
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery of new tetrazines for bioorthogonal reactions with strained alkenes via computational chemistry†
Michal Májek * and
Matej Trtúšek
* and
Matej Trtúšek
Comenius University Bratislava, Faculty of Natural Sciences, Department of Organic Chemistry, Mlynská Dolina, Ilkovičova 6, 842 15 Bratislava, Slovakia. E-mail: michal.majek@uniba.sk
First published on 31st January 2024
Abstract
Tetrazines are widely employed reagents in bioorthogonal chemistry, as they react readily with strained alkenes in inverse electron demand Diels–Alder reactions, allowing for selective labeling of biomacromolecules. For optimal performance, tetrazine reagents have to react readily with strained alkenes, while remaining inert against nucleophiles like thiols. Balancing these conditions is a challenge, as reactivity towards strained alkenes and nucleophiles is governed by the same factor – the energy of unoccupied orbitals of tetrazine. Herein, we utilize computational chemistry to screen a set of tetrazine derivatives, aiming to identify structural elements responsible for a better ratio of reactivity with strained alkenes vs. stability against nucleophiles. This advantageous trait is present in sulfone- and sulfoxide-substituted tetrazines. In the end, the distortion/interaction model helped us to identify that the reason behind this enhanced reactivity profile is a secondary orbital interaction between the strained alkene and sulfone-/sulfoxide-substituted tetrazine. This insight can be used to design new tetrazines for bioorthogonal chemistry with improved reactivity/stability profiles.
Introduction
Biological systems are an important target of molecular science research, containing biomacromolecules, and being the host of fast, selective enzymatic reactions and precise molecular sensing systems. To comprehensively study these complex systems, the capability to perform chemical reactions selectively and in real-time within these systems is essential. However, this is a difficult task due to the inherent hostility of the biological environment towards typical organic reagents. Cells contain high concentrations of free reactive nucleophilic centers like thiols and amines from amino acid residues. This is combined with an aqueous reaction medium containing oxygen and different electrolytes. Bioorthogonal chemistry introduced new reactions that enable selective chemistry in such complex systems.1–6For bioorthogonal reagents, robustness under physiological conditions is crucial. Yet, they need to provide the required reaction at a very high rate, as the local concentrations of the reagents are usually relatively low when compared to a typical organic reaction. Having these requirements in mind, researchers have been successful in developing multiple classes of biorthogonal reactions. The first-generation reactions, such as Staudinger ligation7,8 and copper-catalyzed alkyne–azide cycloaddition,9,10 served as a proof of concept but exhibited significant limitations in real systems. Staudinger ligation relies on phosphorus(III) reagents, unstable in the presence of oxygen, while copper-based catalysts have only limited compatibility with living organisms. The breakthrough came with the second generation of biorthogonal reactions – strain-promoted reactions like azide–alkyne cycloadditions (SPAAC),11,12 alkyne–nitrone cycloadditions (SPANC),13,14 and the tetrazine (s-tetrazine) ligation.15–17 These reactions proceed rapidly while having superior stability of the required reagents at physiological conditions in comparison with the first-generation reactions. Tetrazine ligation has multiple attractive properties concerning the development of systems applicable to living organisms. The reactivity of the tetrazine can be modulated by modifying the substitution pattern on the tetrazine core to achieve the desired reaction rates.18 Moreover, the tetrazine system undergoes significant electronic structure reorganization during the ligation reaction, allowing the design of switchable fluorescent tags, where fluorescence is extinguished or enhanced after the ligation process.19
When designing a new tetrazine reagent suitable for bioorthogonal ligation reactions a balance between their reactivity and stability is crucial. Such tetrazines need to have a high rate constant for the required ligation reaction. Mechanistically, tetrazine ligation reactions are usually inverse-electron demand Diels–Alder reactions (iEDDA).20 Tetrazines serve as the diene component in these reactions and therefore, an efficient interaction of one of their unoccupied orbitals with the HOMO orbital of the dienophile is required to enhance the reaction rate. This can be accomplished by introducing electron-withdrawing functional groups on the tetrazine core.21 Typically ester- and pyridiyl-substituents can be used for this purpose. The presence of electron-withdrawing groups lowers the energy of unoccupied orbitals and promotes the desired iEDDA reaction.
