
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNose to brain targeting of the donepezil nanostructured lipid carrier in situ gel: formulation, in vitro, ex vivo, in vivo pharmacokinetic and pharmacodynamic characterization†
Devika
Sonawane
and
Varsha
Pokharkar
 *
*
Department of Pharmaceutics, Poona College of Pharmacy, Bharati Vidyapeeth (Deemed to be University), Paud Road, Erandwane, Pune 411038, India. E-mail: varsha.pokharkar@bharatividyapeeth.edu; Tel: +919422032985
First published on 13th August 2024
Abstract
Donepezil (DPZ) is a reversible, noncompetitive inhibitor of acetylcholinesterase commonly prescribed against Alzheimer's disease (AD). Its dose-dependent side effects limit its therapeutic benefits. The current study endeavors to design an in situ gel for intranasal delivery of a DPZ nanostructured lipid carrier (DPZ-NLC) to boost pharmacokinetic and pharmacodynamic outcomes. The Box–Behnken design was employed to optimize the NLCs that were produced utilizing a melt emulsification high-pressure homogenization process. Afterward, NLCs were embedded in an in situ gel based on Lutrol F127 and analyzed further. The effects of formulation pharmacodynamics were evaluated in a Wistar rat model with trimethyl tin (TMT) induced neurodegeneration. The batch of the optimized DPZ in situ gel had a spherical shape, with a mean particle size of 112.5 ± 2.44 nm. It showed a high drug entrapment of 98.7 ± 4.01% and an in vitro drug release of 89.51 ± 2.94%. With a Cmax value of 193.41 ± 26.4 ng mL−1 and a Tmax value of 2 hours, the drug's significant therapeutic concentration in the CNS following intranasal (IN) administration was demonstrated by in vivo single-dose pharmacokinetic investigation. The Drug Targeting Efficiency (DTE) of 213.123% and the Drug Targeting Potential (DTP) of 66.27% were greater for the constructed DPZ in situ gel, indicating superior brain targeting efficiency through NLCs. The outcomes showed that as compared to the neurodegeneration control group, the DPZ in situ gel treatment group dramatically reduced the escape latency and path length. The DPZ in situ gel demonstrated superior anti-AD potency to DPZ-sol, as revealed by biochemical and histological investigations. Its potential for managing AD is suggested by the favorable outcomes of the developed and enhanced intranasal DPZ in situ gel.
1. Introduction
The prevalence of neurodegenerative diseases has increased worldwide. Neurodegenerative disorders include Parkinson's disease, Alzheimer's disease (AD), frontal lobe dementia, spinocerebellar ataxias, Huntington's disease, and amyotrophic lateral sclerosis.1 AD is one of the most prevalent neurodegenerative illnesses brought on by dementia.2 Globally, 50 million people suffer from dementia, according to the Alzheimer's International Report 2022.3 Neurons in the brain deteriorate as a result of neurodegenerative diseases like Alzheimer's disease (AD). Loss of neurons, synapses, and abnormal protein aggregation formation are all associated with it.4 A cholinergic deficiency (cholinesterase), which is associated with memory loss and cognitive dysfunction, is one of the most obvious pathogenic markers of AD. Free radical production is a major contributing part, according to another hypothesis about the genesis of AD. The reversible cholinesterase inhibitor is shown to have high selectivity for centrally activated cholinesterase.5,6 Traditional FDA-approved Alzheimer's disease medicines include acetylcholinesterase inhibitors (AchE) and N-methyl-D-aspartate (NMDA) receptor antagonists, which reduce symptoms but have no impact on the disease progress. For the treatment of mild to severe Alzheimer's-related dementia, the FDA has approved donepezil (DPZ), as one of the most widely used AchE inhibitors.7For oral delivery, DPZ is presently available in the market as tablets or capsules (5 or 10 mg day−1). A nonlinear, non-competitive mechanism is used by DPZ to inhibit acetylcholinesterase. In cholinergic synapses, DPZ increases the level of acetylcholine.3 Anorexia, diarrhea, nausea, and stomach bleeding have been reported as side effects of DPZ, even though it showed adequate oral absorption.8 Oral administration of DPZ is limited by its hydrophilicity (freely soluble in water), which restricts its access to the brain and produces strong cholinergic side effects. Although it was not as hepatotoxic as its tacrine predecessor, DPZ showed signs of hepatotoxicity and underwent first-pass metabolism, which once again limited its oral distribution. For this reason, a non-oral delivery system needs to be developed to treat AD while preventing/reducing harmful side effects.
Out of the several non-oral methods (parenteral, transdermal, and rectal), the intranasal route seems to be the most secure and non-invasive way to deliver medications to the brain.9,10 This delivery method is directed to the brain via this route, which not only gets around barriers like the blood–brain barrier (BBB) but also prevents drug exposure to the GI tract and minimizes/eliminates side effects. The formulation will cross either the olfactory or trigeminal neural pathways upon nasal administration, revealing a strong connection between the brain and the nasal mucosa.11 It prevents systemic delivery consequently improving absorption and makes site-specific targeting easier.12 Therapeutic carriers for intranasal brain administration have been described by researchers13–15 and some of these carriers include nanostructure lipid carriers,16 liposomes,17 polymeric nanoparticles,18 solid lipid nanoparticles,19 microemulsions,20 nanobilosomes,21 and in situ gels.22 For the delivery of DPZ, intranasal delivery systems of microemulsion,23 liposomes,24 polymeric nanoparticles,25in situ gels,26 and other delivery methods have all been reported.
Nasal administration has been seen to have minimal residence time in the nasal cavity and a shorter mucociliary clearance time (15–20 minutes), which results in reduced absorption of drugs.9 Consequently, mucoadhesive characteristics are needed for delivery methods to avoid drug removal together with mucus. Due to its long-lasting adhesive properties, mucoadhesive formulations are capable of efficiently delivering medication to the administration site. Among the traditional mucoadhesive dosage forms, in situ gels may be the most successful due to their ease of administration in the liquid form, which turns into gel at the injection site.27 In recent years, in situ nasal gels have gained interest for systemic drug delivery because of pros such as their basic design, ease of use, enhanced physiochemical stability, simplicity of employment, exact dosage, extended nasal residence time, significant bioavailability, sustained drug release, and potentially improved therapeutic efficacy.28 Therefore, to improve DPZ's potential to reach the brain, it is necessary to formulate a formulation that strengthens its biopharmaceutical properties. To reduce how quickly the drug exits from the nasal cavity and expand its duration there, the DPZ-NLCs were disseminated into an in situ gel that contains a gelling agent.
Carrier molecules may be used to facilitate therapeutic transit over the mucosal membrane in addition to aiding nasal retention. Due to their favorable permeability through the lipophilic nasal membrane and biological properties, lipid-based formulations have attracted a lot of interest. The next generation of lipid nanoparticles, like NLCs, improves the chemical stability of molecules that are susceptible to light, hydrolysis, and oxidation.29 Furthermore, the liquid lipid content of the NLC leads to flaws in the lipid matrix. Less drug intake and greater drug loading result from this. Compared to polymeric nanoparticles, NLCs are safer, more stable, more reproducible, have a higher trapping efficiency than solid lipid nanoparticles, and offer specified and prolonged drug release.30 Additionally, the amount of degradants that accumulate in the brain is minimal because the majority of lipids utilized to create lipid nanoparticles are physiologically compatible and biodegradable at developing concentrations.
In light of the above, the goal of the current study was to create an in situ donepezil gel that was packed into a nanostructured lipid carrier for enhanced intranasal delivery of DPZ to the brain. With the use of design expert software and the Box–Behnken design (BBD), NLCs were constructed via the melt-emulsification high-pressure homogenization process. Gellan gum and Lutrol F-127, two gelling agents, were mixed with NLCs of DPZ to develop an in situ gel system. Particle size, entrapment efficiency, zeta potential, and PDI were evaluated as physiochemical parameters of the formulations. In addition to in vitro drug release studies, an ex vivo permeability study was performed on the optimized formulation. By avoiding the use of organic solvents, the drug's delivery system minimizes toxicity or unfavorable effects and ensures safety. It is noteworthy that following intranasal administration of an optimized formulation, pharmacokinetic parameters like maximum drug concentration (Cmax), time to reach Cmax (Tmax), area under curve (AUC), and neuro-pharmacokinetic parameters including Drug targeting index (DTI), drug targeting potential (DTP), and drug targeting efficiency (DTE) were examined. The drug, lipids, and other excipients are all crucial components that come from natural sources. All the essential components, such as drugs, lipids, and other excipients, originate from natural sources. By omitting organic solvents, the drug delivery technology reduces toxicity or unfavorable effects, ensuring safety.
2. Materials and methods
2.1. Materials
A complimentary sample of DPZ was offered by Cipla Ltd, Mumbai (India). Gattefosse Pvt. Ltd, Mumbai (India), provided Transcutol, Compritol 88 ATO, Precirol ATO 5, Capryol 90, GMS, and Labrafil M1994. From Sigma-Aldrich (St. Louis, MO, USA) and BASF, India, Poloxamer 188, polyethylene glycol, propylene glycol, tween 80, gellan gum, and Lutrol F-127 (Poloxamer 407) were obtained. From Lipoid GmbH (Ludwigshafen, Germany), phosphate 50PG and phosphate 53MCT were received. Capmul oil was provided by the Abitec Corporation, Mumbai (India). All of the substances used in the study were analytical-grade chemicals and solvents. All of the solvents used in HPLC were of HPLC grade, and the water used in the process was purified using a Millipore Milli-Q system (Millipore Corporation, Burlington, MA, USA).2.2. Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 methanol:water (v/v) solution. The solubility of DPZ was then determined by measuring it at 268 nm with a UV-Visible spectrophotometer (V-630, Jasco, Japan).
:50 methanol:water (v/v) solution. The solubility of DPZ was then determined by measuring it at 268 nm with a UV-Visible spectrophotometer (V-630, Jasco, Japan).
According to existing research and formulation aspects, the Box–Behnken design is regarded as the most suitable method for evaluating a second-order polynomial model with a quadratic response surface. In the fewest number of trials possible, this approach delivers process optimization with just 17 runs total, which includes 5 repeated center points. The following computer-generated non-linear, polynomial model quadratic equation describes the three-factor, three-level design.
