
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA sustainable metal-free and additive-free olefination route to N-heteroazaarenes from methyl-substituted heterocycles and amines†
Hongyi Cuiac,
Chunyan Zhang *b,
Yuqi Jia and
Guoying Zhang*a
*b,
Yuqi Jia and
Guoying Zhang*a
aState Key Laboratory of Coal Conversion, Institute of Coal Chemistry, Chinese Academy of Sciences, Taiyuan, 030001, P. R. China. E-mail: zhanggy@sxicc.ac.cn
bTaiyuan University of Technology, Taiyuan, 030001, P. R. China. E-mail: qustzcy@163.com
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 31st January 2024
Abstract
A green and sustainable metal-free, additive-free olefination approach is proposed for the facile synthesis of various unsaturated N-heteroazaarenes from simple methyl-substituted heteroarenes and amines. The developed protocol employs only air as the sole oxidant and provides a useful strategy for obtaining various E-selective conjugated heterocyclic olefins. This provides a useful strategy for application in generating grams of a variety of unsaturated N-heteroazaarenes (up to 20.33 grams) and the synthetic imaging agents of STB-8 (2.40 gram) with high regioselectivity in one pot.
Introduction
Unsaturated N-heteroaromatic compounds are widely utilized as intermediates during the synthesis of various natural products and functional materials, as well as in the pharmaceutical industry, and they are also common structural entities in several bioactive natural products (Scheme 1a).1 As such, the development of efficient methods to obtain conjugated N-heteroaromatic olefins is of great significance to chemical and medicinal applications, and has attracted significant attention over the past decades.2 For example, many named reactions,3 such as olefin metathesis,4 and catalytic cross-coupling reactions5 have been developed to access such heteroaromatic molecules, and these approaches commonly involve the reactions of methyl N-heteroaromatics with aldehydes, benzyl alcohols, or N-benzylidene-4-methylbenzenesulfonamides.6 However, the requirement for multi-step functional group manipulations, the use of stoichiometric amounts of oxidants, and the unavoidable generation of significant amounts of undesired waste products are particular shortcomings of these reactions. | ||
| Scheme 1 State of the art and the reaction described here. | ||
The deamination of primary amines is another important transformation that can be employed to assemble unsaturated N-heteroaromatic structures.7 This has been achieved by the aggressive oxidation of aliphatic amines using oxidants, although the potential scope of the reaction is limited due to its low selectivity and its tendency toward overoxidation. Recently, Gong et al. and Susanta et al. discovered that E-2-styrylquinolines could be prepared from 2-methylquinolines by reaction with primary amines in the presence of a stoichiometric amount of TBHP (2.0–4.0 eq., Scheme 1b(I)).8 In addition, employing air as the oxidant, Mao described the La-catalyzed olefination of methylquinolines and amines (Scheme 1b(II)),9 while Li![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9 and Sharma10 reported similar reactions with stoichiometric amounts of NH4I or in a [Bmim]BF4 solvent under air (Scheme 1b(III)). However, the use of stoichiometric amounts of oxidants, additives, and metal catalysts in such processes limits the potential scope of this environmentally benign transformation. Therefore, the development of a green and economic system for the sustainable construction of highly E-selective N-heteroaromatic olefins is highly desirable and constitutes a new synthetic challenge.
9 and Sharma10 reported similar reactions with stoichiometric amounts of NH4I or in a [Bmim]BF4 solvent under air (Scheme 1b(III)). However, the use of stoichiometric amounts of oxidants, additives, and metal catalysts in such processes limits the potential scope of this environmentally benign transformation. Therefore, the development of a green and economic system for the sustainable construction of highly E-selective N-heteroaromatic olefins is highly desirable and constitutes a new synthetic challenge.
These results, together with our research on the use of air as a green partner for the coupling reactions and syntheses of unsaturated N-heteroaromatics,11 prompted us to envision that benzylamines could potentially be oxidized in air prior to a direct condensation with methylazaarenes to obtain disubstituted olefins. Methylazaarenes to synthesize unsaturated N-heteroaromatics in the presence of air without metals or other additives has yet to be reported. Thus, we herein present the first protocol for the successful use of air as the sole oxidant of amines, and subsequent reaction with a variety of alkyl-substituted azaarenes via olefination to prepare a range of multiply-substituted alkenes containing the N-heteroaromatic structure (Scheme 1c).
