
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Programmable protein delivery from microgel/hydrogel composites (MHCs) via discrete combinations of multi-state protein-loaded unit ingredients†
Longfei
Chen
 and
Saif A.
Khan
*
and
Saif A.
Khan
*
Department of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, Singapore 117576, Singapore. E-mail: saifkhan@nus.edu.sg
First published on 20th August 2024
Abstract
Therapeutic proteins have drawn increasing attention in the development of advanced drugs and biomedical devices, yet there are outstanding challenges for the delivery of multiple-protein therapies with customized release profiles. Hydrogel-based drug delivery systems (DDS) have been widely investigated, primarily via highly specific chemical modification routes, for programmable topical, injectable, and depot-based protein delivery. In this paper, we propose a microgel/hydrogel composite (MHC) DDS for tunable and programmable multi-protein delivery, which leverages different physical states of proteins (freely dissolved or coacervated) and completely avoids bespoke chemical modifications on the hydrogels. We load model proteins in distinct physical states into dextran-based hydrogel microparticles (microgels) fabricated using microfluidics, after which simple discrete combinations of these microgel ‘unit ingredients’ are packaged into poly(ethylene glycol) hydrogel matrices to formulate the MHC DDS. With discrete combinations of unit ingredients, we demonstrate how these MHC DDSs can achieve both tunable release for a single low-molecular-weight model protein (and ideally, highly similar proteins) and a counterintuitive rate-reversed release of two model proteins that are vastly different in size. Moreover, we show that these MHCs follow Korsmeyer-Peppas kinetic behavior as a function of the discrete combinations packaged, thus highlighting the quantitative tunability of release behaviors. We envision the use of these MHC DDSs as topically applied wound dressings or implantable protein-releasing depots that allow scheduled and programmable multi-protein delivery in biomedical and clinical applications.
Introduction
Therapeutic proteins play an increasingly prominent role in the development of advanced therapies such as immunotherapy1 for the treatment of cancers,2 allergies,3 autoimmune diseases,4 cytokine storms,5 and in the development of regenerative medicines6 and wound healing dressings.7 Immunotherapy involves the precise regulation or modulation (promotion or suppression) of immune systems1,8 and relies heavily on the controlled co-delivery of various therapeutic proteins including antibodies, cytokines, growth factors, chemokines, receptors, etc.1,8,9 Given the intricate roles that proteins play in the biological signalling pathways, effective release scheduling is essential to activate efficient and safe therapeutic effects. This can entail the coordinated co-delivery of multiple proteins, each with distinct functions, at designated times due to their different pharmacokinetic and pharmacodynamic properties.1,10 For example, during the wound healing process, the high-molecular-weight protein fibrinogen first promotes hemostasis, followed by the action of cytokines such as interleukin-1 beta (IL-1β) and interleukin-4 (IL-4) serving pro-inflammatory and anti-inflammatory functions separately, in cooperation with antibodies to neutralize potential pathogens. Thus, to mimic this natural healing process in a therapy would require a drug delivery system (DDS) comprising the drugs (in this case multiple proteins) and a vector/carrier to load, protect, and deliver the drugs, with appropriately designed molecular interactions to allow programmable drug release.11 Commonly used DDSs include lipid nanoparticles, polymer nanoparticles, hydrogels, dendrimers, etc.11 For the above-mentioned example, a protein-releasing DDS for wound healing would entail the initial delivery of fibrinogen, followed by the sequential delivery of pro- and anti-inflammatory cytokines with sustained antibody supply. Thus, achieving optimal therapeutic effects may require intricate programming and design of the release behavior of each protein from the DDS, so as to achieve, for example: (i) synchronized release of proteins that are vastly different in size, (ii) differential release of multiple proteins that are similar in size, (iii) tunable release of a single protein at multiple time scales, and (iv) rate-reversed release where a smaller protein is released slower than a larger protein in opposition to typical size-exclusive diffusion-driven release mechanisms from typical DDS matrices.12 Since the release behavior of a protein is highly dependent on both the material and formulation of the DDS and the interactions between the protein and the DDS matrix,12 it is typically challenging to realize the types of programmable release listed above for multi-protein co-delivery, requiring careful design of the DDS matrix material as well as the multi-protein formulation within.Hydrogels, a category of