
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStrain-based design, direct macrocyclization, and metal complexation of thiazole-containing calix[3]pyrrole analogues†
Keita
Watanabe
a,
Kotaro
Shibata
a,
Tomoya
Ichino
*b,
Yuki
Ide
 b,
Tomoki
Yoneda
a,
Satoshi
Maeda
bc and
Yasuhide
Inokuma
*ab
b,
Tomoki
Yoneda
a,
Satoshi
Maeda
bc and
Yasuhide
Inokuma
*ab
aDivision of Applied Chemistry, Faculty of Engineering, Hokkaido University, Kita 13, Nishi 8, Kita-ku, Sapporo, Hokkaido 060-8628, Japan. E-mail: inokuma@eng.hokudai.ac.jp
bInstitute for Chemical Reaction Design and Discovery (WPI-ICReDD), Hokkaido University, Kita-21, Nishi 10, Kita-ku, Sapporo, Hokkaido, 001-0021, Japan. E-mail: tichino@eis.hokudai.ac.jp
cDepartment of Chemistry, Faculty of Science, Hokkaido University, Kita 10, Nishi 8, Kita-ku, Sapporo, Hokkaido, 060-8610, Japan
First published on 3rd May 2024
Abstract
The coordination chemistry of ring-contracted porphyrinoids, such as subporphyrins and calix[3]pyrroles, has been largely unexplored owing to the synthetic difficulty of their free-base analogues. Here, we report strain-based molecular design and high-yield synthesis of thiazole-containing calix[3]pyrrole analogues for metal complexation. The artificial force induced reaction and StrainViz analysis methods were used to perform a conformational search and evaluate/visualize the ring strain. The results indicated that the thiazole-containing analogues are less strained than the parent calix[3]pyrrole, while incorporation of imidazole or oxazole unexpectedly leads to an increase in the total strain. Calix[1]furan[2]thiazole was obtained in 60% yield by the direct macrocyclization between α-bromoketone and bis(thioamide), whereas the meso-N(sp2)-bridged analogue, which was calculated to be 5.1 kcal mol−1 more strained, was only obtained in a 2% yield. Calix[1]furan[2]thiazole was converted to calix[1]pyrrole[2]thiazole to investigate metal complexation. Through the reaction with Et2Zn, calix[1]pyrrole[2]thiazole bound a Zn(II) ion in a tridentate fashion adopting a cone conformation, giving a water/air stable organozinc complex that catalyzes polymerization of lactide. Conversely, Ag(I) and Pd(II) ions coordinated to the partial cone conformation of calix[1]pyrrole[2]thiazole in a bidentate fashion. Strain-based molecular design expands the synthetic access to contracted porphyrinoids and provides the opportunity to take advantage of their rich coordination chemistry.
Introduction
Contracted porphyrins, such as subphthalocyanines and subporphyrins, have recently attracted much attention owing to their bowl-shaped structures, 14π-aromatic characters, and visible absorption/emission properties.1–3 Since the discovery of boron(III)–subphthalocyanine by Meller and Ossko in 1972,4 boron-templated synthesis has played a central role in developing their analogues by core modification and peripheral functionalization.5 The boron template is almost essential for the synthesis of triphyrin(1.1.1)-type contracted porphyrinoids, because the boron-free analogues have not yet been obtained from the direct macrocyclization of pyrrole monomers. While most subphthalocyanines and subporphyrins are obtained as boron(III)-complexes, removal of the boron atom remains unsuccessful. The unavailability of boron-free contracted porphyrins has hampered the development of their coordination chemistry, although metal complexation has endowed other porphyrinoids with various properties, such as catalysis,6 photosensitization,7 and coordination-driven self-assembly.8Recent advances have demonstrated that boron-free subporphyrin9 and calix[3]pyrrole (1),10 a porphyrinogen-type congener of subporphyrin, stably exist without any template atoms, which has provided the opportunity to explore their metal complexes. However, the low synthetic yields of the free-base analogues still hinder further investigations. Suzuki–Miyaura cross coupling gives the subporphyrin free-base only in 4% yield, and synthesis of 1 requires five steps from a commercial precursor with a total yield of 4%. Simple, high-yield synthetic routes for boron-free contracted porphyrinoids are still being pursued in this research area.
