
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-healing amino acid-bearing acrylamides/n-butyl acrylate copolymers via multiple noncovalent bonds†
Ryo Kudoa,
Sadaki Samitsu b and
Hideharu Mori*a
b and
Hideharu Mori*a
aDepartment of Organic Material Science, Graduate School of Organic Materials Science, Yamagata University, 4-3-16, Jonan, Yonezawa City, Yamagata Prefecture 992-8510, Japan. E-mail: h.mori@yz.yamagata-u.ac.jp
bNational Institute for Materials Science, 1-2-1, Sengen, Tsukuba, 305-0047, Japan
First published on 6th March 2024
Abstract
Four amino acid-bearing acrylamides, N-acryloyl-L-threonine (AThrOH), N-acryloyl-L-glutamic acid (AGluOH), N-acryloyl-L-phenylalanine (APheOH), and N-acryloyl-L, L-diphenylalanine (APhePheOH), were selected for copolymerization with n-butyl acrylate (nBA) to develop amino acid–based self-healable copolymers. A series of copolymers comprising amino acid-bearing acrylamides and nBA with tunable comonomer compositions and molecular weights were synthesized by free radical and reversible addition–fragmentation chain-transfer copolymerization. Self-healing and mechanical properties originated from the noncovalent bonds between the carboxyl, hydroxyl, and amide groups, and π–π stacking interactions among the amino acid residues in the side chains were evaluated. Among these copolymers, P(nBA-co-AGluOH) with suitable comonomer compositions and molecular weights (nBA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :AGluOH = 82:18, Mn = 18300, Mw/Mn = 2.58) exhibited good mechanical properties (modulus of toughness = 17.3 MJ m−3) and self-healing under ambient conditions. The multiple noncovalent bonds of P(nBA-co-AGluOH)s were also efficient in improving the optical properties with an enhanced refractive index and good transparency.
:AGluOH = 82:18, Mn = 18300, Mw/Mn = 2.58) exhibited good mechanical properties (modulus of toughness = 17.3 MJ m−3) and self-healing under ambient conditions. The multiple noncovalent bonds of P(nBA-co-AGluOH)s were also efficient in improving the optical properties with an enhanced refractive index and good transparency.
Introduction
Self-healing materials that can heal damages have emerged as promising materials for sustainable societies and numerous industrial applications.1–4 In addition to extrinsic self-healing materials utilizing a healing agent,5,6 a variety of intrinsic self-healing materials have been developed, which relied mainly on covalent dynamic bonds (e.g., urea bond,7,8 trithiocarbonate unit,9 disulfide bond,10,11 Diels–Alder reaction,12–14 and transesterification15,16) and noncovalent reversible interactions (e.g., hydrogen bonds,17–19 ionic interactions,20 π–π interactions,21–23 and metal–ligand coordination24–26). The incorporation of such self-healing units into polymeric materials with sophisticated designs is crucial for achieving desirable self-healing properties, mechanical properties, and targeted functions. For instance, various supramolecular hydrogels showing essential functions, e.g., self-healing, stimuli-responsive, biocompatible, antibacterial, antioxidant, and tissue-adhesive functions, have been developed for biomedical and clinical applications.27,28 The capability to utilize conventional and industrially applicable processes that can be extended to future large-scale production is desirable.Self-healing can be found in living organisms, which originated from physical and chemical changes in response to environmental change.4 Peptides and amino acids, which are constitution units for proteins, have attracted considerable attention as bio-mimics and critical units for self-healing polymers and gels, demonstrating various advantages, such as nontoxicity, biodegradability, and hydrophilicity.29 These systems can be governed by abundant units, such as hydrogen bonds, electrostatic and hydrophobic interactions, leading to supramolecular self-assembly (e.g., β-sheets). Because amino acids with the general form RCH(NH2)COOH have an amino group, a carboxyl group, and other functional groups depending on the structure of the R group, their derivatives have been intensively employed as constituent units for various self-healing materials. For example, various self-healable hydrogels and shape memory polymer networks have been developed utilizing amino acid derivatives and polymers, such as chitosan/acryloyl-phenylalanine,30 poly(aspartic acid),31 poly(γ-glutamic acid),32,33 poly(L-glutamic acid)/ureido-pyrimidinone,34 acryloyl-6-aminocaproic acid,35 N-acryloyl glycinamide,36 N-acryloyl glycinamide/N-acryloyl serine methyl ester37 and N-acryloyl alanine.38 These hydrogels and polymer networks mainly relied on chemical crosslinking, occasionally combined with physical crosslinking originating from amino acid-derived hydrogen bonds. Interestingly, N-acryloyl glycinamide was found to form supramolecular hydrogels directly via physical crosslinking owing to dual amide motifs.36 In addition, side-chain type (e.g., triblock and multiblock copolymers with N-acryloyl-L-phenylalanine39) and main-chain type (e.g., multiblock copolymers with oligopeptides,40 polydimethylsiloxane with L-phenylalanine unit41) polymers have been explored, which have amino acid/peptide units that act as physical crosslinking motifs in the side chain and main chain. Furthermore, low-molecular-weight amino acid/peptide derivatives (e.g., phenylalanine,42 nucleo-tripeptides43) have been employed as self-healing gelators.44
n-Butyl acrylate (nBA) is one of the most widely utilized monomers for producing rubbers and elastomers owing to the low glass transition temperature (Tg) of the resulting poly(nBA). Hence, it has been widely employed as a flexible component for self-healing elastomers, gels, and networks. For instance, carboxylic acid-containing monomers (e.g., acrylic acid45,46 and acrylic acid/vinylimidazole47) and hydroxyl group-containing monomers (e.g., dopamine acrylamide48,49 and N-(hydroxymethyl)acrylamide50) have been copolymerized with nBA to form self-healable polymers. Their healing and physical properties were mainly governed by noncovalent interactions (e.g., hydrogen bonds, ionic interactions, and metal coordination) from the functional units and the flexibility from nBA units. The hydrophobic association of n-butyl groups of nBA units in copolymer chains has been occasionally utilized to form a physical hydrogel, depending on the environment (e.g., in aqueous solution).51 A variety of functional monomers, such as (2-acetoacetoxy)ethyl methacrylate,52 epoxy- and urea-containing methacrylates,53 1-vinylimidazole,54 2,2,2-trifluoroethyl methacrylate,55 have also been utilized for nBA-based self-healable polymeric materials. Other attractive features of nBA-containing copolymers include excellent biocompatibility, stability, and good adhesion; therefore, nBA has been utilized in biocompatible gels,56–58 networks,59,60 and copolymers.61,62