water-absorbing soft materials synthesized by crosslinking hydrophilic backbone polymers, represent state-of-the-art matrix materials for drug delivery systems, accommodating a diverse range of drug entities including small-molecule active pharmaceutical ingredients (APIs), therapeutic proteins, nucleic acids, and various types of nanoparticles.1,12,13 The crosslinked network microstructure of hydrogel is meshed at microscopic scales and this enables the encapsulation or immobilization of drug entities; based on the disparity between hydrogel mesh size and the hydrodynamic diameter of the drug entity.12 The substantial water absorption of hydrogels in aqueous media induces volumetric swelling which expands the network microstructure and enlarges the mesh size, facilitating the movement and subsequent (primarily) diffusion-driven release of encapsulated drug entities.12–14 Generally, the average mesh size at the equilibrium swollen state dictates the release mechanism and behavior of a certain drug from a certain hydrogel12–14 – if a drug has a hydrodynamic diameter exceeding the swollen mesh size, the network impedes diffusion, resulting in hindered release, or even immobilization; conversely, for drug molecules smaller than the swollen mesh size, a rapid burst release occurs. Typical mesh size varies between 5 and 80 nm for common hydrogels, and are dependent on the choice and concentrations of backbone polymers, crosslinking mechanisms, and the density and distribution of crosslinks.12,13 This complexity is typically self-limiting, in that once a particular hydrogel fabrication formulation is selected, the mesh size is effectively “locked in”, and cannot be varied in a fine-tuned way to enable the programmable release of multiple molecules of varying size; this naturally poses a significant design challenge due to the broad range of hydrodynamic sizes exhibited by proteins.12,13 For example, small signalling proteins such as cytokines and growth factors (∼3–4 nm)15a typically exhibit rapid, burst release profiles while the release of high-molecular-weight proteins such as antibodies (∼10–12 nm)15b is typically much slower. Hence, strategies that effectively modulate or deviate from the generic diffusion-driven release mechanism assume significance in tackling the challenges facing programmed co-delivery of multiple proteins of varying size.
In pursuit of more customizable multi-protein release mechanisms from hydrogel DDS, recent research has delved into formulation strategies that exploits intermolecular interactions between protein molecules and the hydrogel network or associated excipients.12,16 Approaches such as coacervation involving polyelectrolyte electrostatic interactions between charged proteins and oppositely charged polymers,17 physical chain entanglement utilizing pendent poly(ethylene glycol) (PEG) chains on PEGylated proteins and free PEG components,18 embedding protein crystals,19 and utilization of specific ligand-tag bio-affinities20 have all been investigated for achieving sustained and non-burst release of various low-molecular-weight proteins. Notably, recent work by Appel and colleagues has advanced the field by combining multiple formulation strategies in a shear-thinning injectable liposomal nanocomposite hydrogel (LNH) protein depot, yielding a set of differential release profiles for a single low-molecular-weight protein and a synchronized co-release profile for two proteins of disparate sizes.20 In their work, three distinct release modes were attained for the low-molecular-weight interleukin-12 (IL-12), via surface modifications on composite liposomes: a rapid passive diffusion-driven burst release mode without any surface modification, an ‘intermediate’ slow mode employing electrostatic hindrance between positively charged IL-12 and negatively charged liposomal surface, and an even slower mode enabled by the affinity between His-tagged IL-12 and nickel (Ni) or cobalt (Co) based surface ligands (nitrilotriacetic acid, NTA). Thus, the authors successfully demonstrated differential release of IL-12, with a 50%-released time ranging from 4 days (diffusion-driven mode) to 6 days (electrostatically hindered mode) and ultimately 10 days (affinity hindered mode). Furthermore, they showcased synchronized release of a large antibody, immunoglobulin G (IgG), in its inherent slow diffusion-driven mode and IL-12 slowed by electrostatic interactions. Nevertheless, despite such progress in designing hydrogels to enable programmable multi-protein co-delivery, we note the inherent limitation that each ‘designer’ release profile for a particular protein necessitates a specific, bespoke modification of the hydrogel matrix. In the above example of Appel et al., each new release profile entails the fabrication of a hydrogel matrix with a designated degree of surface modification for either the electrostatically hindered or affinity hindered mode20 that, once fabricated, would preclude the other two modes and, crucially, is hard to be further fine-tuned after fabrication.
