochemical performance. The morphological evaluation of pristine graphite and GC-2 was investigated by SEM after 300 cycles at 0.5C. As shown in Fig. 4c and d, the surface of the GC-2 electrode after cycling is still smooth with almost no dead lithium visible, whereas the pristine graphite has a rough surface decorated with dead lithium. This suggests that the carbon layer can greatly mitigate lithium deposition during cycling and improve cycling stability. To further evaluate the viability of the GC-2 in practical applications, we assembled 1 A h pouch cells coupling with a LiCoO2 cathode to cycle at a high rate of 5C. As shown in Fig. 4e, the pouch cell shows excellent capacity retention of 87% after 1200 cycles at 5C while pristine graphite is 81% (Fig. S9a†). As shown in Fig. S9b,† the constant current charge ratio of GC-2‖LiCoO2 at 5C is 94.18%, higher than the graphite (91.72%). As a result, GC-2 has a high-power capability and high cycling stability, and its practical application illustrates the feasibility of the coating modification.
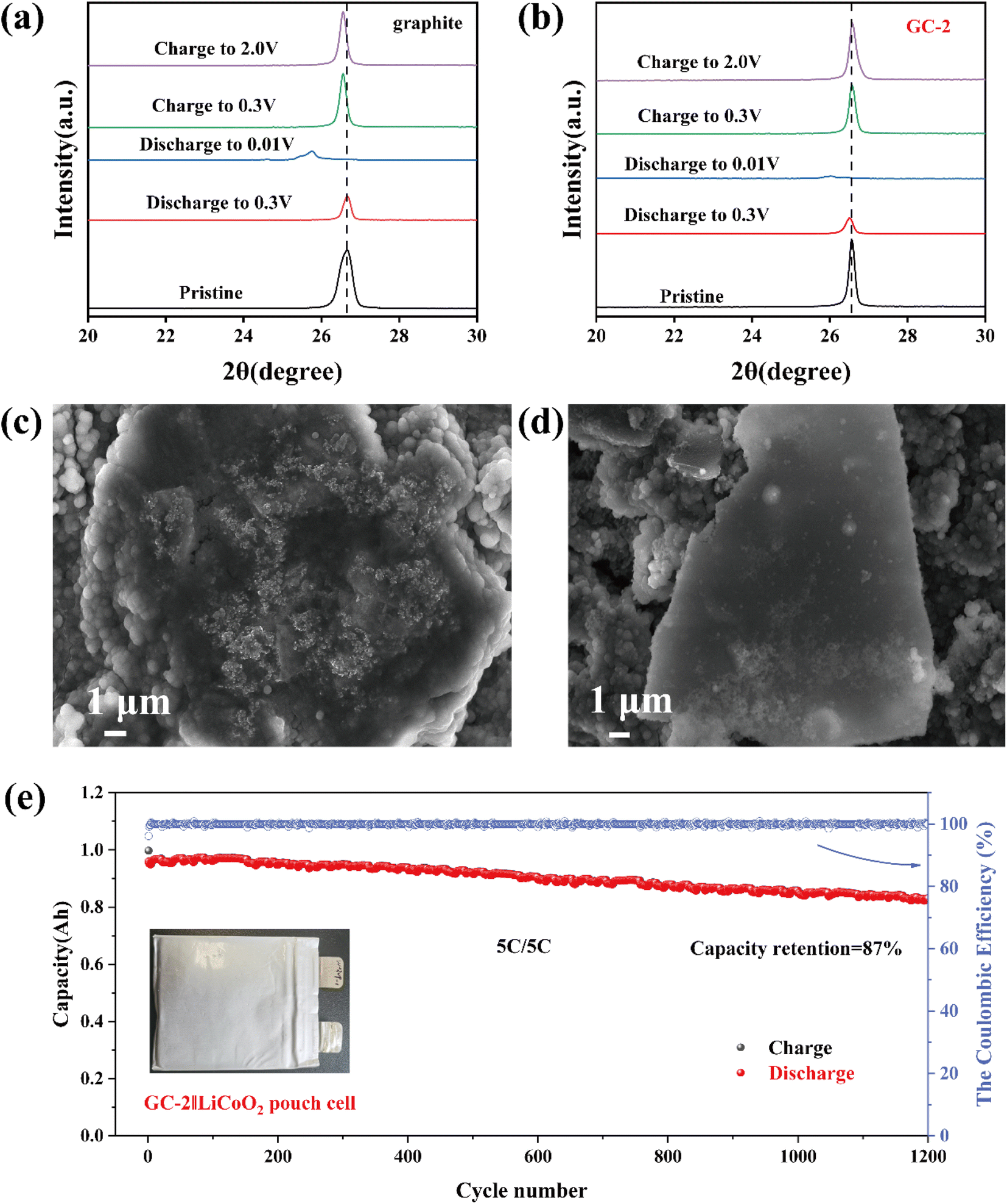 | ||
| Fig. 4 Ex situ XRD of the pristine graphite (a) and GC-2 (b) during the first charge/discharge process. The SEM images of pristine graphite (c) and GC-2 (d) electrodes after 300 cycles at 0.5C. (e) GC-2‖LiCoO2 pouch cell cycles at 5C. | ||
In order to evaluate the low-temperature performance of the anodes, DOL (1,3-dioxolane) with low melting point was used as solvent to minimize the impact of electrolytes.38 In this work, LiTFSI (1 M) dissolved in DOL was used as the electrolyte. Firstly, EIS was tested at 0 °C (Fig. S11a†) and −20 °C (Fig. 5a). GC-2 exhibits smaller