Here, we described a straightforward and efficient approach for developing self-healing copolymers by incorporating amino acid units into nBA-based elastomers via free-radical copolymerization (Fig. 1). Four acrylamides bearing different amino acids, N-acryloyl-L-threonine (AThrOH),63,64 N-acryloyl-L-glutamic acid (AGluOH),65,66 N-acryloyl-L-phenylalanine (APheOH)67 and N-acryloyl-L,L-diphenylalanine (APhePheOH)68 were selected attempting to tune intra- and intermolecular noncovalent interactions (e.g., hydrogen bonds, electrostatic interactions, and π–π stacking) and their flexibilities. AThrOH has a hydroxyl group and a carboxyl group, AGluOH has two carboxyl groups, and APheOH has a carboxyl group and a phenyl group, in addition to an amide group in each monomer unit. APhePheOH contains two amide groups, two phenyl group, and one carboxyl groups. The core of our strategy is the selection of suitable amino acid-bearing acrylamides, which can act as self-healing sites via noncovalent interactions, combined with nBA, which can contribute to tuning the flexibility. By exploiting this unique combination of naturally originating hydrophilic building blocks derived from amino acids and nonionic nBA as a hydrophobic unit, we demonstrated tunable physical, optical, and self-healing properties. The unique intrinsic features of the copolymers, e.g., good transparency, tunable refractive indexes and softness, can be applied for ophthalmic optics in future applications. The introduction of chirality originated from amino acid units is another attractive feature of the copolymers, which can extend to advanced materials, such as aggregation-induced circular dichroism and circularly polarized luminescence materials.69–71
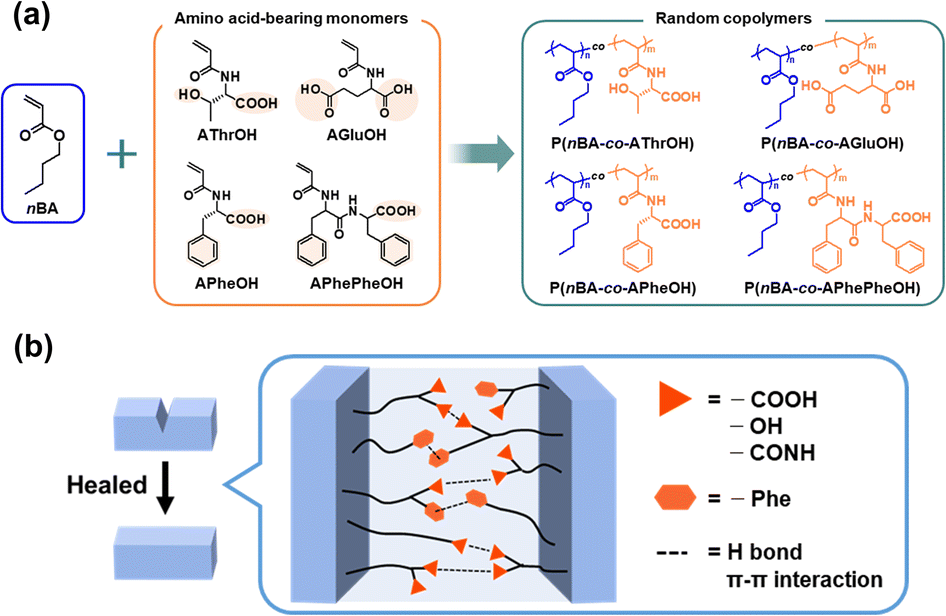 | ||
| Fig. 1 (a) Synthesis and (b) postulated healing process of amino acid–based random copolymers. | ||
Results and discussion
Copolymer synthesis
Amino acid–based random copolymers were initially synthesized by free radical copolymerization of nBA and acrylamides bearing different amino acids (AThrOH, AGluOH, APheOH, and APhePheOH) with AIBN under the appropriate conditions (Table S1 and Scheme S1†). The chemical structure and comonomer composition were verified via proton nuclear magnetic resonance (1H NMR) spectroscopy (Fig. S1–S6†), and the molecular weight and dispersity were verified by size exclusion chromatography (SEC) measurement of the methylated sample (Fig. S7–S10†). For instance, the copolymerization of nBA and AThrOH was performed with various comonomer feed ratios [nBA]0/[AThrOH]0 = 50:150, 100:100, 150:50, 175:25 at a constant monomer-to-initiator ratio [nBA + AThrOH]0/[AIBN]0 = 200:1 in ethanol at 60 °C for 24 h. Targeted P(nBA-co-AThrOH)s with various tunable comonomer compositions (AThrOH content = 10–65 mol%, as determined by 1H NMR, Fig. S4†) with similar molecular weights (Mn,SEC = 18400–30800, Mw/Mn = 2.57–3.17, Fig. S8†) were obtained in good polymer yields of 81–90%, after the reprecipitation from hexane. Similarly, P(nBA-co-AGluOH)s with tunable AGluOH contents (8–49 mol%) with reasonable molecular weights and polymer yields (Mn,SEC = 15100–29400, Mw/Mn = 2.06–2.60, yield = 74–98%), as illustrated in Fig. S7.† The nBA content in P(nBA-co-AGluOH)s (51–92%) is higher than the feed ratio ([nBA]0/[AGluOH]0 = 50:150–175:25), indicating a preferable insertion of nBA during the copolymerization. For the synthesis of P(nBA-co-APheOH) and P(nBA-co-APhePheOH), the copolymerization was conducted at [AIBN]/[nBA]/[amino acid-bearing monomer] at 1/150/50 at 60 °C in DMF, affording the copolymers with targeted molecular weights and comonomer compositions (Mn,SEC = 27200–46400, Mw/Mn = 2.51–2.83, nBA content = 86–88 mol%, and yield = 68–73%).
All copolymers bearing different amino acids (AThrOH, AGluOH, APheOH, and APhePheOH) were dissolved in DMF and DMSO (Tables S2–S4†). Owing to the amphiphilic nature, these copolymers were soluble in specific solvents (e.g., THF, chloroform, methanol, and basic water), dependent on the nature of the amino acid units and composition. For instance, P(nBA-co-AGluOH) with 82 mol% nBA content was soluble in THF an methanol, while insoluble in neutral water. These copolymers exhibited no detectable swelling and degradation (Fig. S11†), owing to the linear (Fig. 1) chain structures consisted of carbon–carbon backbone without cross-linking/network formation. Note that the threonine- and glutamic acid-based homopolymers (PAThrOH63 and PAGluOH65) were soluble in water, independent of the pH value. In contrast, the phenylalanine- and diphenylalanine-based homopolymers (PAPheOH67 and PAPhePheOH68) were only soluble in basic water (pH > 12) but insoluble in acidic and neutral water, owing to pH-dependent degree of the ionization.
Fig. 2 shows attenuated total reflection Fourier transform infrared (ATR FT-IR) spectra of the copolymers with different amino acid units. Characteristic absorption bands due to the carbonyl (1730 cm−1) and C–H stretching vibrations (2775–3020 cm−1) are clearly observed for all copolymers. ATR FT-IR spectra of the copolymers reveal characteristic broad absorption at 3100–3420 cm−1 (Fig. 2b), corresponding to an amide hydrogen bonded N–H.72,73 Among four copolymers, P(nBA-co-AGluOH) and P(nBA-co-AGluOH) exhibit remarkable broad absorbance in the region below 3400 cm−1, suggesting the presence of multiple hydrogen bonds. In all copolymers, characteristic absorption of the amide I is detected at approximately 1650 cm−1, which reflects the secondary structure.40 In addition to the clear peak attributed to ester carbonyl band at 1730 cm−1, a broad shoulder absorbance is detected at approximately 1700 cm−1, depending on the copolymer. Similar tendency was observed for ATR FT-IR spectra of the homopolymers (PAThrOH, PAGluOH, PAPheOH, and PAPhePheOH, Fig. S12†). These results suggest the present of different intramolecular (or intermolecular) interactions, originated from the nature of amino acids, which may affect the thermal, mechanical, and self-healing properties of the copolymers.