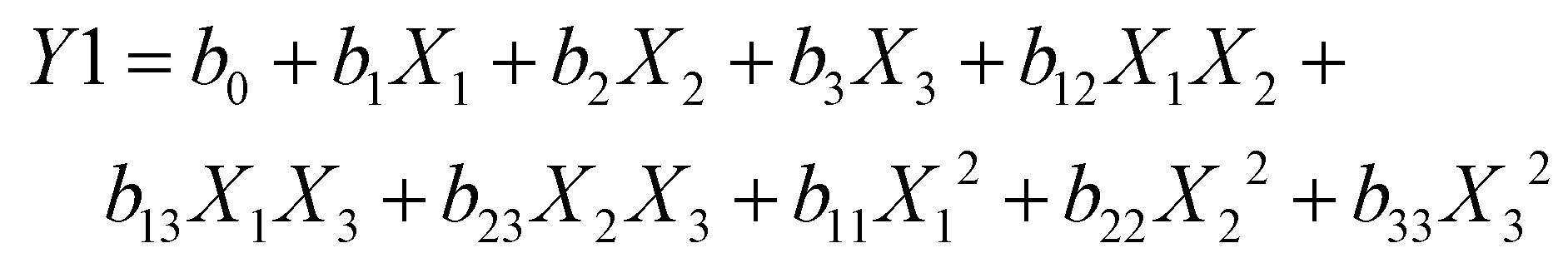
The objective of the design was to determine the relationship between the independent factors (results), which include the solid lipid:liquid lipid ratio (A,%w/w), surfactant (B,%w/v), and HPH cycle (C), and the responses, that comprise the particle size (Y1), drug loading (Y2), and entrapment efficiency (Y3), all of which are listed in Table 1. The three alternative levels low (−1), medium (0), and high (+1) were utilized to assess variables (A), (B), and (C). The Design Expert software (version 13.0.11.0) was used to transfer data using multiple batches that were prepared. Following an evaluation of the coefficient's strength and mathematical sign (positive or negative), the generated polynomial equation was utilized to get the result.
| Variables | Levels | ||
|---|---|---|---|
| Independent variables | −1 | 0 | 1 |
| A = Solid lipid: liquid lipid ratio | 60:40 |
70:30 |
80:20 |
| B = Surfactant (%w/v) | 0.5 | 1 | 1.5 |
| C = HPH cycle | 5 | 10 | 15 |
| Dependent variables | |||
| Y1 = Particle size (nm) | Minimize | ||
| Y2 = Drug loading (%) | Maximize | ||
| Y3 = Entrapment efficiency (%) | Maximize | ||
:25, a surfactant concentration of 0.9%, and a 12 HPH cycle. The results were once more analyzed after the preparation of this new suggested combination formulation.32 (Table S2 and a detailed description of point prediction have been provided in the ESI.†)
2.2.6.1. Particle size, zeta potential, and morphological study. Using a nanoparticle analyzer (SZ-100, Horiba Scientific, Japan), the average particle size, PDI, and zeta potential were measured. The NLC dispersion samples were measured using the light scattering method at 25 ± 2 °C at a 90-degree angle after being diluted ten times with distilled water and placed in a quartz cuvette. Transmission electron microscopy with a 100 kV accelerating voltage (TEM, JEM-1200EX JEOL, Tokyo, Japan) was used to examine the morphological characteristics of optimized NLCs. A droplet of the sample was placed on a copper grid, any excess fluid was drained off, the sample was dried under an infrared lamp, and it was then studied under a microscope.
2.2.6.2. Determination of % drug loading and % entrapment efficiency. The entrapment efficiency of NLCs is calculated by examining the unentrapped drug, which indicates the percentage of total drugs entrapped in NLCs to the total drugs used in their development. The supernatant was produced by centrifuging a specific volume (5 ml) of optimal formulation dispersion at 18
000 rpm for around 20 minutes at 20 °C using a centrifuge (5424R, Eppendorf AG, Germany).19 Afterwards, the supernatant was subjected to an HPLC analysis once passing through a syringe filter (0.45 μm). A UV detector active at 273 nm was used for the detection.
2.2.6.3. Differential scanning calorimetry (DSC) analysis. A DSC (Mettler DSC 822E, Toledo, and Giessen, Germany) was then used to record the thermograms of pure DPZ, Precirol ATO5, Transcutol, Poloxamer 188, and lyophilized optimized NLCs under a nitrogen environment using a STARe SW 16.20 programmer. A reference was an aluminum pan that was sealed and left empty. The 5 mg sample was heated in an inert environment at a rate of 10 °C min−1 from 20 °C to 300 °C which was packed in a perforated aluminum pan.
2.2.6.4. X-ray powder diffraction analysis (XRPD). An X-ray diffractometer (PW 1729, Philips, Netherlands) that operated at a voltage of 40 kV and a current of 40 mA was used to record the XRPD patterns of pure DPZ, lipids, and lyophilized optimized NLCs. At two theta inclinations ranging from 10 to 90°, the samples were subjected to Cu-Kα irradiation.
000 to 14000 kDa. Before being tied at both ends, 1 ml of each DPZ solution (DPZ-sol.) and optimized NLCs (equivalent to 5 mg of DPZ) were specifically inserted in a dialysis bag. The packed dialysis bag was then immersed in 100 mL saline phosphate buffer (pH 7.4, matching the CSF pH) and shaken at 37 ± 0.5 °C at a constant speed of 50 rpm. The receiver compartment's aliquots (2 mL) were taken out and replaced with the new medium at equal intervals to maintain the sink condition, which lasted up to 24 hours. To assess the concentration of released DPZ, a developed and validated High-Performance Liquid Chromatography (HPLC) method was employed. Various models were used to investigate the release kinetics using the model-fitting method. Using the maximum feasible value for the determination of coefficient (R2), the best possible kinetic model was identified (Table S3†).
2.2.8.1. Characterization of the in situ gel. Physical appearance and pH. The gel's appearance, color, and homogeneity were physically assessed. The pH of the gel was determined using a pH meter (Systronics 754, India).
Critical ionic concentration (CIC) for the gellan gum phase transition. In test tubes, 1 mL of the in situ gel was combined with various quantities of simulated nasal fluid (1.29 mg mL−1 KCl, 7.45 mg mL−1 NaCl, and 0.32 mg mL−1 CaCl2·2H2O) to determine the CIC.35 Test tubes were then turned upside down to check the gel production. The CIC is the minimal volume of simulated nasal fluid needed to produce a gel.36
Gelling time. By adding 1 mL of the gel with a certain volume of simulated nasal fluid, the in situ gel's formation time was measured.
Viscosity. A Brookfield Viscometer (HADV-II, Brookfield Engineering Labs, Inc., MA, USA) was used to measure the viscosity of the in situ gel both before and after gelation. Viscosity measurements were made in triplicate using spindle numbers 18 and 96 at 25 °C in the sol and gel phases at a speed of 10 rpm for 10 seconds.
Expansion coefficient (S%). The volume was calculated by using 1 mL of the generated in situ gel and 0.25 mL of simulated nasal fluid in a calibrated cylinder with a total volume of 1.25 mL (V1). After the addition of 2 mL of nasal fluid, the total volume (VT) of the gel was measured. The gel volume (VG = VT-2) was considered to calculate the expansion coefficient.35
Mucoadhesion testing. In situ gels’ mucoadhesive strength was assessed using a texture analyzer (Brookfield Engineering Labs, Inc., MA, USA, Texture Pro CTV 1.3 Build 14) with a mucoadhesive holder. 10 mm × 10 mm of goat nasal mucosa were allowed to acclimatize in 20 mL of simulated nasal fluid for 10 minutes at 32 ± 2 °C. In situ gel (20 mg) was applied to the mucoadhesive holder. At a pace of 0.5 mm s−1, the probe moved downward until it reached the membrane. The probe was retracted to a distance of 20 mm at a rate of 0.5 mm s−1 after 30 seconds of 1 N contact force. The Texture Pro CTV1.3 Build 14 software can be used to directly record the maximum detachment force (Fmax in grams).36
Drug content determination. The drug content was determined by dispersing 1 mL of the in situ gel in 20 mL of distilled water with continuous shaking until the in situ gel dissolved completely. The volume was adjusted to 50 mL using distilled water and the concentration of the drug was determined by UV analysis.38
Nasal ciliotoxicity studies. The local butchery provided the freshly excised goat nasal membrane tissue, which was used in research on nasal ciliotoxicity. After cleaning the membrane segment with saline solutions, it was separately treated with optimized in situ gel and phosphate buffer at pH 6.4 (as a negative control). The tissue samples were rinsed with distilled water and stained with hematoxylin and eosin after 8 hours. To find out if any changes occurred to the membrane that had been treated, the slices were looked under an optical microscope (Olympus, CX21FS1, Japan).22
2.2.10.1. Selection of animals and acclimatization. The Poona College of Pharmacy's Institutional Animal Ethics Committee (IAEC) authorized an experimental protocol based on the recommendations of the Committee for the purpose of control and supervision of experimental Animals (IAEC/PCP/PCT07/2021-2022). Male Wistar rats weighing 200–250 g were bought from the National Institute of Biosciences for the in vivo experiments. The animals were given a week to acclimate to the experiment's temperature and humidity conditions before the experiment. Throughout the study, the animals were kept in group housing in plastic cages that maintained constant temperatures (24 ± 1 °C), relative humidity (55 ± 10%), and 12 hour light/dark cycles. Rats were given regular pelleted food from Pranav agro-industries (Sangli, Maharashtra, India), as well as filtered water; however, they received nothing the night before the trial. Throughout this acclimation period, the animal's health was periodically examined.
2.2.10.2. Animal grouping and administration of medication. Four groups of eighteen animals each were formed from all of the animals. There are six sampling points for each group, with three animals per sample point. The DPZ drug solution and DPZ in situ gel containing 0.20 mg of DPZ, were given to groups I and II; a micropipette was used to administer the medication intranasally (i.n.) in each nostril (20 μL). The DPZ drug solution through the tail vein was given to Group III, and DPZ-NLC dispersion orally was given to Group IV, which is comparable to 0.20 mg of DPZ respectively. Before the administration of the formulation, the animals were anesthetized using pentobarbital sodium (35–50 mg kg−1) intraperitoneally.
2.2.10.3. Blood collection, separation, and processing procedure for other organs. Pentobarbital sodium overdoses were used to mercifully kill the rats at intervals of 0.5, 1, 2, 4, 6, and 12 hours. Blood was then collected via heart punctures into Eppendorf tubes coated with EDTA. After centrifuging the blood at 5000 rpm for 15 minutes, the drugs were analyzed using an HPLC bioanalytical method that has been developed and validated.37 Blood was drawn from rats at the same time that the organs (intestine, kidneys, liver, lungs, and spleen) were slaughtered. To remove any sticking tissue or fluid, each organ was cleansed twice with normal saline before being weighed. Each organ was kept in a cold, homogenized normal saline solution diluted 1
:5 by its weight in normal saline, after being homogenized on ice. Separate treatment was given to each organ. Each organ was homogenized and centrifuged for 15 minutes at 5000 rpm at 4 °C. A portion of the supernatant was taken and stored at −21 °C until HPLC screening.
2.2.10.4. HPLC chromatographic method. Chromatographic analysis was performed on a Jasco HPLC 4000, LC-Net II/ADC HPLC system with a UV-4075 UV/Vis detector and a Rheodyne injector (7725i) with a PU-4180 RHPLC pump using the ChromNav software version (2.02.05). Prior to the drug's HPLC quantification, the method was established and validated. The Hypersil gold C-18 column (250 mm × 4.6 mm, 5 μm) was utilized for evaluation and segregation. The mobile phase in an isocratic elution mode was composed of methanol, Milli-Q water (pH 2.8; adjusted with 0.05% v/v orthophosphoric acid; 45
:55 v/v), and a few drops of TEA. The flow rate was set at 1.0 mL min−1. The volume of the injection was 20 μL. The eluent was checked for the presence of DPZ throughout a 20-minute runtime at 273 nm. Before usage, the mobile phase was processed for 15 minutes of ultrasonication followed by filtration through a 0.22 μm membrane filter. In the drug concentration range of 20–200 ng mL−1, the method was linear, and the retention time was 6.2 minutes, with a correlation coefficient of 0.9861. The proposed method had a 96–100% accuracy (recovery).
The DPZ was determined by centrifuging 0.5 mL of blood sample for 15 minutes at 5000 rpm (5424R, Eppendorf AG, Germany) to extract the plasma. Plasma was extracted by the liquid–liquid extraction technique to obtain DPZ. After adding 0.8 mL of methanol to 0.2 mL plasma and vortexing for another 10 minutes, and centrifuged for 10 minutes at 5000 rpm to separate the organic layer. At room temperature, the organic layer (methanol) from every tube was dried. The dried organic solvent samples were thoroughly cleaned before being individually reconstituted with mobile phase (100 μL) and subjected to an HPLC analysis to determine the presence of DPZ.
