DOI:
10.1039/D4PM00113C
(Paper)
RSC Pharm., 2024,
1, 765-774
Biopharmaceutical advancement of efonidipine hydrochloride ethanolate through amorphous solid dispersion of a Parteck SLC mesoporous silica polymer
Received
16th April 2024
, Accepted 30th May 2024
First published on 5th June 2024
Abstract
Amorphous solid dispersion is the most efficient method for improving the solubility and release of poorly water-soluble crystalline drug molecules. Efonidipine hydrochloride ethanolate (EFE) shows solubility-limited oral bioavailability (BCS class II). The present investigation aimed to improve the solubility, bioavailability, and therapeutic efficacy of EFE using Parteck® SLC mesoporous silica based amorphous solid dispersion (EFESD). EFESD was prepared by employing a solvent evaporation method. An optimized composition (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) of solid dispersion was subjected to in vitro, ex vivo, and in vivo characterization. The solubility of EFE in the EFESD form was found to be 5- and 4-fold improved in distilled water and phosphate buffer (pH 6.8), respectively. An ex vivo permeability study performed using different parts of the Wistar rat small intestine (the duodenum, jejunum, and ileum) using a non-everted sac method showed 2-fold improvements in the permeability of EFE in EFESD. Moreover, in vivo pharmacokinetic and pharmacodynamic studies performed using male Wistar rats showed 1.41- and 2.10-fold increase in the area under the curve and Cmax, respectively, along with the improved anti-hypertensive activity of EFE in EFESD. Thus, amorphous solid dispersion with a novel applied Parteck® SLC 500 mesoporous silica formulation is an effective strategy to improve the biopharmaceutical properties of EFE.
:1 ratio) of solid dispersion was subjected to in vitro, ex vivo, and in vivo characterization. The solubility of EFE in the EFESD form was found to be 5- and 4-fold improved in distilled water and phosphate buffer (pH 6.8), respectively. An ex vivo permeability study performed using different parts of the Wistar rat small intestine (the duodenum, jejunum, and ileum) using a non-everted sac method showed 2-fold improvements in the permeability of EFE in EFESD. Moreover, in vivo pharmacokinetic and pharmacodynamic studies performed using male Wistar rats showed 1.41- and 2.10-fold increase in the area under the curve and Cmax, respectively, along with the improved anti-hypertensive activity of EFE in EFESD. Thus, amorphous solid dispersion with a novel applied Parteck® SLC 500 mesoporous silica formulation is an effective strategy to improve the biopharmaceutical properties of EFE.
1. Introduction
Approximately 70% of new chemical entities have water solubility issues.1,2 The bioavailability of poorly soluble drugs in the oral dose form is controlled by the low solubility and GIT dissolution rate.3 The amorphization with molecular dispersion of the drug increases the surface area and wettability and hence increases the solubility, dissolution rate, and bioavailability of poorly soluble (BCS classes II and IV) drugs.4–6
Efonidipine hydrochloride ethanolate (EFE) is a calcium channel antagonist used to treat hypertension and angina pectoris.7–10 EFE is lipophilic with a logP value of 5.35 and a pKa (basic) of 2.33.11 It is insoluble in water (<10 μg mL−1) but soluble in organic solvents such as DMSO, methanol, ethanol, and acetonitrile.12,13 As an API, this molecule has a very poor water solubility; hence, to boost its bioavailability, its solubility and oral absorption must be improved.
In solid dispersion, a poorly water-soluble drug is dispersed in a highly soluble hydrophilic matrix in the solid state to enhance its solubility, stability, and oral bioavailability.14–21 This involves several mechanisms, including particle size reduction, molecular dispersion, enhanced stability and porosity, and transformation from a crystalline to an amorphous state.17,18 With urea as a third ingredient and hydroxypropyl methylcellulose acetate succinate as a polymeric carrier, Otsuka et al. prepared microwave-assisted EFE solid dispersion. Its bioavailability was eight times better in beagle dogs. According to Cheng et al., flow through the cell system of an open-loop system can be used to forecast how well an EFE solid dispersion with hydroxypropyl methylcellulose acetate succinate performs in vivo. Wet-milling technology was used by Song et al. to create EFE nanosuspension, which increased bioavailability by 2.2 times. Rajput et al. used the hot melt extrusion process to create an amorphous solid dispersion of EFE using Eudragit EPO with enhanced dissolution. However, each method has its limitations.
Recent decades have seen the development of mesoporous silica (MS) as a novel, promising substitute for the conventional techniques used to create amorphous formulations. The ability of MS materials to efficiently entrap drug molecules in mesopores and prevent recrystallization due to finite-size effects is attributed to their distinctive properties of nanoscale mesopores with high pore volumes and high surface areas.22 Consequently, amorphous drugs trapped in mesopores can speed up the pace of drug dissolution and create drug supersaturation in aqueous environments, providing a higher level of bioavailability than crystalline drugs. Therefore, MS materials have garnered significant attention as effective delivery systems for therapeutic compounds that are poorly soluble in water.
The current study aims to explore the advantages of Parteck® SLC 500 mesoporous silica-based amorphous formulation for EFE to improve its biopharmaceutical properties. Solid dispersion of EFE (EFESD) with mesoporous silica could prove useful in enhancing the solubilization of EFE in the GIT and hence enhancing its concentration at the site of absorption.23 In the present investigation, EFESD was prepared with mesoporous silica in different ratios using a solvent evaporation method. The EFESD was further optimized based on its saturation solubility in distilled water and phosphate buffer (pH 6.8) and subsequently characterized by DSC, XRD, FTIR, and FESEM techniques. Moreover, both EFE and EFESD were evaluated for intrinsic dissolution rate, ex vivo permeability in different parts of the Wistar rat intestine, and in vivo pharmacokinetics and pharmacodynamics for the assessment of bioavailability and antihypertensive activity in Wistar rat, respectively.
2. Materials and methods
2.1. Materials
EFE was a kind gift from Ajanta Pharma Ltd (Mumbai, India). Parteck® SLC 500 mesoporous silica was a gift sample from Merck Life Sciences Pvt. Ltd (Mumbai, India). Methanol was purchased from Merck Life Sciences (Pvt. Ltd) (Mumbai, India). The additional excipients utilised were all of the pharmaceutical grade standards. The chemical solvents and reagents utilised were of analytical grade.
2.2. Preparation of EFE solid dispersion (EFESD)
EFESDs were prepared using a solvent evaporation method. Briefly, EFE was mixed with mesoporous silica in different ratios (1:1, 1:2, and 1:3 w/w) and dissolved in 5 mL of methanol to yield dispersion. The methanolic dispersion was turned into a dry mass by evaporating it at 50 °C in a rotary evaporator (Heidolph, Germany). This dry mass was crushed with a mortar and pestle before being sieved through an 18 mm mesh size to create a powder of a uniform size. Before further research, the prepared EFESDs were kept in tightly closed containers at 25 °C and shielded from light.24
2.3. Characterization of EFESD
2.3.1. Saturation solubility.
In distilled water and phosphate buffer (pH 6.8), the saturation solubility of EFE in pure form and EFESD was measured. To facilitate solubilization, an excess amount of sample was added to 5 mL of the appropriate solvent and mixed in firmly closed vials using a vortex mixture (Spinix, India) at the highest frequency for 10 min. The samples were kept at room temperature in a shaking water bath for 48 hours (New Brunswick Scientific, Germany). The resultant samples were centrifuged at 10000 rpm for 10 min. The supernatant was collected, filtered through a 0.22 μm membrane filter, suitability diluted with the respective solvent, and analysed spectrophotometrically (Jasco V730, UV-visible spectrophotometer, Japan) at 252 nm to estimate the amount of EFE. The results of the triplicate measurements were reported as means ± standard deviation (SD).
2.3.2. Residual solvent content determination.
The amount of total residual solvent in solid dispersions was determined using a thermal gravimetric analyser (TGA-60WS0; Shimadzu Corporation, Japan). Thermal gravimetric analysis was performed by heating a weighed amount of the sample in a nitrogen atmosphere from 25 °C to 80 °C at a rate of 2 °C min−1, and the loss of weight as a function of temperature was recorded.