Recent studies have suggested that the synthetic difficulty arises from the unusual increase in the ring strain during the formation of tripyrrolic macrocycles from linear precursors.11 Although synthetic and theoretical evaluation of the calix[n]furan[3 − n]pyrrole (n = 0–3) system indicates that the ring strain increases as the number of NH units increases, the relationship between the ring strain and synthetic accessibility is still unclear. Here, we report the strain-based molecular design and efficient synthesis of thiazole-containing, boron-free calix[3]pyrrole analogues, calix[1]pyrrole[2]thiazole (2) and calix[1]furan[2]thiazole (5), as well as the derivatization of 2 to metal complexes. For the design of less-strained macrocycles, we used the Artificial Force Induced Reaction (AFIR)12 method-based conformational search and visualization of the macrocyclic ring strain using the StrainViz analysis,13 which was developed by Justi and co-workers for the evaluation of the strain of cycloparaphenylenes. Contrary to our semi-empirical expectation, replacement of two pyrrole units in calix[3]pyrrole 1 with oxazole or imidazole rings resulted in a significant increase in the total strain, while the thiazole-containing analogues were less strained. The validity of the theoretical expectations was confirmed through the synthesis of 5 and its meso-N(sp2)-bridged analogue 10. While 5 was obtained in 60% yield, 10, which was calculated to be 5.1 kcal mol−1 more strained, was only obtained in 2% yield. Calix[3]pyrrole-related ligand 2 switched its conformation between the partial cone and the cone to chelate various metal ions to provide a moisture-stable organozinc catalyst for rac-lactide polymerization and coordination assembly, demonstrating the unique coordination chemistry of the contracted calixpyrrole.
Results and discussion
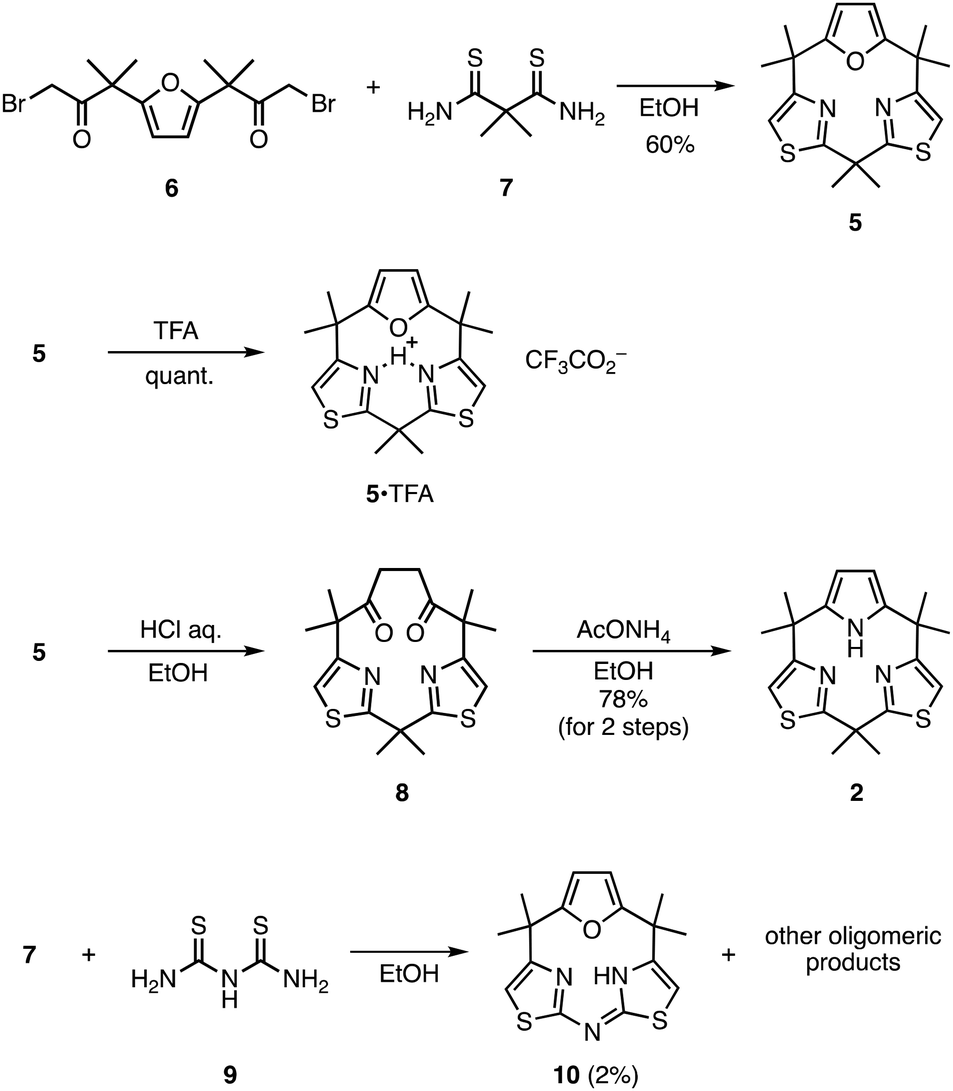
To design a less-strained calix[3]pyrrole analogue for metal complexation, we theoretically evaluated several heteroarene-embedded macrocycles 2–5 (see the ESI† for the computational details). On the basis of the semi-empirical trends obtained for calix[n]furan[3 − n]pyrrole, we expected that replacing the two pyrrole units in 1 with thiazole, imidazole, or oxazole rings would reduce the total strain owing to the decreasing number of inner NH protons, since the strain-induced ring expansion reaction occurred for n = 0 and 1, but not for n = 2, 3. Furthermore, the iminic nitrogen atoms in these heteroarenes would contribute to form metal complexes. To evaluate the ring strain, the lowest energy conformations of 1–5 were obtained by a searched using the AFIR method, and they were further optimized by the density functional theory (DFT) calculations at the M06-2X/6-311+G(2d,p) level (Fig. 1a). The optimized structure of calix[3]pyrrole (1) adopted a partial cone conformation, as observed in its crystal structure. The dihedral strain (11.7 kcal mol−1) greatly