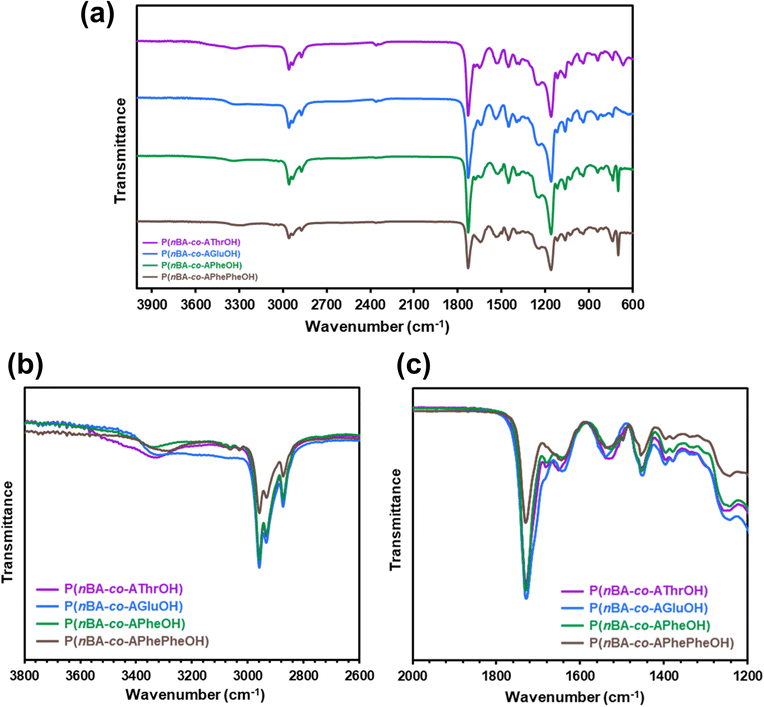 | ||
| Fig. 2 (a) ATR FT-IR spectra of amino acid-based copolymers (nBA content = approximately 80 mol%) and magnification in the region of (b) N–H band and (c) carbonyl band. | ||
Thermal and mechanical properties
The thermal properties of the P(nBA-co-AGluOH)s with different nBA contents (52–92 mol%) were investigated by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). As illustrated in Fig. 3a, all P(nBA-co-AGluOH)s exhibited high thermal stability (5% mass loss temperature Td5> 250 °C under nitrogen conditions), and the Td5 value increased with increasing nBA content, e.g., Td5 = 280 °C for the copolymer with highest nBA content (92 mol%). The glass transition temperature (Tg) value of P(nBA-co-AGluOH)s decreased from 55.1 °C to −20.5 °C when the nBA content increased from 51 to 92 mol% (Fig. 3b). The same trends were seen for P(nBA-co-AThrOH)s, which revealed an increase in the Td5 value from 187 to 213 °C and a decrease in the Tg value from 134.1 to −37.8 °C with increasing nBA content from 35 to 90 mol% (Fig. S13 and Table S5†).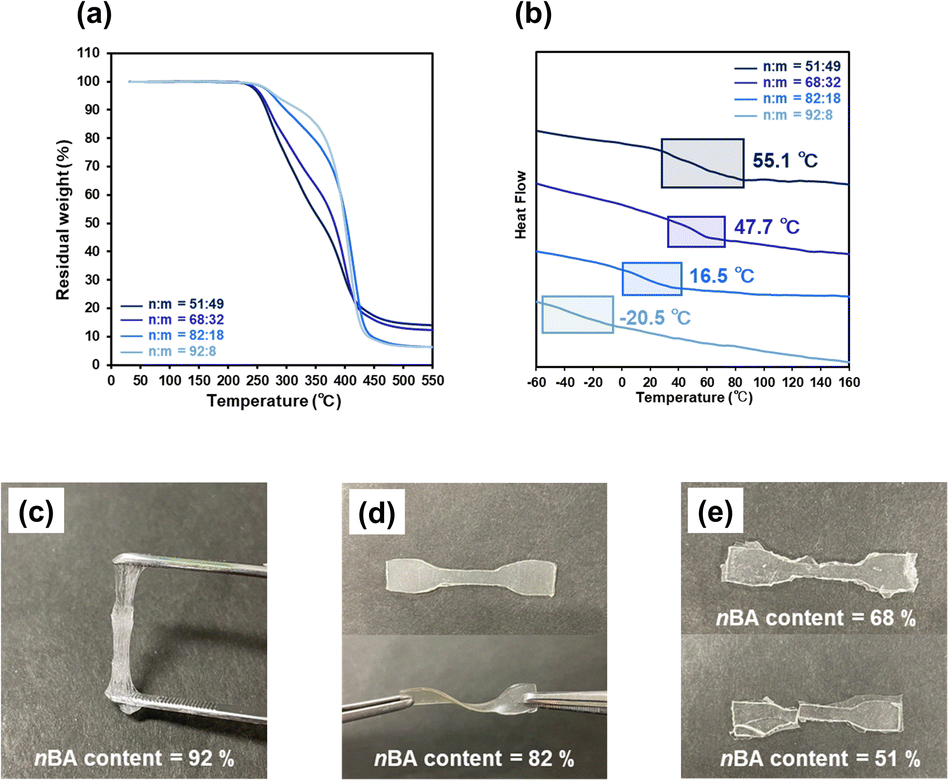 | ||
| Fig. 3 (a) TGA traces (b) DSC traces of P(nBA-co-AGluOH)s (nBA content = 51–92 mol%) and (c–e) appearance of P(nBA-co-AGluOH) (nBA content = 51–92 mol%). | ||
The dog bone-shaped species were prepared from P(nBA-co-AGluOH)s having different comonomer compositions utilizing a Teflon mold at 100–120 °C by hot-press for 1–3 min (Table S6†), followed by cooling at an ambient condition, for tensile test. P(nBA-co-AGluOH) with 92 mol% nBA was viscous with fluid properties, making it difficult to maintain the molded shape (Fig. 3c). In contrast, rubbery samples were obtained from P(nBA-co-AGluOH) with 82 mol% nBA content, which has substantial mechanical properties and flexibility and was bent and twisted without breaking (Fig. 3d). A further decrease in the nBA content (e.g., less than 70 mol%) led to the formation of glassy and fragile samples broken by shearing a small amount of stress (Fig. 3e), implying that a higher AGluOH content led to higher noncovalent interactions and lower nBA, which caused the copolymer to behave as a brittle plastic. Hence, as expected, the comonomer composition significantly influenced the thermal and mechanical properties of P(nBA-co-AGluOH)s. This implied that the noncovalent interactions of the amino acid units (e.g., hydrogen bonds and electrostatic interactions derived from the AGluOH unit in this case) were crucial factors in manipulating the intermolecular interactions between the copolymer chains.
The thermal and physical properties of the four copolymers bearing different amino acids (AThrOH, AGluOH, APheOH, and APhePheOH) were compared, and the results are summarized in Table 1. Copolymers with similar molecular weights and comonomer compositions (Mn,SEC = 18300–46400, and nBA content = 80–88 mol%) were selected to clarify the effects of noncovalent interactions derived from the amino acid unit. When the aromatic amino acid units were incorporated into the side chains, thermally stable copolymers were obtained (Td5 = 309 and 288 °C for the copolymers with APheOH and APhePheOH), which were higher than those with aliphatic amino acid units (Td5 = 200 and 270 °C for the copolymers with AThrOH and AGluOH), as illustrated in Fig. 4a. The Tg values of the copolymers with AThrOH, AGluOH, and APhePheOH units ranged between 27.3–16.5 °C. In contrast, a lower Tg value (−5.1 °C) was seen for P(nBA-co-APheOH), as depicted in Fig. 4b. Slightly lower Tg (16.5 °C) of P(nBA-co-AGluOH), compared to those of P(nBA-co-AThrOH) and P(nBA-co-APhePheOH) (27.3 and 21.5 °C) may be attributed to the presence of flexible alkyl chain in AGluOH unit. These copolymers possess easy processability that are moldable at a certain temperature, which can be tuned by selecting the nature of the amino acid units and their composition.