2.3.3. FTIR.
FTIR spectra of EFE, mesoporous silica, and EFESD samples were recorded using an FTIR spectrometer (Jasco, Japan) to study the molecular interactions. The samples were pulverized and mixed with potassium bromide (KBr) in a 1:1 ratio and subsequently scanned from 3600 to 600 cm−1 wave numbers at a resolution of 4 cm−1 to obtain the FTIR spectra.
2.3.4. DSC.
A calorimeter with an intracooler was used to record the DSC thermograms of EFE, mesoporous silica and EFESD (DSC 821e, Mettler-Toledo, Greifensee, Switzerland). Approximately 5 mg of sample was heated on an aluminium pan at a rate of 10 °C min−1 from 25 to 300 °C and nitrogen gas under a constant flow rate of 20 lb per in2.
2.3.5. XRD.
Using a Philips PW 1729 X-ray generator (United States) equipped with a copper target, a voltage of 40 kV, and a current of 30 mA, the XRD patterns of EFE, mesoporous silica, and EFESD were measured. Over an angle range of 5°–40°, the scanning rate was maintained at 10° min−1. To record the XRD patterns, the materials were ground up and put into an aluminium sample container.25
2.3.6. FESEM analysis.
The surface morphology of EFE, mesoporous silica, and EFESD was investigated using FESEM analysis (Ultra-Plus, Zeiss, Germany). Before the sample was coated with gold using a Zeiss Ion Sputter (E-1030) in a vacuum (6 Pa) at a rate of 6 nm per minute to conduct electricity, the sample was fastened to a brass holder with double-sided adhesive tape.
2.3.7. HPLC analytical method.
The HPLC system (Jasco, Japan) equipped with a PU-1580 Intelligent HPLC pump, UV-Visible detector, and a Chromatopak peerless basic C-18 column (250 mm × 4.6 mm, 5 μm) coupled with a guard column (RP-18, 33 mm Kromasil, 5 μm) was used for the chromatographic separation at ambient temperature. The mobile phase (methanol:water, 90:10 v/v) was filtered through a 0.45 μm membrane filter and injected at a flow rate of 1.0 mL min−1 for isocratic elution. A primary stock solution of EFE was prepared by dissolving 5 mg of EFE in 5 mL of methanol (1000 μg mL−1). For calibration, different concentrations of 0, 50, 100, 250, 500, 1000, 2500, and 5000 ng mL−1 of EFE were prepared using the stock solution. The injection volume of the sample was 20 μL. The eluent was analyzed at 252 nm using a UV-visible detector. The retention time of EFE was found to be 5.18 min. The data obtained were processed using the Jasco Borwin version 1.5, LC-Net II/ADC system. The regression equation and R2 value of EFE for the calibration curve were observed to be y = 32.334x + 3319.2 and 0.9959, which were used for drug content, in vitro intrinsic dissolution study, and ex vivo permeability studies.
2.3.8. Drug content.
A precisely weighed sample of EFESD was dissolved in 10 mL of methanol and stirred to produce a transparent solution. A 0.22 mm syringe filter was used to filter the resulting solution after it had been properly diluted with phosphate buffer (pH 6.8), and the concentration of EFE was determined using HPLC at 252 nm. Triplicates of the drug content study were completed.26
2.3.9.
In vitro intrinsic dissolution study.
The intrinsic dissolution study helps to understand the effect of the pure drug surface area on its rate of dissolution. The pellets for the dissolution study were prepared by compressing EFE (150 mg) and EFESD (equivalent to 150 mg EFE) separately at 6 MPa for 60 seconds using a 13 mm punch and die set. The pellet was placed in a molten beeswax mold, allowing only one face to be exposed to the dissolution media (exposed surface area: 1.32 cm2). The dissolution study was performed using a USP type II dissolution test apparatus (Electrolab, India) in 900 mL of distilled water, 0.1 N HCl (pH 1.2), and phosphate buffer (pH 6.8) containing 0.05% w/v SLS maintained at 37 ± 0.2 °C under constant stirring (50 rpm). At predetermined intervals, the aliquots were removed and replaced with appropriate fresh dissolution media. To estimate the amount of EFE, after being filtered through a 0.22 μm syringe filter, the collected samples were tested using HPLC at 252 nm. The dissolution rate study was performed in triplicate. The intrinsic dissolution rate was calculated by plotting the cumulative amount of drug that was dissolved per unit surface area of the pellet against time.27
2.3.10. Animal studies/biological studies.
The selection of male Wistar rats weighing 200–250 g was procured from Global Animal Supplier Pvt. Ltd, Pune, India. All animal experiments were carried out with the prior approval of the Institutional Animal Ethics Committee of Poona College of Pharmacy, Bharati Vidyapeeth Deemed University (PCP/IAEC/2021-22/2-40), registered under the Committee for the Purpose of Control and Supervision of Experiments on Animals, Government of India. All experiments were performed as per the U. K. Animals Act (1986) and its accompanying recommendations, as detailed in the ARRIVE guidelines.
2.3.12. HPLC bioanalytical method.
The HPLC system used for the bioanalysis of EFE was similar to that used in Section 2.3.6. For calibration, different concentrations of 0, 50, 100, 250, 500, 1000, 2500, and 5000 ng mL−1 of EFE in rat plasma were prepared using a stock solution. The injection volume of the sample was 20 μL. The eluent was analyzed at 252 nm using a UV-visible detector. The plasma (200 μL) is diluted with an equal volume of methanol as the extracting solvent. The mixture was vortexed and centrifuged at 4 °C for 20 minutes at 10000 rpm. The supernatant was collected, filtered, and analyzed using HPLC. The retention time of EFE was found to be 5.29 min. The regression equation and R2 value of EFE for the calibration curve were observed to be y = 42.151x + 1747 and 0.9986. The data obtained were processed using the Jasco Borwin version 1.5, LC-Net II/ADC system.
2.3.13.
In vivo pharmacokinetic studies.
For an in vivo pharmacokinetic study, the animals were divided into two groups (EFE and EFESD), each group comprising six animals. Both groups received a 10 mg kg−1 dose of EFE. The male Wistar rats were anesthetized with ether, and blood samples of 1 mL were collected in 4 mL EDTA tubes at 0.25, 0.5, 1, 2, 3, 4, 6, 8, and 12 hours through the retro-orbital puncture. The blood sample was transferred to 2 mL of Eppendorf tube and centrifuged at 3000 rpm for 15 minutes at 4 °C. Plasma was collected in another Eppendorf tube and stored at −20 °C for further analysis. The EFE concentration in plasma was determined using a validated HPLC bioanalytical method.29
2.3.14.
In vivo pharmacodynamic studies.
Healthy male Wistar rats (thirty) weighing 200–250 g were procured from Global Animal Supplier Pvt. Ltd, Pune, India. The experimental protocol was approved by the IAEC of BVDU, Poona College of Pharmacy, Pune as per CPCSEA guidelines (approval no.: PCP/IAEC/2022/3-02). The animals were divided into five groups, each group comprising six animals.
Group I (control group): this group received distilled water for four weeks (1 mL kg−1) (once a day).
Group II (diseased group): this group received distilled water for four weeks (1 mL kg−1) (once a day).
Group III (standard group): this group received 10 mg kg−1 of captopril dispersed in 1 mL of water for four weeks (once a day).
Group IV (EFE group): this group received 10 mg kg−1 of EFE dispersed in 1 mL of water for four weeks (once a day).
Group V (EFESD group): this group received solid dispersion equivalent to 10 mg kg−1 of EFE in 1 mL of water for four weeks (once a day).
Animals were anesthetized at the end of treatment by an intraperitoneal injection of 1.25 g kg−1 urethane. A small incision was made on the left side of the animal's abdominal cavity to expose the left kidney. The renal artery was occluded for 4 hours with a renal bulldog clamp. The jugular vein was cannulated for the administration of the test drug. To measure blood pressure, the carotid artery was cannulated and connected to the blood pressure transducer of the PowerLab eight-channel recorder (AD Instruments Pty Ltd, PowerLab 8/30, India). The renal bulldog clip was removed once the blood pressure stabilized. Then, 1/10th dose of the administered doses of EFE and EFESD were given through the jugular vein, and mean atrial blood pressure (MABP), systolic blood pressure (SBP), and diastolic blood pressure (DSP) were measured at different time intervals (5, 15, 30, and 60 min). MABP was measured in the normal control groups without clamping the renal artery. Captopril (10 mg kg−1) was given intravenously as a standard. Changes in blood pressure in the treated groups were compared to a negative control.30
2.3.15. Statistical analysis.
The GraphPad Prism 8 program (GraphPad Software Inc., version 8.0, USA) was used to statistically analyze the recorded data. The information is displayed as mean ± SD. Two-way ANOVA was used to analyze the statistical significance of the group differences, and Tukey's test was used to confirm the results at a significance level of p < 0.05.