charge transfer impedance at both low temperatures demonstrated faster interface dynamics. Rate performance of anodes was tested at −20 °C from 0.05C to 0.5C (Fig. 5b). As the current density increases, the capacity difference between GC-2 and pristine graphite becomes larger, GC-2 has a smaller polarization, providing superior low-temperature charging performance. Besides, GC-2 demonstrates excellent capacity retention and high capacity over pristine graphite in low-temperature cycling tests (Fig. 5c). After 100 cycling tests at 0.2C in −20 °C, GC-2 anodes exhibit the higher capacity of 291.52 mA h g−1 compared to the pristine graphite of 123.42 mA h g−1. The morphology of pristine graphite and GC-2 after 100 cycling at −20 °C was evaluated by SEM. The structure of pristine graphite showed structural deficiencies and a large amount of lithium precipitated on the surface of graphite (Fig. S12c†). While the XRD spectra of pristine graphite after low-temperature cycling also proved the precipitation of lithium (Fig. S12a†). The structure of GC-2 remains intact after cycling and no lithium precipitation is observed (Fig. S12b and d†). This indicates that the carbon coating graphite effectively enhance the structural stability of anodes and prevent the severe lithium precipitation on graphite at low-temperature operating. As shown in Fig. 5e and f, we tested the charge/discharge capability of the samples at different temperatures. At 30 °C, the capacity of pristine graphite is 354.5 mA h g−1, which is similar to the GC-2 of 367.75 mA h g−1. When the temperature drops from 0 to −20 °C, the capacity of GC-2 decreased from 322 mA h g−1 to 292.18 mA h g−1, while graphite decreased from 278.7 mA h g−1 to 189.29 mA h g−1. As the temperature decreases, GC-2 demonstrates very small capacity drop compared to pristine graphite.