contributed to the total strain of 13.4 kcal mol−1 (Fig. 1c). The StrainViz analysis revealed that the C(pyrrole-α)–C(meso) bond had the maximum strain (Fig. 1b). Calix[1]pyrrole[2]thiazole (2) exhibited a cone conformation with a NH⋯N distance of 2.11 Å, and the dihedral strain was significantly reduced compared with that of 1. The total strain of 2 was 2.9 kcal mol−1 lower than that of 1, suggesting that 2 is more easily accessible. Contrary to our empirical expectation, the lowest energy conformation of imidazole derivative 3 had three inner NH protons (Fig. S4†). Because of the increased dihedral strain, the total strain of 3 was 2.2 kcal mol−1 larger than that of 1. Although oxazole derivative 4 showed a cone conformation similar to that of 2, the total strain was comparable to that of 3 owing to the large contribution of the dihedral strain around the C(meso)–C(pyrrole-α) bonds. When the pyrrole unit in the less-strained 2 was replaced with furan, the angle strain was reduced by 0.6 kcal mol−1, but the dihedral strain increased by 1.0 kcal mol−1, resulting in 2 and 5 having similar total strain energies. According to the theoretical calculations, thiazole-containing derivatives 2 and 5 are the most feasible among the evaluated compounds 1–5. Given the synthetic availability of furan precursor 6,14 we investigated direct macrocyclization synthesis of 5 using a Hantzsch-type thiazole formation reaction.15 | ||
| Fig. 1 (a) Optimized structures of the lowest energy conformations of calix[3]pyrrole (1) and its analogues 2–5, and 10. (b) Visualization of the total ring strain of 1–5 and 10 using the StrainViz analysis. (c) Calculated total, bond, angle, and dihedral strain energies of 1–5 and 10 (in kcal mol−1). | ||
To our delight, the condensation reaction between 6 and 2,2-dimethylpropanebis(thioamide) (7)16 in refluxing ethanol gave calix[1]furan[2]thiazole (5) in 60% yield (Scheme 1), along with a double sized macrocycle, calix[2]furan[4]thiazole, in 16% yield (compound 14 in ESI†). The 1H NMR spectrum of 5 in CDCl3 showed time-averaged C2v molecular symmetry, featuring four singlet signals at 6.78, 5.88, 1.93, and 1.63 ppm assignable to thiazole, furan and two different methyl protons, respectively.
 | ||
| Scheme 1 Synthesis of thiazole-containing calix[3]pyrrole analogues. | ||
Although strained calix[3]pyrrole analogues typically undergo strain-induced ring expansion reactions under acidic conditions,11 macrocycle 5 was sufficiently stable to exist in the presence of trifluoroacetic acid (TFA) in CH2Cl2. Vapor diffusion of hexane into a dichloromethane/TFA solution of 5 gave single crystals of the protonated form 5·TFA (Fig. 2). X-ray diffraction analysis revealed that 5·TFA adopted a cone conformation that is commonly observed for less-strained calix[3]pyrrole analogues.10 The C–N–C bond angles of the thiazole units were 116.9° and 112.5°, indicating that the former was the N-protonated moiety.