Unfortunately, this also leads to decreased stability of the tetrazine reagent in physiological conditions, making electron-poor tetrazines susceptible to attack by nucleophiles, leading to their decomposition in aqueous conditions.22 Even worse, such tetrazines react readily with exposed nucleophilic centers present on biomacromolecules, leading to unselective labeling of biomacromolecules with the tetrazine reagent. Due to their high reactivity, thiol groups present in molecules containing cysteine are the biggest issue.23
Multiple strategies have been employed to increase the reactivity of dienes for bioorthogonal applications in iEDDA reactions while preserving their stability in biological systems. Due to the low stability of electron-poor tetrazines, this often necessitated shifting away from tetrazines to alternative scaffolds, such as triazines, where sluggish reactivity in iEDDA reactions becomes a problem.24,25 In this case, the reactivity could be successfully increased by alkylating one of the nitrogens to generate a positively charged species – N1-alkyl-1,2,4-triazinium salts.26 Another approach is to decrease the electron density on the triazine core by the coordination of heavy metal ions to the triazine core.27,28 Yet the reactivity of triazines in iEDDA reactions with strained alkynes and trans-alkenes is generally significantly lower than that of similar tetrazines. Thus, the search for an optimal diene system for bioorthogonal iEDDA reactions continues (Scheme 1).
 | ||
| Scheme 1 Bioorthogonal iEDDA reactions using tetrazines or triazines as dienes with strained alkenes as dienophiles. | ||
Computational chemistry has emerged as an efficient tool in this pursuit, enabling rapid screening of potential structures and providing insights into the operative reaction mechanism.29 DFT computational models, extensively applied by Houk and coworkers, have identified multiple new promising reagents.30–32 The simplest approach to the theoretical study of iEDDA reactions relies only on the energies of FMO (frontier molecular orbitals). Unfortunately, while computationally very simple to achieve, this approach does not take into account all of the interactions that occur during the reaction – such as steric effects or electrostatic forces. To obtain a precise prediction of reactivity of the new iEDDA reagents, activation energy has to be calculated, which requires the localization of the transition state(s). This can be coupled with more advanced methods, such as distortion–interaction analysis, which allows us to gain insight into the steric and electronic effects during the reaction, or molecular dynamics simulations.33
The current pool of tetrazines tested in bioorthogonal iEDDA reactions included systems having various substituents on the tetrazine core, notably esters, pyridines, alkyl chains, and amines. Yet, there is still a significant number of substituents that have not been tested for their suitability in this type of application. Our aim is to identify new suitable candidates within the previously unexplored substituent space. To help with this task, we composed a set of different tetrazines to be subjected to our study. We aim to find a new system in which increased reactivity in the desired iEDDA reaction would not translate into increased susceptibility towards nucleophilic attack and thus degradation in biological environments. So far, there have been few reports of substituted tetrazines where the reactivity in iEDDA reactions has been uncoupled from the reactivity towards nucleophiles. Recently, Svatunek and Mikula have uncovered the underlying mechanism responsible for this feature using distortion–interaction analysis.34 We followed a similar approach on our set of tetrazines.