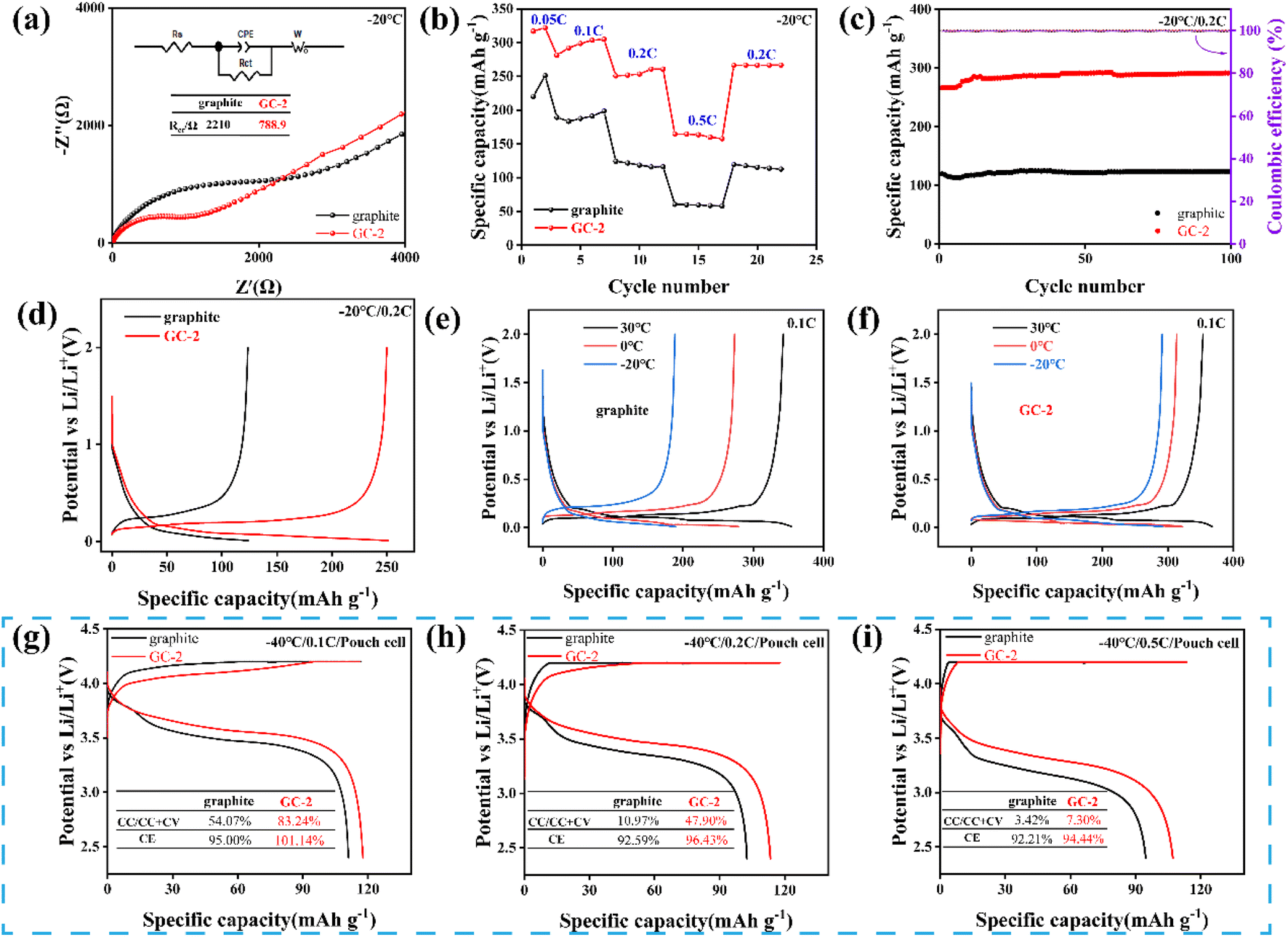 | ||
| Fig. 5 Low-temperature performance of pristine graphite and GC-2. Half cells of (a) EIS Nyquist curves at −20 °C, (b) rate performance from 0.05 to 0.5C (1C = 370 mA h g−1), (c) cycle performance at 0.2C, (d) charge/discharge curves at 0.2C, (e and f) charge/discharge curves at 0.1C from 30 °C to −20 °C. Pouch cell charge/discharge curves under −40 °C at (g) 0.1C, (h) 0.2C, (i) 0.5C. | ||
In addition, the charging capability of the pouch cells were tested at −40 °C. As shown in Fig. 5g, the GC-2‖LiCoO2 full cell still has a charge capacity of 116.3 mA h g−1 at 0.1C, while the constant current charge ratio reaches 83.24%, compared to 54.07% for the pristine graphite. When the current density increases from 0.2C to 0.5C, the constant current charge ratio of GC-2‖LiCoO2 decreases from 47.90% to 7.30%, while graphite‖LiCoO2 decreases from 10.97% to 3.42% (Fig. 5h and i). This indicates that GC-2 has a smaller polarization and a longer constant current zone than pristine graphite. Even at low-temperature, GC-2 possesses higher charging/discharging efficiencies, predicting better cycling capacity. This is due to the fast charge transfer and more diffusion paths at the interface of coated graphite (Fig. S13†), suggesting that GC-2 also has faster surface dynamics at low-temperature, which prevents lithium from being deposited on its surface, thus avoiding the growth of lithium dendrites and allowing it to be charged safely at low-temperature.