3. Results and discussion
3.1. Preparation of EFESD
The EFESD was prepared by applying a solvent evaporation method using mesoporous silica as a carrier to improve the EFE solubility and dissolution. The use of mesoporous silica in solid dispersion offers several characteristics, including ordered pore network, size tunability, surface functionality, optically transparent properties, and low toxicity, allowing control over drug loading and release kinetics. Moreover, the electronegative nature of mesoporous silica aids in the absorption of other substances with improved solubility and dissolution.23–31
3.2. Characterization of EFESD
3.2.1. Saturation solubility.
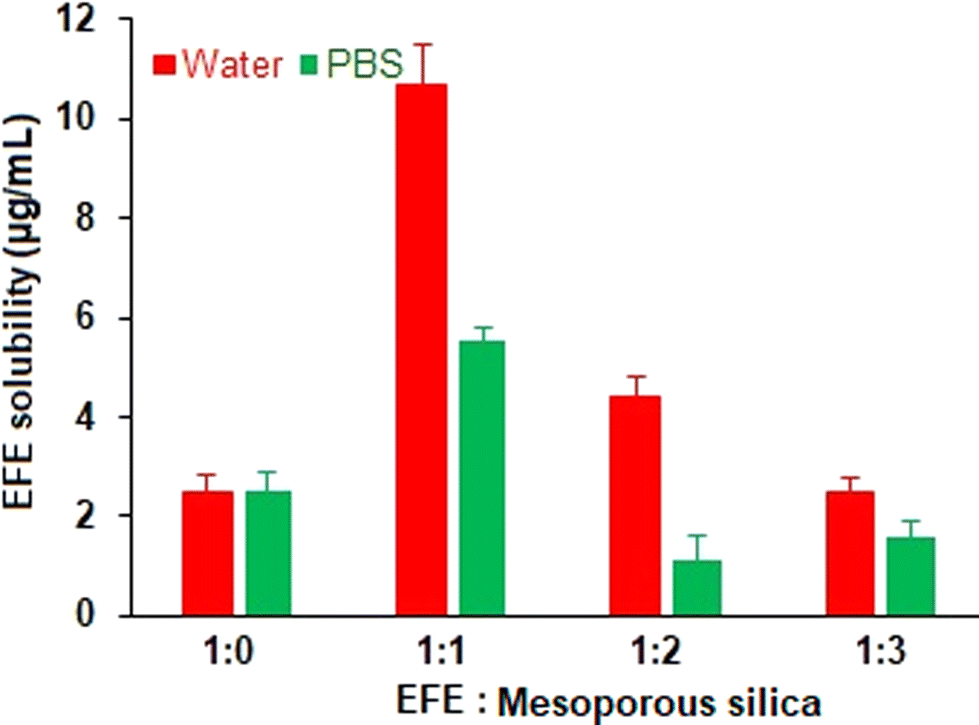
The saturation solubility of EFE and EFESD in different ratios was determined for 48 hours. EFESD showed high saturation solubility at a 1:1 ratio compared to the other ratios. Moreover, a 5-fold and 4-fold increase in EFE solubility from EFESD was observed in water and phosphate buffer (pH 6.8), respectively, compared to pure EFE (Fig. 1) attributed to the disordered amorphous structure of the solid dispersion.32 Considering the improvement in solubility, the EFESD with a 1:1 ratio was optimized and subjected to further evaluation. The total residual solvent in the EFESD was below 0.22% (w/w), justifying the use of methanol to obtain EFESD.
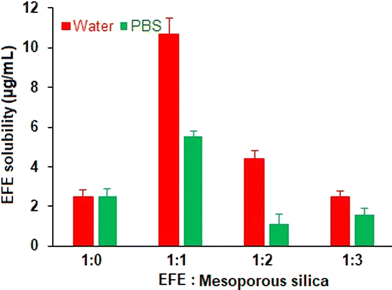 |
| | Fig. 1 Saturation solubility of EFE and EFESD in distilled water and phosphate buffer (pH 6.8), respectively. | |