2.2.10.5. Pharmacokinetic and neuro-pharmacokinetic parameter estimation. The pharmacokinetic parameters were calculated using the WinNonlin program 8.3.4.295 from the plots of plasma DPZ concentration vs. time. 8-version graph pad prism was used for the statistical analysis. When computing pharmacokinetic parameters like Cmax, Tmax and AUC with non-compartment modeling was taken into account. DTI and neuropharmacokinetic characteristics, such as %DTE and %DTP were studied. The formulation's bioavailability following intranasal administration was compared to that of the DPZ solution.39
2.2.11.1. Morris water maze. The protocol was approved by the Institutional Animal Ethical Committee (IAEC) of Poona College of Pharmacy in Pune, India (IAEC/PCP/PCT06/2021-2022). For the experiment, male Wistar rats weighing 200–250 g were employed. Four groups (n = 8) of rats were created: group 1 served as the control group, group 2 as the neurodegeneration control group, group 3 as the standard DPZ-solution (DPZ-sol.) group, and group 4 as the DPZ in situ gel group. Rat's spatial learning and memory were tested using a radial pool (model number VJMWM-01 R, dimensions: 150 cm in circumference and 45 cm in height). Four different colored visual cues are included in the instrument's four quadrants. These cues remained constant throughout the experiment. The maze water was kept at a temperature of 24 ± 2 °C with a water level of up to 40 cm. There was a secret platform erected 2 cm under the water's surface. Four distinct cues were utilized to train the animals to find the platform in the Morris water maze over five days. Assessments were carried out for the path length (the distance travelled) and escape latency (the time it took to reach the platform). On day 7, 8 mg kg−1 trimethyltin chloride (TMT) was injected intraperitoneally (i.p.) into every rat group except the control group to cause neurodegeneration.40 The appropriate treatment was given from day 14th to day 35th. The Morris Water Maze Test was used to assess the study's spatial memory element on 6th, 14th, and 35th day. On the 36th day, the animals were euthanized to analyze biochemical and histopathology parameters.
2.2.11.2. Biochemical parameters. The animals were kindly slaughtered on the 36th day of the study. Following an isotonic saline rinse, the brain was cut into slices and dried with filter paper.41 For the biochemical tests, the right brain was utilized whereas the left brain was used for histopathology which was preserved in 10% neutral buffer formalin. The brain was homogenized using the established technique in a Tris–HCl buffer 7.4 pH.42 The brain samples were examined for acetylcholine esterase (AchEs),43 malondialdehyde (MDA),44 nitric oxide (NO),45 glutathione S-transferase (GSH)46 and catalase45 were all tested in the brain samples.
2.2.11.3. Histopathological evaluation. The hippocampus was removed from the brain tissues and stored for 72 hours at room temperature in 10% neutral buffer formalin. The samples were divided into 5 μm pieces with a microtome and put in paraffin molds. The sections were deparaffinized, rehydrated in a succession of gradient alcohols, and then stained with hematoxylin and eosin (H&E).47 At a 10× magnification, the alterations that took place in the rat brain were compared to those in the vehicle control group.
2.2.11.4. Statistical evaluation. The statistical study was completed using Graph pad prism demo V8. Two-way Analysis of variance (ANOVA) is followed by Bonferroni's multiple comparison tests. Every value is displayed as mean ± SD. A statistical comparison was carried out at P < 0.05.
3. Results
3.1. Preliminary screening for excipient and experimental parameter selection
The most suitable excipient and experimental parameters, comprising lipids (solid and liquid), surfactant concentration, solvent, stirring speed (rpm), stirring duration, and sonication time (min), were determined by an initial screening study. DPZ showed maximum solubility in Precirol ATO5 of all the solid lipids examined. Transcutol HP showed the highest DPZ solubility among the liquid lipids evaluated. Preciprol ATO 5 and Transcutol HP (as a solid lipid and liquid lipid) were selected for the DPZ NLC formation as they showed excellent physical compatibility with no phase separation. To assess physical compatibility, stability, and drug loss, lots of ratios amongst solid and liquid lipids were investigated. Regarding encapsulation efficiency and stability, a 75:25 mixture of Precirol ATO5 and Transcutol HP was discovered to be ideal.
3.2. Drug-excipients compatibility study
Using an FTIR spectrophotometer, the drugs, lipids, and surfactant compatibility were assessed. The FTIR spectrum of DPZ is shown in Fig. 1, which exhibited the characteristic DPZ peaks. The bands at 3588 cm−1, 3373.8 cm−1 and 3265.86 cm−1 (OH stretch), 2937 cm−1 (aliphatic CH2 stretch), 1683.5 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O carbonyl stretch), and 1453 cm−1 (aromatic CC stretches), and 1118 (CH bending) were all present in the pure drug DPZ (Fig. 1a).19Fig. 1b, c and d describe the spectra for precirol, transcutol and poloxamer, respectively. The optimized NLCs (Fig. 1e) and DPZ loaded in situ gel (Fig. 1f) both exhibit the DPZ's characteristic bond stretching peak. No peak was found to be absent from the in situ gel spectra, with a few minor peak shifts, and displayed identifiable peaks in positions that matched those of the individual peaks. In the optimized formulation, as the drug's peak intensity decreases (Fig. 1e), it is probable that the drug was disseminated in the melting lipid. Peaks were observed at 3568.6 cm−1, 3363.25 cm−1, 3270.68 cm−1 (OH stretch), 2864.74 cm−1 (CH stretch), 1729.83 cm−1 (CO carbonyl stretch), 1453 cm−1 and 1121.12 cm−1 (aromatic CC stretch and CH bending) in the optimized NLC formulation. The bands at 3833.79 cm−1, 3731.58 cm−1, 3589.84 cm−1, 3356.5 cm−1 (aromatic OH stretch), 2937.06 cm−1, 2527.26 cm−1 (aliphatic CH stretch), 2019.1 cm−1, 1684.52 cm−1, 1603.2 cm−1, 1505.17 cm−1, 1315.21 cm−1 and 1202.04 cm−1 for the CO carbonyl stretch, aromatic CC stretch and CH bending were all present in the in situ gel spectra, respectively (Fig. 1f). In the FTIR spectrum, these specific drug peaks could also be seen in optimized NLCs and a physical blend of DPZ. The findings demonstrated that the drug, lipid, and surfactant did not interact chemically.
O carbonyl stretch), and 1453 cm−1 (aromatic CC stretches), and 1118 (CH bending) were all present in the pure drug DPZ (Fig. 1a).19Fig. 1b, c and d describe the spectra for precirol, transcutol and poloxamer, respectively. The optimized NLCs (Fig. 1e) and DPZ loaded in situ gel (Fig. 1f) both exhibit the DPZ's characteristic bond stretching peak. No peak was found to be absent from the in situ gel spectra, with a few minor peak shifts, and displayed identifiable peaks in positions that matched those of the individual peaks. In the optimized formulation, as the drug's peak intensity decreases (Fig. 1e), it is probable that the drug was disseminated in the melting lipid. Peaks were observed at 3568.6 cm−1, 3363.25 cm−1, 3270.68 cm−1 (OH stretch), 2864.74 cm−1 (CH stretch), 1729.83 cm−1 (CO carbonyl stretch), 1453 cm−1 and 1121.12 cm−1 (aromatic CC stretch and CH bending) in the optimized NLC formulation. The bands at 3833.79 cm−1, 3731.58 cm−1, 3589.84 cm−1, 3356.5 cm−1 (aromatic OH stretch), 2937.06 cm−1, 2527.26 cm−1 (aliphatic CH stretch), 2019.1 cm−1, 1684.52 cm−1, 1603.2 cm−1, 1505.17 cm−1, 1315.21 cm−1 and 1202.04 cm−1 for the CO carbonyl stretch, aromatic CC stretch and CH bending were all present in the in situ gel spectra, respectively (Fig. 1f). In the FTIR spectrum, these specific drug peaks could also be seen in optimized NLCs and a physical blend of DPZ. The findings demonstrated that the drug, lipid, and surfactant did not interact chemically.
