
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceManganese-oxide-supported gold catalyst derived from metal–organic frameworks for trace PCl3 oxidation in an organic system†
Qianyi Zhao ,
Qiang Geng and
Guoqiang Huang*
,
Qiang Geng and
Guoqiang Huang*
School of Chemical Engineering and Technology, Tianjin University, China. E-mail: hgq@tju.edu.cn
First published on 30th January 2024
Abstract
Polysilicon is widely used in the field of semiconductors and solar energy. Trichlorosilane feedstocks that are used to produce polysilicon in the mainstream production process contain PCl3 impurities that have adverse effects on the quality of the polysilicon. Traditional methods for dephosphorization cannot achieve the effect of complete removal, whereas oxidizing PCl3 to POCl3 in the presence of oxygen for removal via adsorption is a promising and appealing route for establishing a dephosphorization process; it has a high phosphorous removal rate due to the strong Lewis-base property of POCl3 in comparison with PCl3. In this work, we synthesized an active catalyst with an active interface between Au nanoparticles (NPs) and a manganese-oxide support (Mn3O4) by calcination of a corresponding composite, where Au NPs were embedded uniformly in a metal–organic framework (MOF). The catalyst shows a significantly active catalytic performance for trace PCl3 oxidation in an organic system that is an imitation of a trichlorosilane system, with a 99.13% yield of POCl3 in an 80 °C and 0.6 MPa reaction environment. The structure–performance–mechanism analysis shows that the possible reaction and catalytic mechanism is PCl3 oxidation by interface lattice oxygens, which bridge the Au NPs and the support, in a Mars van Krevelen (MvK) process; this process was promoted by the interaction between the Au NPs and Mn3O4 in terms of charge transfer and chemical potential changes. This work provides an effective way to dephosphorize trichlorosilane feedstocks in the polysilicon industry and gives guidance for constructing an efficient catalyst via the study of the structure and mechanism.
1 Introduction
Phosphorus trichloride (PCl3) is widely present in the trichlorosilane feedstocks that are used to produce polysilicon in the industrial process via an improved Siemens method. In most factories, this method is used as a chief means of polysilicon production.1 Crude trichlorosilane is obtained via a preliminary process and subsequently reduced by hydrogen to polysilicon in a large reduction furnace.2,3 Existence of trace PCl3 in a trichlorosilane feedstock would cause P-doping on the polysilicon, which causes devastating damage to the resistivity and quality of the polysilicon product, on account of the valence shell of phosphorus having one extra electron; this would subsequently have a high probability of becoming a free electron when a silicon atom in the polysilicon crystal structure is replaced by a phosphorus atom.4–6 Thus, phosphorus is called a donor impurity. According to the Chinese National Standard, electronic-grade polysilicon requires a donor impurity concentration of less than 0.15 ppb, while solar-grade polysilicon requires there to be less than 1.40 ppb. The two standards are both strict for the purity of trichlorosilane feedstocks. The removal of PCl3 from trichlorosilane is therefore imperative.To date, considerable efforts have been made in the field of trace PCl3 removal from trichlorosilane. At present, the methods of removing PCl3 in the industrial process mainly include distillation, adsorption and reactive distillation. Distillation makes use of the difference in relative volatility between components to make them separate from each other, and currently plays a dominant part in purification of trichlorosilane.7 However, the boiling point of PCl3 is similar to that of trichlorosilane, such that huge energy consumption is needed in the distillation column to achieve the purification requirement, which will increase the cost and cause environmental pollution.8 Adsorption and reactive distillation are relatively effective methods for PCl3 removal.9–11 They take advantage of the Lewis base property of PCl3, in that it easily coordinates with a Lewis acid to achieve PCl3 removal. Unfortunately, the Lewis base property of PCl3 is not very strong, which results in PCl3 being unable to interact with a Lewis acid strongly and form stable compound, finally causing an excessive reaction time and insufficient dephosphorization.12 Part of pentavalent phosphorus compound such as POCl3 are strong Lewis base so they can be transformed into stable compound by combined with Lewis acid and achieve the requirement of dephosphorization. Therefore, making use of oxidation–reduction reactions to turn PCl3 into a pentavalent phosphorus compound is highly desirable. Oxygen (O2) has been widely used as a relatively ideal oxidizing agent for industrial systems. However, the majority of studies of the PCl3 oxidation process under oxygen aim to produce POCl3, such that the reactant PCl3 in these processes was present in large quantities, which is not in accordance with the situation of trace PCl3 removal from trichlorosilane.13 Preliminary research has shown that trace PCl3 can hardly be oxidized to POCl3 under oxygen in the trichlorosilane system. In this situation, POCl3 can be formed from trace PCl3 and O2 in the trichlorosilane system when the reaction is catalyzed by diverse transition-metal oxides. Materials with MoO2, Co3O4, MnO2 and Mn3O4 as active components have been reported to be developed for catalyzing the oxidation–reduction reaction between PCl3 and O2 in the trichlorosilane system.14 It has been confirmed in our preliminary experiment that among these transition-metal oxides, Mn3O4 has the best catalytic activity. However, the oxidation of PCl3 catalyzed by manganese species alone did not exhibit excellent performance. In recent years, metal-oxide-supported Au catalysts that consist of Au nanoparticles (NPs) deposited on metal oxides have attracted enormous interest because of their high catalytic activity for various aerobic oxidation reactions.15–18 Research has shown that catalysts such as Au/CoOx, Au/Fe2O3, Au/CeO2 and Au/TiO2 exhibit remarkably enhanced activity for oxidation reactions in comparison with isolated Au particles and single metal-oxide supports.19–23 It has been demonstrated that gold species loading on the support promotes the interaction between the Au and support, which leads to enhanced reduction of surface lattice oxygen and the activation of gaseous oxygen, thus contributing to reactant oxidation.22,24,25 Based on the above discoveries, it is logical to assume that catalysts with high activity toward the oxidation of PCl3 could be created by supporting Au NPs on the reducible metal oxide Mn3O4.
To obtain a catalyst with superior catalytic performance, the selection of the synthesis method is consequently a key step. Common supported-Au catalyst preparation methods mainly include impregnation, deposition–precipitation, and sol-immobilization.26–30 But generating catalysts with a uniform distribution of Au NPs and strong interactions between the Au and support, which is beneficial to the catalytic activity, is not straightforward when using these traditional methods.31–33 Liao et al. engineered an active catalyst for the CO oxidation reaction with an even distribution of Au–Pd alloy on the support and an active interface between the metal and cobalt oxide supports via calcination of a composite of NPs encapsulated in a metal–organic framework (MOF).23,34 This method provides an effective way to synthesize a catalyst with an even distribution of metal NPs on the metal-oxide support and an active interface between the metal and support material for improved catalytic performance.35
Motivated by the above, a PCl3-oxidation catalyst with high catalytic activity was prepared in this study, wherein Au NPs were loaded on a Mn3O4 support via the synthetic strategy developed by Liao and co-workers. In this strategy, the metal ions used as a metal-NP precursor were encapsulated into the pores of a MOF during the self-assembly process of the MOF, and subsequently reduced to metal NPs embedded in the framework of the MOF. The metal-oxide support is formed through the calcination of the MOF composite, which consists of the metal–organic framework and metal NPs homogeneously distributed on it. In this work, as graphically illustrated in Scheme 1, Au ions are trapped in the framework of Mn-MIL-100 (MIL stands for Materiaux de l’Institut Lavoisier), which is based on a trimeric building unit composed of manganese metal nodes and a trimesic acid (H3BTC) linker.36 Then the Au NPs are formed in the framework of Mn-MIL-100 by reducing the Au ions. The MOF composites were subsequently calcined under air and converted into the final structure of Au NPs embedded in manganese oxide (Mn3O4) cages (Aux/Mn3O4; (x) represents the mass fraction of Au, (x) = 4.13–10.52).