Results and discussion
Initially, phenylmethanamine (1a) and 2-methylpyrazine (2a) were selected as model substrates to investigate the feasibility of the olefination reaction (Table 1). To our delight, an excellent yield of 2-styrylquinoline (3a) was obtained with a high E-selectivity in N-methyl-2-pyrrolidone (NMP) and in the presence of air (6 bar) at 160 °C for 24 h, along with traces of byproduct, most likely the Z isomer, were detected by GC-MS analysis (entry 1). This result indicated that our proposed sequential oxidative olefination reaction of methylazaarenes and amines is indeed possible in a metal-free manner. Among the screened solvents, NMP was the most effective. When NMP was replaced with dimethylacetamide (DMAc, entry 2), a lower product yield was obtained, although the reaction enantioselectivity was good. Other solvents, such as anisole and xylene, produced the desired adduct in a moderate yield but with a poor regioselectivity (entries 3 and 4). In addition, upon reducing the air pressure to atmospheric pressure, only a trace amount of the desired product was detected under the current reaction conditions (entry 5). Furthermore, 3a was obtained in a relatively low yield and selectivity when air was replaced with oxygen (entries 6 and 7). In this case, trace amounts of benzoic acid were detected by gas chromatography-mass spectrometry (GC-MS), and this was attributed to the peroxidation of benzylamine in the presence of O2. Control reactions demonstrated that no desired adduct was formed under a nitrogen atmosphere or at a lower reaction temperature (entries 8 and 9). Interestingly, the olefination reaction was relatively unaffected by the addition of two equivalents of water, thereby indicating the potential stability, flexibility, and applicability of this reaction system.
|
|||
|---|---|---|---|
| Entry | Variation from standard conditions | Yield (%) | 3a/5a |
| a Reaction conditions: 1a (1.0 mmol), 2a (2.0 mmol), NMP (2.0 mL), air (6 bar), 160 °C, 24 h. The yield of 3a and the ratio of 3a/5a were determined by GC analysis using n-hexadecane as an internal standard. | |||
| 1 | None | 98 | >20:1 |
| 2 | DMAc instead of NMP | 28 | >20:1 |
| 3 | Anisole instead of NMP | 40 | 13:1 |
| 4 | Xylene instead of NMP | 62 | 10:1 |
| 5 | Air (1 bar) | <5 | — |
| 6 | O2 (1 bar) | 44 | 16:1 |
| 7 | O2 (6 bar) | 27 | 15:1 |
| 8 | N2 (6 bar) | 0 | — |
| 9 | <140 °C | 0 | — |
| 10 | H2O (2 eq.) was added | 93 | >20:1 |

With the optimized conditions in hand (Table 1, entry 1), we investigated the substrate scope of alkyl amine 1 (Table 2). As indicated, various benzylic-type amine derivatives bearing electron-withdrawing or electron-donating groups reacted smoothly with 2-methylpyrazine to produce the corresponding unsaturated N-heteroarene products in good to excellent yields with excellent E-selectivities. Generally, amines bearing electron-withdrawing groups gave slightly higher yields than those bearing electron-donating groups (i.e., 3b–3i vs. 3j–3q). Importantly, substrates bearing various substituents at the para-, meta-, and ortho positions also underwent smooth olefination under the standard conditions. However, the orientation of the benzylamine substituents had a clear impact on the efficacy of the transformation, affording 3d–3e, 3h, 3l, and 3n in decreasing yields but high selectivities. Notably, olefination reactions carried out using halo-substituted benzylamines proceeded smoothly to generate high yields of the corresponding unsaturated N-heteroarenes (3j–3p), which can be employed in further transformations to synthesize complex unsaturated N-heteroarenes. In addition, trifluoromethyl substituent survived well during the standard process, and product 3q was produced in high yields. Furthermore, 1-naphthaldehydes (1r) was employed to obtain the corresponding product in excellent yield. Remarkably, when the heteroarenes thiophen-2-ylmethanamine and pyridin-2-ylmethanamine were subjected to the transformation, good yields of the desired adducts (3s–3t) were isolated with high E-selectivities. In addition to benzyl-type amines, an aliphatic amine (1u) was also found to tolerate this procedure to give an acceptable yield of 3u in the presence of air (10 bar) over a prolonged reaction time.