4. Conclusions
In summary, the carbon coated graphite prepared by CVD method can meet the charging demand under extreme conditions. The introduction of the carbon layer on graphite can realize the transformation of graphite from anisotropic to isotropic, increase the diffusion channel of lithium-ion, and prevent direct contact between the electrolyte and graphite to ensure the structural stability during the cycling process. The intrinsic defects of the carbon layer provide surface-controlled fast reaction kinetics, attracting more lithium-ion, improving mass transfer kinetics, and mitigating the capacity decay caused by the formation of dead lithium. Both half-cell and pouch cell exhibit excellent stable cycling at high rates, and pouch cell shows 87% capacity retention after 1200 cycles at a high rate of 5C. In particular, due to the rapid migration of lithium-ion at the interface and rapid diffusion in anodes, the GC-2 displays excellent low-temperature resistance and provides 163 mA h g−1 capacity at 0.5C and −20 °C. At the same time, GC-2‖LiCoO2 pouch cell still maintains its leading edge at low-temperature. We confirm that the GC-2 anode has excellent high-power capability, especially at low-temperature. The excellent electrochemical performance of GC-2 demonstrates its potential for application under extreme conditions.Data availability
Data will be made available on request.Author contributions
Pengfei Xiao: conceptualization, data curation, formal analysis, investigation, methodology, validation, visualization, writing – original draft. Zhongming Wang: methodology and formal analysis. Kecheng Long: formal analysis. Jixu Yang: formal analysis, investigation, and methodology. Xinsheng Liu: formal analysis and methodology. Canhui Ling: formal analysis. Libao Chen: funding acquisition, supervision and project administration. Lin Mei: supervision, and project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.References
- V. Viswanathan, A. H. Epstein, Y. M. Chiang, E. Takeuchi, M. Bradley, J. Langford and M. Winter, Nature, 2022, 601, 519–525 CrossRef CAS PubMed.
- W. Cai, Y.-X. Yao, G.-L. Zhu, C. Yan, L.-L. Jiang, C. He, J.-Q. Huang and Q. Zhang, Chem. Soc. Rev., 2020, 49, 3806–3833 RSC.
- S. Deng, D. Li, X. Liu, Z. Cai, W. Wei, J. Chen and L. Zhang, J. Anim. Feed Sci., 2020, 29, 132–142 CrossRef.
- Y. Cho, K. S. Lee, S. Piao, T.-G. Kim, S.-K. Kang, S. Y. Park, K. Yoo and Y. Piao, RSC Adv., 2023, 13, 4656–4668 RSC.
- L. Zhang, H. Tan, H. Zhu, K. Yang, W. Li and L. Sun, Dalton Trans., 2024, 53, 3611–3620 RSC.
- K. Subramanyan, S. Chen, N. Li, T. Ma, Y. Liu, S. Chandrasekaran and V. Aravindan, Electrochim. Acta, 2023, 437, 141505 CrossRef CAS.
- L. Li, D. Zhang, J. Deng, Y. Gou, J. Fang, H. Cui, Y. Zhao and M. Cao, Carbon, 2021, 183, 721–734 CrossRef CAS.
- L. Chang, Y. Lin, K. Wang, R. Yan, W. Chen, Z. Zhao, Y. Yang, G. Huang, W. Chen, J. Huang and Y. Song, RSC Adv., 2023, 13, 440–447 RSC.
- S. S. Zhang, InfoMat, 2019, 2, 942–949 CrossRef.
- Z. Zhang, D. Zhao, Y. Xu, S. Liu, X. Xu, J. Zhou, F. Gao, H. Tang, Z. Wang, Y. Wu, X. Liu and Y. Zhang, Chem. Rec., 2022, 22, 202200127 CrossRef PubMed.
- S. Li, K. Wang, G. Zhang, S. Li, Y. Xu, X. Zhang, X. Zhang, S. Zheng, X. Sun and Y. Ma, Adv. Funct. Mater., 2022, 32, 202200796 Search PubMed.
- W. Qi, J. G. Shapter, Q. Wu, T. Yin, G. Gao and D. Cui, J. Mater. Chem. A, 2017, 5, 19521–19540 RSC.
- K. Pongpanyanate, S. Roddecha, C. Piyanirund, T. Phraewphiphat and P. Hasin, RSC Adv., 2024, 14, 2354–2368 RSC.
- A. Nulu, V. Nulu and K. Y. Sohn, RSC Adv., 2024, 14, 2564–2576 RSC.
- Y. Wen, K. He, Y. Zhu, F. Han, Y. Xu, I. Matsuda, Y. Ishii, J. Cumings and C. Wang, Nat. Commun., 2014, 5, 4033 CrossRef CAS PubMed.
- J.-H. Shim and S. Lee, J. Power Sources, 2016, 324, 475–483 CrossRef CAS.
- B. Guo, Q. Liu, E. Chen, H. Zhu, L. Fang and J. R. Gong, Nano Lett., 2010, 10, 4975–4980 CrossRef CAS PubMed.