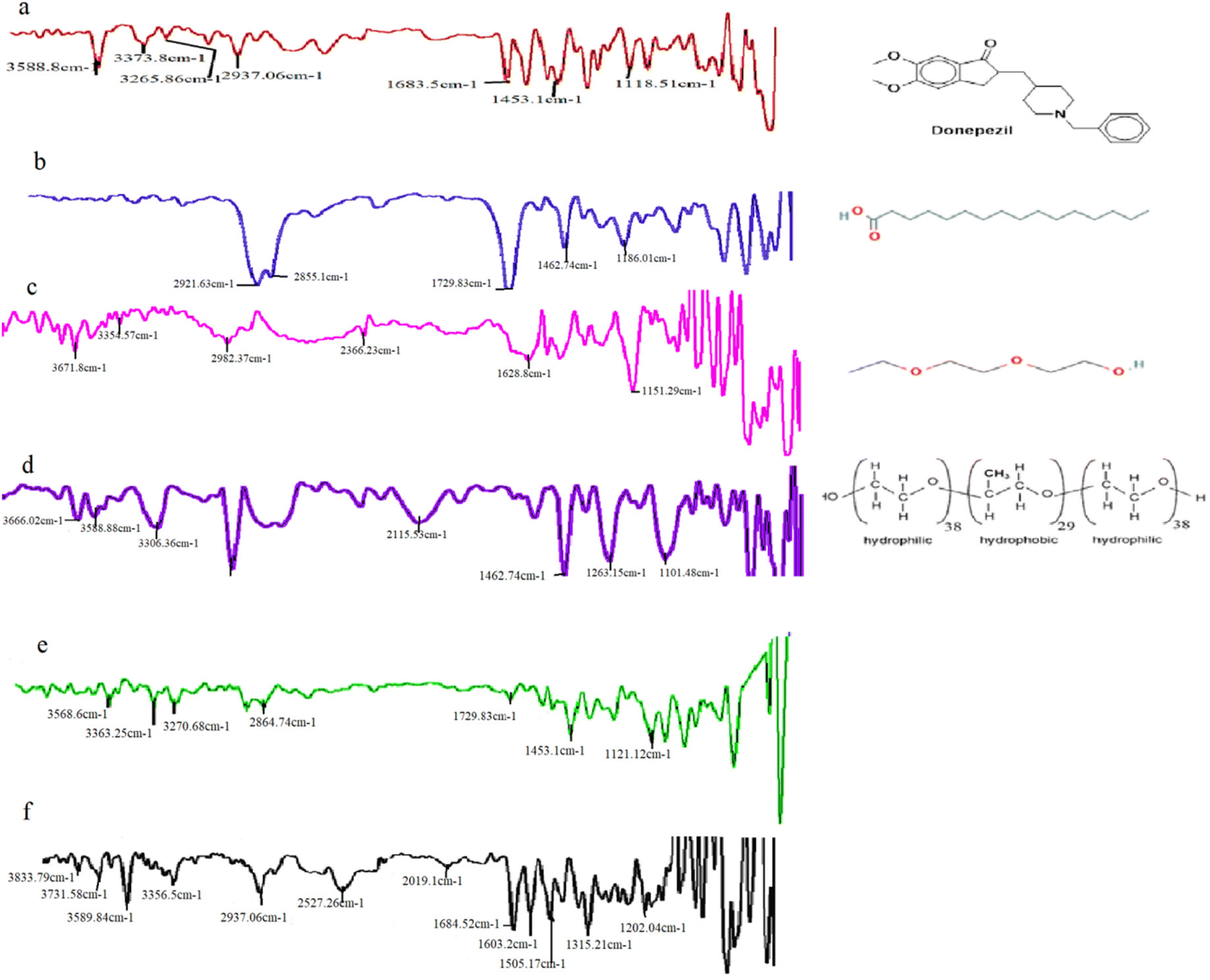 | ||
| Fig. 1 FTIR spectra of plain drug (a) Donepezil; (b) Precirol; (c) Transcutol; (d) Poloxamer; (e) optimized NLCs; and (f) in situ gel. | ||
3.3. Utilization of Box–Behnken design, preparation, and optimization DPZ filled NLCs
The melt emulsification-high pressure homogenization technique was adopted to develop DPZ-loaded NLCs. In Table 2, the findings of the preliminary and pre-optimization batches are used to list three levels of each independent variable (factor). Three components (factor) and three levels were chosen to be accomplished via the Box–Behnken design. Five center points were in the 17 runs that were obtained. Each of these batches’ responses such as particle size (Y1), drug loading (Y2), and percentage of drug entrapment efficiency (Y3) was evaluated. To obtain the projected values and outcome, each outcome was entered into the Box–Behnken design function of the Design Expert software (version 13.0.11.0).| Batch code | Independent constraints | Dependent constraints | |||||||
|---|---|---|---|---|---|---|---|---|---|
| SL:LL (A) |
Surf (%) (B) | HPH cycle (C) | Particle size (nm) Y1 | Drug Loading (%) Y2 | Entrapment efficiency (%) Y3 | ||||
| Actual | Predicted | Actual | Predicted | Actual | Predicted | ||||
| DPZ-NLC1 | 80:20 |
1 | 5 | 178.0 ± 2.05 | 152.5 | 37.3 ± 1.35 | 42.76 | 97.2 ± 3.0 | 98.10 |
| DPZ-NLC2 | 80:20 |
0.5 | 10 | 119.0 ± 8.04 | 144.4 | 9.60 ± 0.44 | 9.60 | 87.0 ± 3.3 | 87.00 |
| DPZ-NLC3 | 60:40 |
1 | 5 | 175.0 ± 4.08 | 200.4 | 12.5 ± 0.28 | 17.88 | 88.0 ± 4.9 | 88.90 |
| DPZ-NLC4 | 60:40 |
1 | 15 | 141.0 ± 6.94 | 166.4 | 40.2 ± 2.00 | 34.89 | 97.4 ± 2.9 | 96.50 |
| DPZ-NLC5 | 70:30 |
1 | 10 | 123.0 ± 2.05 | 123.0 | 57.9 ± 2.02 | 57.99 | 98.5 ± 3.1 | 98.50 |
| DPZ-NLC6 | 80:20 |
1.5 | 10 | 134.5 ± 1.31 | 159.9 | 34.5 ± 1.79 | 34.53 | 95.0 ± 3.1 | 95.00 |
| DPZ-NLC7 | 70:30 |
1.5 | 15 | 166.0 ± 4.10 | 166.0 | 39.5 ± 0.23 | 44.96 | 95.0 ± 7.5 | 95.90 |
| DPZ-NLC8 | 70:30 |
0.5 | 15 | 143.0 ± 4.18 | 134.0 | 9.80 ± 0.67 | 15.18 | 78.9 ± 3.5 | 79.80 |
| DPZ-NLC9 | 70:30 |
1 | 10 | 123.0 ± 2.05 | 123.0 | 57.9 ± 2.04 | 57.99 | 98.5 ± 3.5 | 98.50 |
| DPZ-NLC10 | 80:20 |
1 | 15 | 276.0 ± 2.44 | 250.5 | 52.2 ± 0.96 | 46.89 | 97.7 ± 4.7 | 96.80 |
| DPZ-NLC11 | 70:30 |
0.5 | 5 | 680.0 ± 8.49 | 680.0 | 40.5 ± 1.47 | 35.15 | 97.2 ± 4.5 | 96.30 |
| DPZ-NLC12 | 70:30 |
1.5 | 5 | 387.0 ± 4.18 | 387.0 | 9.20 ± 0.71 | 3.82 | 74.0 ± 1.8 | 73.10 |
| DPZ-NLC13 | 60:40 |
1.5 | 10 | 141.4 ± 2.85 | 115.9 | 51.2 ± 2.18 | 51.25 | 98.0 ± 3.0 | 98.00 |
| DPZ-NLC14 | 60:40 |
0.5 | 10 | 178.0 ± 3.39 | 152.5 | 41.6 ± 1.05 | 41.63 | 93.8 ± 2.6 | 93.80 |
| DPZ-NLC15 | 70:30 |
1 | 10 | 123.0 ± 2.05 | 123.0 | 57.9 ± 3.12 | 57.99 | 98.5 ± 4.0 | 98.50 |
| DPZ-NLC16 | 70:30 |
1 | 10 | 123.0 ± 2.05 | 123.0 | 57.9 ± 3.12 | 57.99 | 98.5 ± 4.0 | 98.50 |
| DPZ-NLC17 | 70:30 |
1 | 10 | 123.0 ± 2.05 | 123.0 | 57.9 ± 3.12 | 57.99 | 98.5 ± 4.0 | 98.50 |
 | (1) |
The model's F-value of 16.47 suggested that it was significant (p < 0.0001). It was significant (ANOVA, p = 0.0206), as indicated by an insignificant lack of fit. The significant model terms in eqn (1), were B, C, BC, B2, and C2. However negative coefficients of the components B, C, and A2 showed an antagonistic influence on particle size, while positive coefficients of the factors A, AB, AC, BC, B2, and C2 showed a synergistic effect on the particle size. The Adeq precision of 22.4811 indicates that the signal was adequate. So, the design space was navigated using this model.
Three-dimensional plots in Fig. 2a show the effects of different formulation parameters on particle size (Y1). The diameters of the particles for DPZ-NLC formulations with various independent parameters ranged from 680.00 ± 8.49 nm to 119.00 ± 8.04 nm. 3D graph and polynomial equation showed that lipids (A) had a favorable influence on particle size (Fig. 2a). It had a negative association with the surfactant concentration (B). Furthermore, no substantial changes were observed in the HPH cycle (C).
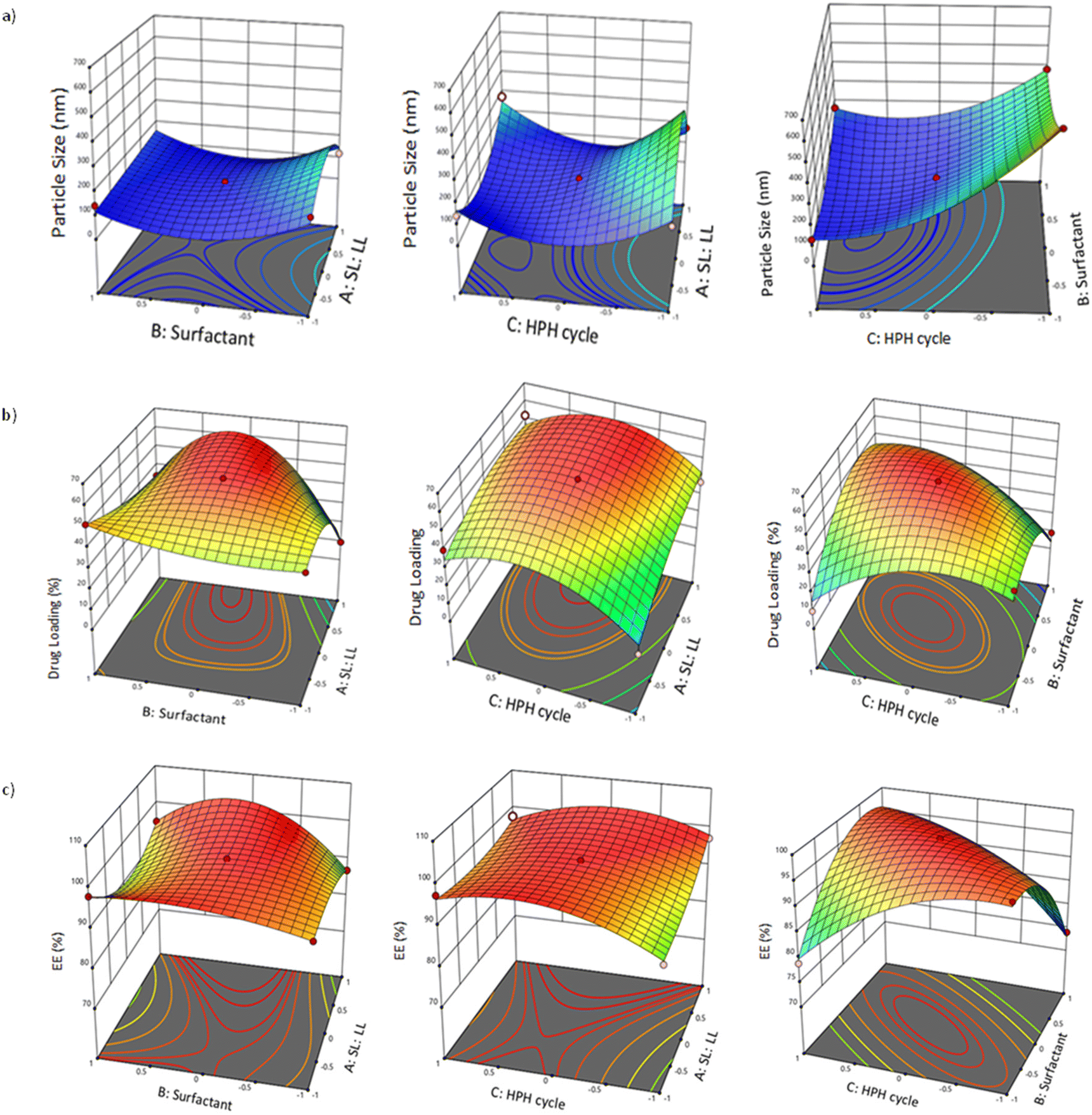 | ||
| Fig. 2 (a) Particle size; (b) drug loading; and (c) entrapment efficiency are the three responses that are represented by a response surface plot with the independent variables. | ||
 | (2) |
The model had a high F-value of 15.25, which implied model significance with ANOVA (p = 0.0504). It was insignificant because of the poor fit. The important model terms in this predicament were BC, B2, and C2. Positive coefficients for A, C, AB, and BC in eqn (2), on the other hand, suggested a synergistic effect, whereas negative coefficients for B, AC, A2, B2, and C2 on drug loading indicated an antagonistic effect. With an Adeq precision of 6.89, a sufficient signal was proposed. Between 9.20 ± 0.71% and 57.99 ± 2.02% was the average drug loading of the DPZ-NLC formulations with various independent variables. For the % drug loading, Fig. 2b shows the 3D surface response graphs.
 | (3) |
The model's F-value of 34.32 indicated that it was significant (p < 0.0001). The significant (p = 0.041) value of the model revealed by the lack of fit is insignificant. A, BC, B2, C2, A2B, and AB2 are essential and effective variables. Positive coefficients for A, C, AB, BC, and A2 were used in eqn (3) to depict the synergistic impact, whereas negative coefficients for B, AC, B2, and C2 were used to demonstrate the antagonistic effect. The center point values of interaction terms AB and BC revealed the maximum level of drug entrapment. The Adeq accuracy of 19.32 suggested an adequate signal. In order to negotiate the design space, this model was used.