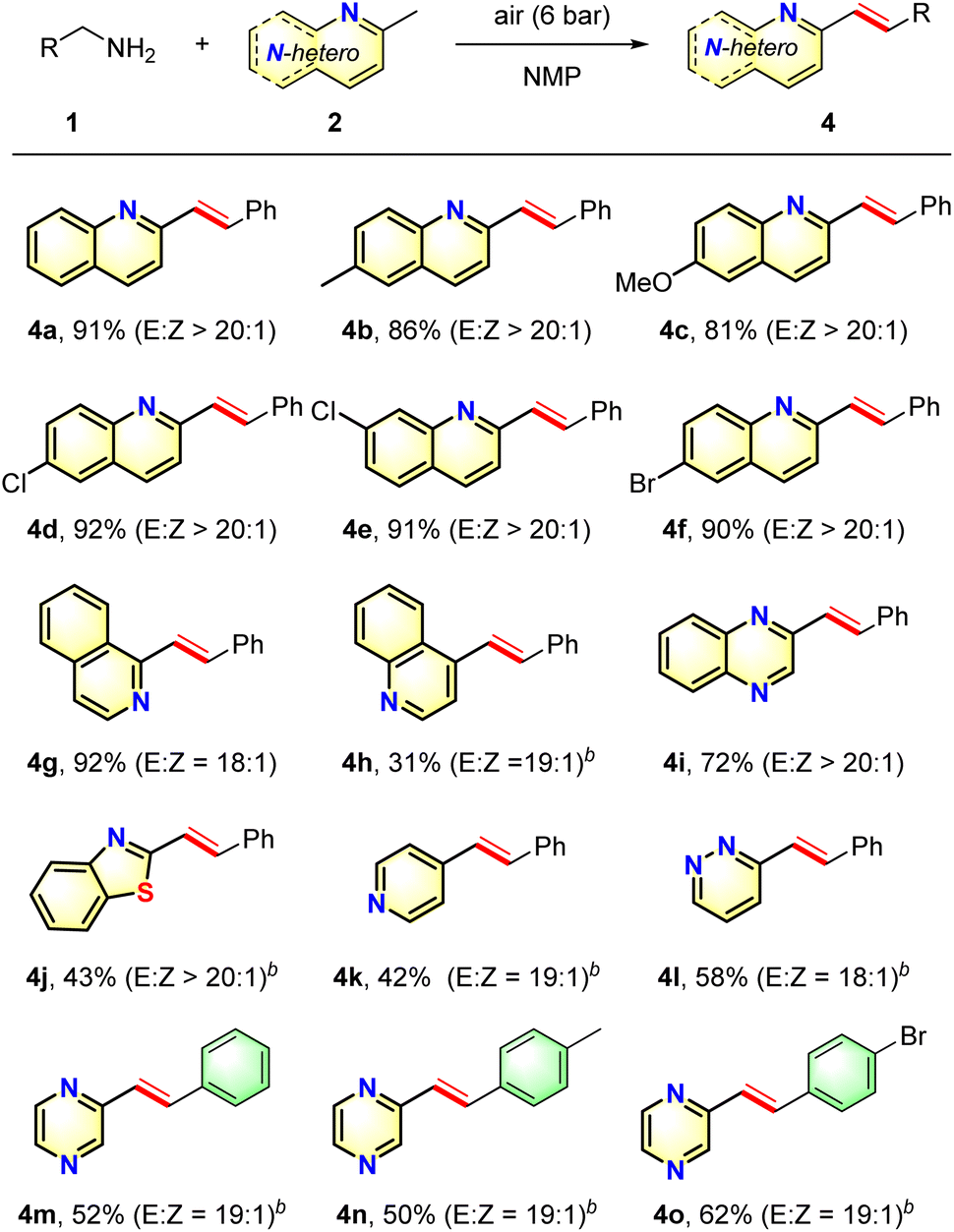
Based on the above results, various N-heteroaromatics were subjected to the oxidation olefination protocol to investigate the scope and generality of the substituted N-heteroarenes (Table 3). Methyl-substituted azaarenes derived from 2-methylquinoline were effective substrates, and reacted smoothly with benzylamine to give the corresponding desired products 4b–4f in good to excellent yields with high selectivities. N-Heteroarenes possessing electron-donating groups gave slightly higher yields than those possessing electron-withdrawing groups, indicating that the olefination step may involve a nucleophilic attack process. In addition, the presence of a halide substituent did not affect the reaction outcome, and these groups remained intact following the transformation, thereby enabling the synthesis of products 4d–4f, which could be employed for further transformations. Furthermore, when the quinoline core was replaced with a quinoxaline or benzo[d]thiazole core, olefination took place to give 4i and 4j in acceptable yields. Although the reaction of 4-methylquinoline afforded 4h in a lower yield, the reaction with 2-methylquinoline proceeded smoothly to give the desired product 4g in an excellent yield. Moreover, substrates 2k–2m were also compatible with this process, furnishing the corresponding products 4k–4m in moderate yields, thereby revealing that a variety of heteroarenes could function as useful substrates for this oxidative olefination transformation. Additionally, the oxidative olefination reactions of 1n and 1o with 2-methylpyrazine resulted in moderate yields of the desired adducts 4n–4o.