Motivated by this limitation, in this work, we investigate the possibility of more general, flexible, and broadly applicable approaches for multi-protein co-delivery with tunable release that involve simple combinations of ‘unit ingredients’ to achieve programmable release, such as to deliver two similar proteins at different rates or two proteins of vastly distinct sizes in a similar or reversed rate (referring to the previously described inflammation-modulation example), without necessitating re-design of the hydrogel matrix. Our approach is conceptually depicted in Fig. 1. The multi-protein loaded hydrogel DDS developed in this study consists of two key components – (i) spherical hydrogel microparticles (hereafter we refer to these microparticles as ‘microgels’) loaded with either dissolved (i.e., solute form) proteins (hereafter we refer to these microgels with a hyphenated prefix ‘sol-’) or protein coacervates (‘coa-’); these microgels are the ‘unit ingredients’ and (ii) combination and packaging of microgel unit ingredients in appropriate mass or number fraction into a bulk carrier hydrogel matrix to achieve a variety of protein release outcomes in a straightforward fashion. This work is grounded in the concept of soft microgel/hydrogel composite (MHC) systems that have been previously developed and demonstrated for small molecule API21 and single protein21c delivery. In a set of studies by Hoare and co-workers, acrylic/acrylamide-based microgels containing bupivacaine were fabricated by precipitation-emulsification polymerization and co-extruded with a chemically crosslinked polysaccharide hydrogel matrix to form the MHC system, while a variety of tuning approaches were investigated, including the fraction of acrylic acid in the microgel formulation,21a,b and the crosslinking density of either microgel or bulk hydrogel.21b In a study presented by Burdick and co-workers, a single therapeutic cytokine, interleukin 10, was released from UV-crosslinked microgels fabricated via microfluidics and embedded in a physically guest–host crosslinked bulk hydrogel matrix, and showed faster release from degradable microgels and slower release from stable non-degradable microgels.21c Most notably, these exemplar works demonstrated the design and synthesis of bespoke MHC systems, where the acrylic acid content or crosslinking density21a,b and degradability achieved by distinct crosslinkers21c were predetermined and fixed after fabrication.