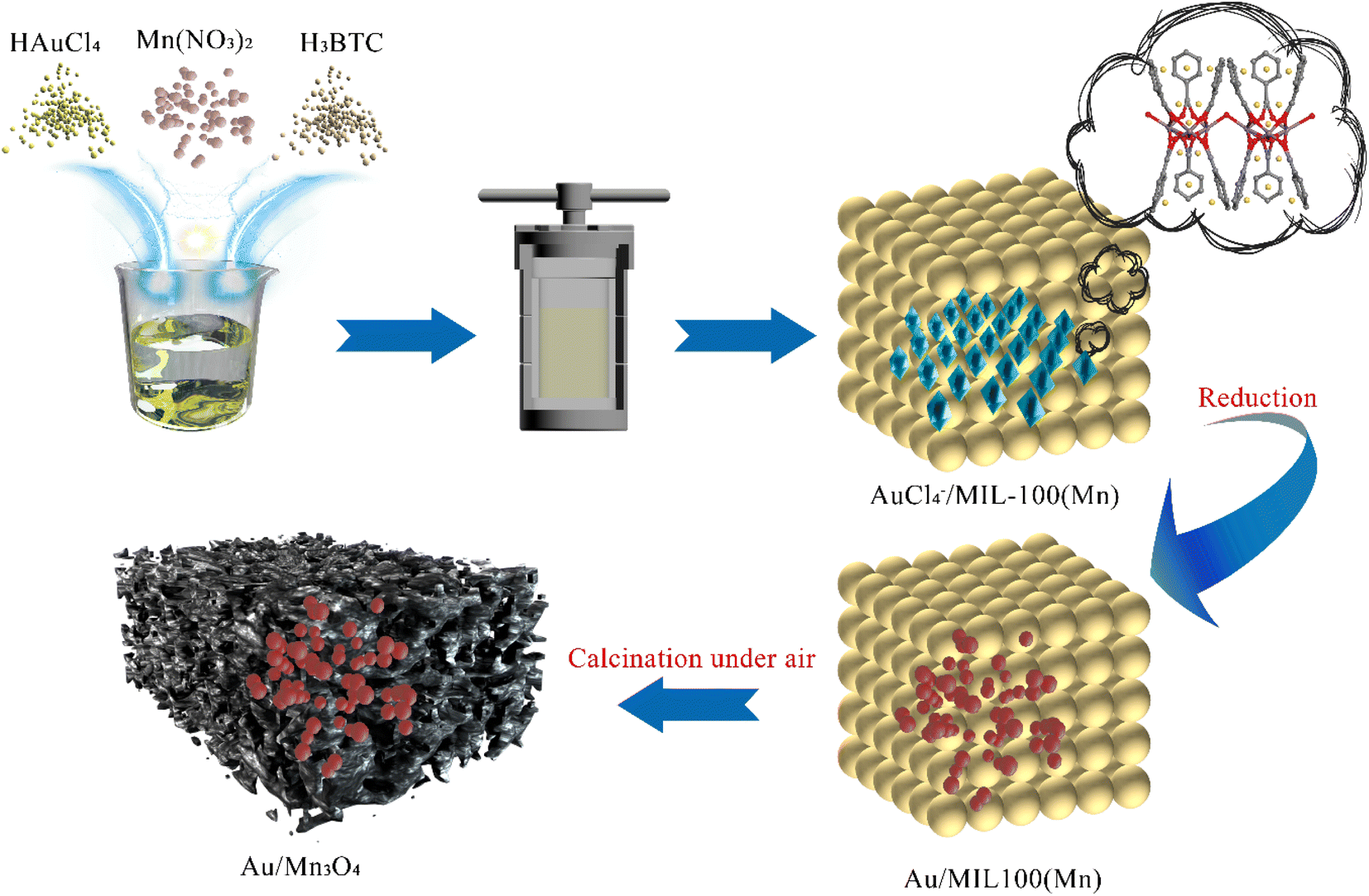 | ||
| Scheme 1 Synthesis of the Mn-MIL-100-derived Au-decorated manganese-oxide support applied for trace PCl3 oxidation under oxygen in an organic system. | ||
In the presence of O2, trace PCl3 oxidation over the Aux/Mn3O4 catalyst was carried out under different conditions to observe the catalytic activity. Considering the toxicity and corrosivity of trichlorosilane, the reaction was conducted in an organic system consisting of PCl3 and n-hexane, which was chosen instead of trichlorosilane due to the similarity in physical properties between trichlorosilane and n-hexane. As expected, the Aux/Mn3O4 catalyst exhibits enhanced catalytic performance in comparison with just Mn3O4. Au NPs have thus been demonstrated to be an effective loaded metal to facilitate the catalytic activity of the single metal oxide. In order to optimize the catalytic activity of the Aux/Mn3O4 catalyst and determine the optimal reaction conditions, a systematic study towards the influence of reaction and catalyst parameters, such as the mass fraction of Au in the Aux/Mn3O4 catalyst, reaction temperature, reaction pressure and catalyst dosage, on the conversion rates of PCl3 was performed. For the purpose of understanding the effects of the catalyst properties on the catalytic activities and investigating the catalytic mechanism of PCl3 oxidation on the Aux/Mn3O4 catalyst, a thorough characterization of the Aux/Mn3O4 catalyst and its precursors and a DFT study of the mechanism of PCl3 oxidation on the Aux/Mn3O4 catalyst has also been carried out.
Herein, an Au/Mn3O4 catalyst was synthesized, derived from a metal–organic framework, and has significantly active catalytic performance for PCl3 oxidation in an organic system. The physicochemical characterization of the as-prepared catalyst was investigated in detail, revealing the structure–performance relationships between the POCl3 yield and the electronic and geometric effects contributed by the Au NPs. Meanwhile, the optimal process conditions and reaction mechanism were studied for better industrial applications.
2 Materials and methods
2.1 Materials
Trimesic acid (H3BTC), lauric acid and chloroauric acid were purchased from Heowns. Manganese(II) nitrate tetrahydrate (Mn(NO3)2·4H2O), sodium borohydride, n-hexane and methanol were purchased from Aladdin. Ammonium molybdate, vitamin C, L-antimony potassium tartrate and sulfuric acid, used to detect the content of pentavalent phosphorus, were purchased from Macklin. PCl3 was purchased from Adamas-Beta. Pure oxygen was supplied by Air Liquid.2.2 Synthesis of Aux/Mn3O4 catalysts
Mn-MIL-100 was synthesized via a modified solvothermal method.37 Briefly, 0.72 mmol of Mn(NO3)2·4H2O, 2.85 mmol of H3BTC and 13.5 mmol lauric acid were dissolved in 75 mL of methanol at a stirring speed of 300 rpm for 40 min. The as-prepared solution was then transferred into an autoclave (100 mL) and maintained at 125 °C for 10 h. The resultant brown suspension was then centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm and the sediment was collected. The collected particles were subsequently washed with methanol 2 times, followed by vacuum drying at 60 °C for 4 h. The manganese-oxide cages used as the control group were produced by calcination of Mn-MIL-100 under air for 4 h.
000 rpm and the sediment was collected. The collected particles were subsequently washed with methanol 2 times, followed by vacuum drying at 60 °C for 4 h. The manganese-oxide cages used as the control group were produced by calcination of Mn-MIL-100 under air for 4 h.
For the Au/Mn-MIL-100 composites, in which Au NPs are embedded in the framework of Mn-MIL-100, various masses of chloroauric acid were melted into the manganese(II) nitrate tetrahydrate-containing solution at a stirring speed of 300 rpm for 40 min. The as-prepared solution was then transferred into an autoclave (100 mL) and maintained at 125 °C for 10 h. The resultant brown suspension was then centrifuged at 12000 rpm and the sediment was collected. The collected particles were washed with methanol 2 times, then re-suspended in 60 mL of methanol. 0.2 g of sodium borohydride was added into the brown suspension and stirring was maintained for 2 h at room temperature. The suspension was centrifuged and the collected particles were washed with methanol twice, followed by vacuum drying for further use. The acquired solid powder was further calcined under air at 500 °C to obtain catalysts supported by manganese oxide.
2.3 Characterization
The morphology of all samples was observed through scanning electron microscopy (SEM) conducted on an FEI Apreo S instrument. The X-ray diffraction (XRD) patterns were acquired using a Rigaku SmartLab SE X-ray diffractometer. N2 adsorption–desorption isotherms were measured at −196 °C with a Micromeritics ASAP2460 sorption analyzer. The Au content measurement of all samples was performed using inductively coupled plasma atomic emission spectroscopy (ICP-AES) on a Agilent 725 instrument. X-ray photoelectron spectroscopy (XPS) was performed using a Thermo ESCA LAB Xi+ spectrometer to analyze the state of the gold and manganese species in the samples, and the results were calibrated using the C 1s peak at 284.8 eV. Fourier-transform infrared (FT-IR) spectra were collected on a Thermo Nicolet iS20 spectrometer. Thermogravimetric analysis (TGA) of the samples, at temperatures ranging from 25–800 °C, was performed using a Henven HTG-4 analyzer.2.4 PCl3 oxidation over Aux/Mn3O4
The oxidation of trace PCl3 under an oxygen environment in an organic system was carried out in a 100 mL autoclave. A trace amount of PCl3 was injected into 100 mL of n-hexane under nitrogen to obtain the reaction liquid. Then a given amount of reaction liquid and catalyst were added into the batch reactor. After purging with oxygen three times, the reactor was pressurized to a specific pressure using oxygen and then heated at a desired temperature under suitable stirring. After a set reaction time, the reaction was finished and the reactor was cooled down to room temperature. The liquid in the reactor after reaction was then subjected to ultrasonic treatment for 20 min and the solid catalyst was removed from the system by centrifugation. 1 mL of the residual liquid was then taken and extracted using 100 mL of deionized water. The chromogenic agent was made up of sulfuric acid solution (5 mol L−1), 3 wt% ammonium molybdate solution, 5.4 wt% vitamin C solution and 0.136 wt% L-antimony potassium tartrate solution with a volume ratio of 3:2:2:1. The content of phosphoric acid in aqueous phase formed from the hydrolyzation of POCl3 in the above residual liquid was determined by spectrophotometry, using ammonium molybdate as the active component in the chromogenic agent, on a ultraviolet spectrophotometer (BINGLIN, λ = 891 nm). The POCl3 content in the system after the reaction was quantified using an ammonium molybdate standard curve and the corresponding conversion relation of the concentration. The yield of POCl3 was defined as the moles of POCl3 generated after reaction divided by the moles of PCl3 added initially, as shown in eqn (1):
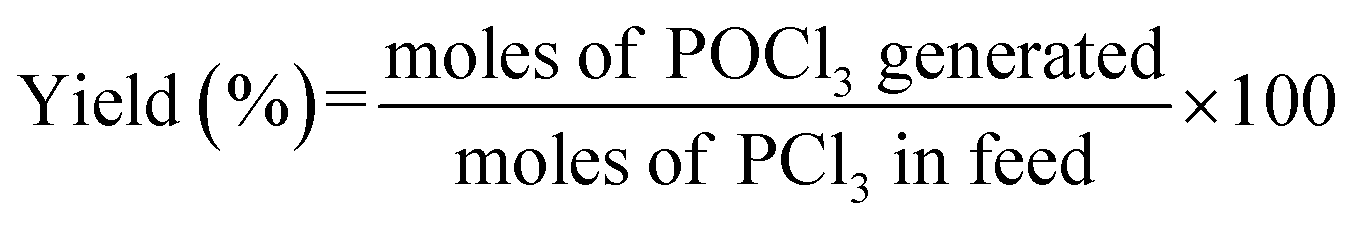 | (1) |
2.5 Computational details
All the calculations are performed in the framework of density functional theory (DFT) with the projector augmented plane-wave method, as implemented in the Vienna ab initio simulation package.38 The generalized gradient approximation proposed by Perdew, Burke, and Ernzerhof is selected for the exchange–correlation potential.39 The long-range van der Waals interaction is described by the DFT-D3 approach.40 The cut-off energy for the plane wave is set to 450 eV. The energy criterion is set to 10−5 eV in the iterative solution of the Kohn–Sham equation. A vacuum layer of 15 Å is added perpendicular to the sheet to avoid artificial interaction between the periodic images. The Brillouin zones for energy calculation of Mn2O3 and Au10@Mn2O3 are sampled with a Gamma-centered k-mesh of 3 × 3 × 1. All the structures are relaxed until the residual forces on the atoms have declined to less than 0.03 eV Å−1.3 Results and discussion
3.1 Characterization of Aux/Mn3O4 catalysts
Different weights of the Au precursor were added into the system during the process of Mn-MIL-100 synthesis, simultaneously to the trimesic acid, manganese nitrate, and lauric acid selected as the precursors, so that the Au precursor could be encapsulated into the pores of Mn-MIL-100. Then the Au precursor was reduced into metal NPs by addition of NaBH4, forming MOF composites in which Au NPs were embedded in the framework of Mn-MIL-100 (Aux/MIL-100; (x) represents the mass fraction of Au). As shown in Fig. S1,† Mn-MIL-100 consists of smooth and uniform octahedral particles with an average size of ca. 500 nm, matching well with its trimeric building units. Scanning electron microscopy (SEM) images of Au/Mn-MIL-100 are shown in Fig. 1A, showing that the structure of Mn-MIL-100 turns out to be slightly collapsed once the Au NPs are embedded in it and Au/Mn-MIL-100 exists in an irregular octahedron configuration, which indicates that the loading of Au NPs causes minor disruption to the structure of Mn-MIL-100. Fig. 1B shows the element distributions of C, O, Mn, and Au species via scanning electron microscopy with energy dispersive X-ray spectroscopy (SEM-EDX) of Au/Mn-MIL-100, confirming the successful loading and even distribution of Au species inside the Mn-MIL-100. | ||