Fig. 2c displays the 3D surface response graphs for the percentage of entrapment efficiency. With DPZ-NLC formulations, the average Entrapment efficiency of the various independent variables ranged from 98.50 ± 3.17% to 74.00 ± 1.88%. High drug Entrapment efficiency is necessary to maintain the high concentration of therapeutic drugs at the site of action. To keep a high therapeutic drug concentration at the site of action, more significant entrapment efficiency is required.
3.4. Data optimization and validation of response surface
The independent variables were refined using point prediction to obtain the optimal loading capacity, entrapment efficiency, and particle size. Plots of the predicted versus actual response and residual versus predicted plots are used as diagnostic tools to identify process control. The measured values appeared to be within the acceptable range, as shown in Fig. 3, indicating that all formulation and process variables were closely within control. | ||
| Fig. 3 Plots illustrating the responses for particle size, drug loading, and entrapment efficiency are displayed in the process diagnostics between residual versus predicted (a, b, and c) and predicted versus actual (d, e, and f). All of the above responses were consistent with the range of independent process factors. | ||
3.5. Evaluation of optimized formulation
 | ||
| Fig. 4 TEM image of optimized formulation DPZ-NLCs. | ||
 | ||
| Fig. 5 (a) DSC spectra of plain drug Donepezil (a), Precirol (b), Transcutol (c), Poloxamer (d), and optimized NLCs (e). (b) XRD spectra of plain drug Donepezil (a), Precirol (b), Poloxamer (c), and optimized NLCs (d). | ||
 | ||
| Fig. 6 (a) In vitro drug release in phosphate-buffered saline pH 7.4 of optimized DPZ-NLC and DPZ sol. (b) Ex vivo permeation investigations on goat nasal mucosa for DPZ sol-based gel, followed by optimized in situ gel. The data are shown as mean ± SD (n = 3). (c) Study on ciliotoxicity of nasal mucosa after treatment with either (a) optimized in situ gel or (b) phosphate buffer solution with a pH of 6.4. | ||
3.6. In situ nanostructured lipid carrier gel preparation
To improve the ease of application and extend the residence duration inside the nasal canal, a mixture of in situ gelling compounds was added to a gelling system together with the NLC dispersion. Some in situ gelling polymers, including lutrol F127, Carbopol 934 P, Carbopol 924 P, and gellan gum were screened for the development of a reliable gelling system.15 At lower concentrations, carbopol 934 P did not gel, nevertheless at a higher concentration (1%) carbopol 924 P formed an extremely stiff gel, whereas 0.8% of gellan gum produced an excellent gel. At basic pH values, carbopol 934 produces a hard gel as opposed to that in a slightly acidic environment like the nasal cavity (pH 6.4).33 The in situ gel contains a final concentration of DPZ-loaded NLC dispersion (5 mL), a solution of 0.8% w/v gellan gum and 10% w/v Lutrol F127 were mixed.| Model | R 2 | Adjusted R2 | Predicted R2 | SD | P-Value | %CV | Remark |
|---|---|---|---|---|---|---|---|
| Response (Y1) particle size | |||||||
| Linear | 0.2268 | 0.0159 | −0.5784 | 149.77 | 0.3993 | 2.05 | Suggested |
| 2FI | 0.3253 | −0.1807 | −2.2673 | 164.05 | 0.7639 | ||
| Quadratic | 0.6907 | 0.1339 | −3.9494 | 140.51 | 0.0206 | ||
| Response (Y2) drug loading | |||||||
| Linear | 0.0807 | −0.1700 | −0.6836 | 19.77 | 0.8096 | 2.3 | Suggested |
| 2FI | 0.3016 | −0.2222 | −1.5203 | 20.21 | 0.5075 | ||
| Quadratic | 0.7199 | 0.2156 | −3.4822 | 16.19 | 0.0504 | ||
| Response (Y3) entrapment efficiency | |||||||
| Linear | 0.0281 | −0.2369 | −0.9709 | 8.52 | 0.9548 | 1.58 | Suggested |
| 2FI | 0.5264 | 0.1712 | −1.0576 | 6.98 | 0.1082 | ||
| Quadratic | 0.8788 | 0.6607 | −0.9389 | 4.46 | 0.0071 | ||
| pH | Critical ionic concentration (%) | Gelling time (S) | Viscosity (cp) | Expansion coefficient (%) | Mucoadhesive strength (dyne per cm2) | |
|---|---|---|---|---|---|---|
| Before gelation | After gelation | |||||
| Values are represented as mean ± SD (n = 3). | ||||||
| 5.3 ± 0.31 | 0.12 ± 0.06 | 23 ± 4.02 | 2.32 ± 1.22 | 48.27 ± 6.40 | 1.2 ± 0.06 | 2940 ± 72 |
3.7. Study of ex vivo permeation
The flux (Jss), apparent permeability coefficient (Papp), and enhancement ratio were calculated as part of the permeability study evaluation (Fig. 6b). Penetration rates and DPZ concentrations were noticeably greater in the DPZ in situ gel than DPZ-Sol after 12 hours (P < 0.05). Compared to DPZ-Sol (4.06 ± 0.81 μg cm−2 h−1), the Jss of DPZ obtained from the in situ gel was substantially greater (8.87 ± 2.61 μg cm−2 h−1) (P < 0.01). Furthermore, it was found that the permeability coefficient of the DPZ-loaded in situ gel was three times greater (12.04 ± 1.89 × 10−3 cm h−1) than that of the DPZ-Sol (4.18 ± 1.16 × 10−3 cm h−1).3.8. In vivo Pharmacokinetic studies
Drug delivery via the IN route has several advantages over traditional drug delivery methods since it enters the brain through the olfactory pathway. In the present investigation, the amount of DPZ in the brain was assessed after intranasal treatment of DPZ in situ gel and DPZ-sol. as well as (DPZ-sol.) intravenous and oral administration of DPZ-NLCs. The WinNonlin 8.3.4.295 software was used to estimate several pharmacokinetic parameters, as shown in Table 5.| P'Kinetic parameter | Type of formulation with the route of administration | |||||||
|---|---|---|---|---|---|---|---|---|
| DPZ-in situ gel -i.n## | DPZ-sol i.n* | DPZ-sol-i.v | DPZ-NLC-oral | |||||
| Brain | Plasma | Brain | Plasma | Brain | Plasma | Brain | Plasma | |
| Values are mean ± SD, n = 3. ##Relative to i.n. DPZ in situ gel, **p < 0.05 versus DPZ-Sol. i.n., *p < 0.05 versus DPZ-sol i.v., #p < 0.05 versus DPZ-NLC oral. | ||||||||
| C max (ng mL−1) | 193 ± 26.4 | 191 ± 34.3 | 152 ± 21.4 | 230 ± 15.3 | 53 ± 4.47 | 129 ± 6.80 | 101 ± 2.26 | 149 ± 10.30 |
| T max (h) | 2 | 4 | 2 | 2 | 0.5 | 0.5 | 2 | 2 |
| AUC0–24 h (ng h mL−1) | 1239 ± 6.7 | 992.7 ± 20.2 | 600 ± 29.8 | 886.3 ± 10.3 | 379.9 ± 2.9 | 648.5 ± 2.5 | 336.3 ± 2.99 | 632 ± 10.6 |
| AUC0–∞ h (ng h mL−1) | 1329 ± 7.1 | 1228 ± 19.1 | 850.8 ± 29.4 | 992.7 ± 10.7 | 391.4 ± 3.1 | 665.5 ± 2.6 | 346.7 ± 1.9 | 707.5 ± 10.4 |
| AUMC0–24 h (ng h2 mL−1) | 6406 ± 17.8 | 5113.9 ± 247.2 | 2695.5 ± 68.9 | 3206.9 ± 37.3 | 1695.6 ± 9.0 | 2727.5 ± 8.2 | 1110.6 ± 12.3 | 2460.5 ± 36.6 |
| AUMC0–∞ h (ng h2 mL−1) | 7882.6 ± 18.7 | 9860.1 ± 48.1 | 8684.7 ± 69.4 | 5191.5 ± 38.2 | 1864.6 ± 9.5 | 2974.2 ± 8.4 | 1266.4 ± 8.3 | 3884.1 ± 35.0 |
| K e (h−1) | 0.27 ± 0.45. | 0.14 ± 0.5 | 0.08 ± 0.2 | 0.15 ± 0.15 | 0.36 ± 0.38 | 0.39 ± 0.17 | 0.33 ± 0.50 | 0.14 ± 0.36 |
| MRT (h)* | 5.1 ± 0.74 | 5.05 ± 2.6 | 4.48 ± 5.72 | 3.61 ± 1.5 | 4.4 ± 0.3 | 4.2 ± 0.2 | 3.3. ± 0.3 | 3.8 ± 2.3 |
T max values of the brain (2 h) and blood (4 h) are different, which could be explained through the brain's preferred transfer from the nose to the brain following intranasal injection. The Cmax of the DPZ in situ gel (193.41 ± 26.46 ng mL−1) was significantly (p < 0.05) higher when administered intranasally than the DPZ-sol. (152.00 ± 21.45 ng mL−1) was administered intranasally and when DPZ-sol was given orally (101.91 ± 2.260 ng mL−1) and intravenously (53.1 ± 4.47 ng mL−1, respectively). This could be because NLCs can be retained better than DPZ-sol. NLCs may have better retention capacities than DPZ-sol, which could explain this. Intranasally administered DPZ in situ gel was found to have an approximately 1.8-fold higher AUC0−α in the brain compared to DPZ-sol. The study determined that the AUC0−α values of DPZ-sol given intravenously and orally were 3.3 and 3.8 times higher, respectively than those of intranasally administered DPZ sol. The reason for this could be the drug's direct administration to the brain through the olfactory route.