Results
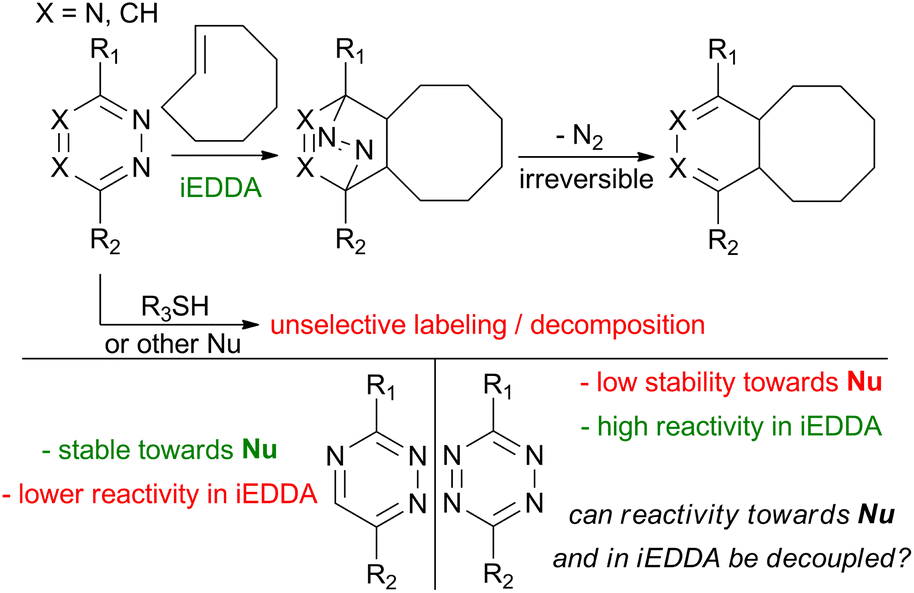
Firstly, we identified which substitution patterns on tetrazine will be the target of this study. When using tetrazine derivatives for biomolecule tagging, one of the substituents on the tetrazine core has to act as a linker. These substituents allow for connection with labels or biomacromolecules. This is accomplished by carboxylic acids and hydroxyl- and amino-groups, which are then used to form an ester- or amide-bond. Importantly, such groups are usually not directly introduced onto the tetrazine core, but they are linked to it via a phenyl- or alkyl-linker.22 In this study, we simplify these linkers as a phenyl- or methyl-substituent. The other substituent on the tetrazine can then be used to modulate its reactivity. We have selected a series of 14 different substituents to study, composed from both electron-withdrawing (EWG) as well as electron donating (EDG) groups (for quantification of the nature of the selected substituents by Hammett constants see ESI†) and atoms from different periods so that also a variance in the size of the substituents is achieved in our set (Fig. 1a).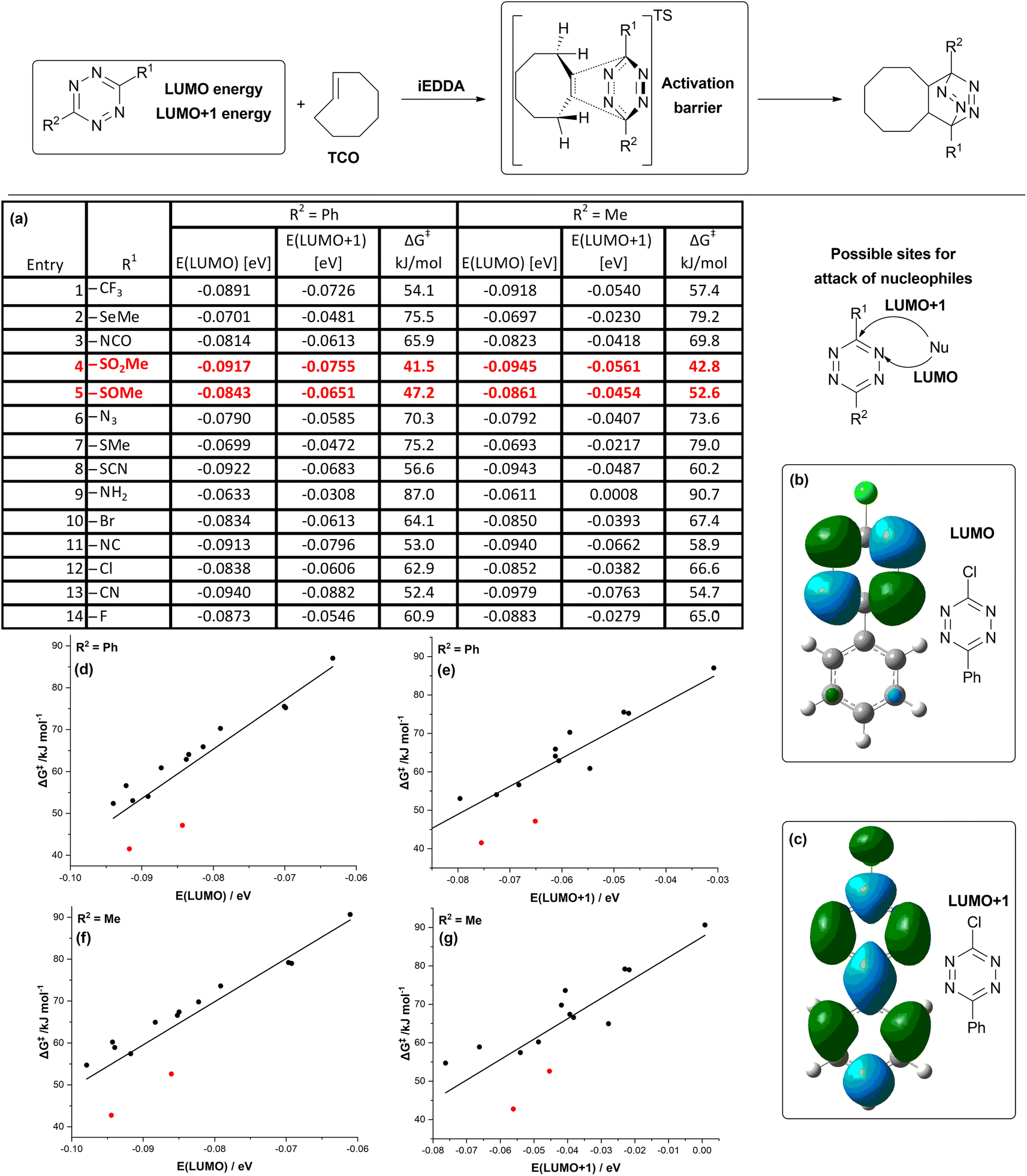 | ||
| Fig. 1 (a) Results of DFT calculations for the set of tetrazine derivatives. Energies of LUMO and LUMO+1 of respective tetrazines are given in eV, free energies of activation for reaction with TCO are given in kJ mol−1. Calculations details are in the ESI;† (b) shape of LUMO orbital of tetrazine TCL; (c) shape of LUMO+1 orbital of tetrazine TCL; (d–g) plots of free energies of activation for reaction of tetrazines with TCO against LUMO and LUMO+1 energies of tetrazines. Derivatives TSO and TSO2 are marked red. | ||
Our strategy involves identifying potential candidates via the correlation of the activation barriers of iEDDA reactions with the energies of the relevant unoccupied orbitals on the tetrazines. To select the appropriate orbitals to be considered in our study, we calculated their shapes (Fig. 1b and c). Previous literature indicates that both carbon atoms and the nitrogen atoms of tetrazine are susceptible to nucleophilic attack.35,36 Given these previous reports on the reactivity of the tetrazines, both LUMO and LUMO+1 orbitals are relevant for the tetrazine decomposition pathway – as the LUMO orbital can be implicated in the mechanism, where an attack on nitrogen occurs, whereas the LUMO+1 orbital is responsible for the reactivity of carbon atoms towards nucleophiles. As the model iEDDA reaction, we chose a reaction with trans-cyclooctene (TCO), a commonly used type of reagent for bioorthogonal applications.37