Several experiments were then carried out to elucidate the mechanism of the olefination reaction (Scheme 2). Initially, a series of reaction conditions were surveyed using a new reaction tube and stirring bar with ultra-pure specimens of 1a, 2a, and NMP. However, it was found that the substrate purity had no significant effect on the reaction, and the metal content was undetectable by ICP-MS analysis (i.e., <5 ppm), thereby indicating that this was not a metal-catalyzed transformation (eqn (1)). Interestingly, only trace amounts of 3a were produced in the presence of TEMPO or 1,1-diphenylethene, indicating that this reaction may involve a radical process (eqn (2)). Several additional experiments were also conducted under standard conditions to gain further insight into the reaction mechanism. More specifically, using 2a as the coupling partner, intermolecular competition experiments were carried out using 1r (4-CF3) and 1 g (4-OCH3) (eqn (3)). Furthermore, applying 1a as the coupling partner, intermolecular competition experiments were conducted using 2b (4-CH3) and 2d (4-Cl) (eqn (4)). In both reactions, the corresponding products 3r and 4d bearing electron-withdrawing functional groups were formed preferentially, further suggesting that the olefination step may involve nucleophilic attack.
 | ||
| Scheme 2 Control and competition experiments. | ||
It was also found that ammonia gas could be collected as a product in an acceptable yield, strongly indicating that ammonia gas forms from the amine benzylamine substrate (Scheme 3, eqn (5)). In addition, in the reaction outlined in eqn (6), phenylmethanimine was detected by GC-MS under standard conditions after 60 min and after a prolonged reaction time of 6 hours. Furthermore, benzaldehyde and N-benzyl-1-phenylmethanimine were also detected in low amounts, which was attributed to imine hydrolysis and the condensation of 1a with the aldehyde (Scheme 3, eqn (6)). Moreover, high yields of 3a were obtained when the reaction was performed using phenylmethanimine as the reaction partner under the standard reaction conditions, while an acceptable yield was obtained when benzaldehyde was employed. Additionally, the replacement of 1a with N-benzyl-1-phenylmethanimine resulted in a moderate product yield (Scheme 3, eqn (7)–(9)). These results therefore reveal that phenylmethanimine, benzaldehyde, and N-benzyl-1-phenylmethanimine may be intermediates in this reaction, as they as they were observed in situ and were found to be suitable partners for producing the desired adducts.
 | ||
| Scheme 3 Mechanistic investigations for the transformation. | ||
Based on the above results in combination with our previous studies,12 a plausible mechanism for this oxidative olefination was proposed, as outlined in Fig. 1. More specifically, using 1a and 2a as the model substrates, benzylamine 1a is initially oxidized to the highly active phenylmethanimine under an air atmosphere. Subsequently, the phenylmethanimine undergoes nucleophilic attack by 2a to produce the desired product 3 and releases a molecule of ammonia gas. It is also possible that phenylmethanimine may be converted to benzaldehyde in the presence of water and subsequently by transformed to an N-benzyl-1-phenylmethanimine intermediate in the presence of benzylamine. The intermediate aldehyde or the imine can then be attacked by 2a to generate product 3a.