| Copolymer | Mna (SEC) | n:mb |
Td5c (°C) | Tgd (°C) | Young's moduluse (MPa) | Maximum strength (MPa) | Maximum strain (%) | Modulus of toughnessf (MJ m−3) |
|---|---|---|---|---|---|---|---|---|
| a This was evaluated by SEC.b 1H NMR (Table S1).c 5 wt% loss temperature.d Glass transition temperature.e Calculated from stress at slight strain (<5%).f Estimated by area under stress–strain until fracture point. | ||||||||
| P(nBA-co-AThrOH) | 18400 |
80:20 |
200 | 27.3 | 148 | 11.0 | 8.8 | 0.54 |
| P(nBA-co-AGluOH) | 18300 |
82:18 |
270 | 16.5 | 97 | 5.2 | 385 | 17.30 |
| P(nBA-co-APheOH) | 46400 |
88:12 |
309 | −5.1 | 5.1 | 0.40 | 997 | 3.95 |
| P(nBA-co-APhePheOH) | 27200 |
86:14 |
288 | 21.5 | 109 | 5.0 | 350 | 14.08 |
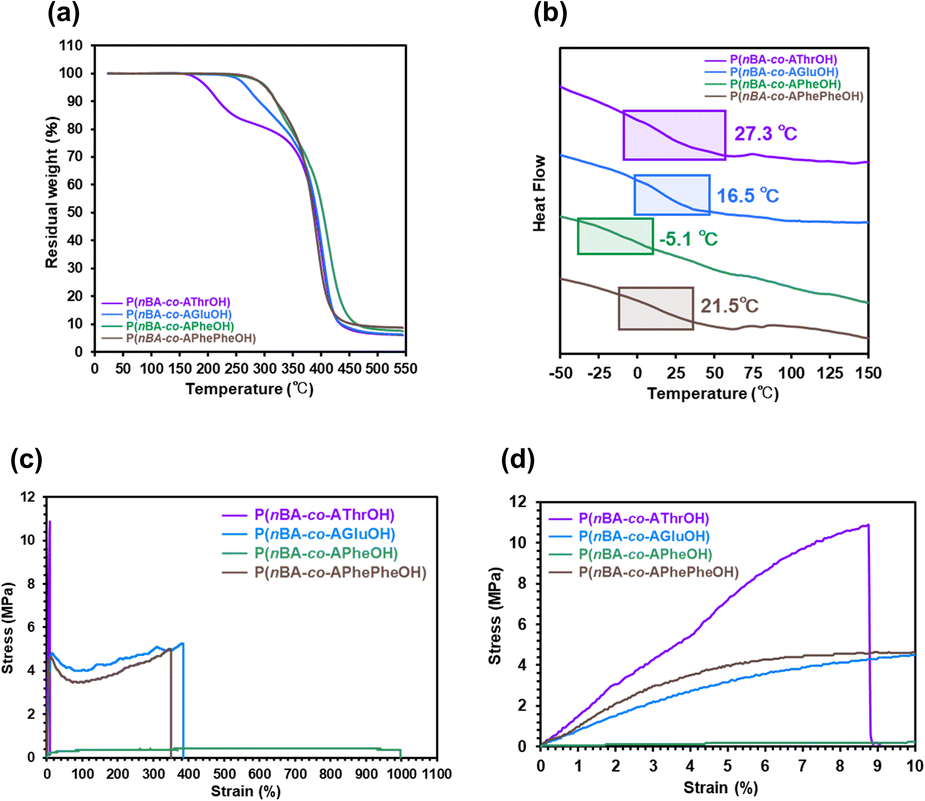 | ||
| Fig. 4 (a) TGA traces (b) DSC traces of amino acid-based copolymers (nBA content = approximately 80 mol%) and (c and d) stress–strain curves of the random copolymers (nBA content = approximately 80 mol%) in (c) 0–1100% strain and (d) 0–10% strain. | ||
The mechanical properties of four amino acid-based copolymers were evaluated by tensile stress–strain tests at room temperature (approximately 23–26 °C, Fig. 4c, d and Table 1). Young's moduli of the copolymers with AThrOH, AGluOH, and APhePheOH units ranged between 97–148 MPa. In contrast, a lower value (5.1 MPa) was observed for P(nBA-co-APheOH), demonstrating the highest strain of more than 900% without fracture. P(nBA-co-AThrOH) exhibited the highest maximum strength (11.0 MPa), whereas failure owing to brittleness was detected when the rupture strain reached 8.8%. Among the four copolymers, P(nBA-co-AGluOH) exhibited a good balance of moderate Young's modulus (97 MPa) and the highest modulus of toughness (17.3 MJ m−3) and was therefore utilized for the self-healing test.
Self-healing properties
Fig. 5a illustrates a preliminary demonstration of the self-healing capability of P(nBA-co-AGluOH) with 82% nBA content. A dog bone-shaped specimen prepared by hot press at 100 °C for 1 min can pull without breaking and then return to the original shape, implying stretchable and good mechanical properties. The specimens were cut into two pieces and manually pressed. The fractured surfaces of P(nBA-co-AGluOH) were rejoined after being pressed together with the cut samples for approximately 30 s under ambient conditions (approximately 25 °C). Similarly, the fractured surfaces of P(nBA-co-AThrOH) were rejoined by pressing at 40 °C for 30 min (Fig. S14†), suggesting that the copolymer with AThrOH demonstrated self-healable property. Still, the self-healable property was less than that with AGluOH. In contrast, P(nBA-co-APheOH) and P(nBA-co-APhePheOH) having aromatic amino acids exhibited no healing capability, even if the healing temperature increased up to 50–60 °C (Fig. S15 and Table S7†), which may be due to the interference of the hydrogen bonds by bulky aromatic units in APheOH and APhePheOH. Further studies were required to determine optimal comonomer compositions and sequences of these aromatic amino acid-based copolymers. Nevertheless, these results suggest that a suitable selection of the amino acid unit and its composition with nBA units are essential for achieving reasonable self-healing and mechanical properties of the nBA/amino acid–based copolymers by tuning suitable noncovalent interactions.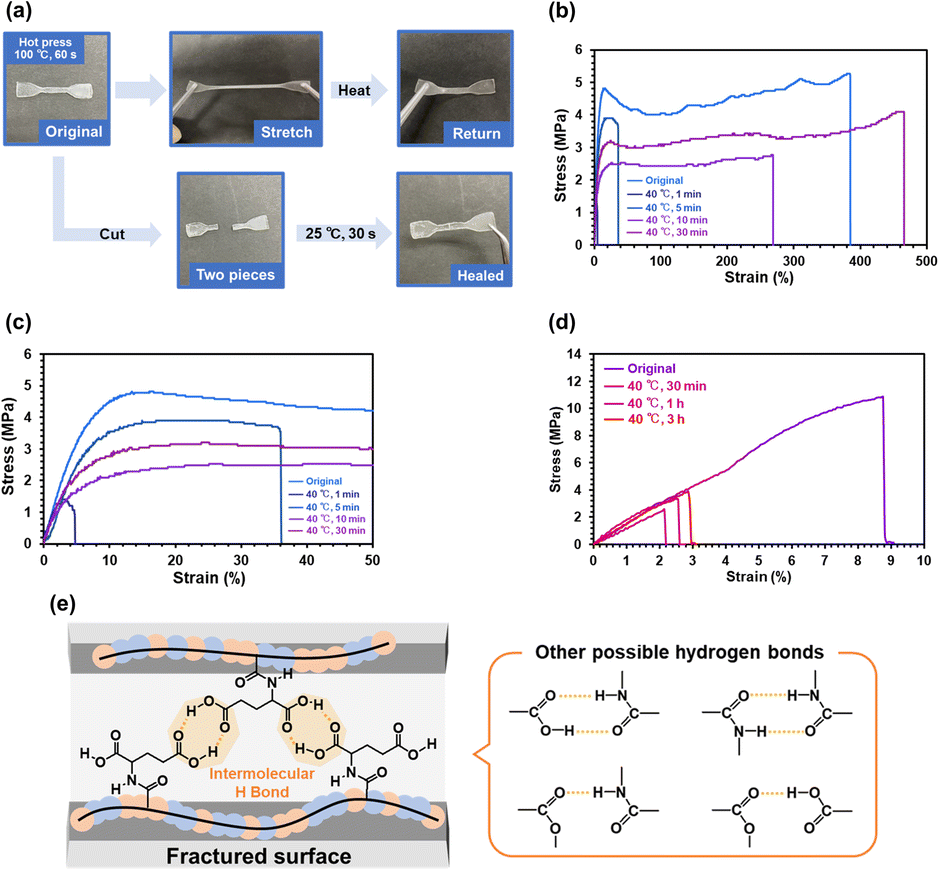 | ||
| Fig. 5 (a) Photos of original, stretched, and recovered P(nBA-co-AGluOH) sample and preliminary healing test. Stress–strain curves of (b and c) P(nBA-co-AGluOH) after healing in (b) 0–500% strain and (c) 0–50% strain, and (d) P(nBA-co-AThrOH) after healing and (e) schematics of healing process of P(nBA-co-AGluOH). | ||
The self-healing efficiencies of P(nBA-co-AGluOH) and P(nBA-co-AThrOH) were evaluated by comparing maximal strength of the pristine specimen and healed ones treated at 40 °C for different compression time (Fig. 5 and Table S8†). Both copolymer samples demonstrated improved self-healing capabilities with increasing compression time. Notably, P(nBA-co-AGluOH) recovered 90% of the maximum strength of the pristine sample after 30 min compression (Fig. 5b and c). In contrast, P(nBA-co-AThrOH) recovered approximately 10%, even after 3 h (Fig. 5d). These results suggest that intermolecular hydrogen bonds between two carboxylic acids in the AGluOH unit, in addition to carboxylic acid/amide and amide/amide hydrogen bonds, are essential for achieving reasonable self-healing ability (Fig. 5e). Obviously, self-healing properties are affected by molecular mobility of the copolymer chain. Therefore, each copolymer having different Tg values possess suitable healing temperature. In this study, the self-healing behaviors of the copolymers having different animo acid units were compared at 40 °C, which are higher than those of Tg values of the copolymers (−5.1–27.3 °C). Further studies on the amino acid–based copolymers, such as the influence on the healing temperature on the healing properties and temperature-dependent rheological behavior, will be reported separately.