 | ||
| Fig. 2 Single crystal X-ray structures (left, top view; right, side view) of (a) 5·TFA, (b) 2, and (c) 10 drawn at the 50% probability level of the thermal ellipsoids (H, white; C, gray; N, blue; O, red; S, yellow; and F, yellow green). | ||
When 5 was treated with aqueous hydrochloric acid, the furan moiety was hydrolyzed to give 1,4-diketone-linked macrocycle 8 quantitatively. The Paal–Knorr reaction of 8 with ammonium acetate gave calix[1]pyrrole[2]thiazole (2) in 78% yield. The total yield of 2 from the known precursors was 30% in three steps, which is markedly higher than those reported for 1 and the subporphyrin free-base. The 1H NMR spectrum of 2 recorded in CDCl3 also exhibited a C2v-symmetric signal pattern. The NH proton was observed at 12.17 ppm, indicating a hydrogen bond with the thiazole units. Similar to 5, macrocycle 2 did not undergo a strain-induced ring expansion reaction under standard conditions using TFA.11 Single crystal X-ray diffraction analysis confirmed that 2 had a cone conformation, which was predicted by an AFIR-based conformational search. The N(thiazole)–N(thiazole) and N(pyrrole)–N(thiazole) distances were 2.820 and 2.702 Å, respectively, suggesting the potential for the cooperative coordination of metal ions using three nitrogen atoms.
The significance of evaluating the ring strain before conducting a synthetic examination was demonstrated by the synthesis of meso-N(sp2)-bridged analogue 10. An AFIR-based conformational search revealed that the most stable conformation of 10 is an uncommon gable-like conformation, in which the N-bridged bis(thiazole) unit is almost planar, but the total strain (15.8 kcal mol−1) is the largest of the series of molecules (Fig. 1). Despite our tremendous efforts, reaction between 7 and 9 gave only a trace amount (2% yield) of the target 10 (Scheme 1), while a calix[6]-type macrocycle was obtained in 32% yield along with other oligomeric products (compound 15 in ESI†). Single crystal X-ray analysis also showed the gable-like conformation of 10 (Fig. 2c). Through the imine–enamine tautomerism, the meso-N atom lost a hydrogen atom, resulting in the formation of an inner NH group. Owing to the intramolecular hydrogen-bonding interaction, the inner NH proton was observed at 18.67 ppm in the 1H NMR spectrum recorded in CDCl3. These results demonstrated that the AFIR method is useful for searching for the stable conformations, and the comparison of the calculated total strains can indicate the synthetic feasibility of calix[3]pyrrole derivatives.
In our previous report, we found that absorption spectra of calix[3]pyrrole analogues show characteristically red-shifted absorption as a result of strained ring system.17 Compounds 2 and 5 also exhibited similar broad bands around 300 nm in dichloromethane (Fig. S26†). In the case of N(sp2)-linked 10, a strong absorption band with absorption coefficient of ε = 1.9 × 104 M−1 cm−1 was observed at 339 nm, showing the effect of π-conjugation through the meso-N atom.
The reliable synthetic route of 2 allowed further investigation of its coordination chemistry. Among porphyrin-related ligands, calix[4]pyrroles, a parent tetrapyrrolic macrocycle of 1, have shown their unique metal binding behavior. Depending on the metal ions, calix[4]pyrroles can switch their conformations and the coordination mode of each pyrrole unit from η1- to η5-manners.18 After several attempts using various metal salts, we found that Et2Zn, Pd(MeCN)4(BF4)2, and AgBF4 can form isolable metal complexes with 2.
When 2 was treated with diethylzinc in dry toluene, ethyl zinc complex 11 was obtained as a yellow crystalline solid in 72% yield (Fig. 3). 1H NMR spectroscopy of 11 confirmed that the pyrrole NH proton disappeared, while the ethyl proton signals were observed at 0.54(q) and 1.37(t) ppm. The single crystal X-ray structure of 11 showed a cone-shaped conformation of the ligand that chelated to the ethyl Zn(II) moiety in a monoanionic tridentate manner. Because three meso-Me substituents were located in an almost perpendicular fashion to the macrocycle, the zinc center was sterically protected by the ligand. The UV-vis absorption spectrum of 11 showed that the absorption bands of ligand 2 were slightly red-shifted with broadening upon zinc complexation (Fig. S26†).