Across all cases studied, phenylated (R2 = Ph) tetrazines exhibited a lower activation barrier with TCO compared to respective alkylated (R2 = Me) tetrazines. This is in line with previously reported data, where the tetrazines bearing alkyl linker generally reacted more sluggishly than the tetrazines with phenyl linker.22 As anticipated, the introduction of electron-donating groups on tetrazines increased the activation barriers of the iEDDA reaction. This was most pronounced with amino- alkylsulfanyl- and alkylselenyl-compounds (Fig. 1a; entries 2, 7 and 9). Conversely, electron-withdrawing substituents had the opposite effect. Among these were trifluoromethyl- and nitrile-substituents (Fig. 1a; entries 1 and 13), but interestingly, also thiocyano- and isonitrile-substituents (Fig. 1a; entries 8 and 11). The most activated compounds were sulfoxides and sulfones (Fig. 1a; entries 4 and 5). With this data at hand, we proceeded to plot the activation energy of the iEDDA reaction of tetrazines with TCO against the energies of LUMO orbitals (Fig. 1d and f) and LUMO+1 orbitals (Fig. 1e and g) of tetrazines. This was done in order to identify compounds where uncoupling of their reactivity in iEDDA reactions from their reactivity towards nucleophiles can be expected. For most of the compounds, a linear relationship between reactivity in the iEDDA reaction with TCO with the energy of the LUMO+1 orbital of tetrazines should be observed, as this is the orbital interacting with the dienophile during the course of the reaction. Indeed, this relationship holds true for most of the studied compounds. The same can be said about the energy of the LUMO orbital of tetrazines – the linear relationship between its energy and the predicted reactivity is again seen for most of the derivatives. There are two classes of compounds which do not follow this trend – sulfoxides and sulfones (Fig. 1a; entries 4 and 5). Interestingly, this behaviour of sulfones and sulfoxides is exhibited at all of the four studied relationships (Fig. 1d–g). The predicted reactivity of sulfones and sulfoxides is significantly higher than expected, based on their energies of LUMO and LUMO+1 orbitals. In order to gain further insight into this phenomenon, we subjected these derivatives to a more thorough study via the distortion–interaction analysis. The distortion–interaction model allows for the decomposition of the activation barrier into two components – distortion energy, which is the energy necessary for the deformation of the reactants from their equilibrium geometry into the geometry required by the transition state, and interaction energy, which has a stabilizing effect and comes from the interaction of orbitals of the two reactants in the transition state. This model has been successfully used before to provide explanation for the unexpected reactivity of several bioorthogonal reagents.13,38
For the analysis via the distortion–interaction model, we have chosen three different derivatives from our set. All of them have methyl as one of the substituents (R2 = Me). Reason behind this choice is that methyl group is likely to influence the transition states less than phenyl, which may influence the electronic distribution due to conjugation of its π-electrons. We chose chlorine, sulfoxide and sulfone as the other substituents (Fig. 1a, entries 4, 5 and 12). Sulfone TSO and sulfoxide TSO2 were the substituents on tetrazines, which exhibited the abnormally low activation barrier of the iEDDA reaction.