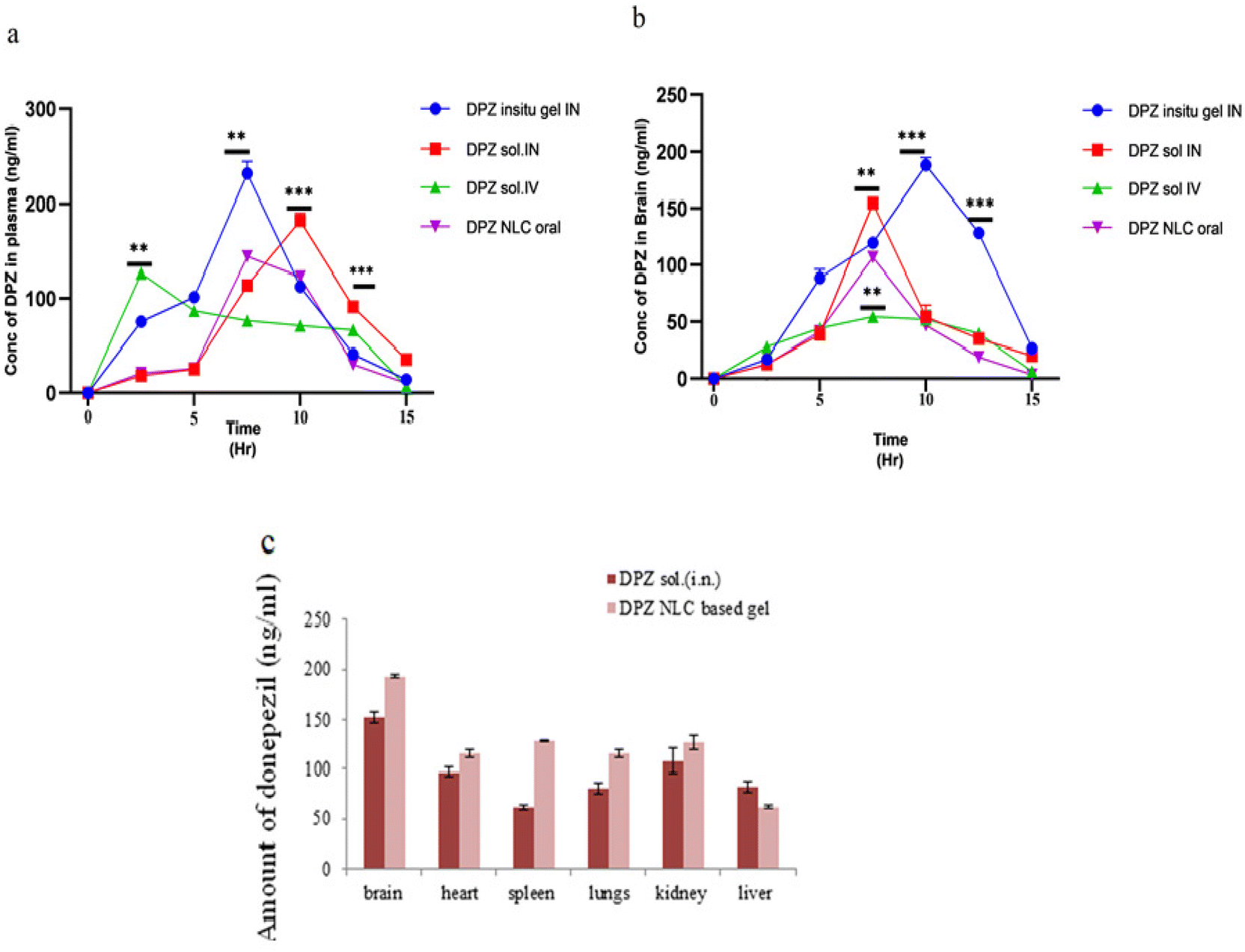 | ||
| Fig. 7 Concentrations of DPZ in the brain (a) and plasma (b) after administration of DPZ- in situ gel i.n., DPZ-sol.i.n., DPZ-sol.i.v., and DPZ-NLC oral (n = 3). (c) Studies on biodistribution using DPZ-sol (i.n.) and DPZ-NLCs based in situ gel (i.n.). | ||
| Formulation/route of administration | Brain/blood ratio at 0.5 h | DTI | DTE (%) | DTP (%) |
|---|---|---|---|---|
| DPZ-in situgel i.n | 1.08 | 2.13 | 213.123 | 66.27 |
| DPZ-Sol i.n | 0.61 | 1.15 | 115.564 | 41.40 |
| DPZ-sol- i.v | 0.67 | — | — | — |
| DPZ-NLC- oral | 0.68 | — | — | — |
3.9. Behavioral, biochemical, and histopathological study of the pharmacodynamic effect
The animal groups treated with DPZ-sol (p < 0.0001) and DPZ in situ gel (p < 0.0001) demonstrated a significant decrease in escape latency (sec) on the 14th and 35th day of the trial when compared to the neurodegeneration control. Compared to the neurodegenerative control group, DPZ in situ gel and DPZ-sol showed a substantial escape latency reduction on day 35th of the study (p < 0.005) (Fig. 8a). The increased residence time in the nasal cavity of the formulation's lipidic NLCs may be the reason for the three times lower escape latency in DPZ in situ gel compared to DPZ-sol. Furthermore, on days 14th and 35th, there was a discernible reduction in path length (cm) compared to the neurodegeneration control group, the DPZ-sol (p < 0.007 and p < 0.0010) and DPZ in situ gel (p < 0.0001 and p < 0.0003) treated groups. The path length travelled on day 35th showed a substantial decline (p < 0.0003) in DPZ-sol (1 mg kg−1) and DPZ in situ gel (1 mg kg−1) (Fig. 8b).
 | ||
| Fig. 8 Morris water maze test: effect of DPZ-sol and DPZ in situ gel on (a) escape latency time (sec) and (b) path length (cm). Two-way ANOVA with Bonferroni's multiple comparison testing after data were expressed as mean ± SEM for n = 8. ###p < 0.001 in comparison to vehicle control, **p < 0.05 and ***p < 0.01 is observed; in comparison to neurodegeneration control. | ||
 | ||
| Fig. 9 The outcomes of DPZ in situ gel on AchE (a), MDA (b), NO (c), GSH (d), and Catalase (e) in the brain. Data (n = 8) are shown as mean ± SEM. Multiple comparison tests using Bonferroni's two-way ANOVA. In contrast to vehicle control ###p < 0.001, **p < 0.01, ***p < 0.001 in comparison with neurodegeneration control. | ||
 | ||
| Fig. 10 Rats with trimethyl tin-induced neurodegeneration: the impact of DPZ-sol and DPZ in situ gel on brain histopathology; H&E stain. (a) The vehicle control group's brain histology reveals undamaged neurons. (b)Neurodegeneration control group brain histopath demonstrating a considerable amount of neurodegeneration (↗). (c) Noteable neurodegeneration (↗) in the brain histology of the donepezil-treated (donepezil-sol) standard group. (d) Brain histology of the group treated with DPZ in situ gel demonstrates substantial avoidance of neurodegeneration (↗). | ||
4. Discussion
Because of their well-established function in brain targeting, non-ionic surfactants were of interest. Since non-ionic surfactants exhibited fewer negative side effects and made it easier for the drug to enter the brain, they were chosen to prepare DPZ-NLC for brain targeting.44 Stabilizers from several categories were evaluated in the concentration range of 0.5–1.5%, including Tween 20, Tween 80, Poloxamer 188, and Cremophor EL. All other stabilizers that cleared the screening, except for Poloxamer 188, produced dispersions with a broad distribution of particle sizes (PDI > 0.6) that became viscous after a day. Poloxamer 188, a non-ionic surfactant, was chosen for NLC development because it offered a narrow particle size range, along with small particles and long-term stability. The components of the final NLC formulation were Precirol ATO 5 (solid lipid), Transcutol HP (liquid lipid/stabilizer), and Poloxamer 188 (surfactant/stabilizer).The DPZ-NLCs were developed and optimized using 33 Box–Behnken design with solid-to-liquid lipid ratio (60:40, 70:30, 80:20), stabilizer concentration (0.5–1.5%), and the high-pressure homogenizer cycle (5–15) acting as an independent variable (Table 1). Particle size (Y1) was selected as the dependent response variable, followed by drug loading (Y2), and drug entrapment efficiency (%) (Y3). First-order, second-order, and quadratic models were all used to fit the responses from these runs, with the quadratic model providing the best fit (p > 0.0001) and least significant fit (p > 0.05). A summary of the analysis of variance and results for the responses for 17 runs of DPZ-NLC is presented in Tables 2 and 3. A variance analysis is also presented for the calculated models for Y1, Y2, and Y3 (Table S1†). As indicated by the polynomial equation, a synergistic effect is indicated by a positive and negative sign revealing an antagonistic effect. Factors of second order represent the nonlinear relationships between the variables and the responses, whereas phrases that contain two factors represent the interactions between the variables and the responses.31 Three three-dimensional plots revealed the interactions between the independent factors and the responses and showed how these plots could be used to analyze the effects of two variables on a single response at a time (Fig. 2).
Particle size (Y1) was predicted to increase in parallel with the percentage of lipids. The initial increase in lipid ratio will give the ideal structure of NLC having a small particle size. In contrast, as the lipid concentration increases further, more solid materials accumulate, which in turn causes the particle size to increase. Both the tendency of lipids at high concentrations to coalesce and the main influence of lipid concentration on the particle size may be explained by a higher dispersion viscosity that results in a slower solute molecule diffusion rate in the outer phase.32 Because Poloxamer 188 reduced the interfacial tension between the lipid and aqueous phases, it affected the particle size. To minimize particle accumulation, poloxamer 188 decreases interfacial tension, and stearic hindrance generated on surfaces of NLCs. As the surfactant concentration crosses a saturation threshold, it results subsequently in the accumulation of more surfactant molecules on the NLC surface, which causes NLCs to increase in size (Fig. 2a). As the HPH cycle's duration increased, the particle size reduced. There is a possibility that increasing the system's energy would have improved the ability of the system to keep particles from aggregating. Particle size was not noticeably affected by the HPH cycle. Very slight differences in the particle size were found as the HPH cycle increased. The interaction factors AB and AC have a synergistic effect on the particle size. The smallest possible particle size was observed by setting all independent parameters to their center values.
The finding showed that there was a comparable increase in drug loading (Y2) when the lipid percentage and HPH cycle increased. Similar observations were made with the early increase in the surfactant and the HPH cycle. The surface response plot exhibits an increase in response followed by a decrease in response with increasing lipid ratio (Fig. 2b). These responses were consistent with how each element affected the effectiveness of trapping. This could be a result of the high energy level of HPH, as it hinders the drugs from losing off the particles which promotes drug loading enhancement. It was shown that in the formulations developed via the center point values of independent components, higher concentrations of drugs remained in the lipid matrix. Additionally, this proportion showed maximum drug loading.
To sustain the high concentration of therapeutic drugs at the site of action, significant drug EE is required. The drug's capacity to separate and dissolve more readily in the aqueous and lipid phases may be enhanced by increasing the concentration of the surfactant. The 3D plot showed that the entrapment efficiency (Y3) significantly increased as the ratio of lipids increased, then somewhat decreased as the ratio grew further. This might be attributed to the solubilization of the drug in the presence of an appropriate surfactant, which reduces interfacial tension and increases the entrapment efficiency once the amount of surfactant is increased in the presence of adequate lipids.50 It came to light that as the lipid percentage and HPH cycle increased, the entrapment efficiency did increase. Conversely, during the HPH cycle, high energy agitation promoted drug entrapment by facilitating the transport of drug molecules through the aqueous phase to the lipid matrix; even though further increases in the HPH cycle might promote the removal of the drug from the matrix. The interaction terms AB and BC's center point values indicated the maximum amount of drug entrapment.
Particle size is an important consideration for administering medications by the nose to the brain, even if PDI simply displays the particle size distribution. The brain can be targeted intranasally with particles smaller than 200 nm, according to the literature.51 The zeta potential, which has an ideal range of +30 mV to −30 mV, serves as an indication of the stability of nanoformulation. The zeta potentials of all batches fell between −12.9 ± 1.0 and −25.1 ± 1.9 mV demonstrating good stability. The reason for the negative zeta potential of NLC may be due to the presence of a free non-ionic surfactant (Poloxamer 188) on its surface. Nevertheless, electric double-layer shifting or fatty acid impurities can cause a reduction in zeta potential. The system is stabilized because of steric hindrance caused by its non-ionic surfactant Poloxamer 188. This can happen as a result of fatty ester group ionization or hydrolysis in Precirol ATO5 (Glyceryl palmistearate).
The TEM results of the formulation indicate that the NLC particles are homogeneous and spherical. The results of the TEM investigation validate the data obtained from the particle size analysis.