 | ||
| Fig. 1 Proposed mechanism and preparation of pharmaceuticals. | ||
To further demonstrate the robustness of our transformation, attempted gram-scale reactions. More specifically, we found that products 3a and STB-8 could be obtained in yields of 88% (20.33 g) and 71% (2.4 g), respectively. Based on this newly developed transformation system, the preparation of 2E10 (ref. 13) and montelukast,14 the brain-imaging agent for amyloid deposits and cysteinyl leukotriene receptor 1, could potentially be carried out by iteration. Furthermore, we expect that montelukast, a compound employed for the treatment of asthma and seasonal allergies, could be obtained via the transformation of 4e.
Conclusions
In conclusion, we presented a sustainable and practical protocol for the oxidative olefination of methyl-substituted heterocycles and amines to obtain 1,2-disubstituted unsaturated N-heteroaromatics using air as an inexpensive, green, and sole oxidant. This green method is compatible with various functional groups and can be employed to selectively construct a range of 1,2-disubstituted unsaturated N-heteroaromatics with high E-selectivities. In contrast to existing routes to these compounds, the developed reaction system was efficiently realized under metal- and additive-free reaction conditions. Further studies are currently underway in our laboratory to gain a detailed mechanistic understanding of this aerobic oxidative olefination, and the application of air as the sole oxidant is also being investigated for other reaction systems.Author contributions
H. Cui: methodology; C. Zhang: funding acquisition, writing original draft, investigation, methodology; Y. Ji: methodology, review and editing; G. Zhang: funding acquisition, conceptualization, supervision, writing – review & editing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors are grateful to the financial support of the National Natural Science Foundation of China (22002067, 22202228), the Fund Program for the Scientific Activities of Selected Returned Overseas Professionals in Shanxi Province (20220052), the Science and Technology Project of Shanxi Province (202103021223457), the State Key Laboratory of Coal Conversion, Institute of Coal Chemistry, CAS (2021BWZ011).References
- (a) S. Matar and L. F. Hatch, in Chemistry of Petrochemical Processes, Gulf Professional Publishing, Woburn, 2001 Search PubMed; (b) G. D. Henry, Tetrahedron, 2004, 60, 6043–6061 CrossRef CAS; (c) J. P. Michael, Nat. Prod. Rep., 2005, 22, 627–646 RSC; (d) T. Laird, Org. Process Res. Dev., 2006, 10, 851–852 CrossRef CAS; (e) L.-C. Campeau and K. Fagnou, Chem. Soc. Rev., 2007, 36, 1058–1068 RSC; (f) M. C. Bagley, C. Glover and E. A. Merritt, Synlett, 2007, 2007, 2459–2482 CrossRef; (g) N.-Y. Kang, H.-H. Ha, S.-W. Yun, Y. Yu and Y.-T. Chang, Chem. Soc. Rev., 2011, 40, 3613–3626 RSC.
- For selected examples, see: (a) M. Murakami and S. Hori, J. Am. Chem. Soc., 2003, 125, 4720–4721 CrossRef CAS PubMed; (b) R. Dumeunier and I. E. Markó, in Modern Carbonyl Olefination: Methods and Applications, ed. T. Takeda, Wiley-VCH, Weinheim, 2004 Search PubMed; (c) K. S. Kanyiva, Y. Nakao and T. Hiyama, Angew. Chem., Int. Ed., 2007, 46, 8872–8874 CrossRef CAS PubMed; (d) Y. Nakao, K. S. Kanyiva and T. Hiyama, J. Am. Chem. Soc., 2008, 130, 2448–2449 CrossRef CAS PubMed; (e) J. Wu, X. Cui, L. Chen, G. Jiang and Y. Wu, J. Am. Chem. Soc., 2009, 131, 13888–13889 CrossRef CAS PubMed; (f) J. J. Mousseau, J. A. Bull and A. B. Charette, Angew. Chem., Int. Ed., 2010, 49, 1115–1118 CrossRef CAS PubMed; (g) F. Xiao, C. Liu, S. Yuan, H. Huang and G.-J. Deng, J. Org. Chem., 2018, 83, 10420–10429 CrossRef CAS PubMed.