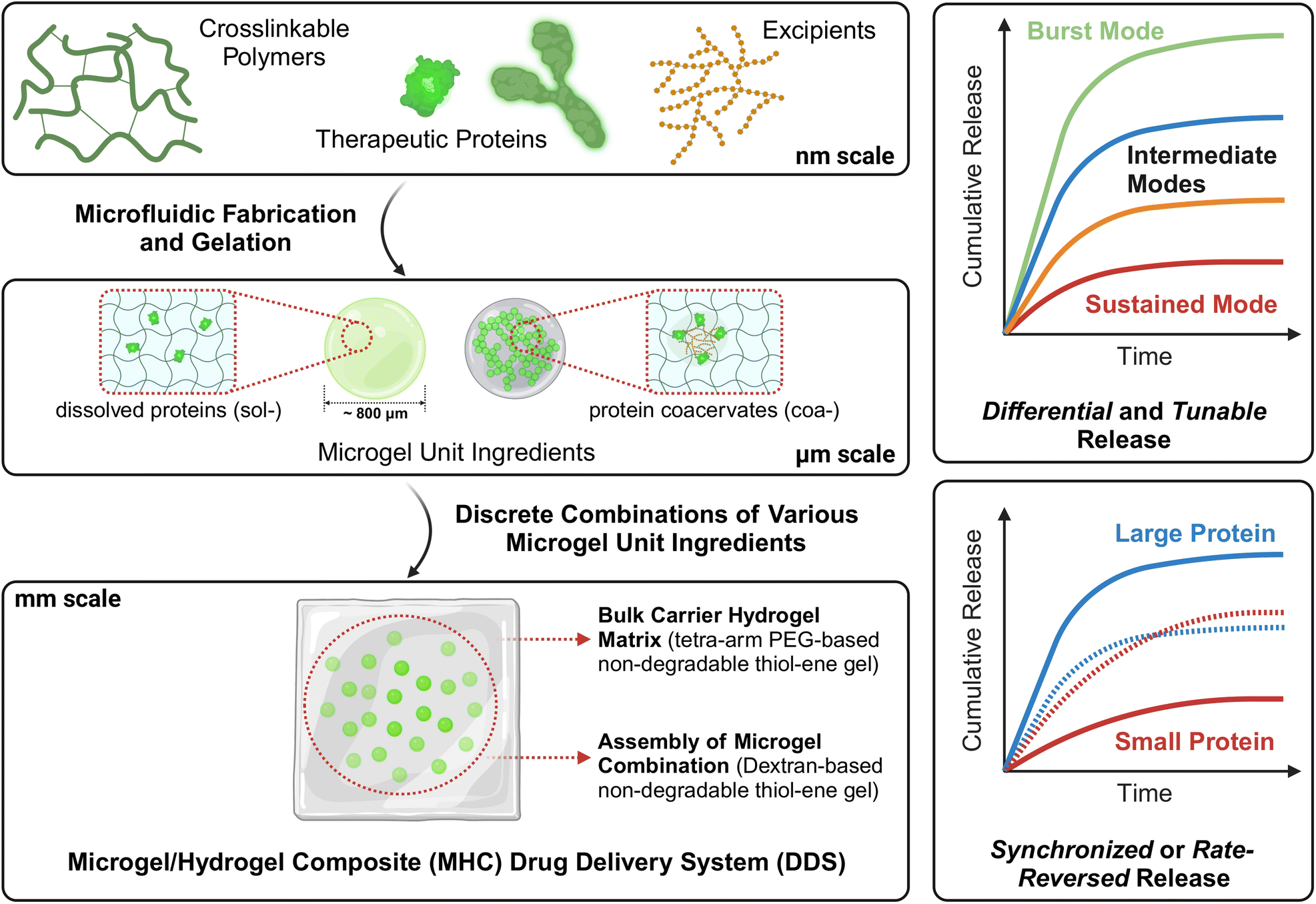 | ||
| Fig. 1 Conceptual schematic of the proposed microgel/hydrogel composite (MHC) drug delivery system (DDS) with embedded discrete combination of “microgel” unit ingredients stepwise (left) – Selection of target therapeutic protein payloads, basis hydrogel materials, and excipients to facilitate formulation strategies (top), is followed by the microfluidic fabrication of hydrogel microparticles (microgels) loaded with proteins in multiple physical states (middle), and lastly discrete combinations of microgel ‘unit ingredients’ are packaged into a bulk carrier hydrogel matrix (bottom). The MHC DDS is designed to achieve complicated release scheduling and programming (right), such as the differential release of multiple similar proteins, tunable release of a single protein at different rates, and synchronized or rate-reversed release of proteins that are vastly different in size. (This figure is designed and created with BioRender: https://app.biorender.com/). | ||
In this work, we extend this concept of microgel/hydrogel composite systems a step further, by developing microgel unit ingredients containing the same protein in different physical states (i.e., in free solute form or in coacervate form). This ultimately enables a wide range of tunability of release behavior that is achieved via simple, discrete combinations of unit ingredients without re-design of the hydrogel matrix. To demonstrate this concept, we use native hen egg white lysozyme (‘nLyz’) as a low-molecular-weight model protein and PEGylated bovine serum albumin (‘pBSA’) as a high-molecular-weight model protein in this study. Starting with the successful syntheses of vinyl sulfone (–VS) and thiol (–SH) grafted dextran polymers through chemical modification, we exploited the commonly used ‘thiol–ene’ click chemistry as the crosslinking mechanism for both microgels and the bulk carrier matrix for its selectivity, efficacy, biocompatibility, and rapid crosslinking rate. We then leverage droplet-based microfluidics to fabricate monodisperse protein-loaded microgels. In particular, we demonstrate the facile generation and release characterization of sol-nLyz and coa-nLyz loaded dextran-based, ‘thiol–ene’ click chemistry mediated microgels, and packaging of multiple unit ingredients into a single PEG-based ‘thiol–ene’ click-crosslinked bulk hydrogel matrix. This packaged MHC is intended to model a topical wound dressing patch or an implantable drug-releasing depot. We then showcase a variety of in vitro release profiles for nLyz combinations as a function of the microgel number fraction between sol-nLyz and coa-nLyz unit ingredients, and a dual protein release with solute-form pBSA (sol-pBSA) in which we observe a rate-reversed release when switching nLyz from the solute state to the coacervate state.
Results and discussion
Multi-state protein-loaded unit ingredients – fabrication and characterization of microgels and packaging into MHC systems