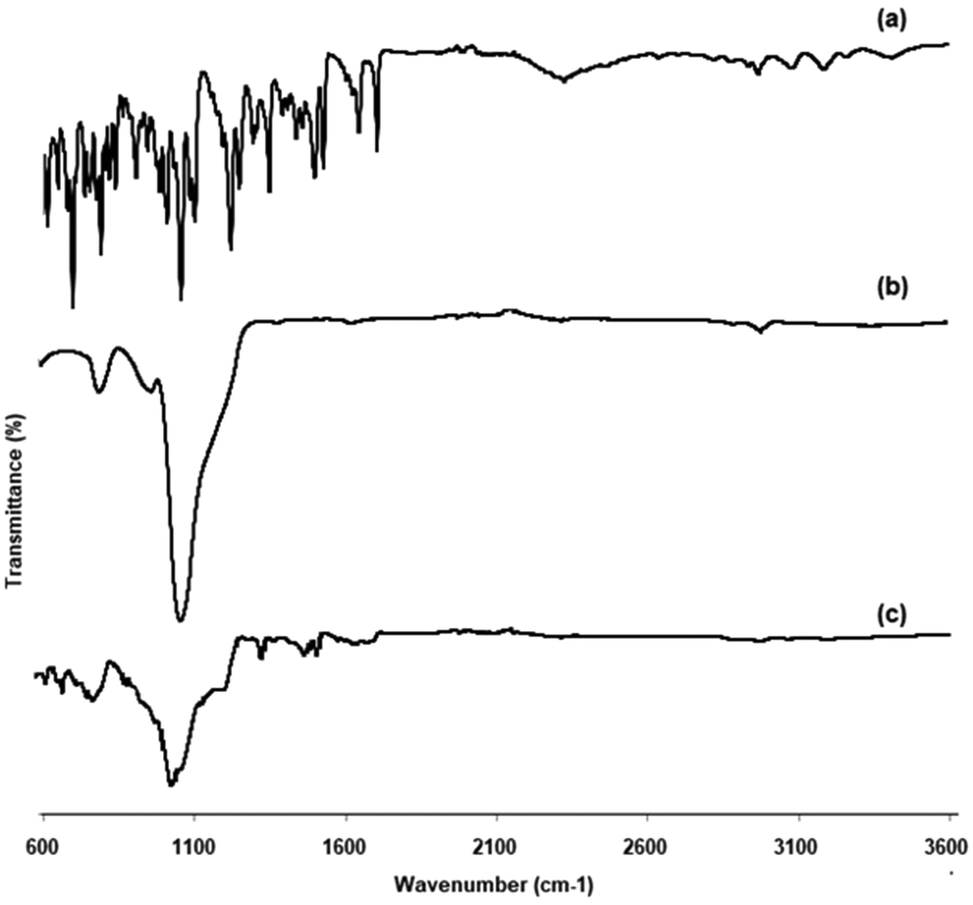
3.2.2. FTIR.
FTIR spectroscopy analysis helps to investigate the intermolecular interaction and compatibility between the drug and carrier. Fig. 2 depicts the FTIR spectra of EFE, mesoporous silica, and EFESD. The principal peaks observed for EFE at specific wavelengths along with functional groups are aromatic C–H (3187.76 cm−1), alkyl C–H (2965.98 cm−1), ester C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (1704.76 cm−1), aromatic nitro (1526.38 cm−1), C–N (1348 cm−1), PO (1101.15 cm−1) and NH stretching at 3415.31 cm−1. The FTIR spectrum of mesoporous silica showed silanol (Si–OH) symmetric stretching at 3396.49 cm−1, Si–O–Si bending at 1053.78 cm−1, and toxiloxan bond at 797.97 cm−1. However, the FTIR spectrum of EFESD exhibited the Si–O–Si bending at 1053.78 cm−1. The presence of a single peak of EFE and mesoporous silica in the FTIR spectrum of EFESD indicates no distinction at the molecular level between the internal structure and conformation of these samples.33
O (1704.76 cm−1), aromatic nitro (1526.38 cm−1), C–N (1348 cm−1), PO (1101.15 cm−1) and NH stretching at 3415.31 cm−1. The FTIR spectrum of mesoporous silica showed silanol (Si–OH) symmetric stretching at 3396.49 cm−1, Si–O–Si bending at 1053.78 cm−1, and toxiloxan bond at 797.97 cm−1. However, the FTIR spectrum of EFESD exhibited the Si–O–Si bending at 1053.78 cm−1. The presence of a single peak of EFE and mesoporous silica in the FTIR spectrum of EFESD indicates no distinction at the molecular level between the internal structure and conformation of these samples.33
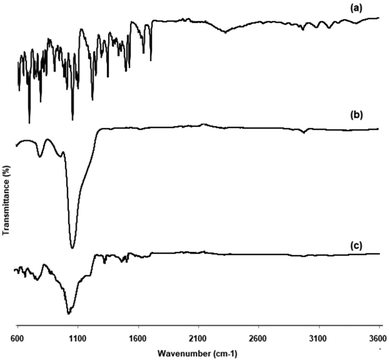 |
| | Fig. 2 FTIR spectra of (a) EFE, (b) mesoporous silica, and (c) EFESD. | |
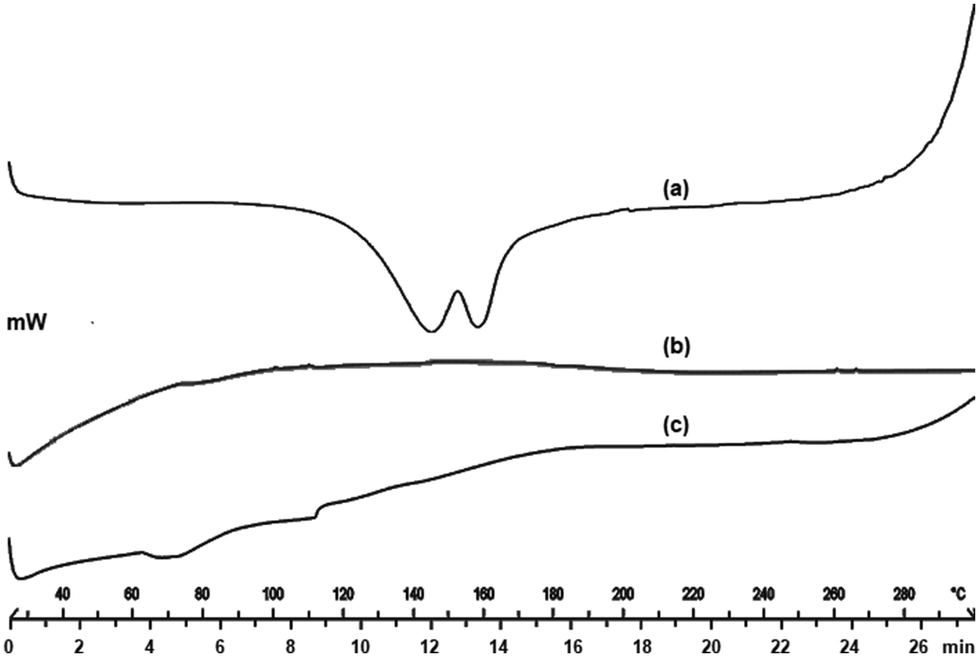
3.2.3. DSC.
DSC is the most preferred thermal technique that provides information on the melting point, glass transition temperature, and energy changes associated with the phase transition, including the crystallization and fusion process. DSC thermogram of EFE showed a double hump with a unique melting endotherm at 140 °C and 160 °C, indicating the volatilization of ethanol, followed by the removal of chloride ion at 140 °C and subsequent breakdown at 160 °C (Fig. 3a). The absence of a peak in the DSC thermogram of mesoporous silica (Fig. 3b) indicates its amorphous nature. The thermal behavior of mesoporous silica is typical of hygroscopic and amorphous materials, with a significant endo-thermal effect. The DSC thermogram of EFESD (Fig. 3c) showed small endothermic peaks at 70 °C and 110 °C attributed to the Tg of EFE with no endothermic drug peak, indicating that the EFE was molecularly dispersed into the pores of mesoporous silica and transformed from crystalline to amorphous nature, which was attributed to the improvement in solubility and dissolution.34
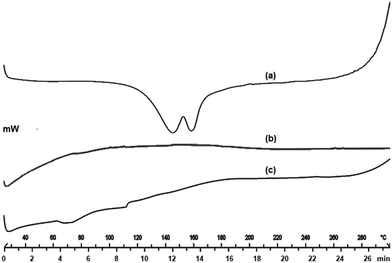 |
| | Fig. 3 DSC thermograms of (a) EFE, (b) mesoporous silica, and (c) EFESD. | |
3.2.4. XRD.
EFE diffractogram showed several peaks from 5° to 40° (2θ values), confirming its crystalline nature (Fig. 4). The mesoporous silica was amorphous, with just two peaks observed at 5.12° and 22.04° (2θ values). The absence of intense EFE peaks, followed by the formation of a halo pattern in the XRD diffractogram of EFESD, indicates the transition from the crystalline to amorphous state as well as the amorphous dispersion of the drug in the polymeric matrix, which agrees with DSC.35
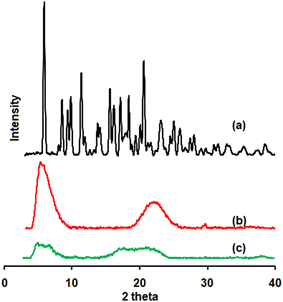 |
| | Fig. 4 Powder X-ray diffractograms of (a) EFE, (b) mesoporous silica and (c) EFESD. | |
3.2.5. FESEM analysis.
The FESEM image of EFE showed flat, irregular crystals with a smooth surface (Fig. 5a and b). Small and irregularly shaped particles were observed in the FESEM images of mesoporous silica (Fig. 5c and d). However, FESEM images of the solid dispersion confirmed significant absorption of EFE within the porous structure of mesoporous silica, which conforms to the conversion of EFE to the amorphous forms (Fig. 5e and f).35–37
 |
| | Fig. 5 FESEM images of (a and b) EFE, (c and d) mesoporous silica and (e and f) EFESD. | |
3.2.6. Drug content.
The quality of pharmaceutical preparations is crucial for maintaining a consistent dose of the drug. The EFESD containing the ratios 1:1, 1:2, and 1:3 showed drug contents of 64.20 ± 1.98, 74.87 ± 1.25, and 62.35 ± 2.15 (values are expressed as mean ± SD, (n = 3)), respectively (Alam et al. 2018; Munir et al. 2022).38,39 The 1:2 ratio showed a higher drug content. However, it did not show solubility improvement compared to the 1:1 ratio. Considering the solubility, a ratio of 1:1 was optimized and used for further analysis.
3.2.7.