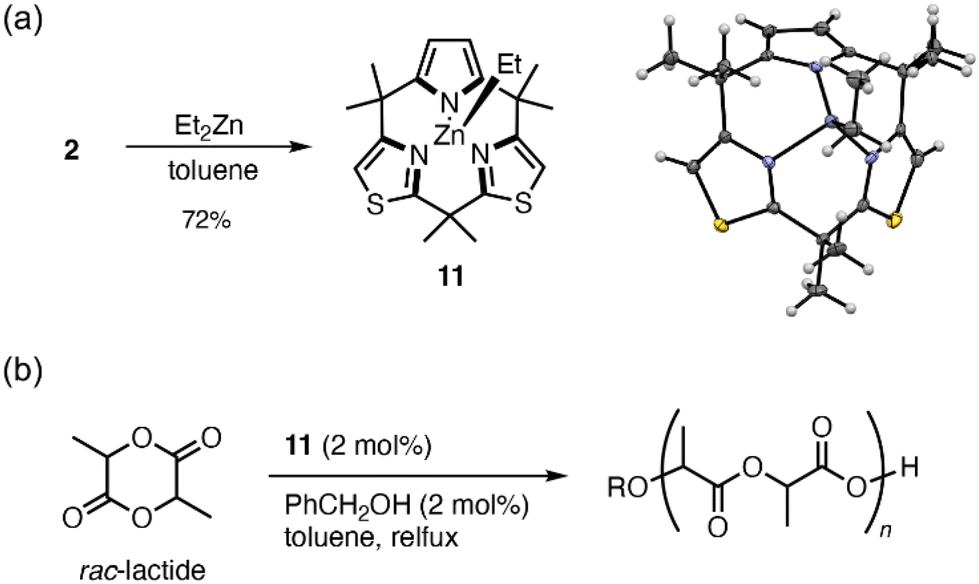 | ||
| Fig. 3 (a) Synthesis and crystal structure of organozinc complex 11. (b) Polymerization of rac-lactide catalyzed by 11. | ||
Organozinc complex 11 showed remarkable stability toward water, alcohol, and air at room temperature, and it can thus be treated in organic solutions without special care.19 When complex 11 was dissolved in wet CDCl3 at 13 mM (containing ≥5.0 equiv. of H2O), hydrolysis was not observed after 1 h at room temperature by 1H NMR spectroscopy, while diethylzinc was immediately decomposed under similar conditions. Similar tests with ≥70 equiv. of methanol, ethanol, and isopropyl alcohol exhibited recovery of ≥97% of 11 after 1 h (Fig. S21†). Whereas, addition of more acidic phenol or benzoic acid (3.0 equiv.) resulted in the considerable demetallation of 11 to give 2. Air oxidation of 11 was not observed after standing a chloroform solution of 11 or the crystalline solids in an aerobic atmosphere for more than 2 days.
Organozinc complex 11 is sufficiently stable to be handled under non-dried aerobic conditions, and it shows catalytic activity for the polymerization of rac-lactide.20 When a toluene solution of rac-lactide was heated to reflux in the presence of 11 and benzyl alcohol (2 mol% each), >95% of the monomer was converted in 30 min to give poly(lactide) with number-average molecular weight of 9.0 kg mol−1 and polydispersity index of 1.4.
Upon complexation with Pd(MeCN)4(BF4)2, macrocycle 2 formed cationic Pd(II) complex 12 adopting a partial cone conformation (Fig. 4). The quantitative complexation behavior was monitored by 1H NMR spectroscopy (Fig. S23†). Upon addition of the Pd(II) salt to a CD3CN solution of 2 at 5.6 mM, new signals assignable to the thiazole and pyrrole protons of 12 appeared at 7.68 and 6.72 ppm, respectively. After the addition of 1.0 equiv. of the salt, the signals of 2 were almost converted to those of 12, indicating its quantitative formation. Vapor diffusion of diethyl ether into an acetonitrile solution of 12 gave diffraction grade single crystals. Crystallographic analysis revealed a partial cone conformation of 12 in which ligand 2 coordinated to the Pd(II) ion in a square planar coordination geometry with two thiazole nitrogen atoms, leaving the pyrrole unit uncoordinated. The pyrrole NH site was hydrogen bonded to a BF4 anion, which was also in a close proximity to two thiazole planes. NCI plot analysis21 suggested that weak but nontrivial anion–π interactions existed between the BF4 anion and thiazole moieties (Fig. S18†). Although such anion binding behavior was not observed in solution, these observations indicated the unique structural and supramolecular properties of calix[3]pyrrole-related ligand 2.
 | ||
| Fig. 4 Transition metal complexes of ligand 2 with partial cone conformations. (a) Chemical structure (left), ORTEP drawing of the crystal structure (center), and anion binding mode in the solid state (right) of Pd-complex 12. (b) Chemical structure (left) and ORTEP drawing (right) of Ag(I)-linked coordination assembly 13. | ||
Complexation with AgBF4 also gave a coordination assembly of 2. 1H NMR titration of 2 with the Ag(I) salt in CD3CN exhibited a gradual shift of the proton signals due to 2, indicating reversible metal coordination with a rapid exchanging rate on the NMR time scale. Job's plot analysis indicated a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexation of 2 with Ag(I) ions in solution at ∼10 mM concentration (Fig. S24†). However, when single crystals of the Ag(I)-complex were grown from the solution, Ag(I)-linked dyad 13 was obtained (Fig. 4). Complex 13 was composed of an Ag(I) ion and two ligands 2 in different conformations. The ligand 2 in the partial cone conformation coordinated to the Ag(I) ion using two N(thiazole) atoms in a similar fashion to 12, while the Ag–C(pyrrole-β) distance of 2.66 Å indicated coordination of the pyrrole unit in an η1 fashion. The other ligand 2 in the cone conformation chelated to the Ag(I) ion with η2-pyrrole–Ag(I) coordination bonds, leaving the thiazole units uncoordinated. When the crystals of 13 were dissolved in CD3CN, the 1H NMR spectrum of an equilibrated solution, which was also observed during titration with Ag(I), was obtained due to the reversible metal coordination. Nevertheless, the formation of complex 13 demonstrated the possibility of coordination self-assembly of 2, which can be used as the principal interactions to construct giant supramolecules.