Chlorine-bearing tetrazine TCL, on the other hand, behaved in line with the trend of reactivity predicted from the energies of LUMO+1 orbitals. It is known that distortion–interaction analysis at transition state geometries can lead to skewed results when used to compare transition states with very different geometries. In our set of compounds, bond lengths in the transition state of the sulfone system (Fig. 2c) vary somewhat from the ones of chloro compound (Fig. 2a), but the difference between the geometry of the transition state of the sulfoxo-tetrazine (Fig. 2b) is similar, so the analysis of this pair (Fig. 2a vs. b) should give meaningful results.39 The outcome of the distortion–interaction analysis shows that both sulfone TSO2 as well as sulfoxide TSO have significantly stronger interaction with the TCO than the chlorinated tetrazine TCL (−71.1 kJ mol−1 and −70.5 kJ mol−1 vs. −62.1 kJ mol−1) in the transition state. The distortion energy of the tetrazine moiety is also lower (42.2 kJ mol−1 and 45.2 kJ mol−1 vs. 51.9 kJ mol−1). This hints that interaction other than the bond-forming process is happening during the course of the reaction of the compounds TSO2 and TSO with TCO, stabilizing the transition state. Lower distortion energy in combination with somewhat longer bonds in transition show state (2.40 Å; 2.341 Å for TSO2 and 2.35 Å; 2.37 Å for TSO vs. 2.32 Å; 2.29 Å for TCL), that the transition state occurs earlier in the reaction for sulfone and sulfoxide than for the TCL.
 | ||
| Fig. 2 Distortion–interaction analyses of reaction of TCO with (a) TCL; (b) TSO; (c) TSO2. Electronic activation energy ΔE‡ is shown with black arrows, distortion energy ΔEdis is shown with blue arrows and interaction energy ΔEint is shown with red arrows. | ||
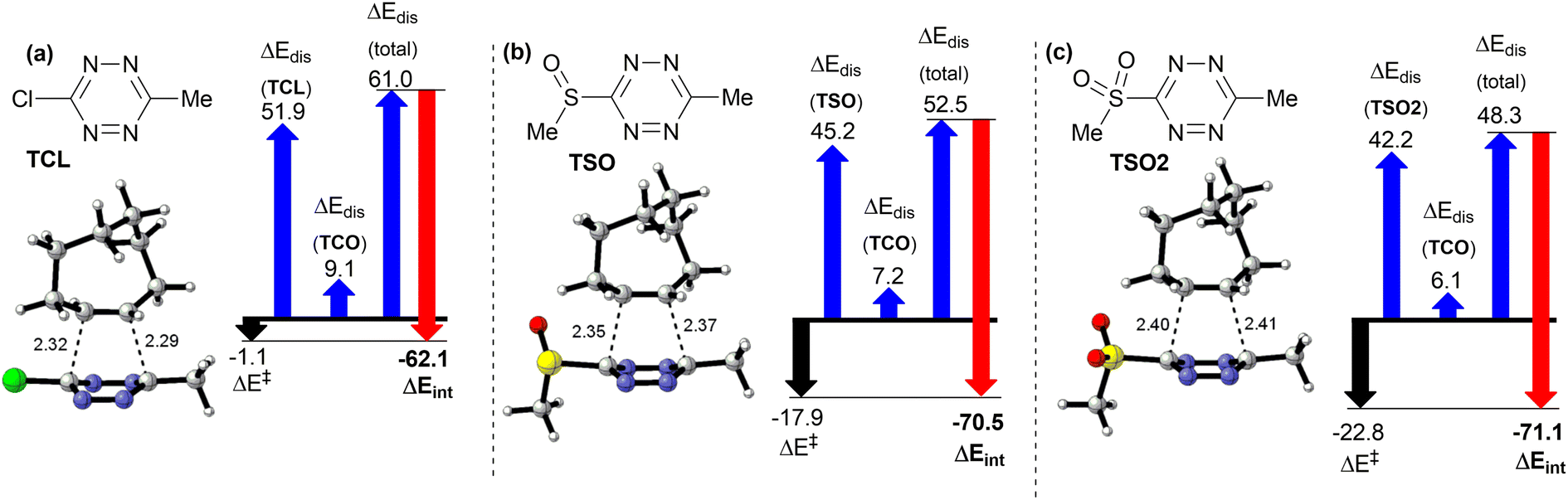
In order to identify the source of this effect, we have calculated the shape of the orbitals interacting together in the reaction – i.e. HOMO orbital of the TCO (Fig. 3a) and the LUMO+1 orbitals of the tetrazines TSO and TSO2 (Fig. 3b and c). Due to the strained nature of the double bond in TCO, its HOMO orbital is not a pure π-bonding orbital centered above and below the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond, but it also contains lobes on adjacent C–H bonds. This leads to a stabilizing interaction with lobes of LUMO+1 orbitals centered on the sulfone- and sulfoxide moiety of the respective tetrazine derivatives TSO2 and TSO (Fig. 3b and c), explaining the increased interaction energy of compounds TSO and TSO2 with respect to the compound TCL. Geometry of the transition state suggests that such secondary orbital interaction indeed occurs, as the C–H bond of TSO moves towards the SO moiety of TSO and TSO2 (2.63 Å for sulfoxide and 2.53 Å for sulfone) during the reaction. Such additional stabilization of the transition state explains the unexpectedly low activation barrier for the derivatives TSO and TSO2 in iEDDA cycloaddition with TCO. Secondary orbital interactions have been reported