| Fig. 1 (A) SEM image, and (B) element distributions of C, O, Mn and Au species via SEM-EDX of Au/Mn-MIL-100. | ||
Fig. 2A shows the X-ray diffraction (XRD) pattern of Au/Mn-MIL-100, which is in accordance with that of Mn-MIL-100 to a great extent, but the peak intensities decrease with the enhancement in the Au mass fraction, verifying that Au NPs do have some effect on the octahedral configuration of Mn-MIL-100. The disruption is aggravated with the enhancement in the Au mass fraction, but the as-prepared Au/Mn-MIL-100 basically maintained the well-constructed framework of MIL-100, with metal NPs embedded in it. The electronic properties of the Au element were explored via X-ray photoelectron spectroscopy (XPS), as exhibited in Fig. 2C. The Au-free Mn-MIL-100 sample shows no Au-related signal in Au 4f XPS, as expected, while obvious bands are observed for the four Au/Mn-MIL-100 samples, whose intensities progressively increase with the rise in the Au mass fraction. Considering the complicated micro-environment of Au species and the properties of Au 4f, each spectrum was deconvolved into five peaks at 83.4, 84.4, 87.4, 88.1 and 89.7 eV. The subordinate peaks at 83.4 and 87.4 eV are associated with the 4f7/2 and 4f5/2 photoelectrons of Au0 with a zero valence state and the peaks at 84.4 and 88.1 eV are attributed to Auδ+ with a positive charge, further proving that Au NPs were successfully embedded in the framework of Mn-MIL-100 and an interaction was simultaneously generated in terms of charge transfer between the Au NPs and Mn-MIL-100.41–44 In addition, the peak at 89.7 eV exhibits a slight shift to higher energy compared to the characteristic peaks of Auδ+ and Au0; this was ascribed to Au*, which represents Au species that replaced a small portion of the metal sites in the trimeric building units of Mn-MIL-100 during the self-assembly process between Mn ions and H3BTC. The replacement results in sparsity of the electron cloud density around these Au species owing to the higher electronegativity of the O element compared to that of the Au element and leads to the higher binding energy of Au*.45 The Au*/(Au* + Au0 + Auδ+) average molar ratio (0.14) is relatively low, indicating that the damage to the structure of Mn-MIL-100 owing to the exchange of Au ions for metal sites during the self-assembly process occurs to a very low degree. Fig. 2B shows the Fourier-transform infrared (FTIR) spectra of Au/Mn-MIL-100. The band at 490 cm−1, which could be associated with the marked Mn–O band in Mn-MIL-100, is shifted to a higher wavenumber (i.e., 527 cm−1) for the Au/Mn-Mn-100 materials.46 This is attributed to free electrons, which were lost by Au species in the process of transition from Au0 to Auδ+, moving around the Mn–O bond and increasing its stretching vibration frequency. The peaks at 718 cm−1 are assigned to the benzene ring of the ligand, while the peaks at 1369 and 1621 cm−1 are ascribed to the C–O stretching of the BTC anions.36,47 The peaks centered at 1570 cm−1 can be attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching of benzene.48 The N2 adsorption/desorption isotherms of Au/Mn-MIL-100 are exhibited in Fig. 2D, displaying typical type I isotherms, which could be attributed to the micropores of Mn-MIL-100. An H4 hysteresis loop could also be observed in some of the isotherms, revealing the presence of internal voids inside the Mn-MIL-100. One of the possible causes for the formation of these internal voids is the substitution of Mn ions in Mn-MIL-100 by Au ions, as mentioned above. The trimeric units that Mn-MIL-100 was based on were constructed through coordination bonds between the d2sp3 hybrid orbitals of the Mn ions and the lone pairs of the six O atoms supplied by H3BTC. Since the orbital hybridization mode of the Au ions is distinct from that of the Mn ions, some of the trimeric units would collapse and be transformed to the coordination structure between the Au ions and O atoms, leading to irregular pore construction and internal voids in Au/Mn-MIL-100. Furthermore, Pang et al indicated that the reduction of chloroauric acid would generate a large number of hydrogen ions and expose Au/Mn-MIL-100 to an acidic environment.49 Liao et al. figured out that another cause for the formation of internal voids is that metal ligands such as H3BTC would protonate rather than coordinate with Mn ions in an acidic environment; Mn ions located in the core of Mn-MIL-100 consequently move outward to the environment and compensate the coordinated Mn ions located in the shell of Mn-MIL-100, which promotes the formation of voids.23
C stretching of benzene.48 The N2 adsorption/desorption isotherms of Au/Mn-MIL-100 are exhibited in Fig. 2D, displaying typical type I isotherms, which could be attributed to the micropores of Mn-MIL-100. An H4 hysteresis loop could also be observed in some of the isotherms, revealing the presence of internal voids inside the Mn-MIL-100. One of the possible causes for the formation of these internal voids is the substitution of Mn ions in Mn-MIL-100 by Au ions, as mentioned above. The trimeric units that Mn-MIL-100 was based on were constructed through coordination bonds between the d2sp3 hybrid orbitals of the Mn ions and the lone pairs of the six O atoms supplied by H3BTC. Since the orbital hybridization mode of the Au ions is distinct from that of the Mn ions, some of the trimeric units would collapse and be transformed to the coordination structure between the Au ions and O atoms, leading to irregular pore construction and internal voids in Au/Mn-MIL-100. Furthermore, Pang et al indicated that the reduction of chloroauric acid would generate a large number of hydrogen ions and expose Au/Mn-MIL-100 to an acidic environment.49 Liao et al. figured out that another cause for the formation of internal voids is that metal ligands such as H3BTC would protonate rather than coordinate with Mn ions in an acidic environment; Mn ions located in the core of Mn-MIL-100 consequently move outward to the environment and compensate the coordinated Mn ions located in the shell of Mn-MIL-100, which promotes the formation of voids.23
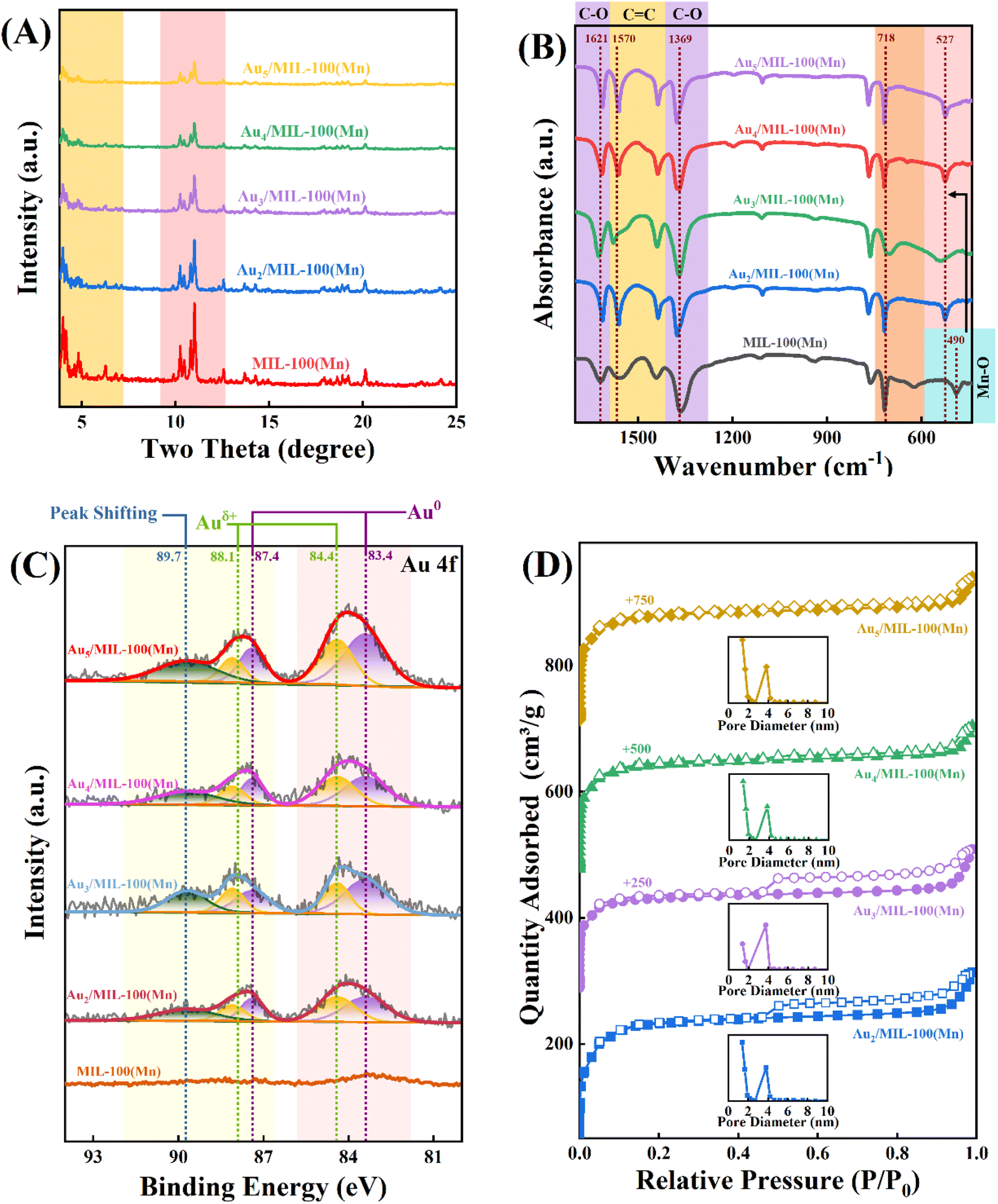 | ||
| Fig. 2 (A) XRD patterns, (B) FT-IR spectra, (C) Au 4f XPS spectra, and (D) N2 adsorption–desorption isotherms (with inset pore-size distributions) of Au/Mn-MIL-100. | ||
Au/Mn3O4 catalysts were obtained through further calcination of Au/Mn-MIL-100 under air. As shown in Fig. 3A, the morphology evolution during pyrolysis was evaluated via SEM images for different calcination temperatures. When the temperature reaches 400 °C, the MOF structure of Au/Mn-MIL-100 converts to small clusters without a fixed form. When the temperature continues to increase, blocky clusters of porous structure, with an average particle size of 500 nm, start to appear at 500 °C and further transform into big clusters at 600 °C and 700 °C. The size of the clusters calcined at 600 °C was five times larger than that of clusters calcined at 500 °C, while the size of the clusters calcined at 700 °C was about forty times larger than that of clusters calcined at 600 °C, indicating that the materials would gradually aggregate when the calcination temperature rises from 500 to 700 °C. To fully recognize the chemical reactions and phase transitions involved during the pyrolysis of Au/Mn-MIL-100, thermogravimetric analysis–mass spectrometry (TGA–MS) was conducted under air. As shown in Fig. 3C, the pyrolysis process in the temperature range from 0–800 °C could be divided into four phases: (i) ethanol that permeated into the pores of Au/Mn-MIL-100 evaporates (0 °C < T < 220 °C); (ii) the H3BTC ligand loses some of its benzene rings, leaving the Mn atom bonded to the two oxygen atoms of a carboxy group (220 °C < T < 320 °C); (iii) the H3BTC ligand loses all of its benzene rings, leading to bond-breaking between Mn and O and the release of CO2 (320 °C < T < 430 °C); (iv) Au/Mn3O4 compounds form, which are stable within 800 °C.50 The XRD results of the Au/Mn3O4 materials obtained by calcination at different temperatures are shown in Fig. 3B. The intensity of the peak at 38.3° that is ascribed to Au NPs first increases and then decreases with the rise in calcination temperature, revealing an increase in size of the Au particles with the calcination temperature increasing from 500 to 600 °C; then the Au particles and Mn3O4 support were gradually encapsulated in a carbon matrix when the temperature continued to increase. Thus, different calcination temperatures were used to optimize the interface between the metal NPs and oxide with 500 °C being selected for further testing.