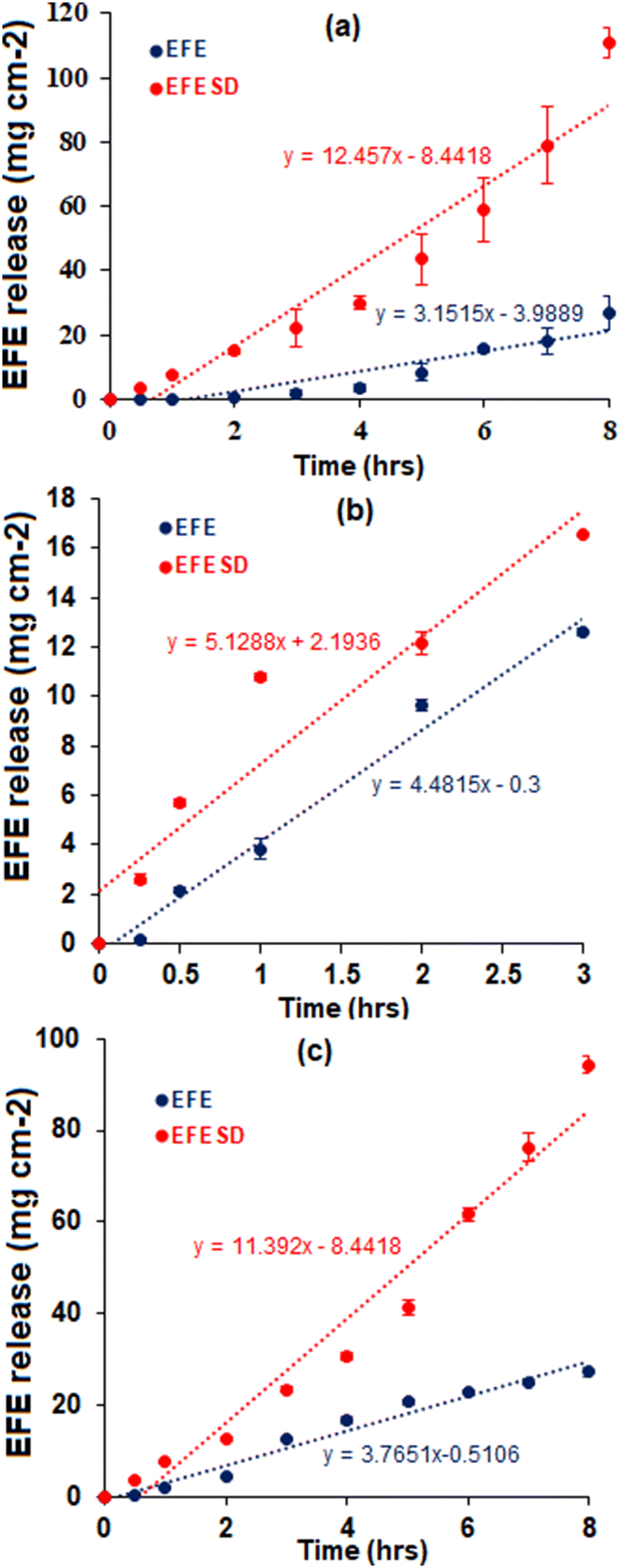
In vitro dissolution rate study.
The dissolution profiles of EFE and EFESD in different dissolution media are shown in Fig. 6. The rates of dissolution for EFE and EFESD in distilled water, 0.1 N HCl (pH 1.2), and phosphate buffer (pH 6.8) with 0.05% w/v SLS are summarized in Table 1. The solid dispersion showed a 3.54-fold and 3.02-fold increase in the dissolution rate in distilled water and phosphate buffer (pH 6.8) (0.05% w/v SLS), respectively. This was attributed to the entrapment of EFE in the porous structure of mesoporous silica as well as the high surface area exposed to the dissolution media.40,41
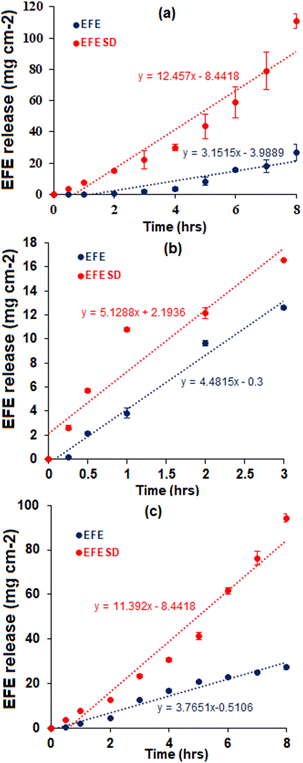 |
| | Fig. 6
In vitro intrinsic dissolution profiles of EFE and EFESD in (a) distilled water, (b) 0.1 N HCl (pH 1.2) and (c) phosphate buffer (pH 6.8) (0.05% w/v SLS). | |
Table 1 Dissolution rate of EFE and EFESD in different mediaa
| Dissolution medium |
Rate of dissolution (mg cm−2 h−1) |
| EFE |
EFESD |
|
Values are expressed as mean ± SD and n = 3.
|
| 0.1 N HCl (pH 1.2) |
4.20 ± 0.26 |
6.23 ± 0.17 |
| Phosphate buffer (pH 6.8) (0.05% w/v SLS) |
3.40 ± 0.11 |
11.79 ± 0.24 |
| Distilled water |
3.36 ± 0.64 |
13.85 ± 0.59 |
3.2.8.
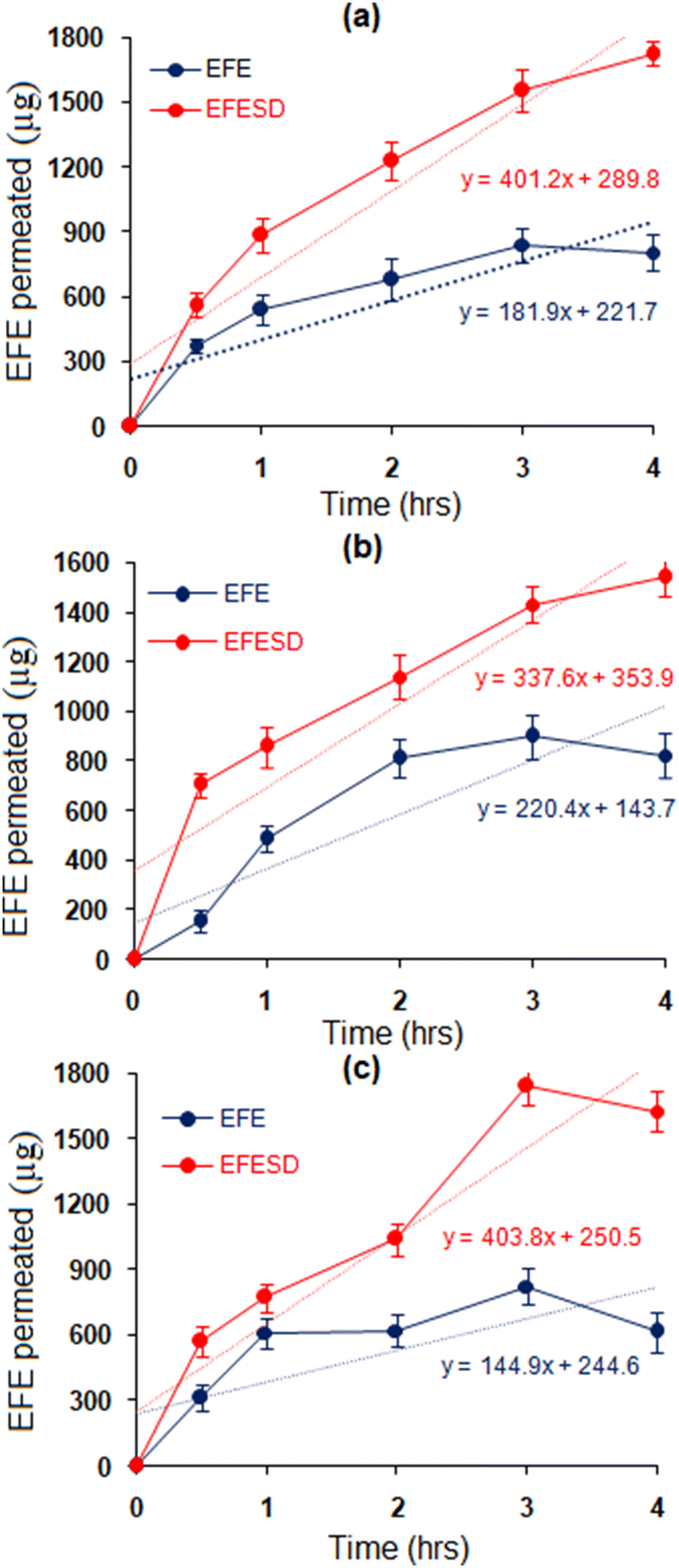
Ex vivo permeation study.