as the driving force behind unexpected reactivity patterns of tetrazines before.40 As this lowering of the activation barrier was not achieved by decreasing the energy of LUMO and LUMO+1 orbitals, we can predict that sulfone- and sulfoxide-substituted tetrazines will exhibit a better ratio between reactivity in iEDDA reactions with TCO to their lability towards nucleophiles, making them an interesting candidate for incorporation in bioorthogonal reagents. Importantly, from the point of future applications, the synthesis of sulfoxide/sulfone-substituted tetrazines is already known.41 In addition to the found secondary orbital interaction, it is possible that the unusual reactivity of TSO and TSO2 is also affected by differences in Pauli repulsion of the reactants, as this has been proposed recently for similar derivatives where reactivity is not driven by differences in orbital energy.42 Our group is currently actively working on the application of sulfoxide/sulfone-substituted tetrazines in bioorthogonal chemistry and further analysis of their reactivity by experimental as well as theoretical means.
C double bond, but it also contains lobes on adjacent C–H bonds. This leads to a stabilizing interaction with lobes of LUMO+1 orbitals centered on the sulfone- and sulfoxide moiety of the respective tetrazine derivatives TSO2 and TSO (Fig. 3b and c), explaining the increased interaction energy of compounds TSO and TSO2 with respect to the compound TCL. Geometry of the transition state suggests that such secondary orbital interaction indeed occurs, as the C–H bond of TSO moves towards the SO moiety of TSO and TSO2 (2.63 Å for sulfoxide and 2.53 Å for sulfone) during the reaction. Such additional stabilization of the transition state explains the unexpectedly low activation barrier for the derivatives TSO and TSO2 in iEDDA cycloaddition with TCO. Secondary orbital interactions have been reported as the driving force behind unexpected reactivity patterns of tetrazines before.40 As this lowering of the activation barrier was not achieved by decreasing the energy of LUMO and LUMO+1 orbitals, we can predict that sulfone- and sulfoxide-substituted tetrazines will exhibit a better ratio between reactivity in iEDDA reactions with TCO to their lability towards nucleophiles, making them an interesting candidate for incorporation in bioorthogonal reagents. Importantly, from the point of future applications, the synthesis of sulfoxide/sulfone-substituted tetrazines is already known.41 In addition to the found secondary orbital interaction, it is possible that the unusual reactivity of TSO and TSO2 is also affected by differences in Pauli repulsion of the reactants, as this has been proposed recently for similar derivatives where reactivity is not driven by differences in orbital energy.42 Our group is currently actively working on the application of sulfoxide/sulfone-substituted tetrazines in bioorthogonal chemistry and further analysis of their reactivity by experimental as well as theoretical means.