:1 complexation of 2 with Ag(I) ions in solution at ∼10 mM concentration (Fig. S24†). However, when single crystals of the Ag(I)-complex were grown from the solution, Ag(I)-linked dyad 13 was obtained (Fig. 4). Complex 13 was composed of an Ag(I) ion and two ligands 2 in different conformations. The ligand 2 in the partial cone conformation coordinated to the Ag(I) ion using two N(thiazole) atoms in a similar fashion to 12, while the Ag–C(pyrrole-β) distance of 2.66 Å indicated coordination of the pyrrole unit in an η1 fashion. The other ligand 2 in the cone conformation chelated to the Ag(I) ion with η2-pyrrole–Ag(I) coordination bonds, leaving the thiazole units uncoordinated. When the crystals of 13 were dissolved in CD3CN, the 1H NMR spectrum of an equilibrated solution, which was also observed during titration with Ag(I), was obtained due to the reversible metal coordination. Nevertheless, the formation of complex 13 demonstrated the possibility of coordination self-assembly of 2, which can be used as the principal interactions to construct giant supramolecules.
Conclusions
In conclusion, a conformational search and strain visualization using the AFIR and StrainViz methods allowed the rational design of less-strained thiazole-containing calix[3]pyrrole analogues, 2 and 5. The total strain energy and synthetic yield were found to be almost inversely proportional. Hence, strain calculations are more important than semi-empirical design for considering the feasible synthetic targets. The scalable synthetic route for 2 enabled investigation of metal complexes of the calix[3]pyrrole-type ligand. Formation of the moisture-stable organozinc complex 11 demonstrated the unique properties of ligand 2 that can protect the metal center within the narrow cavity. Complexation with Pd(II) and Ag(I) ions showed the conformational flexibility of 2 during metal coordination and the possibility of coordination self-assembly to produce giant supramolecules. The present results will lead to various types of contracted porphyrins without template atoms through strain-based design, allowing further synthetic functionalization and metal complexation. Moreover, as a novel class of ligands, boron-free calix[3]pyrrole-type macrocycles will produce a range of organometallic complexes, metal clusters, and coordination polymers, which will contribute to advance in the field of inorganic chemistry.Data availability
The ESI contains synthetic procedures, computational details, crystallographic data, and NMR spectra.Author contributions
Y. Inokuma designed the study, supervised the project, and wrote the first version of the draft. K. W., K. S., Y. Ide, and T. Y. performed experimental work. T. I. and S. M. did computational analyses.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by a JSPS Grant-in-Aid for Scientific Research (B) (No. 22H02058) and JST FOREST Program (No. JPMJFR211H), of which Y. Inokuma is the principal investigator. This work was also partly supported by Toyota Riken Scholar and NOASTEC Foundation to T. Y. The Institute for Chemical Reaction Design and Discovery (ICReDD) was established by World Premier International Research Initiative (WPI), MEXT, Japan.References
- G. Lavarda, J. Labella, M. V. Martínez-Díaz, M. S. Rodríguez-Morgade, A. Osuka and T. Torres, Recent advances in subphthalocyanines and related subporphyrinoids, Chem. Soc. Rev., 2022, 51, 9482–9619 RSC.
- (a) C. G. Claessens, D. González-Rodríguez, M. S. Rodríguez-Morgade, A. Medina and T. Torres, Subphthalocyanines, Subporphyrazines, and Subporphyrins: Singular Nonplanar Aromatic Systems, Chem. Rev., 2014, 114, 2192–2277 CrossRef CAS PubMed; (b) Y. Inokuma and A. Osuka, Subporphyrins: emerging contracted porphyrins with aromatic 14π-electronic systems and bowl-shaped structures: rational and unexpected synthetic routes, Dalton Trans., 2008, 2517–2526 RSC; (c) M. Pawlicki and L. Latos-Grażyński, Aromaticity Switching in Porphyrinoids, Chem. – Asian J., 2015, 10, 1438–1451 CrossRef CAS PubMed.