As the fundamental component of proposed MHC systems, microgels were fabricated with a microfluidic water-in-oil (W/O) droplet generation system as presented in Fig. 2. Three types of microgel unit ingredient were fabricated as shown in Fig. 3A: (i) solute form nLyz-loaded microgel (‘sol-nLyz’), (ii) coacervate form nLyz-loaded microgel with free coacervating agent (‘coa-nLyz’), and (iii) solute form pBSA-loaded microgel (‘sol-pBSA’). All three types of microgels were dextran-based, spherical in shape, with a diameter of ∼800 μm. Both sol-nLyz and sol-pBSA microgels appeared transparent and clear while coa-nLyz microgels exhibited visible turbidity and opaque patches, indicating the formation of coacervates. In the subsequent MHC fabrication, these microgels were packaged into a transparent, colourless PEG-based bulk matrix hydrogel as shown in Fig. 3B (blank) and Fig. 3C (packaged for subsequent release studies). Detailed information regarding microgel formulation and characterization are provided as ESI Table S2,† and the experimental procedures are described in the relevant section below. | ||
| Fig. 2 Simplified schematic of the microfluidic microgel fabrication system, which involves three aqueous streams of the hydrogel precursor solutions and protein (Stream X, Y, and Z), a cross-junction for pre-mixing of these aqueous precursors, a 50 mm transferring segment, a T-junction to generate water-in-oil (W/O) emulsion droplets, and a coiled in-line mixing segment of extended length of 2880 mm length to enhance internal mixing of droplets. The fabricated microgels are printed into an immobilizing bed for gelation and curing (not shown). (This figure is designed and created with BioRender: https://app.biorender.com/). | ||
 | ||
| Fig. 3 Sample images of fabricated microgel unit ingredients and proposed MHC systems. (a) Optical microscope images of sol-nLyz (left), coa-nLyz (middle), and sol-pBSA (right) microgels under visible light (top) and in darkfield mode under UV-exposure (bottom). (b) An image of empty PEG-based bulk carrier hydrogel matrix of square slab shape, with approximately 12 mm side length and 2.4 mm thickness (not shown here). (c) A set of images of fabricated MHC with some discrete combinations of sol-nLyz and coa-nLyz unit ingredients, during the in vitro release study inside sterilized cosmetic jars. | ||
In terms of crosslinking mechanism, both dextran-based microgels and the PEG-based matrix material exploited a non-degradable, thiol–ene click chemistry between vinyl sulfone (–VS) moiety and thiol (–SH) group functionalized with dithiothreitol. This click chemistry can crosslink under mild physiological conditions to preserve loaded proteins. The resulting gels are non-degradable – an aspect that was used in this work to understand key differences between the intrinsic release properties of the distinct formulations used, which would be independent of any disintegration of the hydrogel matrix. That being said, we note that non-degradable hydrogels may not be suitable for injectable or implantable DDSs, and the incorporation of degradability is an important direction to be addressed in future work.