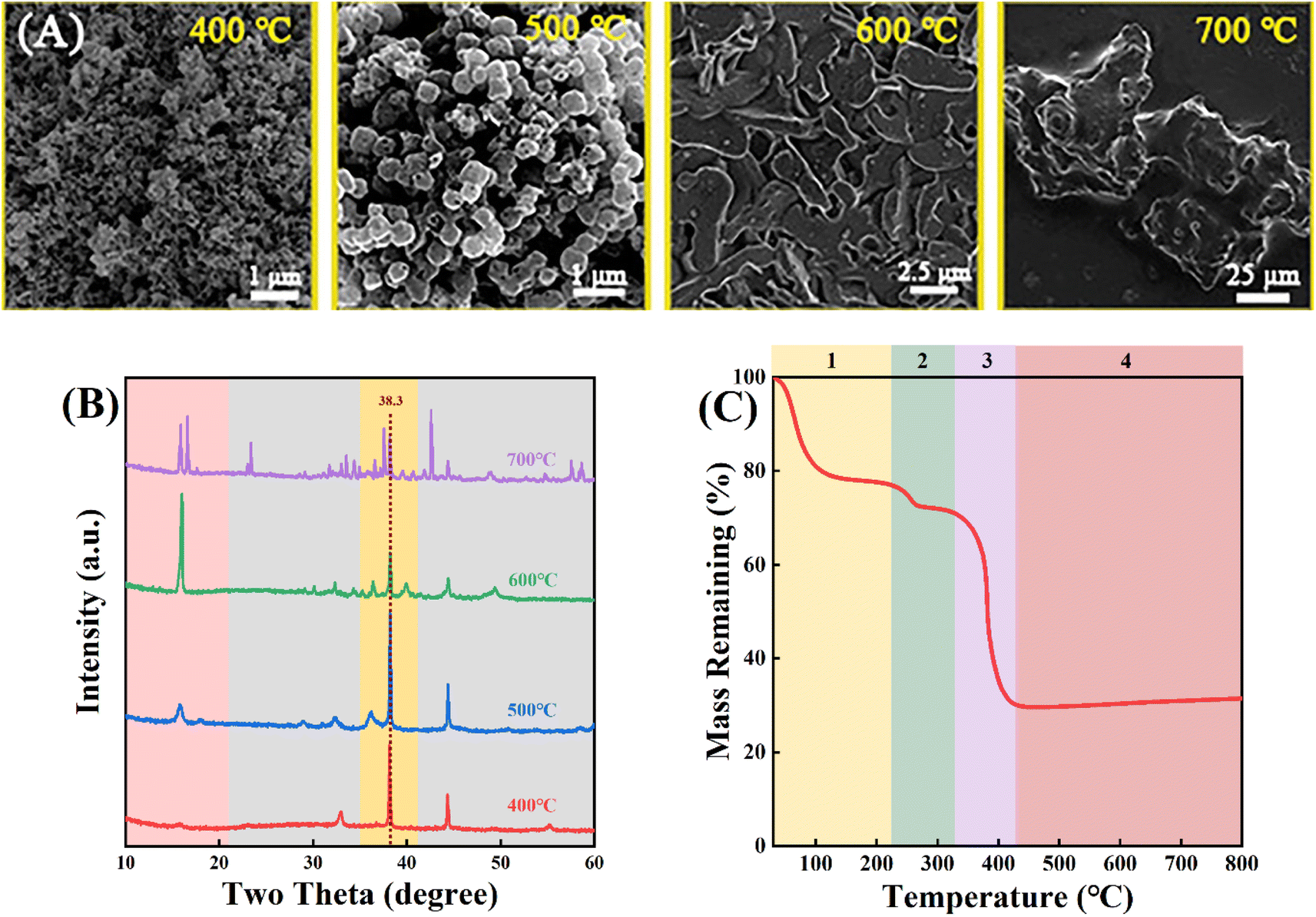 | ||
| Fig. 3 (A) SEM images and (B) XRD patterns of Au2/Mn-MIL-100 calcined at different temperatures (400, 500, 600 and 700 °C) under air. (C) Weight loss of Au2/Mn-MIL-100 calcined under air. | ||
The mass fraction of Au(x) in the calcined sample was quantified via ICP-MS. Fig. 4A shows SEM images of the as-prepared Au4.13/Mn3O4 calcined at 500 °C. The elemental distribution of O, Mn, and Au species (Fig. 4B) shows overlap of the four species, indicating the formation of manganese oxide and a uniform distribution of Au NPs on the Mn3O4 support. In addition, the sparse elemental mapping of C shows that most of the C element from the H3BTC linker escaped in the form of CO2 during the pyrolysis process, revealing that the quantity of carbon matrix that the catalysts prepared by calcination at 500 °C were embedded in was at a relatively low level. To confirm the advantages of the method for the preparation of Aux/Mn3O4 catalysts derived from Mn-MIL-100, the Au–Mn3O4 material was synthesized via an impregnation method for comparison. The SEM images of the Au–Mn3O4 (Fig. S2A†) show that its morphology is particles with a relatively large size, and the corresponding selected area element distribution analysis (Fig. S2B†) shows a situation of local accumulation of the Au element, indicating that the Au element was dispersed on the material unevenly, further demonstrating that the synthesis method using MOFs as a precursor could promote distribution uniformity. The XRD patterns of Aux/Mn3O4 in Fig. 5A show that the diffraction peaks of Au/Mn3O4 derived from Mn-MIL-100 are in accordance with the standard values of Mn3O4 (JCPDS No. 24-0734) and the intensity of the characteristic Au peak located at 38.3° progressively increases with the rise in Au mass fraction, further confirming the successful synthesis of the Mn3O4 support and the increase in size of the Au NPs embedded in the metal oxide with the increase in the Au mass fraction.51 The N2 adsorption–desorption isotherm (Fig. 5B) can be designated as a type-II isotherm, revealing a mesoporous and macroporous structure of the as-prepared Aux/Mn3O4 catalysts. The specific areas, total pore volumes and pore sizes of the Aux/Mn3O4 catalysts are summarized in Table 1. The average specific surface area of Aux/Mn3O4 is 8.0 m2 g−1, which is significantly reduced compared to that of Au/Mn-MIL-100 (i.e. 736 m2 g−1) due to the pyrolysis of its precursor. Although MIL-100, with a high specific surface area, has been extensively researched because of its high catalytic activity for various reactions, in this study its high specific area was taken advantage of to make the distribution of Au NPs uniform, and it was converted into a manganese-oxide support, which was the catalytically active ingredient for the PCl3 oxidation reaction afterwards.52,53 Thus, Mn-MIL-100 was used as a template to separate the Au clusters rather than as a material for the catalyst. During the process of pyrolysis, the H3BTC linker was sacrificed to make the Mn ions move close to each other and generate the interface with the Au NPs. XPS measurements were carried out to figure out the electronic properties of the Mn and Au elements in the Aux/Mn3O4 catalysts. In the Mn 2p spectra, as presented in Fig. 5C, four peaks with electron binding energies of 641.4, 643.3, 653.0 and 654.6 eV are observed after deconvolution and can be attributed to Mn2+ and Mn3+, further confirming the formation of the Mn3O4 support.54 In the Au 4f spectra exhibited in Fig. 5D, the Au species in Aux/Mn3O4 show binding energies of 83.4, 84.4, 87.4 and 88.1 eV, which can be ascribed to Au0 with a zero valence state and Auδ+ with a minor positive charge, indicating the interaction in terms of charge transfer between the Au NPs and Mn3O4 support at their generated interface. The Auδ+/(Au0 + Auδ+) molar ratio was obtained from the analysis of the corresponding peak area. An increasing–decreasing trend is observed for the Auδ+/(Au0 + Auδ+) molar ratio with the increase in the Au mass fraction (Fig. 6), reaching the maximum value of 0.585 when the Au mass fraction is 6.25. It can be inferred that although the amounts of Auδ+ and Au0 increase with the elevation in the total Au content, the degree of increase in the Auδ+ amount is less than the increase in the Au0 amount when the Au mass fraction exceeds 6.25.