- (a) G. Wittig and G. Geissler, Adv. Cycloaddit., 1953, 580, 44–57 CAS; (b) B. E. Maryanoff and A. B. Reitz, Chem. Rev., 1989, 89, 863–927 CrossRef CAS; (c) Q.-Q. Li, Z. Shah, J.-P. Qu and Y.-B. Kang, J. Org. Chem., 2018, 83, 296–302 CrossRef CAS PubMed; (d) W. S. Wadsworth and W. D. Emmons, J. Am. Chem. Soc., 1961, 83, 1733–1738 CrossRef CAS; (e) J. Clayden and S. Warren, Angew. Chem., Int. Ed., 1996, 35, 241–270 CrossRef CAS; (f) D. J. Peterson, J. Org. Chem., 1968, 33, 780–784 CrossRef CAS; (g) L. F. v. Staden, D. Gravestock and D. J. Ager, Chem. Soc. Rev., 2002, 31, 195–200 RSC; (h) A. K. Simlandy and S. Mukherjee, J. Org. Chem., 2017, 82, 4851–4858 CrossRef CAS PubMed.
- (a) T. M. Trnka and R. H. Grubbs, Acc. Chem. Res., 2001, 34, 18–29 CrossRef CAS PubMed; (b) R. H. Grubbs, in Handbook of Metathesis, WileyVCH, Weinheim, 2003 Search PubMed.
- (a) R. F. Heck and J. P. Nolley, J. Org. Chem., 1972, 37, 2320–2322 CrossRef CAS; (b) M. J. D. Pires, D. L. Poeira, S. I. Purificação and M. M. B. Marques, Org. Lett., 2016, 18, 3250–3253 CrossRef CAS PubMed; (c) J. A. Carmona, V. Hornillos, P. Ramírez-López, A. Ros, J. Iglesias-Sigüenza, E. Gómez-Bengoa, R. Fernández and J. M. Lassaletta, J. Am. Chem. Soc., 2018, 140, 11067–11075 CrossRef CAS PubMed; (d) K. Ferré-Filmon, L. Delaude, A. Demonceau and A. F. Noels, Coord. Chem. Rev., 2004, 248, 2323–2336 CrossRef; (e) J. P. G. Rygus and C. M. Crudden, J. Am. Chem. Soc., 2017, 139, 18124–18137 CrossRef CAS PubMed; (f) S. H. Nazari, J. E. Bourdeau, M. R. Talley, G. A. Valdivia-Berroeta, S. J. Smith and D. J. Michaelis, ACS Catal., 2018, 8, 86–89 CrossRef CAS.
- (a) C. Chaudhari, S. M. A. H. Siddiki and K.-i. Shimizu, Tetrahedron Lett., 2013, 54, 6490–6493 CrossRef CAS; (b) M. Staderinia, N. Cabezasa, M. L. Bolognesi, J. C. Menéndez, D. d. Q. O. y. Farmacéutica and F. d. Farmacia, Synlett, 2011, 17, 2577–2579 Search PubMed; (c) E. Liang, J. Wang, Y. Wu, L. Huang, X. Yao and X. Tang, Adv. Synth. Catal., 2019, 361, 3619–3623 CrossRef CAS; (d) Z. Jamal, Y.-C. Teo and G. S. Lim, Tetrahedron, 2016, 72, 2132–2138 CrossRef CAS; (e) S. Yaragorla, G. Singh and R. Dada, Tetrahedron Lett., 2015, 56, 5924–5929 CrossRef CAS; (f) D. Pi, K. Jiang, H. Zhou, Y. Sui, Y. Uozumi and K. Zou, RSC Adv., 2014, 4, 57875–57884 RSC; (g) B. Qian, P. Xie, Y. Xie and H. Huang, Org. Lett., 2011, 13, 2580–2583 CrossRef CAS PubMed.