The precise concentration of the drug towards the site of absorption can only be assured through adequate drug loading and entrapment efficiency. The addition of liquid lipid (transcutol HP) to solid lipid (Precirol ATO5) leads to an increase in entrapment efficiency. The greater entrapment efficiency might have been attributed to a decrease in particle crystallinity, which is accountable for the formulation's improved stability.52 Greater drug accumulation in the nanostructured lipid carrier is correlated with higher lipid concentrations, resulting in a reduction of drug exclusion into external phases. This ultimately leads to higher drug entrapment efficiency.53 It is crucial to provide optimal drug loading and entrapment efficiency to guarantee the desired drug concentration at the site of absorption.
In vitro drug release, XRPD, and DSC tests were conducted on the developed NLCs as part of the NLC characterization process. Even though their intensities differed, some of the peaks in the X-ray diffraction pattern of the improved formulation were in the same location or very close to those in the pure DPZ. There might be variations in the intensity of certain peaks as a consequence of high-pressure homogenization being performed with high pressure. The procedures that were utilized to amorphize donepezil were freeze-drying and high-pressure homogenization. Based on in vitro drug release studies, the drug's persistent effect might be attributed to the drug trapped in the NLCs’ matrix, whereas the drug's burst release could be caused by the unbound drug on the surface of the optimized formulation. As a result, it was found that the Higuchi model, with R2 = 0.977, presented the best fit (Table S3†).
Details for pH, CIC, gelling time, viscosity of gel (before and after gelation), expansion coefficient, and mucoadhesive strength are presented in Table 4. The pH of nasal formulations should be between 4 and 6, as an acidic or alkaline pH may irritate the nasal passages.54 Its ease of administration relies significantly on the viscosity of the formulation. If there is any toxicity of the formulation's components to the nasal mucosa, it is determined via the nasal ciliotoxicity study. The untreated nasal tissue exhibited an intact pseudo-epithelial layer and a basement membrane containing glandular cells. An additional indication for the safety of the in situ gel is a lack of toxicity to the basement membrane, glandular cell nucleus, or epithelial layer in nasal tissue exposed to the DPZ-loaded in situ gel (Fig. 6c). The in situ gel based on NLCs administered via nasal administration is therefore regarded as safe. The enhancement ratio of ex vivo permeation studies indicates that the DPZ-loaded in situ gel was 3.05 times higher than that of DPZ-Sol. The ability to enhance DPZ penetration through the nasal mucosa was therefore made possible by NLCs infused in situ gel. This might be the result of NLCs’ enhanced mucoadhesive properties and the DPZ that is encapsulated being more lipophilic superior to previously reported literature results.35 The NLCs’ ability to enhance mucosal membrane penetration for small particle sizes was made possible by Transcutol HP.55
In vivo, pharmacokinetic and biodistribution studies were performed over 12 hours. Nasal cavity roofs are located very close to the brain (skull base) and contain nerves that extend to the brain. Drugs are transported to the brain via the IN pathway using the olfactory and trigeminal nerves. The trigeminal pathway or olfactory system can both mediate IN transfer.56 Increase in the amount of DPZ in situ gel in the brain after intranasal NLC treatment compared to intravenous injection suggests that lipidic nanocarriers such as NLCs, have the potential to target the nose and brain. The nasal mucosa's vast surface area for absorption, lipophilic nature, and the nano size of the NLCs (112.5 ± 2.44 nm) contribute to increased permeability. The lipids used for NLCs also had an occlusive effect, which kept the mucosa moist and increased the permeability. The rheological properties of these NLCs are improved and give a mucoadhesive character when they are introduced in an in situ gelling system.33 In the upper nasal mucosa, this allows for longer residence times and increased formulation adherence. As an activator of skin penetration, transcutol's cationic properties enable the NLCs to adhere to the negatively charged nasal epithelium.35 Rather than using a regular DPZ solution, these possible pathways could account for the higher concentration and penetration of DPZ in some brain regions.
In a range of relevant organs, such as the intestine, kidneys, liver, lungs, and spleen, Fig. 7c displays the biodistribution of DPZ in situ gel and DPZ-Sol. DPZ in situ gel is found in the brain, heart, and kidneys as opposed to free DPZ (DPZ sol.), which builds up in the heart and kidneys. The distribution of DPZ in situ gel throughout the brain is amazing, especially when compared to lipid NLCs and Poloxamer 188. The increased distribution in the brain might be the result of interactions between Precirol, Transcutol, and Poloxamer 188 and the DPZ. There was no visible accumulation of any samples in the spleen. A small amount of NLCs dispersed in situ gel may circumvent the BBB, which could lead to a smaller amount of liver extraction. The BBB was extensively penetrated and neuroprotection was delivered at much higher levels by the in situ gel. Improved brain delivery can be attributed to NLC in situ gel, minimum size, zeta potential charge, and biocompatible excipients.
An animal model of AD was used in the experiments to assess the pharmacological effect. For this study, the TMT-induced AD rat model was selected.41 Specifically, to return to its original platform, it crossed the pool and traveled a longer path, basically like the TMT-treated rat would have predicted. This kind of behavior indicates memory loss. Their reference ability and working memory for the short term were constant, and there was no noticeable difference in the amount of time it took them to learn across the four-day testing period. Rats given DPZ in situ gel showed the same behavioral pattern as the control group. This suggests a considerable difference between DPZ sol and DPZ in situ gel's memory-enhancing ability. The reduction in escape latency associated with the intranasal DPZ in situ gel as compared with the intranasal DPZ sol provides evidence for brain targeting through the nose-to-brain pathways. In the literature, previously reported results were also addressed.24,57
The DPZ sol and DPZ in situ gel treatment regimens had considerably (p < 0.001) increased AchE levels than the neurodegeneration control group. Further evidence of DPZ's antioxidant efficacy comes from a significant drop in levels of MDA and an increase in GSH for comparison with the neurodegenerative control group, suggesting that oxidative stress decreases in the brain. Consistent with the results, administering DPZ in situ gel to animals enhanced their memory for spatial information.24,26 The neuroprotection was confirmed through histopathological analysis. These results suggest that the NLCs in situ gel application probably raised in absorption of the drug to the brain.
5. Conclusion
The potential of employing an in situ gel to transport DPZ-NLCs to the brain was examined in this work. The melt emulsification high-pressure homogenization process was used to successfully develop DPZ-NLCs with a 112.5 nm particle size and an entrapment effectiveness of 98.7%. Spherical, nanosized DPZ-NLCs were produced with an ideal ratio of Transcutol to Precirol using the Box–Behnken design optimization. Both in vitro and in vivo investigations were used to assess the effectiveness of this delivery strategy. Once the DPZ in situ gel was delivered through the nasal canal, better absorption in the brain was observed. When compared to DPZ sol. given intravenously (i.v.) and intranasally (i.n.), biodistribution and pharmacokinetic studies performed in Wistar rats showed that the DPZ in situ gel had a more notable nose-to-brain direct transport bypassing the blood–brain barrier (BBB). The BBB was circumvented by the DPZ-NLCs due to their small size, which allowed them to enter the brain simply by the olfactory and trigeminal nerve systems. By circumventing mucociliary clearance, the DPZ-NLCs can be incorporated into gelling systems that react to a variety of triggers, including ions and temperature, to improve the residence time and ease of application. The DPZ in situ gel's intranasal delivery helped to maintain therapeutic concentration for an extended period, first-pass metabolism, and minimize or eradicate the drug's GIT-associated adverse effects. When it came to pharmacokinetics and pharmacodynamics, in vivo research on rats revealed that in situ gelling NLCs performed better than the intravenous method. The study led to the conclusion that brain targeting may be successful with NLCs in a simple, safe, scalable, and effective manner. To improve other therapeutics’ ability to pass through the nasal route and penetrate the blood–brain barrier, this strategy could be applied. To promote outcomes, further research is needed to build up a link between these preliminary findings and clinical data.Abbreviations
| AD | Alzheimer's disease |
| DPZ | Donepezil |
| NLC | Nanostructured lipid carrier |
| DoE | Design of expert |
| DPZ-OPT | Donepezil optimized batch |
| SEM | Scanning electron microscopy |
| TEM | Transmission electron microscopy |
| DPZ sol. | Donepezil solution |
| IN | Intranasal |
| IV | Intravenous |
| C max | Maximum drug concentration |
| T max | Time to reach Cmax |
| AUC | Area under the curve |
| DTI | Drug targeting index |
| DTP | Drug targeting potential |
| DTE | Drug targeting efficiency |
Author contributions
Devika Sonawane: conceptualization, methodology, resources, formal analysis, writing – original draft, and review & editing. Varsha Pokharkar: supervision and review & editing. All authors have read and agreed to the published version of the manuscript.Ethical statement
All animal experiments were performed with the prior approval of the Poona College of Pharmacy's Institutional Animal Ethics Committee (IAEC), which authorized an experimental protocol (IAEC/PCP/PCT06/2021-2022), (IAEC/PCP/PCT07/2021-2022) based on the recommendations of the Committee for the Purpose of Control and Supervision on Experimental Animals, Government of India. All experiments were performed as per the U. K. Animals Act (1986) and its accompanying recommendations, as detailed in the ARRIVE guidelines.Data availability
The data that support the findings of this study are available upon reasonable request.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors are thankful to the All India Council for Technical Education (AICTE) and the Government of India, respectively, for the National Doctoral Fellowship (NDF) to Devika Sonawane. The authors would like to extend their gratitude to Cipla and Gattefosse for providing gift samples for research.References
- S. Tiwari, A. Venkata, A. Kaushik, Y. Adriana and M. Nair, Alzheimer's disease diagnostics and therapeutics market, Int. J. Nanomed., 2019, 14, 5541–5554 CrossRef CAS PubMed.
- D. Xiaoguang, W. Xinyi and G. Meiyu, Alzheimer’s disease hypothesis and related therapies, Transl Neurodegener, 2018, 7(1), 1–7, DOI:10.1186/s40035-018-0107.
- Alzheimer’s association, 2020 Alzheimer’s disease facts and figures, Alzheimer’s Dementia, 2020, 16, 391–460, DOI:10.1002/alz.12068.
- R. H. Swerdlow, Pathogenesis of Alzheimer's disease, Clin. Interventions Aging, 2007, 2(3), 347–359 CAS.
- P. P. Liu, et al., History and progress of hypotheses and clinical trials for Alzheimer's disease, Signal Transduction Targeted Ther, 2019, 4, 1, DOI:10.1038/s41392-019-0063-8.
- M. Agrawal, et al., Nose-to-brain drug delivery: An update on clinical challenges and progress towards approval of anti-Alzheimer drugs, J. Controlled Release, 2018, 281, 139–177, DOI:10.1016/j.jconrel.2018.05.011.
- L. C. Espinoza, et al., Formulation strategies to improve nose-to-brain delivery of donepezil, Pharmaceutics, 2019, 11, 1–16 CrossRef PubMed.
- P. Sozio, et al., Transdermal donepezil on the treatment of Alzheimer's disease, Neuropsychiatr. Dis. Treat., 2012, 8, 361–368 CAS.
- S. Gänger and K. Schindowski, Tailoring formulations for intranasal nose-to-brain delivery: A review on architecture, physico-chemical characteristics and mucociliary clearance of the nasal olfactory mucosa, Pharmaceutics, 2018, 10, 3 CrossRef PubMed.
- D. Lee and T. Minko, Nanotherapeutics for nose-to-brain drug delivery: An approach to bypass the blood brain barrier, Pharmaceutics, 2021, 13(12), 2049 CrossRef CAS PubMed.