In vitro short-term release behavior from microgels
Next, to have a basic understanding of the release behavior of each individual microgel unit ingredient, Fig. 4 provides the in vitro short-term (∼2 h) cumulative release profiles of all three types of unit ingredients fabricated. The short-term cumulative release of sol-nLyz microgels showed a typical burst behavior as more than 70% of the protein payload was released in a duration of ∼10 min and reached a plateau at 90% of payload released after ∼30 min. However, the coacervate counterpart showed a near-linear hindered release profile, with less than 5% of the protein payload released within first 10 min and below 20% of payload released in the entire measurement span of 2 h. This was due to the strong intermolecular electrostatic interaction between a high-sulfate-content coacervating agent (Dex40k-SO3Na) and the positively charged nLyz at physiological pH, resulting in the formation of coacervates that immobilized protein molecules into a solid state. By introducing proteins in such immobilized coacervated state, an additional step would be necessary to ‘liberate’ the proteins from immobilized state into dissolved state; only then, the proteins can be released by the passive diffusion. Hence, limited dissociation of coacervates and the consequent strong suppression of free diffusion-driven release of nLyz was observed, in accordance with results from prior literature.17 Lastly, sol-pBSA microgels showed similar burst release with a relatively slower rate compared to sol-nLyz microgels. Around 30% of the pBSA payload was released within the initial 10 min and reached a plateau of less than 70% of the total payload released after approximately 1 h. This difference between sol-nLyz and sol-pBSA can be rationalized by comparing the typical mesh size of fabricated dextran-based microgels, which was around 5.0 nm (see Table S1†) and the hydrodynamic diameters of model proteins. Since nBSA has a hydrodynamic diameter (Dh,nBSA ≈ 8.1 nm)22a larger than the typical mesh size while nLyz has a hydrodynamic diameter (Dh,nLyz ≈ 3.8 nm)22b slightly smaller than the typical mesh size, the passive diffusion-driven release of nLyz is expected to be faster than that of pBSA. In general, the primary factor affecting mesh size of the gels is the crosslinking density. In practice, higher concentrations of the crosslinking polymerlead to higher crosslinking densities, yielding a smaller mesh size that can further hinder the diffusive protein release.23a A related factor is the degree of modification of the polymer, which relates to the number of crosslinkable moieties present. This is in particular more important for polymers such as PEG, that have relatively low numbers of crosslinking moieties to begin with, and where factors such as the number of polymer arms become important, while it is less significant for richly modified polymers such as dextran (or polysaccharides in general), which typically contain excess residual moieties unreacted and buriedd inside the polymer globules.23 | ||
| Fig. 4 Short-term microgel-only cumulative release profiles of sol-nLyz (solid dots), coa-nLyz (hollow circles), and sol-pBSA (solid boxes) microgels, at sink condition with nsample = 3 for each type of microgels. | ||
Tunable release of nLyz from MHCs via discrete combinations of sol-nLyz and coa-nLyz microgels