The permeation study of EFE and EFESD was performed using the non-everted sac method in three different intestinal sections (duodenum, jejunum, and ileum) of rats to determine site-dependent absorption. Further, the data obtained were used to plot the amount of EFE permeated against time (Fig. 7). The rates of permeation and apparent permeation coefficients of EFE and EFESD are summarized in Table 2. EFESD showed a 2-fold increase in the rate of permeation and apparent permeability coefficient in a mixture of KRS (pH 7.4) and isopropyl alcohol (70:30 v/v) compared to EFE. The improved solubility of EFE and the advantageous partitioning through the intestinal barrier were attributed to this. No discernible difference in penetration was observed across the three intestinal regions, indicating non-specific absorption of EFE through the small intestine.42
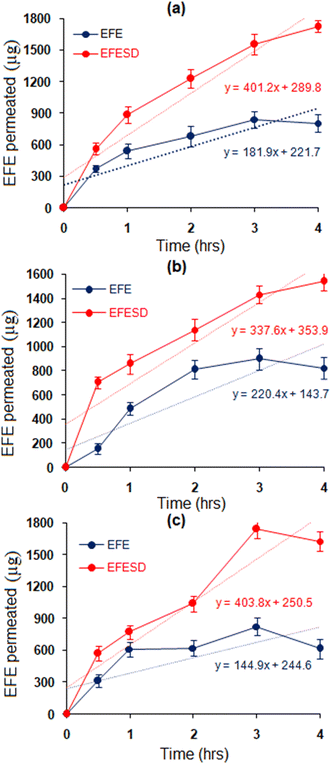 |
| | Fig. 7
Ex vivo permeation study using non-everted rat intestine sections, viz. (a) duodenum, (b) jejunum and (c) ileum. | |
Table 2 Permeation rate and apparent permeability coefficients of EFE and EFESD in different sections of the rat intestinea
| Parameters |
Duodenum |
Jejunum |
Ileum |
| EFE |
EFESD |
EFE |
EFESD |
EFE |
EFESD |
|
Values are expressed as mean ± SD and n = 3.
|
| Rate of permeation (μg h−1) |
144.44 ± 12.30 |
403.86 ± 6.06 |
207.84 ± 19.57 |
337.60 ± 16.08 |
181.97 ± 2.04 |
401.78 ± 12.05 |
| Apparent permeability coefficients (cm h−1) |
109.52 ± 12.13 |
321.54 ± 14.32 |
144.14 ± 18.74 |
274.97 ± 16.63 |
120.73 ± 9.80 |
283.94 ± 13.90 |
3.2.9.
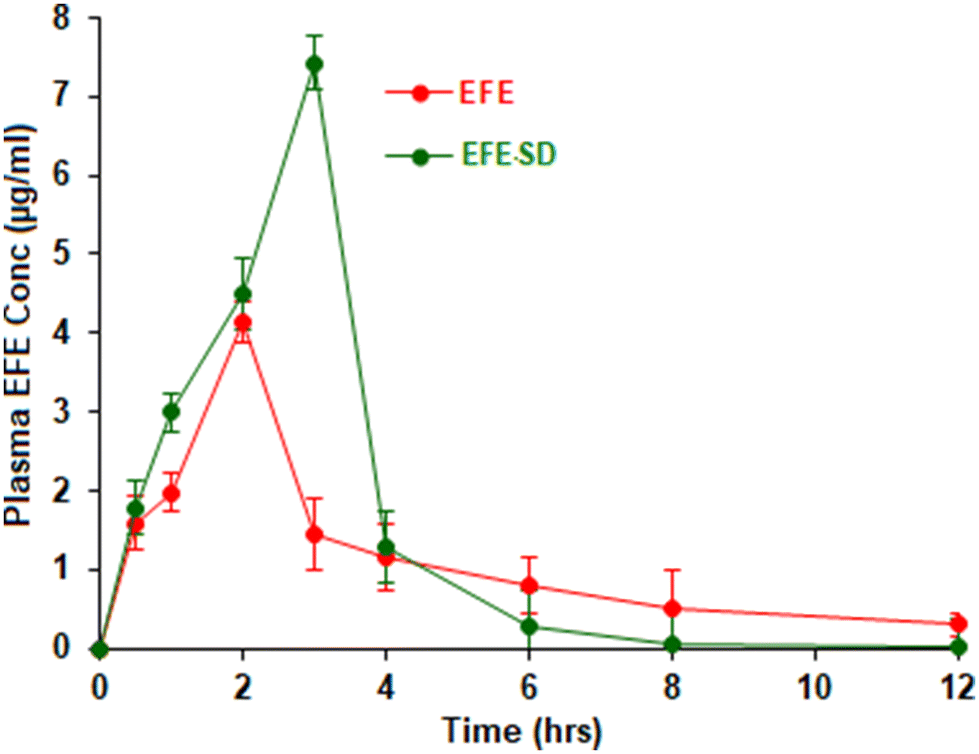
In vivo pharmacokinetic studies.
The in vivo pharmacokinetic parameters observed for EFE and EFESD after oral administration are summarized in Table 3. The oral bioavailability of EFE and EFESD was assessed from the plasma EFE concentration–time profile (Fig. 8). The EFESD showed a 2.10, 1.5, and 1.41-fold increase in Cmax, Tmax, and AUC, respectively, compared to pure EFE. This might be attributed to the improved solubility, dissolution, and ultimately bioavailability associated with an amorphous form of the drug in EFESD.43 The EFESD showed lower MRT compared to pure EFE attributed to improvement in solubility, dissolution and absorption, which led to the rapid removal of the drug from systemic circulation.
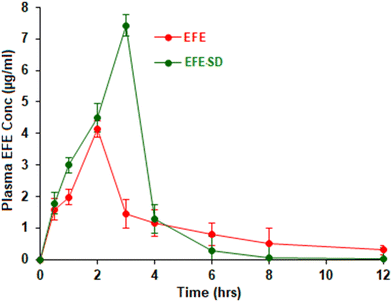 |
| | Fig. 8 Plasma concentration–time profile of EFE and EFESD. | |
Table 3 Pharmacokinetic parameters of EFE and EFESDa
| Parameters |
EFE |
EFESD |
|
Values are expressed as mean ± SD and n = 3.
|
|
C
max (ng mL−1) |
3547.56 ± 61.96 |
7453.73 ± 83.46 |
|
T
max (hours) |
2 |
3 |
| AUC (ng h mL−1) |
11883.52 ± 121.97 |
16804.75 ± 132.56 |
| MRT (hours) |
5.25 ± 0.56 |
2.27 ± 0.74 |
3.2.10.
In vivo pharmacodynamic studies.
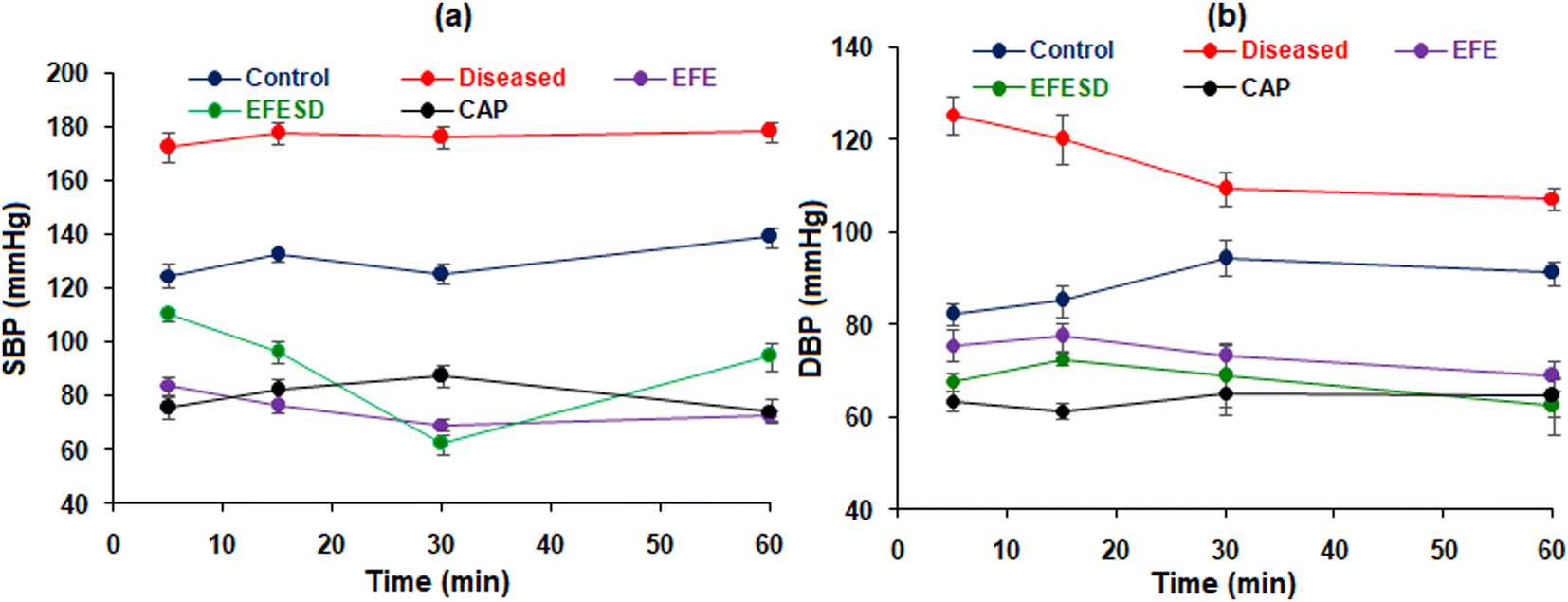
When the renal artery is blocked for up to 4 hours, kidney ischemia occurs, which triggers the renin–angiotensin system and increases blood pressure. In male Wistar rats, acute renal hypertension was created by clamping the left renal artery for four hours; once the vessels were reopened, accumulated renin was released into the bloodstream. The removal of the renal bulldog clip resulted in a significant increase in MABP, SBP, and DBP in the disease control group. The MABP, SBP, and DBP of all groups at different time intervals are shown in Table 4 and Fig. 9. Administration of captopril (1 mg kg−1) in the standard group of animals showed a significant reduction in MABP, SBP, and DBP.44,45 After an hour, there was a rapid reduction in MABP, which could cause reflex tachycardia and an increase in sympathetic tone. EFESD shows enhanced antihypertensive activity compared to pure EFE. The pharmacodynamic data were analysed using two-way ANOVA, and the results were found to be statistically significant (p < 0.05).