- (a) T. A. Turney and G. A. Wright, Chem. Rev., 1959, 59, 497–513 CrossRef; (b) S. S. Rawalay and H. Shechter, J. Org. Chem., 1967, 32, 3129–3131 CrossRef CAS; (c) N. A. Noureldin and J. W. Bellegarde, Synthesis, 1999, 1999, 939–942 CrossRef; (d) S. Sobhani, S. Aryanejad and M. F. Maleki, Helv. Chim. Acta, 2012, 95, 613–617 CrossRef CAS; (e) M. Largeron and M.-B. Fleury, J. Org. Chem., 2000, 65, 8874–8881 CrossRef CAS PubMed; (f) J. K. Crandall and T. Reix, J. Org. Chem., 1992, 57, 6759–6764 CrossRef CAS; (g) S. Chiampanichayakul, M. Pohmakotra, V. Reutrakula, T. Jaipetchb and C. Kuhakarn, Synthesis, 2008, 2008, 2045–2048 CrossRef.
- (a) S. Hazra, V. Tiwari, A. Verma, P. Dolui and A. J. Elias, Org. Lett., 2020, 22, 5496–5501 CrossRef CAS PubMed; (b) L. Gong, L.-J. Xing, T. Xu, X.-P. Zhu, W. Zhou, N. Kang and B. Wang, Org. Biomol. Chem., 2014, 12, 6557–6560 RSC.
- (a) D. Mao, X. Zhu, G. Hong, S. Wu and L. Wang, Synlett, 2016, 27, 2481–2484 CrossRef CAS; (b) P. R. Thorve and B. Maji, Org. Lett., 2021, 23, 542–547 CrossRef CAS PubMed; (c) X. Li, B. Huang, J. Wang, Y. Zhang and W. Liao, J. Chem. Res., 2021, 45, 903–910 CrossRef CAS; (d) C. Dong, Y. Wang, S. Chen, Y. Zhang, C. Wang, J. Li and Y. Zhou, Eur. J. Org Chem., 2023, 26, e202300194 CrossRef CAS.
- R. Sharma, M. Abdullaha and S. B. Bharate, J. Org. Chem., 2017, 82, 9786–9793 CrossRef CAS PubMed.
- (a) G. Zhang, T. Irrgang, T. Dietel, F. Kallmeier and R. Kempe, Angew. Chem., Int. Ed., 2018, 57, 9131–9135 CrossRef CAS PubMed; (b) G. Zhang, L. Yang, Y. Wang, Y. Xie and H. Huang, J. Am. Chem. Soc., 2013, 135, 8850–8853 CrossRef CAS PubMed; (c) L. Yang, G. Zhang and H. Huang, Adv. Synth. Catal., 2014, 356, 1509–1515 CrossRef CAS; (d) G. Zhang, H. Yu, G. Qin and H. Huang, Chem. Commun., 2014, 50, 4331–4334 RSC; (e) F. Wang, D. Ding, C. Zhang and G. Zhang, Org. Chem. Front., 2024 10.1039/D3QO01890C.
- (a) A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328–3435 CrossRef CAS PubMed; (b) S. Hazra, M. Deb and A. J. Elias, Green Chem., 2017, 19, 5548–5552 RSC; (c) X.-F. Wu, J.-L. Gong and X. Qi, Org. Biomol. Chem., 2014, 12, 5807–5817 RSC; (d) U. P. N. Tran, G. Oss, M. Breugst, E. Detmar, D. P. Pace, K. Liyanto and T. V. Nguyen, ACS Catal., 2019, 9, 912–919 CrossRef CAS; (e) B. Wang and H. N. C. Wong, Bull. Chem. Soc. Jpn., 2018, 91, 710–719 CrossRef CAS.
- Q. Li, J. Min, Y.-H. Ahn, J. Namm, E. M. Kim, R. Lui, H. Y. Kim, Y. Ji, H. Wu, T. Wisniewski and Y.-T. Chang, ChemBioChem, 2007, 8, 1679–1687 CrossRef CAS PubMed.
- (a) S. Pu, Q. Liu, Y. Li, R. Li, T. Wu, Z. Zhang, C. Huang, X. Yang and J. He, Front. Pharmacol, 2019, 18, 1070 CrossRef PubMed; (b) A. R. Khan, C. Misdary, N. Yegya-Raman, S. Kim, N. Narayanan, S. Siddiqui, P. Salgame, J. Radbel, F. De Groote, C. Michel, J. Mehnert, C. Hernandez, T. Braciale, J. Malhotra, M. A. Gentile and S. K. Jabbour, J. Asthma, 2022, 59, 780–786 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra00189c |
| This journal is © The Royal Society of Chemistry 2024 |