 | ||
| Fig. 4 (A) SEM image, and (B) element distribution of C, O, Mn and Au species via SEM-EDX of Aux/Mn3O4. | ||
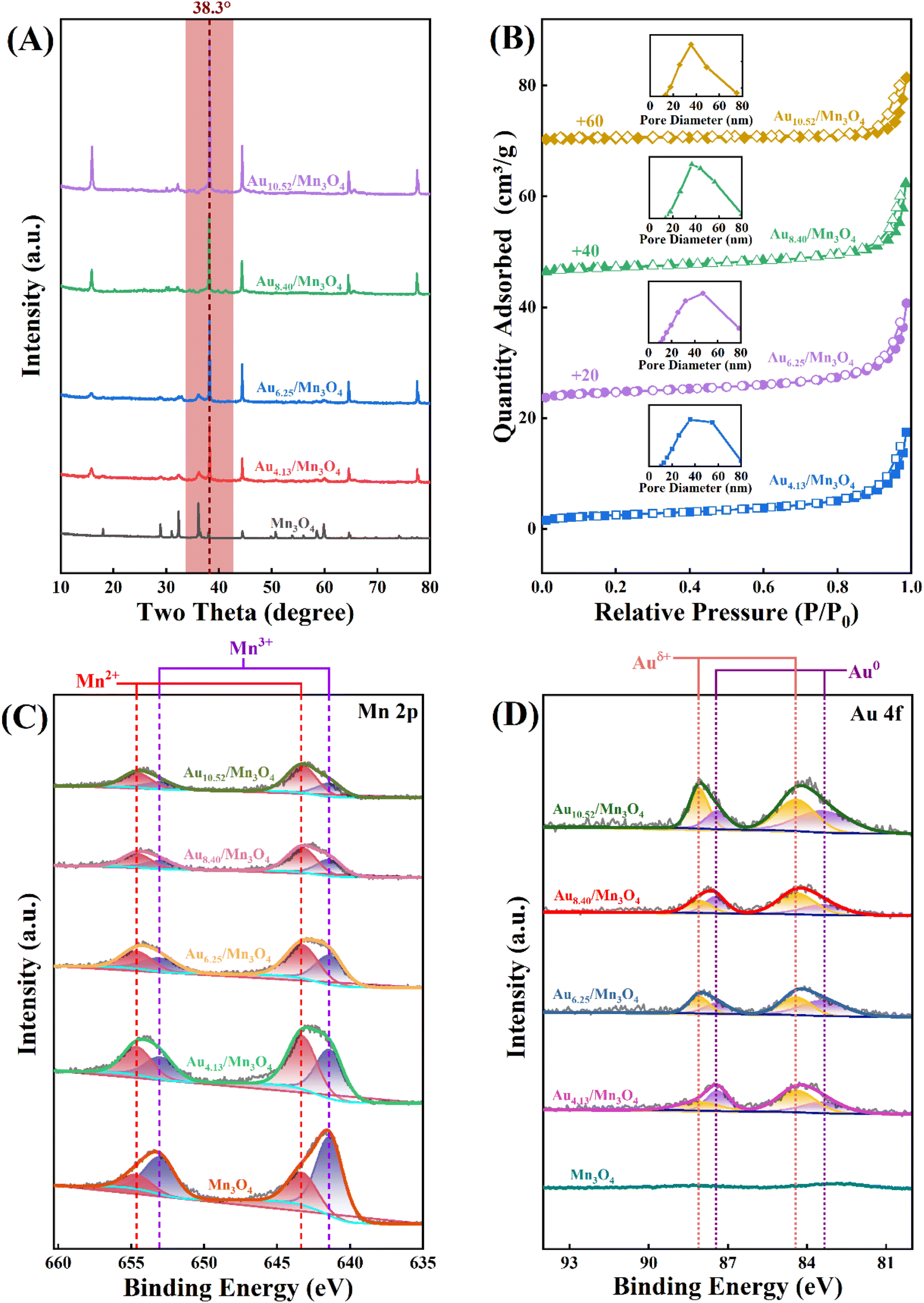 | ||
| Fig. 5 (A) XRD patterns, (B) N2 adsorption–desorption isotherms (with inset pore-size distributions), (C) Mn 2p XPS spectra, and (D) Au 4f XPS spectra of Aux/Mn3O4. | ||
| Specific surface area (m2 g−1) | Pore volume (cm3 g−1) | Pore size (nm) | |
|---|---|---|---|
| Au4.13/Mn3O4 | 8.7 | 0.026 | 13 |
| Au6.25/Mn3O4 | 9.3 | 0.027 | 13 |
| Au8.40/Mn3O4 | 7.7 | 0.026 | 14 |
| Au10.52/Mn3O4 | 6.4 | 0.046 | 31 |
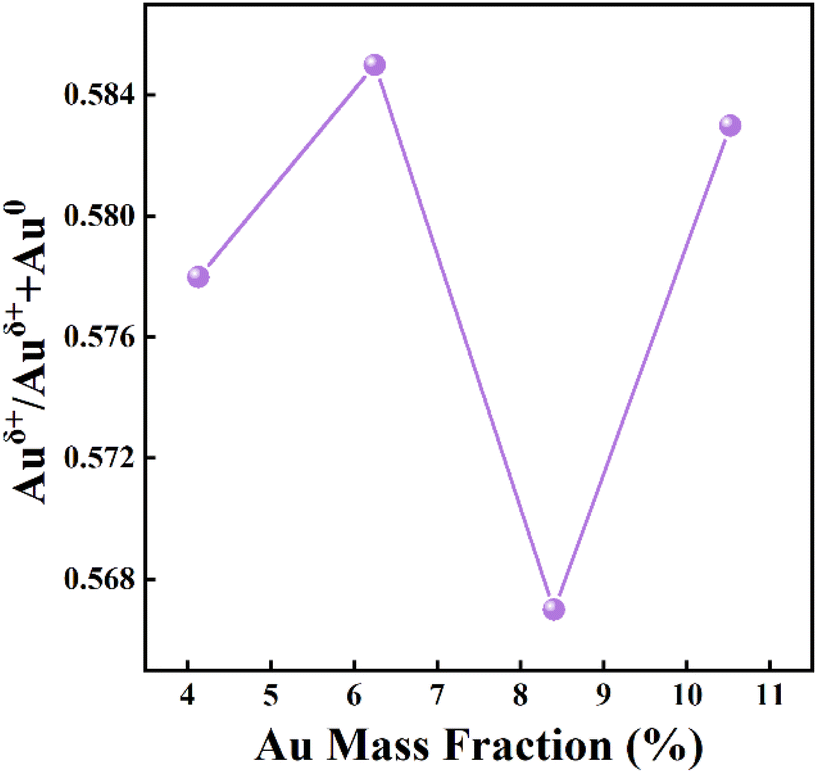 | ||
| Fig. 6 The influence of Au mass fraction on the Auδ+/(Au0 + Auδ+) molar ratio of the Aux/Mn3O4 catalysts. | ||
3.2 PCl3 oxidation over Aux/Mn3O4 catalysts
Trace PCl3 was oxidized to POCl3 by O2 over the as-prepared Aux/Mn3O4 catalysts; this was done in an autoclave, where O2 was introduced into the system and used for pressure control. The yield of POCl3 after the reaction was chosen for evaluating the catalytic activity of the catalysts. Fig. 7A presents the POCl3 yield of the Au/Mn3O4 catalysts with different Au mass fractions. It can be clearly seen that the POCl3 yield was dramatically enhanced from 70.58% to 89.56% once Au was loaded on the support, revealing that the presence of Au species gave a significant improvement in the activity of the catalysts, verifying that the catalytic performance of the as-synthesized catalysts derived from MOFs was greatly enhanced in comparison to that of the traditional catalyst Mn3O4. The yield of POCl3 then showed a trend of first increasing and then decreasing with the rise in Au content, where Au6.25/Mn3O4 gave the maximum POCl3 yield of 97.58%. It is widely accepted that a close relationship between the metal loading on a support and its catalytic performance can be ascribed to the geometric and electronic effects on the interface of the metal NPs and their support. In terms of the geometric effects, a higher Au content creates more active sites at the beginning, but the size of the Au NPs embedded in the metal oxide becomes too large as the Au content increases, leading to a decrease in the number of active sites instead. From the aspect of the electronic effects, the trend of the Auδ+/(Au0+Auδ+) molar ratio is in accordance with the trend of the catalytic performance with the rise in Au content, revealing that the catalytic activity was mainly influenced by the relative content of Auδ+. To summarize, the origin of the influence of Au content on the POCl3 yield was intrinsically derived from the synergistic results of geometric and electronic effects. Fig. 7B shows the yield of POCl3 over Aux/Mn3O4 catalysts calcined at different temperatures (400, 500, 600 and 700 °C), where the best catalytic activity was obtained with the catalyst calcined at 500 °C. This can be attributed to the aggregation of catalyst particles and the encapsulation of Au NPs into the carbon matrix as the calcination temperature exceeds 500 °C, leading to a significant reduction in active sites.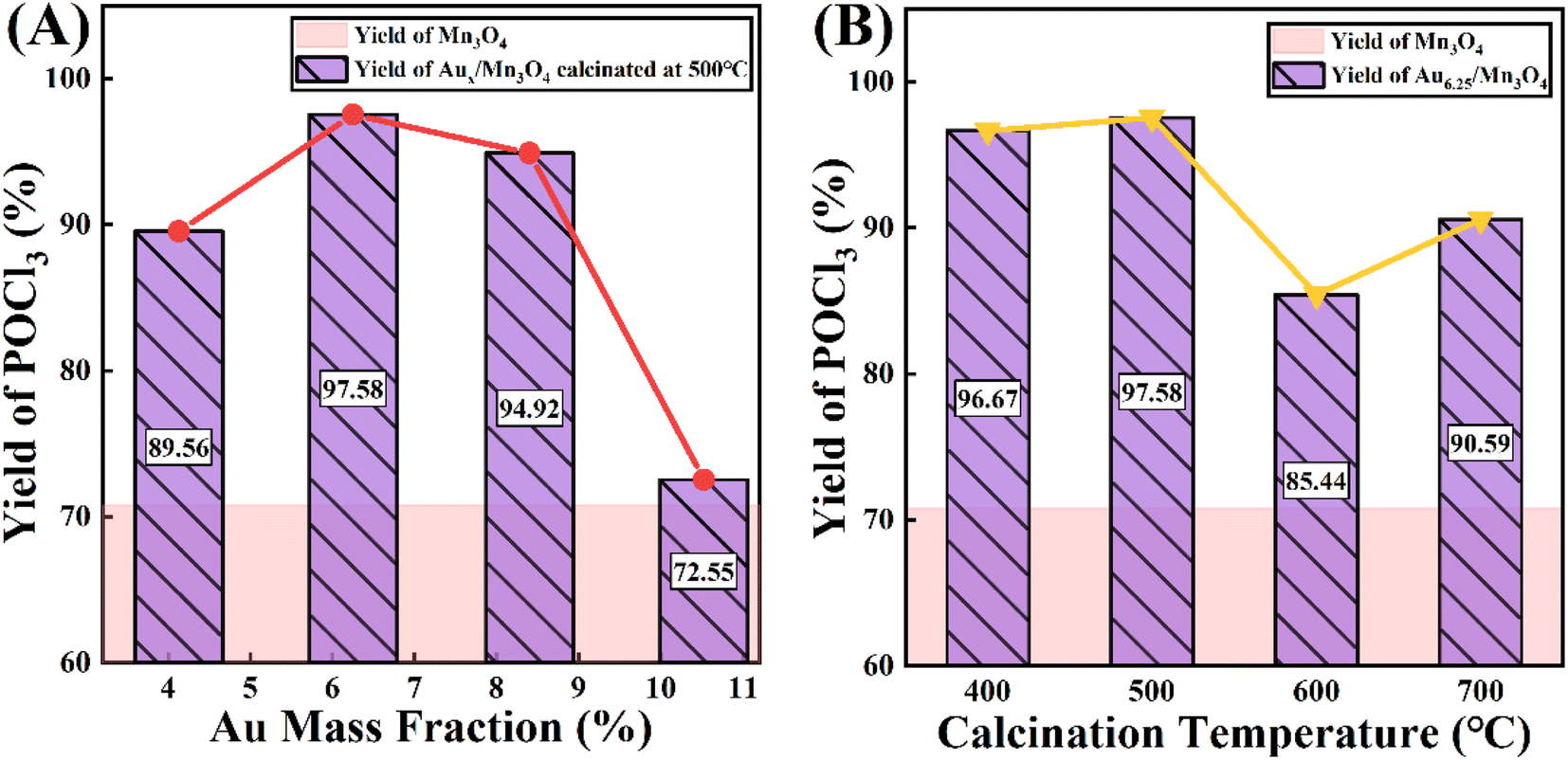 | ||
| Fig. 7 The influence of (A) Au mass fraction and (B) calcination temperature on catalytic performance of Au/Mn3O4 in terms of the yield of POCl3 in the PCl3 oxidation reaction. | ||