- (a) D. González-Rodríguez, T. Torres, M. M. Olmstead, J. Rivera, M. Á. Herranz, L. Echegoyen, C. A. Castellanos and D. M. Guldi, Photoinduced Charge-Transfer States in Subphthalocyanine-Ferrocene Dyads, J. Am. Chem. Soc., 2006, 128, 10680–10681 CrossRef PubMed; (b) S. Shimizu, Recent Advances in Subporphyrins and Triphyrin Analogues: Contracted Porphyrins Comprising Three Pyrrole Rings, Chem. Rev., 2017, 117, 2730–2784 CrossRef CAS PubMed; (c) A. Osuka, E. Tsurumaki and T. Tanaka, Subporphyrins: A Legitimate Ring-Contracted Porphyrin with Versatile Electronic and Optical Properties, Bull. Chem. Soc. Jpn., 2011, 84, 679–697 CrossRef CAS.
- A. Meller and A. Ossko, Phthalocyaninartige Bor-Komplexe, Monatsh. Chem., 1972, 103, 150–155 CrossRef CAS.
- (a) C. G. Claessens, D. González-Rodríguez and T. Torres, Subphthalocyanines: Singular Nonplanar Aromatic CompoundsSynthesis, Reactivity, and Physical Properties, Chem. Rev., 2002, 102, 835–853 CrossRef CAS PubMed; (b) Y. Inokuma, J. H. Kwon, T. K. Ahn, M.-C. Yoon, D. Kim and A. Osuka, Tribenzosubporphines: Synthesis and Characterization, Angew. Chem., Int. Ed., 2006, 45, 961–964 CrossRef CAS PubMed; (c) N. Kobayashi, Y. Takeuchi and A. Matsuda, meso-Aryl Subporphyrins, Angew. Chem., Int. Ed., 2007, 46, 758–760 CrossRef CAS PubMed; (d) Z. Li, L. Zhang, Q. Wu, H. Li, Z. Kang, C. Yu, E. Hao and L. Jiao, Boron-Templated Synthesis of B(III)-Submonoazaporphyrins: The Hybrids of B(III)-Subporphyrins and B(III)-Subporphyrazines, J. Am. Chem. Soc., 2022, 144, 6692–6697 CrossRef CAS PubMed.
- (a) B. Meunier, Metalloporphyrins as versatile catalysts for oxidation reactions and oxidative DNA cleavage, Chem. Rev., 1992, 92, 1411–1456 CrossRef CAS; (b) S. Fukuzumi, Y.-M. Lee and W. Nam, Mechanisms of Two-Electron versus Four-Electron Reduction of Dioxygen Catalyzed by Earth-Abundant Metal Complexes, ChemCatChem, 2018, 10, 9–28 CrossRef CAS; (c) C.-M. Che, V. K.-Y. Lo, C.-Y. Zhou and J.-S. Huang, Selective functionalisation of saturated C–H bonds with metalloporphyrincatalysts, Chem. Soc. Rev., 2011, 40, 1950–1975 RSC; (d) L. Feng, K.-Y. Wang, E. Joseph and H.-C. Zhou, Catalytic Porphyrin Framework Compounds, Trends Chem., 2020, 555–568 CrossRef CAS.
- (a) J. R. Darwent, P. Douglas, A. Harriman, G. Porter and M.-C. Richoux, Metal phthalocyanines and porphyrins as photosensitizers for reduction of water to hydrogen, Coord. Chem. Rev., 1982, 44, 83–126 CrossRef CAS; (b) R. Bonnet, Photosensitizers of the porphyrin and phthalocyanine series for photodynamic therapy, Chem. Soc. Rev., 1995, 24, 19–33 RSC; (c) J. Karges, Clinical Development of Metal Complexes as Photosensitizers for Photodynamic Therapy of Cancer, Angew. Chem., Int. Ed., 2022, 61, e202112236 CrossRef CAS PubMed; (d) A. R. Sekhar, Y. Chitose, J. Janoš, S. I. Dangoor, A. Ramundo, R. Satchi-Fainaro, P. Slavíček, P. Klán and R. Weinstain, Porphyrin as a versatile visible-light-activatable organic/metal hybrid photoremovable protecting group, Nat. Commun., 2022, 13, 3614 CrossRef CAS PubMed.