 | ||
| Fig. 3 (a) HOMO orbital of TCO; (b) LUMO+1 orbital of TSO; (c) LUMO+1 orbital of TSO2. | ||
Computational details
Conformation analysis was performed using Spartan program package.43 quantum chemical calculations were performed using Gaussian G16 RevC.01 software package.44 Geometry optimization of minima involved were performed at M062X level of theory and 6-31G(d) basis set for optimisation of geometries. This combination has been successfully used before to obtain transition state geometries of pericyclic reactions of tetrazine.15,45 Single point calculations were performed using 6-311+g(2d,p) basis set. All calculations were done in vacuo. Frequency calculations were performed on the minima in order to ascertain the type of stationary point as well as to obtain thermochemistry. The nature of the transition states was investigated by running an IRC calculation, in order to prove that the correct transition state was found. Thermodynamic properties were calculated at 298 Kelvin. A quasiharmonic correction was applied during the entropy calculation by setting all positive frequencies that are less than 100 cm−1 to 100 cm−1 using script GoodVibes.46Conclusions
We have predicted the reactivity of a series of substituted tetrazines in iEDDA reaction with trans-cyclooctene. For most of the tetrazines, their reactivity in such reaction was linearly correlated with the energy of their LUMO and LUMO+1. Tetrazines bearing sulfoxide and sulfone moiety were an exception to this trend, as their calculated activation barrier for the iEDDA reaction with trans-cyclooctene was significantly lower than what would be expected based on the energy of their LUMO and LUMO+1 orbitals. This unexpected enhancement of reactivity could be explained by distortion–interaction analysis. The underlying reason behind this phenomenon is a stabilizing secondary orbital interaction between the sulfoxide/sulfone moiety and the trans-cyclooctene. This makes sulfoxide/sulfone-substituted tetrazines an interesting candidate for applications in bioorthogonal chemistry, as they should exhibit increased reactivity in the iEDDA reactions with trans-cyclooctene without being more susceptible to decomposition by nucleophiles (e.g. thiols).Author contributions
MM designed the project, analysed the data, prepared and edited the manuscript; MT performed the DFT calculations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by VEGA grant no. 1/0633/21. Funded by the European Union (ERC, CAPELE, 101078608). Views and opinions expressed are however those of the author(s) only and do not necessarily reflect those of the European Union or the European Research Council. Neither the European Union nor the granting authority can be held responsible for them.Notes and references
- Q. Fu, S. Shen, P. Sun, Z. Gu, Y. Bai, X. Wang and Z. Liu, Chem. Soc. Rev., 2023, 52, 7737–7772 RSC.
- R. E. Bird, S. A. Lemmel, X. Yu and Q. A. Zhou, Bioconjugate Chem., 2021, 32, 2457–2479 CrossRef CAS PubMed.
- S. S. Nguyen and J. A. Prescher, Nat. Rev. Chem., 2020, 4, 476–789 CrossRef CAS PubMed.
- S. L. Scinto, D. A. Bilodeau, R. Hincapie, W. Lee, S. S. Nguyen, M. Xu, C. W. am Ende, M. G. Finn, K. Lang, Q. Lin, J. P. Pezacki, J. A. Prescher, M. S. Robillard and J. M. Fox, Nat. Rev. Methods Primers, 2021, 1, 30 CrossRef CAS PubMed.
- N. K. Devaraj, ACS Cent. Sci., 2018, 4, 952–959 CrossRef CAS PubMed.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed.
- C. Bednarek, I. Wehl, N. Jung, U. Schepers and S. Braese, Chem. Rev., 2020, 120, 4301–4354 CrossRef CAS PubMed.
- E. Saxon and C. R. Bertozzi, Science, 2000, 287, 2007–2010 CrossRef CAS PubMed.
- L. Li and Z. Zhang, Molecules, 2016, 21, 1393 CrossRef PubMed.
- Q. Wang, T. R. Chan, R. Hilgraf, V. V. Fokin, B. Sharpless and M. G. Finn, J. Am. Chem. Soc., 2003, 125(11), 3192–3193 CrossRef CAS PubMed.
- E. G. Chupakhin and M. Y. Krasavin, Chem. Heterocycl. Compd., 2018, 54, 483–501 CrossRef CAS.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- M. Nakajima, D. A. Bilodeau and J. P. Pezacki, RSC Adv., 2020, 10, 29306–29310 RSC.
- C. S. Craig, J. Moran and J. P. Pezacki, Chem. Commun., 2010, 46, 931–933 RSC.
- H. Wu and N. K. Devaraj, Acc. Chem. Res., 2018, 51, 1249–1259 CrossRef CAS PubMed.
- N. K. Devaraj, R. Weissleder and S. A. Hilderbrand, Bioconjugate Chem., 2008, 19, 2297–2299 CrossRef CAS PubMed.
- M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518–13519 CrossRef CAS PubMed.
- M. R. Karver, R. Weissleder and S. A. Hilderbrand, Bioconjugate Chem., 2011, 22, 2263–2270 CrossRef CAS PubMed.
- G. Beliu, A. J. Kurz, A. C. Kuhlemann, L. Behringer-Pliess, M. Meub, N. Wolf, J. Seibel, Z.-D. Shi, M. Schnermann, J. B. Grimm, L. D. Lavis, S. Doose and M. Sauer, Commun. Biol., 2019, 2, 261 CrossRef PubMed.
- J. Sauer and D. Lang, Angew. Chem., 1964, 76, 603 CrossRef.