Subsequently, four process parameters, namely agitation speed, reaction temperature, reaction pressure and catalyst dosage, were probed to obtain optimum process conditions that enable a maximum yield of POCl3 (Fig. 8). The yield of POCl3 (97.58%) at an agitation speed of 500 rpm was slightly higher than that at 400 rpm and then showed inconspicuous changes with the increase in agitation speed (Fig. 8A). In terms of the reaction temperature (Fig. 8B), an increasing–decreasing trend was observed for the POCl3 yield as the reaction temperature continued to rise, reaching a maximum yield of 99.13% at 80 °C. For the influence of reaction pressure on the reaction, as presented in Fig. 8C, the yield of POCl3 was remarkably enhanced when the pressure increased to 0.6 MPa from 0.5 MPa, reaching 99.13%, which was the maximum value across the whole pressure range. With regard to the catalyst dosage (Fig. 8D), the optimal yield of POCl3 was obtained at a dosage of 40 mg. In conclusion, after comprehensively considering POCl3 yield and economic efficiency, the optimal technological parameters were selected as follows: an agitation speed of 500 rpm, reaction temperature of 80 °C, reaction pressure of 0.6 MPa and catalyst dosage of 40 mg.
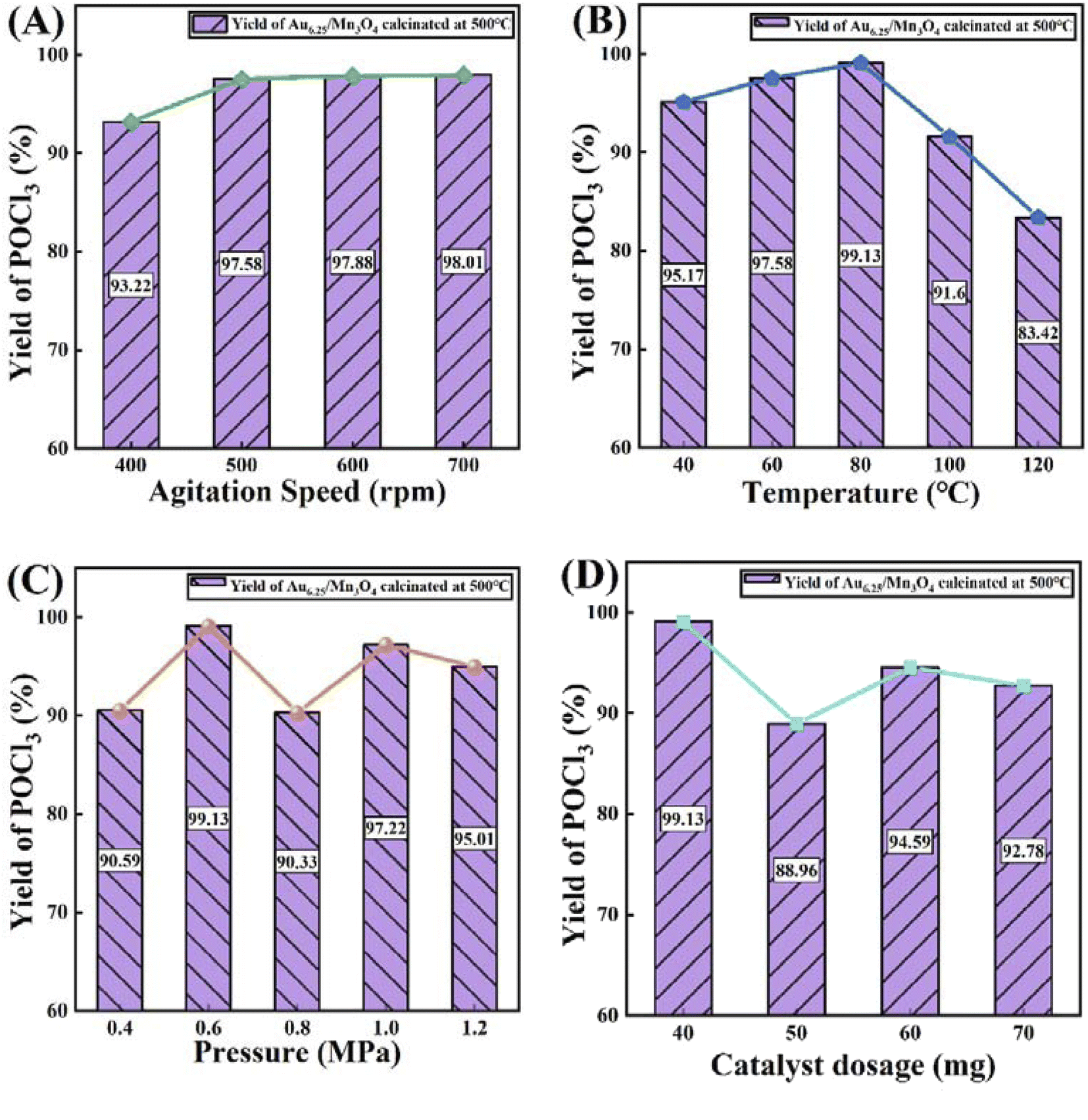 | ||
| Fig. 8 The influence of (A) agitation speed, (B) reaction temperature, (C) reaction pressure and (D) catalyst dosage on catalytic performance of Au6.25/Mn3O4 in terms of the yield of POCl3 in the PCl3 oxidation reaction. | ||
3.3 Reaction mechanism
According to the results shown in Fig. 7 and 8, we hypothesize that the possible explanation for the higher PCl3 oxidation activity of Au NPs supported on Mn3O4 is the supply of oxygen from the lattice of the metal-oxide support in a Mars van Krevelen (MvK) process. Fig. 9 shows the reaction sequence of PCl3 oxidation over the Au/Mn3O4 catalysts, which includes a MvK-style supply of oxygen. First, PCl3 is adsorbed close to the interface between the Au NPs and metal-oxide support. Then lattice oxygen of Mn3O4 forms a new bond with phosphorus, transforming PCl3 into POCl3 and forming an oxygen anion vacancy on the interface at the same time. O atoms produced by the dissociation of O2 that was adsorbed on the interface between the Au NPs and the support compensate the oxygen vacancies; thus, the final result is that PCl3 is oxidized by oxygen over the Au/Mn3O4 catalysts, which maintain their properties after the reaction.16,20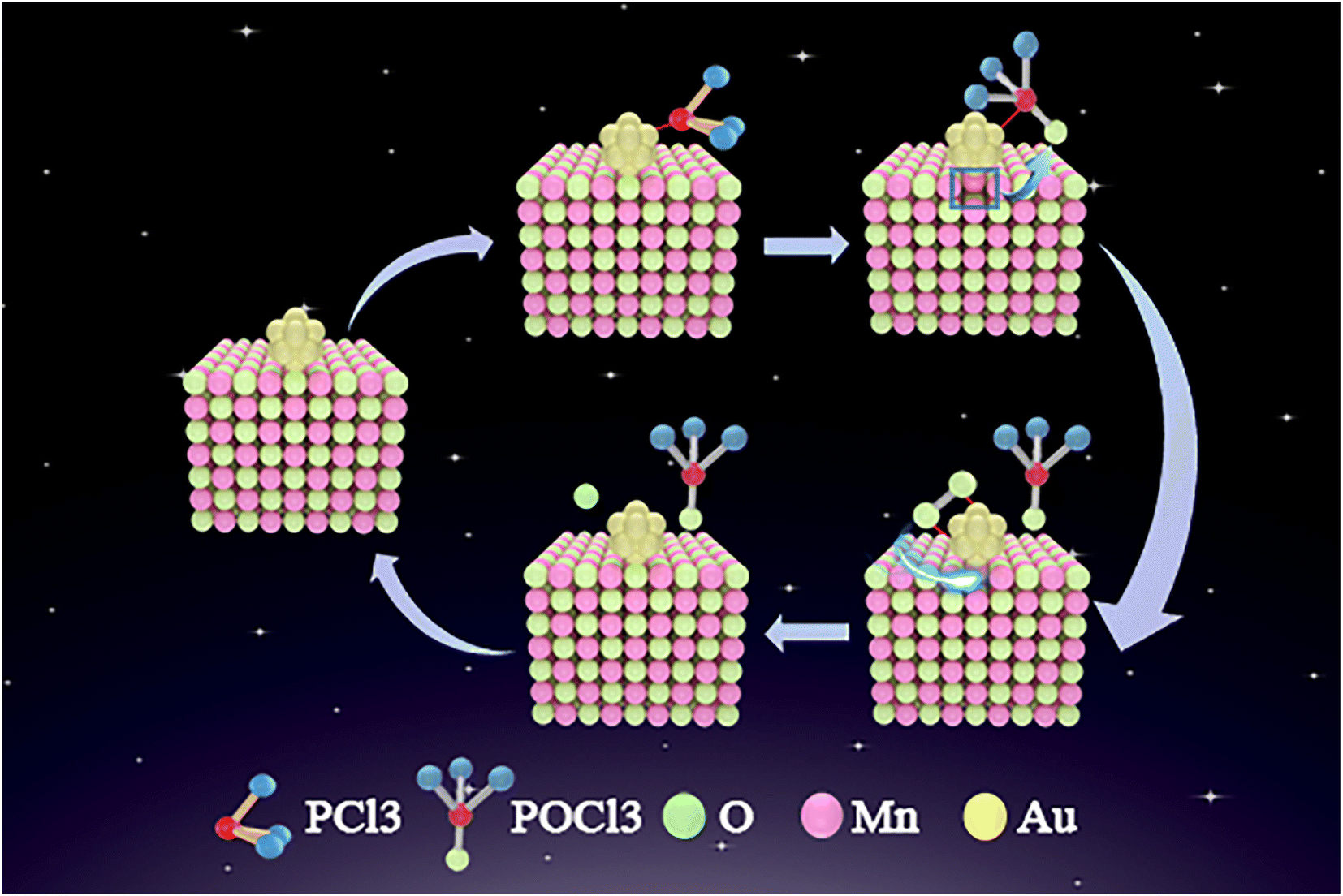 | ||
| Fig. 9 MvK process for oxidation of PCl3 over the interface between the Au NPs and Mn3O4. | ||
To demonstrate this hypothesis, DFT calculations were performed to analyze the thermodynamics of the oxidation of PCl3 by the oxygen from the lattice of the support, generating Au/Mn3O4 with an oxygen vacancy and POCl3.55 The equation is as follows:
| PCl3(g) + Au/Mn3O4 → POCl3 + Au/Mn3O4(vac) |
The oxygen atoms of the Au/Mn3O4 catalysts can be divided into two groups. One is the interface oxygen atoms (Oint) that bridge the Mn atoms with Au; the other is the perimeter oxygen atoms (Operi) that bridge two Mn atoms at the periphery of Au. In order to figure out which group of oxygen atoms is effective in the above process, the variation in Gibbs free energy (ΔG) for the reactions involving Oint and Operi, respectively, were calculated as shown in Fig. 10.