Effect of molecular weight
P(nBA-co-AGluOH)s with different molecular weights and approximately the same comonomer composition (nBA/AGluOH molar ratio = approximately 8/2) were synthesized to evaluate the effect of the molecular weight on the mechanical and self-healing properties (Table S9†). RAFT copolymerization of nBA and AGluOH utilizing a trithiocarbonate-type chain transfer agent (CTA) at different monomer-to-CTA ratios ([nBA + AGluOH]/[CTA] = 100–400) at a constant CTA-to-initiator and a comonomer feed ratio ([CTA]0/[AIBN]0 = 2/1 and [nBA]/[AGluOH] = 3/1) produced P(nBA-co-AGluOH)s with adjustable molecular weights (Mn = 9500–15900). When RAFT copolymerization was conducted at [nBA]/[AGluOH]/[CTA]/[AIBN] = 150/50/2/1–300/100/2/1, P(nBA-co-AGluOH)s having relatively low disparities (Mw/Mn = 1.25–1.36) were obtained, while further increase in the monomer-to-CTA ratio led to the broadening disparity (Mw/Mn = 1.79), as depicted in Fig. 6a. As comparisons, P(nBA-co-AGluOH)s having higher molecular weights (Mn = 18300–21500, Mw/Mn = 2.58–2.73) with (nBA/AGluOH = approximately 8/2 molar ratio) were prepared by free radical copolymerization at different [nBA]/[AGluOH]/[AIBN] ratios (150/50/1 and 300/100/1). P(nBA-co-AGluOH) samples of different molecular weights were utilized to evaluate their thermal and mechanical properties (Table S9† and Fig. 6).
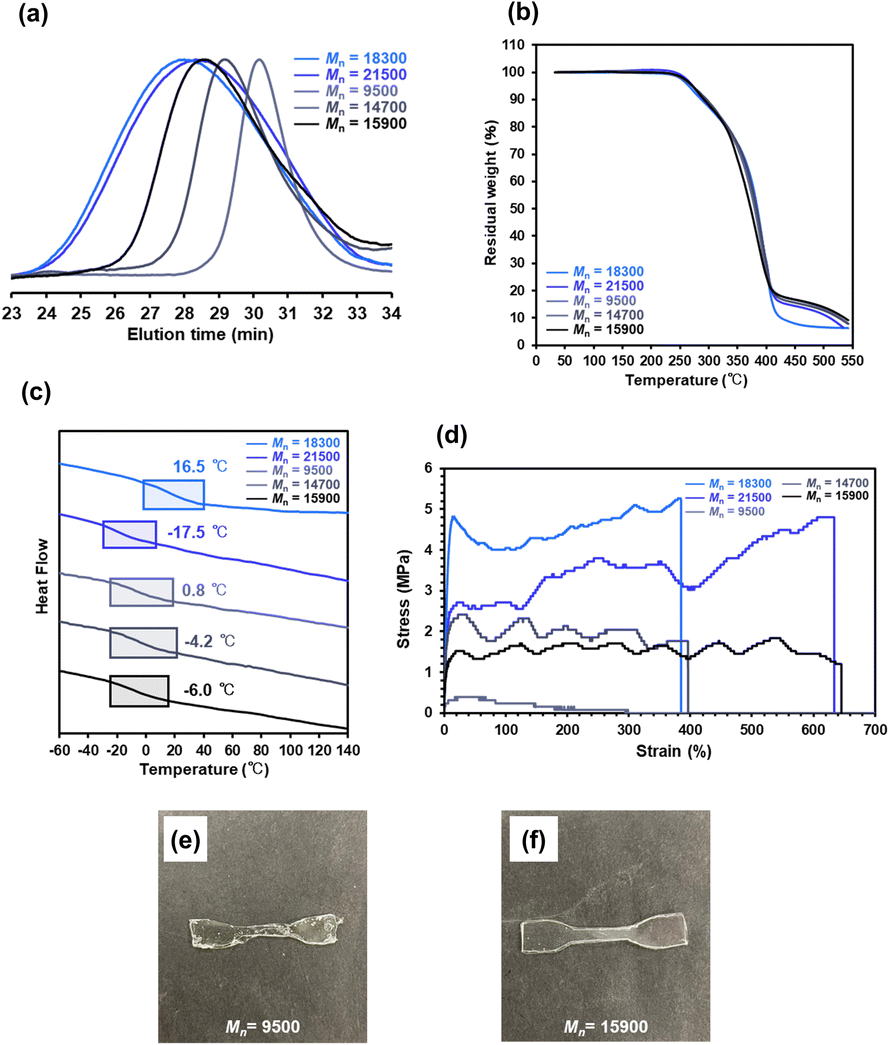 | ||
| Fig. 6 (a) SEC, (b) TGA, and (c) DSC traces, and (d) stress–strain curves of P(nBA-co-AGluOH)s (Mn = 9500–21500) and (e and f) appearance of P(nBA-co-AGluOH)s (Mn = 9500, 15900). | ||
TGA results revealed that P(nBA-co-AGluOH)s displayed similar thermal stabilities (Td5 = 268–273 °C), regardless of the molecular weights. A substantial difference was observed in the Tg values. For instance, the Tg values of P(nBA-co-AGluOH)s synthesized by RAFT copolymerization decrease slightly from 0.8 to −6.0 °C with increasing the molecular weights. This tendency may be due to the higher content of the end-groups and the absence of low-molecular-weight products, which are probably removed during the purification process, resulting in lower polymer yield (e.g., 60% at [nBA]/[AGluOH]/[CTA]/[AIBN] = 150/50/2/1). P(nBA-co-AGluOH)s prepared by free radical copolymerization exhibited similar molecular weight-dependent tendency, demonstrating the higher Tg value (16.5 °C for Mn = 18300) and lower one (−17.5 °C for Mn = 21500). RAFT-synthesized P(nBA-co-AGluOH) with relatively high molecular weights (Mn = 14700 and 15900) was obtained as a rubbery sample with good stability and film-forming ability. Low-molecular-weight P(nBA-co-AGluOH) (Mn = 9500) afforded a sample, but its mechanical properties were relatively poor (Fig. 6e). The RAFT-synthesized P(nBA-co-AGluOH)s (nBA content = approximately 80 mol%) with different molecular weights demonstrated moderate Young's moduli in the range of 5.0–92 MPa. The strain of the RAFT-synthesized P(nBA-co-AGluOH)s increased with increasing molecular weight. The same tendency, where higher molecular weight led to higher strain, was also observed for P(nBA-co-AGluOH) prepared by free radical copolymerization. A substantial increase in the modulus of toughness was observed for P(nBA-co-AGluOH)s designed by RAFT and free-radical copolymerization. Interestingly, P(nBA-co-AGluOH) having the highest molecular weight (Mn = 21500, Mw/Mn = 2.73), exhibited relatively high tensile strength (4.8 MPa) and modulus of toughness (22.28 MJ m−3), yet possessed a strain at break of 634%. In contrast, P(nBA-co-AGluOH) having lowest molecular-weight (Mn = 7800), which was prepared using a dithiocarbamate-type CTA, afforded viscous product with fluid property, even if almost the same nBA content (82%, Table S10 and Fig. S16†). These results imply that the molecular weight has a remarkable effect on the mechanical properties, particularly the flexibility and toughness.