- M. Wang, J. Wang, J. Xiao, N. Ren, B. Pan, C.-s. Chen and C.-h. Chen, ACS Appl. Mater. Interfaces, 2022, 14, 16279–16288 CrossRef CAS PubMed.
- X. Chang, N. Sun, H. Zhou, R. A. Soomro and B. Xu, Chin. Chem. Lett., 2023, 34, 107312 CrossRef CAS.
- H. Li and H. Zhou, Chem. Commun., 2012, 48, 1201–1217 RSC.
- H. Oka, H. Kadoura, N. T. Takahashi and T. Ikawa, J. Power Sources, 2022, 543, 231850 CrossRef CAS.
- H. Lu, X. Chen, Y. Jia, H. Chen, Y. Wang, X. Ai, H. Yang and Y. Cao, Nano Energy, 2019, 64, 103903 CrossRef CAS.
- J. Zhou, K. Ma, X. Lian, Q. Shi, J. Wang, Z. Chen, L. Guo, Y. Liu, A. Bachmatiuk, J. Sun, R. Yang, J. H. Choi and M. H. Rümmeli, Small, 2022, 18, 2107460 CrossRef CAS PubMed.
- W. Cai, C. Yan, Y.-X. Yao, L. Xu, R. Xu, L.-L. Jiang, J.-Q. Huang and Q. Zhang, Small Struct., 2020, 1, 2000010 CrossRef.
- J.-H. Lin and C.-Y. Chen, Surf. Coat. Technol., 2022, 436, 128270 CrossRef CAS.
- Y.-J. Han, J. Kim, J.-S. Yeo, J. C. An, I.-P. Hong, K. Nakabayashi, J. Miyawaki, J.-D. Jung and S.-H. Yoon, Carbon, 2015, 94, 432–438 CrossRef CAS.
- P. Toth, Carbon, 2021, 178, 688–707 CrossRef CAS.
- L. Yang, Y. Zhao, L. Cao, H. Chen and G. Han, Energy Fuels, 2021, 35, 5308–5319 CrossRef CAS.
- P. Du, X. Fan, B. Zhang, L. Cao, J. Ren, X. Ou, X. Guo and Q. Liu, Energy Storage Mater., 2022, 50, 648–657 CrossRef.
- X.-X. He, J.-H. Zhao, W.-H. Lai, R. Li, Z. Yang, C.-m. Xu, Y. Dai, Y. Gao, X.-H. Liu, L. Li, G. Xu, Y. Qiao, S.-L. Chou and M. Wu, ACS Appl. Mater. Interfaces, 2021, 13, 44358–44368 CrossRef CAS PubMed.
- C. Ge, Z. Fan, J. Zhang, Y. Qiao, J. Wang and L. Ling, RSC Adv., 2018, 8, 34682–34689 RSC.
- H. Kim, Y. Son, J. Lee, M. Lee, S. Park, J. Cho and H. C. Choi, Chem. Mater., 2016, 28, 6146–6151 CrossRef CAS.
- X.-L. Wu, Y.-G. Guo, J. Su, J.-W. Xiong, Y.-L. Zhang and L.-J. Wan, Adv. Energy Mater., 2013, 3, 1155–1160 CrossRef CAS.
- X. Dong, Y. Yang, B. Wang, Y. Cao, N. Wang, P. Li, Y. Wang and Y. Xia, Adv. Sci., 2020, 7, 2000196 CrossRef CAS PubMed.
- V. Augustyn, J. Come, M. A. Lowe, J. W. Kim, P.-L. Taberna, S. H. Tolbert, H. D. Abruña, P. Simon and B. Dunn, Nat. Mater., 2013, 12, 518–522 CrossRef CAS PubMed.
- L. Zhang, X. Zhang, G. Tian, Q. Zhang, M. Knapp, H. Ehrenberg, G. Chen, Z. Shen, G. Yang, L. Gu and F. Du, Nat. Commun., 2020, 11, 3490 CrossRef CAS PubMed.
- T. Brousse, D. Bélanger and J. W. Long, J. Electrochem. Soc., 2015, 162, A5185–A5189 CrossRef CAS.
- C. Sun, X. Ji, S. Weng, R. Li, X. Huang, C. Zhu, X. Xiao, T. Deng, L. Fan, L. Chen, X. Wang, C. Wang and X. Fan, Adv. Mater., 2022, 34, 2206020 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra01560f |
| This journal is © The Royal Society of Chemistry 2024 |