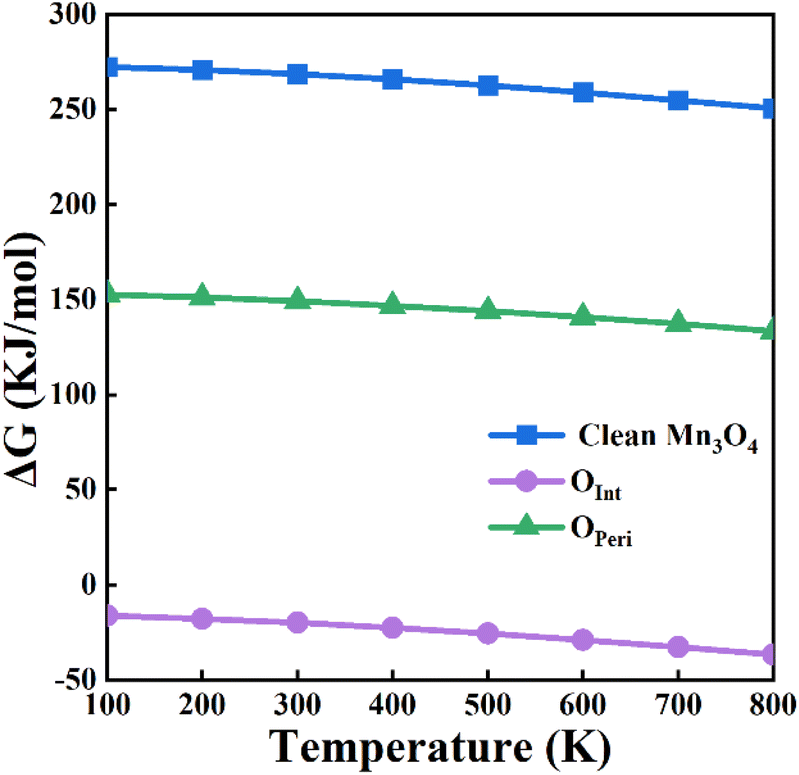 | ||
| Fig. 10 Variation in ΔG for the formation of POCl3 using the lattice oxygens of Mn3O4 and using the interface and perimeter lattice oxygens of the Au10/Mn3O4 model system. | ||
The values of ΔG suggest that PCl3 oxidation by lattice oxygens from clean Mn3O4 is non-spontaneous from 100 to 800 K. Similarly, PCl3 oxidation by Operi is difficult thermodynamically owing to the positive variation in the Gibbs free energy. The variation in ΔG exhibits a trend of slightly decreasing with an increase in temperature, but the trend of overall has a flat manner. The situation is quite different when the Oint atoms are used to participate in the PCl3 oxidation reaction with a MvK-style supply of oxygen. The lattice oxygen atoms at the interface between the Au NPs and Mn3O4 have negative free energies from 100–800 K, revealing that the lattice oxygen atoms of Au/Mn3O4 that are located near the periphery of Au would not participate in the PCl3 oxidation reaction, whereas the lattice oxygen atoms that bridge the Au cluster with the metal-oxide support show great potential for the reaction. An insignificant descending trend in the Gibbs free energy variation for Oint can also be observed as the temperature increases, indicating that the spontaneity of the PCl3 oxidation reaction using Oint as the reactant becomes more intense, but the effect is not that remarkable. The comparison of ΔG for the reaction between the lattice oxygen from clean Mn3O4 and Oint also suggested that the bridging lattice oxygens between the Au clusters and Mn species were activated by the loading of Au; this indicates the generation of an interaction that could change the chemical potential of the lattice oxygen between the Au and metal-oxide support, ensuring the reaction proceeded and improving the catalytic activity of the Au/Mn3O4 catalyst.
The corresponding relationship between the trend in the Auδ+/(Au0 + Auδ+) molar ratio and the increase in the Au mass fraction, and the trend in the POCl3 yield with the increase in the Au mass fraction, indicated that Auδ+ has a certain supporting effect on the PCl3 oxidation reaction. The MvK mechanism gives an insight into the possible role of Auδ+, in that it promotes the dissociation of O2 adsorbed on the Au–support interface. The amount of lattice oxygen on the interface between the Au NPs and Mn3O4 is restricted, such that PCl3 oxidation would be unable to happen when the rate of PCl3 oxidation is faster than that of lattice oxygen supplementation. The rate of lattice oxygen supplementation depends on the rate of oxygen adsorption and dissociation. Owing to the system being exposed to the oxygen-rich and constantly stirred environment, we suppose that a constant supply of oxygen is introduced to the interface of catalyst. The strong interaction in terms of charge transfer between the Au NPs and oxide support promotes the generation of Auδ+, which subsequently accelerates the dissociation of oxygen with as much oxidation as possible of every PCl3 adsorbed on the Au NPs, resulting in the enhancement in PCl3 yield.
The Au/Mn3O4 catalyst engineered via the MOF-mediated method consists of Au NPs deposited on a manganese-oxide support, which is the traditional, industrial, active catalytic component for trace PCl3 oxidation in an organic system. Compared with the traditional Mn3O4 catalyst, Au NPs loaded on the support were strongly capable of capturing the trace reactant PCl3 and activating the lattice oxygen of the metal oxide, enabling PCl3 to be directly oxidized by the lattice oxygen bridging the oxide support and the Au atom that it adsorbs on. The synthesis method, which derives the catalyst from the corresponding MOF composite, could significantly enhance the uniformity of Au NP distribution and induce the formation of an active interface, promoting an increase in the number of reaction active sites and the supplementation of lattice oxygen vacancies in comparison to conventional metal-loading methods. Thus, the as-synthesized catalyst exhibits remarkable progress in catalytic activity compared with the catalysts used in industry or synthesized by conventional methods, with a yield of POCl3 higher than that for Mn3O4 by 28.55 percentage points. In addition, it is inevitable that the cost and the process complexity of the catalyst engineered in this study are higher than for the traditional catalyst, and could be further investigated and modified.
4 Conclusion
To sum up, a high-performance catalyst, Au/Mn3O4, for trace PCl3 oxidation reaction in an organic system was successfully synthesized by using an effective strategy wherein the catalyst was derived from MOFs. This method provides an efficient way to synthesize catalysts with a uniform distribution of Au NPs on a metal-oxide support and strong interaction between them, which could promote the catalytic activity of the as-synthesized catalyst. DFT calculations suggested that a possible catalytic mechanism is the supply of lattice oxygen as a PCl3 oxidant, which is from interface oxygens bridging the Au NPs and Mn, in a MvK process. Structure–performance analysis revealed that the interaction between the Au and support, in terms of charge transfer and chemical potential change, dominantly determined the yield of PCl3. This work provides an effective method, an active catalyst and the best process parameters for the dephosphorization of trichlorosilane feedstocks in the polysilicon industry.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Our research was entirely supported by Tianjin University.References
- C. Ramírez-Márquez, et al., Safety, Economic, and Environmental Optimization Applied to Three Processes for the Production of Solar-Grade Silicon, ACS Sustain. Chem. Eng., 2019, 7(5), 5355–5366 CrossRef.
- A. F. B. Braga, et al., New processes for the production of solar-grade polycrystalline silicon: A review, Sol. Energy Mater. Sol. Cells, 2008, 92(4), 418–424 CrossRef CAS.
- K. Yasuda, K. Morita and T. H. Okabe, Processes for Production of Solar-Grade Silicon Using Hydrogen Reduction and/or Thermal Decomposition, Energy Technol., 2014, 2(2), 141–154 CrossRef CAS.