Optical properties
In addition to the mechanical and self-healing properties, integration with additional functions is desirable for future applications of self-healable materials (e.g., soft robotics, sensors, electronic devices).74,75 Here, we focused on achieving good optical properties, including transparency and improved refractive index, because the simultaneous achievement of a high refractive index with good mechanical and self-healing properties remains a challenge. Based on the Lorentz–Lorenz equation, molar refraction and molecular volume are crucial parameters to determine refractive index.76 Hydrogen bonds have been recently employed to increase the refractive index via decreasing the free volume.77 Here, P(nBA-co-AGluOH)s having different AGluOH contents (49–8 mol%) were employed to achieve good transparency and high refractive indexes via tuning intermolecular hydrogen bonds. As illustrated in Fig. 7, P(nBA-co-AGluOH)s demonstrated good film-forming properties and good transparency in the visible region (>90% at 400 nm), regardless of the comonomer composition. The ellipsometry measurements of P(nBA-co-AGluOH)s indicated typical wavelength-dependent refractive indices (Fig. 7c), which increased from 1.4883 to 1.5089 with decreasing Abbe numbers from 63.5–56.7 by increasing AGluOH content (Table S11†). These values are comparable to those of poly(methyl methacrylate) (refractive index = 1.49 and Abbe number = 58),78 and silicon and acrylic intraocular lenses (refractive index = 1.41–1.55 and Abbe number = 37–58).79 These results implied that the AGluOH having hydrogen bonding ability can efficiently enhance the refractive index, while maintaining good transparency.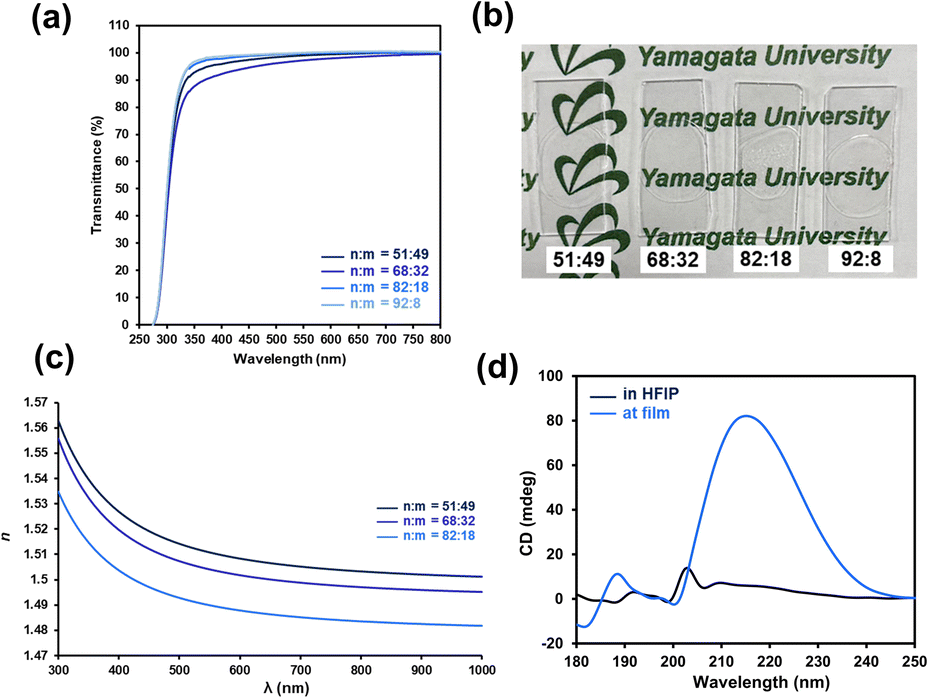 | ||
| Fig. 7 (a) UV-vis transmittance spectra of P(nBA-co-AGluOH)s having different AGluOH contents (49–8 mol%) and (b) photographs of P(nBA-co-AGluOH)s films utilized for transmittance measurements, (c) wavelength-dependent refractive indices determined by ellipsometry, and (d) CD spectra of P(nBA-co-AGluOH) in HFIP (0.03 g L−1) and at the film state on a quartz plate. | ||
Chiroptical properties of P(nBA-co-AGluOH) in solid and solution states were evaluated by circular dichroism (CD) measurement. As shown in Fig. 7d, a positive CD peak at approximately 215 nm appeared in the film state, whereas no remarkable peak was detected in the hexafluoroisopropanol (HFIP) solution, showing aggregation-induced CD effect. The P(nBA-co-AGluOH)s exhibited good transparency, tunable refractive index, aggregation-induced CD effect, in addition to tunable flexibility and self-healing properties. This approach provides a practical and efficient method for the preparation of self-healable polymers with tunable properties by simple radical copolymerization of amino acid-carrying monomers, which can open new avenues for advanced optical materials, such as ophthalmic optics.