- B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Chem. Soc. Rev., 2017, 46, 4895–4950 RSC.
- A. Maggi, E. Ruivo, J. Fissers, C. Vangestel, S. Chatterjee, J. Joossens, F. Sobott, S. Staelens, S. Stroobants, R. Van Der Veken, L. wyffels and K. Augustyns, Org. Biomol. Chem., 2016, 14, 7544–7551 RSC.
- H. E. Murrey, J. C. Judkins, C. W. am Ende, T. E. Ballard, Y. Fang, K. Riccardi, E. R. Guilmette, J. W. Schwartz, J. M. Fox and D. S. Johnson, J. Am. Chem. Soc., 2015, 137, 11461–11475 CrossRef CAS PubMed.
- F.-G. Zhang, Z. Chen, X. Tang and J.-A. Ma, Chem. Rev., 2021, 121, 14555–14593 CrossRef CAS PubMed.
- D. N. Kamber, Y. Liang, R. J. Blizzard, F. Liu, R. A. Mehl, K. N. Houk and J. A. Prescher, J. Am. Chem. Soc., 2015, 137, 8388–8391 CrossRef CAS PubMed.
- Z. V. Šlachtová, S. Bellová, A. La-Venia, J. Galeta, M. Dračínský, K. Chalupský, A. Dvořáková, H. Mertlíková-Kaiserová, P. Rukovanský, R. Dzijak and M. Vrábel, Angew. Chem., Int. Ed., 2023, 62, e202306828 CrossRef PubMed.
- M. Sims, S. Kyriakou, A. Matthews, M. E. Deary and V. N. Kozhevnikov, Dalton Trans., 2023, 52, 10927–10932 RSC.
- V. N. Kozhevnikov, M. E. Deary, T. Mantso, M. I. Panayiotidis and M. T. Sims, Chem. Commun., 2019, 55, 14283–14286 RSC.
- T. Deb, J. Tu and R. M. Franzini, Chem. Rev., 2021, 121, 6850–6914 CrossRef CAS PubMed.
- A. Sengupta, B. Li, D. Svatunek and K. N. Houk, Acc. Chem. Res., 2022, 55, 2467–2479 CrossRef CAS PubMed.
- F. Liu, Y. Liang and K. N. Houk, Acc. Chem. Res., 2017, 50, 2297–2308 CrossRef CAS PubMed.
- F. Liu, Y. Liang and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 11483–11493 CrossRef CAS PubMed.
- P. Yu, Z. Yang, Y. Liang, X. Hong, Y. Li and K. N. Houk, J. Am. Chem. Soc., 2016, 138, 8247–8252 CrossRef CAS PubMed.
- D. Svatunek, M. Wilkovitsch, L. Hartmann, K. N. Houk and H. Mikula, J. Am. Chem. Soc., 2022, 144, 8171–8177 CrossRef CAS PubMed.
- S. D. Schnell, M. Schilling, J. Sklyaruk, A. Linden, S. Luber and K. Gademann, Org. Lett., 2021, 23, 2426–2430 CrossRef CAS PubMed.
- Q. Zhou, P. Audebert, G. Clavier, F. Miomandre and J. Tang, RSC Adv., 2014, 4, 7193–7195 RSC.
- R. Selvaraj and J. M. Fox, Curr. Opin. Chem. Biol., 2013, 17, 753–760 CrossRef CAS PubMed.
- E. M. Bickelhaupt and K. N. Houk, Angew. Chem., 2017, 56, 10070–10086 CrossRef PubMed.
- T. A. Hamlin, D. Svatunek, S. Yu, L. Ridder, I. Infante, L. Visscher and F. M. Bickelhaupt, Eur. J. Org. Chem., 2019, 2–3, 378–386 CrossRef.
- B. J. Levandowski, D. Svatunek, B. Sohr, H. Mikula and K. N. Houk, J. Am. Chem. Soc., 2019, 141, 2224–2227 CrossRef CAS PubMed.
- A. Hamasaki, R. Ducray and D. L. Boger, J. Org. Chem., 2006, 71, 185–193 CrossRef CAS PubMed.
- N. Houszka, H. Mikula and D. Svatunek, Chem. Eur. J., 2023, 29, e202300345 CrossRef CAS PubMed.
- Spartan 14v114, Wavefunction, Inc., Irvine, 2014 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- S.-E. Suh, S. Chen, K. N. Houk and D. M. Chenoweth, Chem. Sci., 2018, 9, 7688–7693 RSC.
- GoodVibes Version 3.2, Zenodo, DOI:10.5281/zenodo.595246.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra08712c |
| This journal is © The Royal Society of Chemistry 2024 |