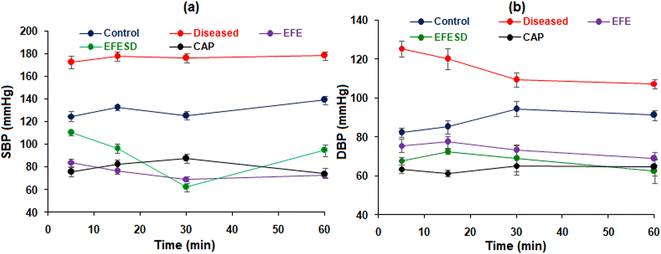 |
| | Fig. 9 (a) Systolic and (b) diastolic blood pressure of the control, diseased, standard, EFE, and EFESD groups. | |
Table 4 Mean arterial blood pressure at different time intervalsa
| Groups |
Mean arterial blood pressure (MABP) in mmHg at different time intervals |
| Treatments (mg kg−1) |
MABP after removing the clip |
5 min |
15 min |
30 min |
60 min |
|
Values are expressed as mean ± SD (n = 6). Bars indicate ***P – 0.0001 and ****P < 0.000 via two-way ANOVA with Tukey's test analysis.
|
| Control |
Distilled water (p.o.) |
— |
85.22 ± 1.13 |
88.51 ± 1.34 |
85.88 ± 1.21 |
92.89 ± 1.45 |
| Disease |
Distilled water (p.o.) |
132.85 ± 2.05**** |
122.00 ± 1.50**** |
126.36 ± 1.22**** |
118.10 ± 1.55**** |
113.35 ± 1.87**** |
| Standard |
1 (iv) |
122.44 ± 2.68**** |
62.97 ± 2.18**** |
60.45 ± 1.07**** |
57.60 ± 1.70**** |
51.84 ± 2.86**** |
| EFE |
4.11 (iv) |
117.01 ± 2.24**** |
83.82 ± 3.13**** |
76.21 ± 2.16**** |
69.24 ± 2.08**** |
72.71 ± 1.89**** |
| EFESD |
4.11 (iv) |
109.67 ± 4.53**** |
73.33 ± 2.22**** |
67.67 ± 1.61**** |
60.54 ± 1.17**** |
71.90 ± 5.84**** |
4. Conclusion
Solid dispersion of poorly water-soluble EFE was prepared using mesoporous silica as a carrier by applying a solvent evaporation method. The significant absorption of EFE within the porous structure of mesoporous silica together with the transition from the crystalline to amorphous form in solid dispersion results in improved solubility, dissolution rate, intestinal permeability, and, ultimately, bioavailability and antihypertensive activity. Therefore, mesoporous silica can be used to prepare solid dispersions of other BCS class-II drugs, potentially improving their physicochemical properties and therapeutic efficacy.
Abbreviations
| GIT | Gastrointestinal tract |
| EFE | Efonidipine hydrochloride ethanolate |
| BCS | Biopharmaceutical classification system |
| DMSO | Dimethyl sulfoxide |
| EFESD | Efonidipine hydrochloride ethanolate solid dispersion |
| CPCSEA | Committee for the purpose of control and supervision on experimental animals |
| IAEC | Institutional animal ethics committee |
| DSC | Differential scanning calorimetry |
| XRD | X-ray diffraction |
| FTIR | Fourier transform infrared |
| FESEM | Field emission scanning electron microscopy |
| EDTA | Ethylene diamine tetraacetic acid |
| ANOVA | Analysis of variance |
| SD | Standard deviation |
| MABP | Mean arterial blood pressure |
| SBP | Systolic blood pressure |
| DBP | Diastolic blood pressure |
| AUC | Area under curve |
| MRT | Mean residence time |
| SLS | Sodium lauryl sulphate |
| HPLC | High performance liquid chromatography |
| KRS | Krebs-Ringer solution |
Ethical statement
All animal experiments were carried out with the prior approval of the Institutional Animal Ethics Committee of Poona College of Pharmacy, Bharati Vidyapeeth Deemed University (PCP/IAEC/2021-22/2-40), registered under the Committee for the Purpose of Control and Supervision of Experiments on Animals, Government of India. All experiments were performed per the U. K. Animals Act (1986) and its accompanying recommendations, as detailed in the ARRIVE guidelines.
Author contributions
Miss Swati Bharati: writing – original draft, drawing figure. Dr V. L. Gaikwad: supervision, editing, and review. Dr C. Bothiraja: supervision, editing, and review.
Conflicts of interest
There are no conflicts to declare.
References
- S. Gupta, R. Kesarla and A. Omri, Formulation Strategies to Improve the Bioavailability of Poorly Absorbed Drugs with Special Emphasis on Self-Emulsifying Systems, Adv. Challenges Pharm. Technol., 2021, 229–242 CAS.
- D. V. Bhalani,
et al., Bioavailability Enhancement Techniques for Poorly Aqueous Soluble Drugs and Therapeutics, Biomedicines, 2022, 10, 1–33 CrossRef PubMed.
- M. S. Alqahtani,
et al., Advances in Oral Drug Delivery, Front. Pharmacol., 2021, 12, 1–21 CrossRef PubMed.
- K. T. Savjani, A. K. Gajjar and K. Savjani, Drug Solubility: Importance and Enhancement Techniques, ISRN Pharm., 2012, 1–10 Search PubMed.
- D. Horter and J. B. Dressman, Influence of physiochemical properties on dissolution of drugs, Adv. Drug Delivery Rev., 1997, 25, 3–14 CrossRef.
- A. A. Enose,
et al., Formulation and Characterization of Solid Dispersion Prepared by Hot Melt Mixing: A Fast Screening Approach for Polymer Selection, J. Pharm., 2014, 1–13 Search PubMed.
- T. Yokoyama, K. Ichihara and Y. Abiko, Efonidipine, a Long-Acting Dihydropyridine Derivative, Attenuates coronary Vasoconstriction Induced by Endothelin-1 in Dogs, Jpn. J. Pharmacol., 1996, 72, 291–297 CrossRef CAS PubMed.
- C. Shudo,
et al., Studies on the hypotensive mechanisms of NZ-105, a new dihydropyridine derivative, in rats and rabbits, Gen. Pharmacol., 1993, 24, 411–418 CrossRef CAS PubMed.
- H. Tanaka and K. Shigenobu, Efonidipine hydrochloride: A dual blocker of L- and T-type Ca2+ channels, Cardiovasc. Drug Rev., 2002, 20, 81–92 CrossRef CAS PubMed.