Conclusion
In summary, we developed a series of amino acid-bearing acrylamide/nBA copolymers with good mechanical and self-healing properties under ambient conditions by selecting suitable amino acid units, comonomer compositions, and molecular weights. Combining noncovalent interactions (e.g., hydrogen bonds, electrostatic interactions, and π–π stacking) from the amino acid motifs and flexibility from nBA can be achieved by copolymerizing charged monomers with a neutral monomer under suitable conditions. The nature of the amino acids (AThrOH, AGluOH, APheOH, and APhePheOH) and amino acid-bearing acrylamide/nBA content substantially affected the thermal, mechanical, and self-healing properties. The molecular weight of the copolymers is also a crucial factor for manipulating the mechanical strength. The AGluOH content in P(nBA-co-AGluOH) efficiently enhanced the refractive indices while maintaining good transparency by tuning the noncovalent interactions. This strategy is regarded as physical and reversible crosslinking via noncovalent interactions derived from naturally originating amino acid motifs, which can contribute to the tuning of thermal, mechanical, and self-healing properties, as well as optical properties involving transparency and refractive index.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
Ryo Kudo: investigation, methodology, writing – original draft. Sadaki Samitsu: investigation, validation, writing – review & editing. Hideharu Mori: conceptualization, supervision, validation, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Toshiaki Ogasawara Memorial Foundation. We thank MANA foundry at NIMS for instrumental support for spectroscopic ellipsometry. We also thank Ryo Yonenuma and Kaori Morikawa at Yamagata University for the help in the monomer synthesis and ellipsometry measurement.Notes and references
- A. Campanella, D. Dohler and W. H. Binder, Macromol. Rapid Commun., 2018, 39, e1700739 CrossRef PubMed
.
- C. Kim and N. Yoshie, Polym. J., 2018, 50, 919–929 CrossRef CAS
- Y. Yang and M. W. Urban, Adv. Mater. Interfaces, 2018, 5, 1800384 CrossRef
- C. E. Diesendruck, N. R. Sottos, J. S. Moore and S. R. White, Angew. Chem., Int. Ed., 2015, 54, 10428–10447 CrossRef CAS PubMed
- S. R. White, N. R. Sottos, P. H. Geubelle, J. S. Moore, M. R. Kessler, S. R. Sriram, E. N. Brown and S. Viswanathan, Nature, 2001, 409, 794–797 CrossRef CAS PubMed
- M. Gragert, M. Schunack and W. H. Binder, Macromol. Rapid Commun., 2011, 32, 419–425 CrossRef CAS PubMed
- H. Ying, Y. Zhang and J. Cheng, Nat. Commun., 2014, 5, 3218 CrossRef PubMed
- Z. Wang, S. Gangarapu, J. Escorihuela, G. Fei, H. Zuilhof and H. Xia, J. Mater. Chem. A, 2019, 7, 15933–15943 RSC
- Y. Amamoto, J. Kamada, H. Otsuka, A. Takahara and K. Matyjaszewski, Angew Chem. Int. Ed. Engl., 2011, 50, 1660–1663 CrossRef CAS PubMed
- J. Canadell, H. Goossens and B. Klumperman, Macromolecules, 2011, 44, 2536–2541 CrossRef CAS
- U. Lafont, H. van Zeijl and S. van der Zwaag, ACS Appl. Mater. Interfaces, 2012, 4, 6280–6288 CrossRef CAS PubMed
- J. Bai, H. Li, Z. Shi and J. Yin, Macromolecules, 2015, 48, 3539–3546 CrossRef CAS
- J. Kötteritzsch, S. Stumpf, S. Hoeppener, J. Vitz, M. D. Hager and U. S. Schubert, Macromol. Chem. Phys., 2013, 214, 1636–1649 CrossRef
- K. K. Oehlenschlaeger, J. O. Mueller, J. Brandt, S. Hilf, A. Lederer, M. Wilhelm, R. Graf, M. L. Coote, F. G. Schmidt and C. Barner-Kowollik, Adv. Mater., 2014, 26, 3561–3566 CrossRef CAS PubMed
- T. Liu, C. Hao, S. Zhang, X. Yang, L. Wang, J. Han, Y. Li, J. Xin and J. Zhang, Macromolecules, 2018, 51, 5577–5585 CrossRef CAS
- J. Tan, C. Li, K. De Bruycker, G. Zhang, J. Gu and Q. Zhang, RSC Adv., 2017, 7, 51763–51772 RSC
- P. Cordier, F. Tournilhac, C. Soulie-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS PubMed
- D. Montarnal, P. Cordier, C. Soulié-Ziakovic, F. Tournilhac and L. Leibler, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7925–7936 CrossRef CAS
- D. Montarnal, F. Tournilhac, M. Hidalgo, J. L. Couturier and L. Leibler, J. Am. Chem. Soc., 2009, 131, 7966–7967 CrossRef CAS PubMed
- T. L. Sun, T. Kurokawa, S. Kuroda, A. Bin Ihsan, T. Akasaki, K. Sato, M. A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 932–937 CrossRef CAS PubMed
- S. Burattini, B. W. Greenland, D. H. Merino, W. Weng, J. Seppala, H. M. Colquhoun, W. Hayes, M. E. Mackay, I. W. Hamley and S. J. Rowan, J. Am. Chem. Soc., 2010, 132, 12051–12058 CrossRef CAS PubMed
- J. Fox, J. J. Wie, B. W. Greenland, S. Burattini, W. Hayes, H. M. Colquhoun, M. E. Mackay and S. J. Rowan, J. Am. Chem. Soc., 2012, 134, 5362–5368 CrossRef CAS PubMed
- L. R. Hart, J. H. Hunter, N. A. Nguyen, J. L. Harries, B. W. Greenland, M. E. Mackay, H. M. Colquhoun and W. Hayes, Polym. Chem., 2014, 5, 3680–3688 RSC
- S. Bode, L. Zedler, F. H. Schacher, B. Dietzek, M. Schmitt, J. Popp, M. D. Hager and U. S. Schubert, Adv. Mater., 2013, 25, 1634–1638 CrossRef CAS PubMed
- W. Weng, J. B. Beck, A. M. Jamieson and S. J. Rowan, J. Am. Chem. Soc., 2006, 128, 11663–11672 CrossRef CAS PubMed
- Y. Zhao, J. B. Beck, S. J. Rowan and A. M. Jamieson, Macromolecules, 2004, 37, 3529–3531 CrossRef CAS
- B. Guo, Y. Liang and R. Dong, Nat. Protoc., 2023, 18, 3322–3354 CrossRef CAS PubMed
- S. Zhuo, Y. Liang, Z. Wu, X. Zhao, Y. Han and B. Guo, Mater. Horiz., 2024, 11, 37–101 RSC
- L. Cai, S. Liu, J. Guo and Y.-G. Jia, Acta Biomater., 2020, 113, 84–100 CrossRef CAS PubMed
- S. Sharma, A. Kumar, Deepak, R. Kumar, N. K. Rana and B. Koch, Int. J. Biol. Macromol., 2018, 116, 37–44 CrossRef CAS PubMed
- H. An, L. Zhu, J. Shen, W. Li, Y. Wang and J. Qin, Colloids Surf., B, 2020, 185, 110601 CrossRef CAS PubMed