- H. Akel and I. Csók Ismai, Progress and perspectives of brain-targeting lipid-based nanosystems via the nasal route in Alzheimer's disease, Eur. J. Pharm. Biopharm., 2020, 148, 38–53, DOI:10.1016/j.ejpb.2019.12.014.
- S. Bahadur, et al., Intranasal nanoemulsions for direct nose-to-brain delivery of actives for cns disorders, Pharmaceutics, 2020, 12, 1–27 CrossRef PubMed.
- N. A. Emad, et al., Recent progress in nanocarriers for direct nose to brain drug delivery, J. Drug Delivery Sci. Technol., 2021, 64, 102642, DOI:10.1016/j.jddst.2021.102642.
- M. M. Wen, et al., Nanotechnology-based drug delivery systems for Alzheimer's disease management: Technical, industrial, and clinical challenges, J. Controlled Release, 2017, 245, 95–107, DOI:10.1016/j.jconrel.2016.11.025.
- S. Cunha, et al., Improving drug delivery for alzheimer's disease through nose-to-brain delivery using nanoemulsions, nanostructured lipid carriers (NLC) and in situ hydrogels, Int. J. Nanomed., 2021, 16, 4373–4390 CrossRef PubMed.
- R. G. Madane and H. S. Mahajan, Curcumin-loaded nanostructured lipid carriers (NLCs) for nasal administration: design, characterization, and in vivo study, Drug Delivery, 2016, 23, 1326–1334 CrossRef CAS PubMed.
- S. G. Antimisiaris, et al., Overcoming barriers by local drug delivery with liposomes, Adv. Drug Delivery Rev., 2021, 174, 53–86, DOI:10.1016/j.addr.2021.01.019.
- N. Rabiee, et al., Polymeric Nanoparticles for Nasal Drug Delivery to the Brain: Relevance to Alzheimer's Disease, Adv. Ther., 2021, 4, 1–24 Search PubMed.
- M. Yasir, et al., Solid lipid nanoparticles for nose to brain delivery of donepezil: formulation, optimization by Box–Behnken design, in vitro and in vivo evaluation, Artif. Cells, Nanomed., Biotechnol., 2018, 46, 1838–1851, DOI:10.1080/21691401.2017.1394872.
- S. Nagaraja, et al., Ion-triggered in situ gelling nanoemulgel as a platform for nose-to-brain delivery of small lipophilic molecules, Pharmaceutics, 2021, 13, 1–20 Search PubMed.
- M. A. Elsheikh, et al., Brain-Targeted Approach to Ameliorate Memory Disorders in a Sporadic Alzheimer's Disease Mouse Model via Intranasal Luteolin-Loaded Nanobilosomes, Pharmaceutics, 2022, 14, 3 CrossRef PubMed.
- A. P. Rajput and S. B. Butani, Resveratrol anchored nanostructured lipid carrier loaded in situ gel via nasal route: Formulation, optimization and in vivo characterization, J. Drug Delivery Sci. Technol, 2019, 51, 214–223, DOI:10.1016/j.jddst.2019.01.040.
- D. Khunt, et al., Role of Omega-3 Fatty Acids and Butter Oil in Targeting Delivery of Donepezil Hydrochloride Microemulsion to Brain via the Intranasal Route: a Comparative Study, AAPS PharmSciTech, 2020, 21, 2 CrossRef PubMed.
- A. K. Al Asmari, et al., Preparation, characterization, and in vivo evaluation of intranasally administered liposomal formulation of donepezil, Drug Des., Dev. Ther., 2016, 10, 205–215 CrossRef CAS PubMed.
- I. Baysal, et al., Donepezil loaded PLGA-b-PEG nanoparticles: their ability to induce destabilization of amyloid fibrils and to cross blood brain barrier in vitro, J. Neural Transm., 2017, 124, 33–45 CrossRef CAS PubMed.
- S. Al Harthi, et al., Nasal delivery of donepezil HCl-loaded hydrogels for the treatment of Alzheimer's disease, Sci. Rep., 2019, 9, 1–20 CrossRef CAS PubMed.
- S. Ul Islam, et al., Intranasal delivery of nanoformulations: A potential way of treatment for neurological disorders, Molecules, 2020, 25, 1–27 Search PubMed.
- F. Gu, H. Fan and C. Wu, Thermosensitive in Situ Nasal Gel of Donepezil Hydrochloride, Acta Pharm., 2020, 70, 411–422 CrossRef CAS PubMed.
- B. A. Aderibigbe, In situ-based gels for nose to brain delivery for the treatment of neurological diseases, Pharmaceutics, 2018, 10, 2 Search PubMed.
- Y. M. Gabal, et al., Effect of surface charge on the brain delivery of nanostructured lipid carriers in situ gels via the nasal route, Int. J. Pharm., 2014, 473, 442–457, DOI:10.1016/j.ijpharm.2014.07.025.
- M. Agrawal, et al., Design and optimization of curcumin loaded lipid nano carrier system using Box–Behnken design, Biomed. Pharmacother., 2021, 141, 111919 CrossRef CAS PubMed.
- M. Masjedi, et al., Brain targeted delivery of sumatriptan succinate loaded chitosan nanoparticles: Preparation, In vitro characterization, and Neuro-pharmacokinetic evaluations, J. Drug Delivery Sci. Technol., 2021, 61, 102179, DOI:10.1016/j.jddst.2020.102179.
- P. Wavikar, R. Pai and P. Vavia, Nose to Brain Delivery of Rivastigmine by In Situ Gelling Cationic Nanostructured Lipid Carriers: Enhanced Brain Distribution and Pharmacodynamics, J. Pharm. Sci., 2017, 106, 3613–3622, DOI:10.1016/j.xphs.2017.08.024.
- A. Rajput, et al., In situ nanostructured hydrogel of resveratrol for brain targeting: in vitro-in vivo characterization, Drug Delivery Transl. Res., 2018, 8, 1460–1470 CrossRef CAS PubMed.
- A. P. Rajput and S. B. Butani, Fabrication of an ion-sensitive in situ gel loaded with nanostructured lipid carrier for nose to brain delivery of Donepezil, Asian J. Pharm., 2018, 12, 293–302 CAS.
- R. M. Farid, et al., Formulation and In Vitro Evaluation of Salbutamol Sulphate In Situ Gelling Nasal Inserts, AAPS PharmSciTech, 2013, 14(2), 712–718 CrossRef CAS PubMed.
- D. Sonawane and V. Pokharkar, Donepezil and Quercetin Simultaneous Estimation in Rat Plasma Using Developed Bioanalytical HPLC Method: Relevance in Pharmacokinetic Studies, Int. J. Drug Delivery Technol., 2023, 13(4), 1538–1543, DOI:10.25258/ijddt.13.4.64.
- R. Patil, D. Pawara, C. Gudewar and A. Tekade, Nanostructured cubosomes in an in situ nasal gel system: an alternative approach for the controlled delivery of donepezil HCl to brain, J. Liposome Res., 2019, 29(3), 264–273, DOI:10.1080/08982104.2018.1552703.
- V. Pokharkar, A. Patil-Gadhe and P. Palla, Efavirenz loaded nanostructured lipid carrier engineered for brain targeting through intranasal route: In vivo pharmacokinetic and toxicity study, Biomed. Pharmacother., 2017, 94, 150–164, DOI:10.1016/j.biopha.2017.07.067.
- G. Choi, et al., Efect of quercetin on learning and memory performance in ICR mice under neurotoxic trimethyltin exposure, Food Chem., 2012, 132, 1019–1024, DOI:10.1016/j.foodchem.2011.11.089.
- J. Li, et al., Puerarin attenuates amyloid-beta-induced cognitive impairment through suppression of apoptosis in rat hippocampus in vivo, Eur. J. Pharmacol., 2010, 649, 195–201, DOI:10.1016/j.ejphar.2010.09.045.
- M. Banayschwartz, et al., Protein content of various regions of rat brain and adult and aging human brain, Age, 1992, 15, 51–54 CrossRef CAS.
- R. Mashoque, et al., Neuroprotective role of Asiatic acid in aluminum chloride-induced rat model of Alzheimer's disease, Front. Biosci., 2018, 10, 262–275 CrossRef PubMed.
- S. Asru, Colorimetric assay of catalase, Anal. Biochem., 1972, 47, 389–394 CrossRef PubMed.
- S. Diddi, et al., Standardization and ameliorative efect of Kalyanaka ghrita in β-amyloid induced memory impairment in Wistar rats, J. Ethnopharmacol., 2023, 300, 115671, DOI:10.1016/j.jep.2022.115671.
- S. Maria and J. Moron, Depierrebengt mannervik. levels of glutathione, glutathione reductase and glutathione s-transferase activities in rat lung and liver, Biochim. Biophys. Acta, 1979, 582, 67–78 CrossRef PubMed.
- F. Pu, et al., Neuroprotective Effects of Quercetin and Rutin on Spatial Memory Impairment in an 8-Arm Radial Maze Task and Neuronal Death Induced by Repeated Cerebral Ischemia in Rats, J. Pharmacol. Sci., 2007, 334, 329–334 CrossRef PubMed.
- S. Md. Bhavna, et al., Preparation, characterization, in vivo biodistribution and pharmacokinetic studies of donepezil-loaded PLGA nanoparticles for brain targeting, Drug Dev. Ind. Pharm., 2014, 40(2), 278–287 CrossRef CAS PubMed.
- I. T. Mendes, et al., Development and characterization of nanostructured lipid carrier-based gels for the transdermal delivery of donepezil, Colloids Surf., B, 2019, 177, 274–281 CrossRef CAS PubMed.
- V. Pokharkar, S. Suryawanshi and V. Dhapte-Pawar, Exploring micellarbased polymeric systems for efective nose-to-brain drug delivery as potential neurotherapeutics, Drug Delivery Transl. Res., 2020, 10, 1019–1031, DOI:10.1007/s13346-019-00702-6.
- A. K. Al Asmari, et al., Preparation, characterization, and in vivo evaluation of intranasally administered liposomal formulation of donepezil, Drug Des., Dev. Ther., 2016, 10, 205–215, DOI:10.2147/DDDT.S93937.
- E. C. Lupe, et al., Development of a Nasal Donepezil-loaded Microemulsion for the Treatment of Alzheimer's Disease: in vitro and ex vivo Characterization, CNS & Neurological Disorder, Drug Targets, 2018, 17, 1, DOI:10.2174/1871527317666180104122347.
- I. Khan, et al., Impact of phospholipids, surfactants and cholesterol selection on the performance of transfersomes vesicles using medical nebulizers for pulmonary drug delivery, J. Drug Delivery Sci. Technol., 2021, 66, 102822, DOI:10.1016/j.jddst.2021.102822.
- M. Agrawal, et al., Stimuli-responsive In situ gelling system for nose-to-brain drug delivery, J. Controlled Release, 2020, 327, 235–265 CrossRef CAS PubMed.
- D. W. Osborne and J. Musakhanian, Skin Penetration and Permeation Properties of Transcutol-Neat or Diluted Mixtures, AAPS PharmSciTech, 2018, 19(8), 3512–3533, DOI:10.1208/s12249-018-1196-8.
- F. Erd, et al., Evaluation of the intranasal delivery route of drug administration for brain targeting, Brain Res. Bull., 2018, 143, 155–170 CrossRef PubMed.
- F. Gu, et al., Preparation, characterization, and in vivo pharmacokinetics of thermosensitive in situ nasal gel of donepezil hydrochloride, Acta Pharm., 2020, 70, 411–422 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4pm00174e |
| This journal is © The Royal Society of Chemistry 2024 |