- (a) I. Beletskaya, V. S. Tyurin, A. Y. Tsivadze, R. Guilard and C. Stern, Supramolecular Chemistry of Metalloporphyrins, Chem. Rev., 2009, 109, 1659–1713 CrossRef CAS PubMed; (b) P. S. Bols and H. L. Anderson, Template-Directed Synthesis of Molecular Nanorings and Cages, Acc. Chem. Res., 2018, 51, 2083–2092 CrossRef CAS PubMed; (c) X. Chi, A. J. Guerin, R. A. Haycock, C. A. Hunter and L. D. Sarson, Self-assembly of macrocyclic porphyrin oligomers, J. Chem. Soc., Chem. Commun., 1995, 24, 2567–2569 RSC; (d) J. A. Faiz, V. Heitz and J.-P. Sauvage, Design and synthesis of porphyrin-containing catenanes and rotaxanes, Chem. Soc. Rev., 2009, 38, 422–442 RSC.
- L. Liu, J. Kim, L. Xu, Y. Rao, M. Zhou, B. Yin, J. Oh, D. Kim, A. Osuka and J. Song, Synthesis of Subporphyrin Free Bases, Angew. Chem., Int. Ed., 2022, 61, e202214342 CrossRef CAS PubMed.
- Y. Inaba, Y. Nomata, Y. Ide, J. Pirillo, Y. Hijikata, T. Yoneda, A. Osuka, J. L. Sessler and Y. Inokuma, Calix[3]pyrrole: A Missing Link in Porphyrin-Related Chemistry, J. Am. Chem. Soc., 2021, 143, 12355–12360 CrossRef CAS PubMed.
- Y. Inaba, Y. Kakibayashi, Y. Ide, J. Pirillo, Y. Hijikata and Y. Inokuma, Strain-Induced Ring Expansion Reactions of Calix[3]pyrrole-Related Macrocycles, Chem. – Eur. J., 2022, 28, e202200056 CrossRef CAS PubMed.
- (a) S. Maeda, K. Ohno and K. Mororkuma, Systematic exploration of the mechanism of chemical reactions: the global reaction route mapping (GRRM) strategy using the ADDF and AFIR methods, Phys. Chem. Chem. Phys., 2013, 15, 3683–3701 RSC; (b) S. Maeda and Y. Harabuchi, Exploring paths of chemical transformations in molecular and periodic systems: An approach utilizing force, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2021, 11, e1538 CAS.
- C. E. Colwell, T. W. Price, T. Stauch and R. Jasti, Strain visualization for strained macrocycles, Chem. Sci., 2020, 11, 3923–3930 RSC.
- H. Fujita, S. Arai, H. Arakawa, K. Hamamoto, T. Kato, T. Arai, N. Nitta, K. Hotta, N. Hosokawa, T. Ohbayashi, C. Takahashi, Y. Inokuma, I. Tamai, S. Yano, M. Kunishima and Y. Watanabe, Drug–drug conjugates of MEK and Akt inhibitors for RAS-mutant cancers, Bioorg. Med. Chem., 2024, 102, 117674 CrossRef CAS PubMed.
- A. Hantzsch and J. H. Weber, Ueber Verbindungen des Thiazols (Pyridins der Thiophenreihe), Ber. Dtsch. Chem. Ges., 1887, 20, 3118 CrossRef.
- S. H. J. D. Beuleleer and H. O. Desseyn, Thermal analysis of dithiomalonoamide complexes, J. Therm. Anal., 1996, 47, 135–142 CrossRef.
- K. Watanabe, R. Saha, Y. Inaba, Y. Manabe, T. Yoneda, Y. Ide, Y. Hijikata and Y. Inokuma, Absorption Spectra of Calix[3]pyrrole Analogues as Probes for Contracted Macrocycles, J. Porphyrins Phthalocyanines, 2023, 27, 157–163 CrossRef CAS.
- H. Ruppert, L. M. Sigmund and L. Greb, Calix[4]pyrroles as ligands: recent progress with a focus on the emerging p-block element chemistry, Chem. Commun., 2021, 57, 11751 RSC.
- H. Irving and R. J. P. Williams, The stability of transition-metal complexes, J. Chem. Soc., 1953, 3192–3210 RSC.
- (a) O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Controlled Ring-Opening Polymerization of Lactide and Glycolide, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed; (b) R. Petrus and P. Sobota, Magnesium and zinc alkoxides and aryloxides supported by commercially available ligands as promoters of chemical transformations of lactic acid derivatives to industrially important fine chemicals, Coord. Chem. Rev., 2019, 396, 72–88 CrossRef CAS; (c) G. Sachdeva, Y. Bamal, A. Ladan, O. S. Tiwari, V. Rawat, P. Yadav and V. P. Verma, Calixarene-Metal Complexes in Lactide Polymerization: The Story so Far, ACS Omega, 2023, 8, 13479–13491 CrossRef CAS PubMed.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, NCIPLOT: A Program for Plotting Noncovalent Interaction Regions, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2326011–2326017. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qi00684d |
| This journal is © the Partner Organisations 2024 |