- S. Dai, X. Zhou, S. Wang, J. Ding and N. Yuan, Nanoscale, 2018, 10, 19360–19366 RSC
- R. Yang, X. Liu, Y. Ren, W. Xue, S. Liu, P. Wang, M. Zhao, H. Xu and B. Chi, Acta Biomater., 2021, 127, 102–115 CrossRef CAS PubMed
- Q. Wang, Z. Shi, Y. Shou, K. Zhang, G. Li, P. Xia, S. Yan and J. Yin, ACS Biomater. Sci. Eng., 2020, 6, 1715–1726 CrossRef CAS PubMed
- A. Phadke, C. Zhang, B. Arman, C.-C. Hsu, R. A. Mashelkar, A. K. Lele, M. J. Tauber, G. Arya and S. Varghese, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4383–4388 CrossRef CAS PubMed
- X. Dai, Y. Zhang, L. Gao, T. Bai, W. Wang, Y. Cui and W. Liu, Adv. Mater., 2015, 27, 3566–3571 CrossRef CAS PubMed
- S.-n. Nishimura, D. Sato and T. Koga, Gels, 2023, 9, 829 CrossRef CAS PubMed
- T. Koga, K. Tomimori and N. Higashi, Macromol. Rapid Commun., 2020, 41, 1900650 CrossRef CAS PubMed
- M. Hendrich, L. Lewerdomski and P. Vana, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 2809–2819 CrossRef CAS
- T. Koga, T. Morishita, Y. Harumoto, S.-n. Nishimura and N. Higashi, Mater. Adv., 2021, 2, 7851–7860 RSC
- S. Tazawa, A. Shimojima, T. Maeda and A. Hotta, J. Appl. Polym. Sci., 2018, 135, 45419 CrossRef
- D. Zaguri, S. Shaham-Niv, P. Chakraborty, Z. Arnon, P. Makam, S. Bera, S. Rencus-Lazar, P. R. Stoddart, E. Gazit and N. P. Reynolds, ACS Appl. Mater. Interfaces, 2020, 12, 21992–22001 CrossRef CAS PubMed
- K. Baek, A. D. Noblett, P. Ren and L. J. Suggs, ACS Appl. Bio Mater., 2019, 2, 2812–2821 CrossRef CAS PubMed
- N. Zanna, A. Merlettini and C. Tomasini, Org. Chem. Front., 2016, 3, 1699–1704 RSC
- W. Wang, Z. Liu, Z. Guo, J. Zhang, C. Li, S. Qiu, X. Lei and Q. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 50812–50822 CrossRef CAS PubMed
- R. K. Bose, N. Hohlbein, S. J. Garcia, A. M. Schmidt and S. van der Zwaag, Phys. Chem. Chem. Phys., 2015, 17, 1697–1704 RSC
- Y. Hou, Y. Peng, P. Li, Q. Wu, J. Zhang, W. Li, G. Zhou and J. Wu, ACS Appl. Mater. Interfaces, 2022, 14, 35097–35104 CrossRef CAS PubMed
- J. Li, H. Ejima and N. Yoshie, ACS Appl. Mater. Interfaces, 2016, 8, 19047–19053 CrossRef CAS PubMed
- C. Kim, H. Ejima and N. Yoshie, RSC Adv., 2017, 19288–19295 RSC
- L. Laysandra, C.-H. Chuang, S. Kobayashi, A.-N. Au-Duong, Y.-H. Cheng, Y.-T. Li, M. M. Mburu, T. sono, T. Satoh and Y.-C. Chiu, ACS Appl. Polym. Mater., 2020, 2, 5432–5443 CrossRef CAS
- S. Chen, G. Ma, X. Duan, H. Zhuo, J. Xu and H. Chen, ACS Appl. Polym. Mater., 2023, 5, 6390–6398 CrossRef CAS
- S. Wang, L. Li, Q. Liu and M. W. Urban, Macromolecules, 2022, 55, 4703–4709 CrossRef CAS
- Y. Zhou, G. Chen, S. Yan, C. Ni, L. Yu and X. Li, Prog. Org. Coat., 2022, 172, 107098 CrossRef CAS
- W. Wang, F. Wang, C. Zhang, J. Tang, X. Zeng and X. Wan, Chem. Eng. J., 2021, 404, 126358 CrossRef CAS
- S. Wang and M. W. Urban, Adv. Sci., 2021, 8, 2101399 CrossRef CAS PubMed
- S. S. Halacheva, D. J. Adlam, E. K. Hendow, T. J. Freemont, J. Hoyland and B. R. Saunders, Biomacromolecules, 2014, 15, 1814–1827 CrossRef CAS PubMed
- D. Cao, Y. Zhang, Z. Cui, Y. Du and Z. Shi, Mater. Sci. Eng., C, 2017, 70, 665–672 CrossRef CAS PubMed
- M. Prasitsilp, T. Siriwittayakorn, R. Molloy, N. Suebsanit, P. Siriwittayakorn and S. Veeranondha, J. Mater. Sci.: Mater. Med., 2003, 14, 595–600 CrossRef CAS PubMed
- C. A. Schoener, H. N. Hutson and N. A. Peppas, AIChE J., 2013, 59, 1472–1478 CrossRef CAS
- A. Mayer, T. Roch, K. Kratz, A. Lendlein and F. Jung, Acta Biomater., 2012, 8, 4253–4259 CrossRef CAS PubMed
- G. Rodriguez, M. Fernandez-Gutierrez, J. Parra, A. Lopez-Bravo, N. G. Honduvilla, J. Bujan, M. Molina, L. Duocastella and J. San Roman, Biomacromolecules, 2010, 11, 2740–2747 CrossRef CAS PubMed
- J. Cui, K. Trescher, K. Kratz, F. Jung, B. Hiebl and A. Lendlein, Clin. Hemorheol. Microcirc., 2010, 45, 401–411 CAS
- K. Shoji, M. Nakayama, T. Koseki, K. Nakabayashi and H. Mori, Polymer, 2016, 97, 20–30 CrossRef CAS
- M. P. Bueno, C. A. Cativiela, J. A. Mayoral and A. Avenoza, J. Org. Chem., 1991, 56, 6551–6555 CrossRef CAS
- T. Koseki, R. Kanto, R. Yonenuma, K. Nakabayashi, H. Furusawa, S. Yano and H. Mori, React. Funct. Polym., 2020, 150, 104540 CrossRef CAS
- S. Y. Park, S.-Y. Kim, T. Kim, H. Ahn and I. Chung, Polym. Adv. Technol., 2019, 30, 872–878 CrossRef CAS
- H. Mori, M. Matsuyama, K. Sutoh and T. Endo, Macromolecules, 2006, 39, 4351–4360 CrossRef CAS
- R. Yonenuma, A. Ishizuki, K. Nakabayashi and H. Mori, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 2562–2574 CrossRef
- J. Liu, H. Su, L. Meng, Y. Zhao, C. Deng, J. C. Y. Ng, P. Lu, M. Faisal, J. W. Y. Lam, X. Huang, K. S. Wong and B. Z. Tang, Chem. Sci., 2012, 3, 2737–2747 RSC
- H. Tanaka, Y. Inoue and T. Mori, ChemPhotoChem, 2018, 2, 386–402 CrossRef CAS
- Y. Sang, J. Han, T. Zhao, P. Duan and M. Liu, Adv. Mater., 2020, 32, 1900110 CrossRef CAS PubMed
- G. Dado and S. Gellman, J. Am. Chem. Soc., 1994, 116, 1054–1062 CrossRef CAS
- K. Tsang, H. Diaz, N. Graciani and J. Kelly, J. Am. Chem. Soc., 1994, 116, 3988–4005 CrossRef CAS
- W. Zhang, H. Jiang, Z. Chang, W. Wu, G. WWu, R. Wu and J. Li, J. Mater. Sci., 2020, 55, 13543–13558 CrossRef CAS
- Y. J. Tan, G. J. Susanto, H. P. Anwar Ali and B. C. K. Tee, Adv. Mater., 2021, 33, 2002800 CrossRef CAS PubMed
- J.-g. Liu and M. Ueda, J. Mater. Chem., 2009, 19, 8907–8919 RSC
- S. Watanabe, H. Nishio, T. Takayama and K. Oyaizu, ACS Appl. Polym. Mater., 2023, 5, 2307–2311 CrossRef CAS
- G. S. Jha, G. Seshadri, A. Mohan and R. K. Khandal, e-Polymers, 2007, 120 Search PubMed
- M. Vacalebre, R. Frison, C. Corsaro, F. Neri, A. Santoro, S. Conoci, E. Anastasi, M. C. Curatolo and E. Fazio, Polymers, 2023, 15, 1590 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra00800f |
| This journal is © The Royal Society of Chemistry 2024 |