-
L. Kwon and C. Rosendorff, The Medical Treatment of Stable Angina, Chronic Coronary Artery Disease: A Companion to Braunwald's Heart Disease, Elsevier Inc., 2018, pp. 280–302 Search PubMed.
- C. P. Pandya and S. J. Rajput, Forced degradation study of efonidipine HCl ethanolate, characterization of degradation products by LC-Q-TOF-MS and NMR, J. Appl. Pharm. Sci., 2020, 10, 75–99 CAS.
- PubChem, Efonidipine, National Library of Medicine, 2020, accessed November 21, 2022.
- Drugs Controller General of India, Efonidipine, in Wikipedia, 2022, pp. 1–6.
- S. Sareen, L. Joseph and G. Mathew, Improvement in solubility of poor water-soluble drugs by solid dispersion, Int. J. Pharm. Invest., 2012, 2, 1–6 CrossRef.
- R. Malkawi,
et al., Current Trends on Solid Dispersions: Past, Present, and Future, Adv. Pharmacol. Pharm. Sci., 2022, 1–17 Search PubMed.
- M. Kakran, L. Li and R. H. Müller, Overcoming the challenge of poor drug solubility, Pharm. Eng., 2012, 32, 82–89 Search PubMed.
- M. S. Attia,
et al., Solid dispersion as a technical solution to boost the dissolution rate and bioavailability of poorly water-soluble drugs, Indian J. Pharm. Educ. Res., 2021, 55, s327–s339 CrossRef CAS.
- M. Otsuka,
et al., Solid dispersions of efonidipine hydrochloride ethanolate with improved physicochemical and pharmacokinetic properties prepared with microwave treatment, Eur. J. Pharm. Biopharm., 2016, 108, 25–31 CrossRef CAS PubMed.
- X. Cheng,
et al., In Vitro-In Vivo Correlation for Solid Dispersion of a Poorly Water-Soluble Drug Efonidipine Hydrochloride, AAPS PharmSciTech, 2020, 21, 1–16 CrossRef PubMed.
- A. S. Rajput,
et al., RP-HPLC method development and validation for the quantification of Efonidipine hydrochloride in HME processed solid dispersions, Future J. Pharm. Sci., 2020, 6, 1–9 CrossRef.
- K. Bukara,
et al., Oredered mesoporous silica to enhance the bioavailability of poorly water-soluble drugs: Proof of concept in man, Eur. J. Pharm. Biopharm., 2016, 108, 220–225 CrossRef CAS PubMed.
- K. Bukara,
et al., Invivo performance of Fenofibrate formulated with ordered mesoporous silica versus 2-marketed formulation: a comparative bioavailability study in beagle dogs, J. Pharm. Sci., 2016, 105, 2381–2385 CrossRef CAS PubMed.
- S. Chaudhari and A. Gupte, Mesoporous Silica as a Carrier for Amorphous Solid Dispersion, Br. J. Pharm. Res., 2017, 6, 1–19 Search PubMed.
- B. Daravath,
et al., Solubility and dissolution enhancement of flurbiprofen by solid dispersion using hydrophilic carriers, Braz. J. Pharm. Sci., 2017, 53, 1–10 Search PubMed.
- S. Shamsuddin,
et al., Development and evaluation of solid dispersion of spironolactone using fusion method, Int. J. Pharm. Invest., 2016, 6, 63–68 CrossRef PubMed.
- A. Sharma and C. P. Jain, Preparation and characterization of solid dispersions of carvedilol with PVP K30, Res. Pharm. Sci., 2010, 5, 49–56 CAS.
- R. J. Chokshi,
et al., Improving the dissolution rate of poorly water soluble drug by solid dispersion and solid solution – Pros and cons, Drug Delivery, 2007, 14, 33–45 CrossRef CAS PubMed.
- Z. Attari,
et al., Enhanced ex vivo intestinal absorption of olmesartan medoxomil nanosuspension: Preparation by combinative technology, Saudi Pharm. J., 2015, 1–23 Search PubMed.
- S. Bharati, V. Gaikwad and C. Bothiraja, Bioanalytical method development and Validation for determination of efonidipine hydrochloride ethanolate in rat plasma by high-performance liquid chromatography: applications for pharmacokinetic study, Anal. Chem. Lett., 2023, 13(4), 394–402 CrossRef CAS.
- S. S. Sakat,
et al., Antihypertensive effect of aqueous extract of Elaeocarpus ganitrus Roxb. seeds in renal artery occluded hypertensive rats, Int. J. PharmTech Res., 2009, 1, 779–782 Search PubMed.
- M. Alcoutlabi and G. McKenna, Effects of confinement on material behaviour at the nanometre size scale, J. Phys.: Condens. Matter, 2012, 17, 461–524 CrossRef.
- Y. Huang,
et al., Enhanced solubility and bioavailability of apigenin via preparation of solid dispersions of mesoporous silica nanoparticles, Iran. J. Pharm. Res., 2019, 18, 168–182 CAS.
- O. Planinšek, B. Kovačič and F. Vrečer, Carvedilol dissolution improvement by preparation of solid dispersions with porous silica, Int. J. Pharm., 2011, 406, 41–48 CrossRef PubMed.
- K. Krishnamurthy and P. Patel, Solubility Enhancement of Diflunisal by Solid Dispersion Techniques, Int. J. Pharm. Res. Scholars, 2018, 7, 23–29 CrossRef.
- S. C. Arora,
et al., Development, characterization and solubility study of solid dispersions of cefuroxime axetil by the solvent evaporation method, J. Adv. Pharm. Technol. Res., 2010, 1, 326–329 CAS.
- A. Das,
et al., Solubility and Dissolution Enhancement of Etoricoxib by Solid Dispersion Technique Using Sugar Carriers, ISRN Pharm., 2011, 1–8 Search PubMed.
- Z. Xi,
et al., Evaluation of the solid dispersion system engineered from mesoporous silica and polymers for the poorly water soluble drug indomethacin: In vitro and in vivo, Pharmaceutics, 2020, 12, 1–19 CrossRef PubMed.
- A. Alam,
et al., Formulation and evaluation of solid dispersion and inclusion complex of poorly aqueous soluble diacerein, JOJ Mater. Sci., 2018, 5, 1–9 Search PubMed.
- M. U. Munir,
et al., Fabrication, In Vitro and In Vivo Evaluation of Non-Ordered Mesoporous Silica-Based Ternary Solid Dispersions for Enhanced Solubility of Flurbiprofen, Pharmaceuticals, 2022, 15, 856–873 CrossRef CAS PubMed.
- P. C. Chiang,
et al., In vitro and in vivo evaluation of amorphous solid dispersions generated by different bench-scale processes, using griseofulvin as a model compound, AAPS J., 2013, 15, 608–617 CrossRef CAS PubMed.
- S. Alshehri,
et al., Enhanced Dissolution of Luteolin by Solid Dispersion Prepared by Different Methods: Physicochemical Characterization and Antioxidant Activity, ACS Omega, 2020, 5, 6461–6471 CrossRef CAS PubMed.
- W. L. Masiiwa and L. L. Gadaga, Intestinal Permeability of Artesunate-Loaded Solid Lipid Nanoparticles Using the Everted Gut Method, J. Drug Delivery, 2018, 1–9 Search PubMed.
- L. F. Yin,
et al., In vitro and in vivo studies on a novel solid dispersion of repaglinide using polyvinylpyrrolidone as the carrier, Drug Dev. Ind. Pharm., 2012, 38, 1371–1380 CrossRef CAS PubMed.
- D. Villarreal, R. H. Freeman and J. O. Davis, Pathogenesis of one-kidney, one-clip hypertension in rats after renal denervation, Am. J. Physiol.: Heart Circ. Physiol., 1984, 16, 61–66 CrossRef PubMed.
- S. A. Nurfaradilla, F. C. Saputri and Y. Harahap, Effects of Hibiscus Sabdariffa Calyces Aqueous Extract on the Antihypertensive Potency of Captopril in the Two-Kidney-One-Clip Rat Hypertension Model, Evidence-Based Complementary Altern. Med., 2019, 1–8 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Vinod
Gaikwad
a,
Vinod
Gaikwad