- J. R. Lowney and H. S. Bennett, Effect of donor impurities on the conduction and valence bands of silicon, J. Appl. Phys., 1982, 53, 433–438 CrossRef CAS.
- H. S. Bennett and J. R. Lowney, Effect of donor impurities on the density of states near the band edge in silicon, J. Appl. Phys., 1981, 52, 5633–5642 CrossRef CAS.
- B. Ceccaroli and O. Lohne, Solar Grade Silicon Feedstock. 2003 Search PubMed.
- E. Díez, et al., Distillation assisted heat pump in a trichlorosilane purification process, Chem. Eng. Process., 2013, 69, 70–76 CrossRef.
- H. Zhang, et al., Design optimization and control of dividing wall column for purification of trichlorosilane, Chem. Eng. Sci., 2022, 257, 117716 CrossRef CAS.
- M. Tzou and M. Midland, Phosphorus removal from chlorosilane, European Pat., EP0878477B1, 2002 Search PubMed.
- K. Uehara, T. Kubota and M. Osima, Method for purifying chlorosilanes, European Pat., EP2036858A3, 2009 Search PubMed.
- N. Naoki, et al., Method for Purifying Chlorosilanes, WO2011018875A1, 2011.
- G. Ghetti, Process and System for the Purification of Trichlorosilane and Silicon Tetrachloride, US Pat., US201213606953, 2012 Search PubMed.
- J. Gallus-olender and B. Franc, The Identification of Oxidation and Decomposition Products of Phosphorus Trichloride by Infrared Spectroscopy, Spectrosc. Lett., 1975, 8(8), 551–560 CrossRef CAS.
- L. V. Bezgubenko, et al., Nucleophilic catalysis of phosphorus trichloride oxygen oxidation, Heteroat. Chem., 2008, 19(4), 408–411 CrossRef CAS.
- C. Kunick and I. Ott, Metal Complexes as Protein Kinase Inhibitors, Angew. Chem., Int. Ed., 2010, 49(31), 5226–5227 CrossRef CAS PubMed.
- D. Widmann, et al., How Temperature Affects the Mechanism of CO Oxidation on Au/TiO2: A Combined EPR and TAP Reactor Study of the Reactive Removal of TiO2 Surface Lattice Oxygen in Au/TiO2 by CO, ACS Catal., 2016, 6(8), 5005–5011 CrossRef CAS.
- M. Haruta, Size- and support-dependency in the catalysis of gold, Catal. Today, 1997, 36(1), 153–166 CrossRef CAS.
- T. V. W. Janssens, et al., Insights into the reactivity of supported Au nanoparticles: combining theory and experiments, Top. Catal., 2007, 44(1–2), 15–26 CrossRef CAS.
- O. Casanova, S. Iborra and A. Corma, Biomass into Chemicals: Aerobic Oxidation of 5-Hydroxymethyl-2-furfural into 2,5-Furandicarboxylic Acid with Gold Nanoparticle Catalysts, ChemSusChem, 2009, 2(12), 1138–1144 CrossRef CAS PubMed.
- M. Sankar, et al., Role of the Support in Gold-Containing Nanoparticles as Heterogeneous Catalysts, Chem. Rev., 2020, 120(8), 3890–3938 CrossRef CAS PubMed.
- B. Sarkodie, et al., Boosting the efficiency of low-loaded Au on spongy Fe2O3 via interfacial ferric hydroxide for low-temperature CO oxidation, Mater. Chem. Phys., 2022, 288, 126407 CrossRef CAS.
- Z. Qiu, et al., The catalytic performance of isolated-dispersed Au on nanosized CeO2 for CO preferential oxidation in H2-rich stream, Appl. Surf. Sci., 2019, 481, 1072–1079 CrossRef CAS.
- Y. Liao, et al., Engineering a homogeneous alloy-oxide interface derived from metal-organic frameworks for selective oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid, Appl. Catal., B, 2020, 270, 118805 CrossRef CAS.
- I. N. Remediakis, N. Lopez and J. K. Nørskov, CO Oxidation on Rutile-Supported Au Nanoparticles, Angew. Chem., 2005, 117, 1858 CrossRef.
- N. Lopez, On the origin of the catalytic activity of gold nanoparticles for low-temperature CO oxidation, J. Catal., 2004, 223(1), 232–235 CrossRef CAS.
- M. Haruta, Catalysis of Gold Nanoparticles Deposited on Metal Oxides, CATTECH, 2002, 6, 102–115 CrossRef CAS.
- M. Haruta, et al., Low-Temperature Oxidation of CO over Gold Supported on TiO2, α-Fe2O3, and Co3O4, J. Catal., 1993, 144, 175–192 CrossRef CAS.
- S. Tsubota, et al., Preparation of Highly Dispersed Gold on Titanium and Magnesium Oxide, Stud. Surf. Sci. Catal., 1991, 63, 695–704 CrossRef CAS.
- D. Andreeva, et al., Au/α-Fe2O3 catalyst for water-gas shift reaction prepared by deposition-precipitation, Appl. Catal., A, 1998, 169(1), 9–14 CrossRef CAS.
- A. Villa, et al., Nitrogen functionalized carbon nanostructures supported Pd and Au–Pd NPs as catalyst for alcohols oxidation, Catal. Today, 2010, 157(1–4), 89–93 CrossRef CAS.
- H. S. Oh, et al., Selective Catalytic Oxidation of CO: Effect of Chloride on Supported Au Catalysts, J. Catal., 2002, 210(2), 375–386 CrossRef CAS.
- C. K. Costello, et al., Nature of the active site for CO oxidation on highly active Au/γ-Al2O3, Appl. Catal., A, 2002, 232(1–2), 159–168 CrossRef CAS.
- F. Moreau, G. C. Bond and A. O. Taylor, Gold on titania catalysts for the oxidation of carbon monoxide: control of pH during preparation with various gold contents, J. Catal., 2005, 231(1), 105–114 CrossRef CAS.
- Y. T. Liao, et al., De Novo Synthesis of Gold-Nanoparticle-Embedded, Nitrogen-Doped Nanoporous Carbon Nanoparticles (Au@NC) with Enhanced Reduction Ability, ChemCatChem, 2016, 8(3), 502–509 CrossRef CAS.
- Y. Liao, et al., Mesoporous TiO2 Embedded with a Uniform Distribution of CuO Exhibit Enhanced Charge Separation and Photocatalytic Efficiency, ACS Appl. Mater. Interfaces, 2017, 9(49), 42425–42429 CrossRef CAS PubMed.
- H. Reinsch and N. Stock, Formation and characterisation of Mn-MIL-100, CrystEngComm, 2013, 15(3), 544–550 RSC.
- D. Sun, et al., In Situ Construction of MIL-100@NiMn-LDH Hierarchical Architectures for Highly Selective Photoreduction of CO2 to CH4, ACS Appl. Mater. Interfaces, 2022, 14(14), 16369–16378 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented- wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, et al., A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- Z. Zhu, et al., Rational construction of metal–base synergetic sites on Au/Mg-beta catalyst for selective aerobic oxidation of 5-hydroxymethylfurfural, J. Energy Chem., 2021, 62, 599–609 CrossRef CAS.
- Q. Li, et al., Selective oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid over Au/CeO2 catalysts: the morphology effect of CeO2, Catal. Sci. Technol., 2019, 9(7), 1570–1580 RSC.
- J. Zhang, et al., Wet-Chemistry Strong Metal–Support Interactions in Titania-Supported Au Catalysts, J. Am. Chem. Soc., 2019, 141(7), 2975–2983 CrossRef CAS PubMed.
- A. Abad, et al., A Collaborative Effect between Gold and a Support Induces the Selective Oxidation of Alcohols, Angew. Chem., Int. Ed., 2005, 44(26), 4066–4069 CrossRef CAS PubMed.
- G. Greczynski and L. Hultman, A step-by-step guide to perform x-ray photoelectron spectroscopy, J. Appl. Phys., 2022, 132, 011101 CrossRef CAS.
- Z. Xu, et al., A novel γ-like MnO2 catalyst for ozone decomposition in high humidity conditions, J. Hazard. Mater., 2021, 420, 126641 CrossRef CAS PubMed.
- Y. Ha, et al., Mn-MIL-100 heterogeneous catalyst for the selective oxidative cleavage of alkenes to aldehydes, Catal. Commun., 2018, 103, 51–55 CrossRef CAS.
- W. Zhang, et al., Synthesis of Bimetallic MOFs MIL-100(Fe-Mn) as an Efficient Catalyst for Selective Catalytic Reduction of NOx with NH3, Catal. Lett., 2016, 146(10), 1956–1964 CrossRef CAS.
- M. Pang, Q. Wang and H. C. Zeng, Self-Generated Etchant for Synthetic Sculpturing of Cu2O-Au, Cu2O@Au, Au/Cu2O, and 3D-Au Nanostructures, Chem.–Eur. J., 2012, 18(46), 14605–14609 CrossRef CAS PubMed.
- Y. Hu, et al., Two-step pyrolysis of Mn MIL-100 MOFinto MnO nanoclusters/carbon and the effect of N-doping, J. Mater. Chem. A, 2022, 10, 8172 RSC.
- M. Umar, et al., Synthesis and characterization of highly efficient Te-doped Mn3O4 and s-g-C3N4/Te-Mn3O4 nanocomposites as an excellent antimicrobial and photocatalytic agent, Inorg. Chem. Commun., 2023, 157, 111353 CrossRef CAS.
- X. Zhang, et al., MIL-100(Fe) supported Mn-based catalyst and its behavior in Hg0 removal from flue gas, J. Hazard. Mater., 2020, 381, 121003 CrossRef CAS PubMed.
- A. Rapeyko, et al., Polymers from biomass: one pot two-step synthesis of furilydenepropanenitrile derivatives with MIL-100(Fe) catalyst, Catal. Sci. Technol., 2017, 7, 3008 RSC.
- N. Li, et al., Efficient degradation of bentazone via peroxymonosulfate activation by 1D/2D γ-MnOOH-rGO under simulated sunlight: Performance and mechanism insight, Sci. Total Environ., 2020, 741, 140492 CrossRef CAS PubMed.
- M. A. Saqlain, et al., A DFT+U study of the Mars Van Krevelen mechanism of CO oxidation on Au/TiO2 catalysts, Appl. Catal., A, 2016, 519, 27–33 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra08566j |
| This journal is © The Royal Society of Chemistry 2024 |