Based on the different in vitro short-term release behaviors of sol-nLyz microgels and coa-nLyz microgels, a series of discrete combinations of sol-nLyz and coa-nLyz unit ingredients of varying number fractions of sol-nLyz component (NsL) were fabricated into the proposed MHC systems. As Fig. 5A shows, for the ‘limit’ cases of NsL = 1.0 (pure sol-nLyz MHC) and NsL = 0 (pure coa-nLyz MHC), resultant cumulative release profiles suggested similar trends for release behavior from MHCs to the short-term release of unpackaged sol-nLyz and coa-nLyz microgels, respectively (referring to Fig. 4). This similarity confirmed that the chosen PEG-based bulk hydrogel matrix exerted a minimal effect on the release of the nLyz payload. For the cases of discrete combinations, decreasing NsL led to gradual suppression of the burst release behavior, as evidenced by the observation that the nLyz released at the 12-hr time point decreased from 90% for NsL = 1.0 down to ∼50% for NsL = 0.875 and ∼30% for NsL = 0.5. Mass swelling data obtained from measurements on the blank bulk carrier matrix revealed an estimated mesh size of the bulk carrier matrix to be ∼11 nm, which is larger than the hydrodynamic size nLyz (please see ESI† for more details); this suggested that the bulk carrier matrix of MHCs exerted negligible hindrance due to the mesh of network. | ||
| Fig. 5 Long-term cumulative release of MHCs packaged with discrete combinations of sol-nLyz and coa-nLyz unit ingredients. (a) The spectrum of cumulative percentage release profiles of NsL ranging from 1.0 (pure sol-nLyz MHC) to 0 (pure coa-nLyz MHC) at batch condition with nsample = 3 for each combination. (b) The logarithmic plot of release data for Korsmeyer-Peppas kinetic model fitting. (c) Korsmeyer-Peppas kinetic exponent (n, red solid dots) and rate constant (Kr, black solid boxes) plotted as a function of NsL ranging from 0 to 1.0, with n fitted by 4-parameter logistic (4PL) sigmoidal function (red dashed curve) and Kr fitted by exponential growth function (black dashed curve). | ||
To better exploit the available release data obtained from the set of discrete combinations, Korsmeyer-Peppas empirical model was used to fit the release data and extract release kinetics parameters as functions of NsL. Briefly, as eqn (1) indicates, Mt refers to the cumulative released amount of protein at time point t, and the M∞ term is equivalent to the total amount of protein loaded in the MHC such that the ratio of Mt over M∞ is ideally the cumulative fractional release of protein. Then, n refers to the geometry-dependent Korsmeyer-Peppas rate exponent and Kr refers to the Korsmeyer-Peppas rate constant with a unit of [T]−n indicating a relative rate of release kinetics.24 To extract the fitted value of n and Kr from release data, linear regression fitting was performed on the logarithmic plot of fractional cumulative release versus release time in minutes as shown by Fig. 5B.
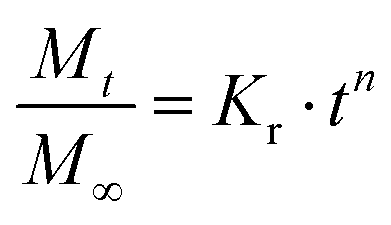 | (1) |
Fig. 5C is a plot of the Korsmeyer-Peppas parameters n and Kr as a function of NsL. These parameters were used to construct the fitted release curves plotted on Fig. 5A for visual comparison. There are several interesting trends in Fig. 5C; firstly, we observed that the rate exponent n increases with decreasing NsL indicating a transition of the release from typical burst release behavior to a near-linear release behavior for the specific ‘packaged’ geometry of proposed MHCs. Furthermore, two regimes of release are apparent in Fig. 5C; both n and Kr are observed to vary primarily in the range of NsL = 0.5–1.0 and appear to plateau in a relatively insensitive regime of NsL < 0.25. These regimes can be rationalized by considering the presence and involvement of coacervating agents in free form, enabling re-association of coacervates dictated by the dynamic association-dissociation equilibrium;25 when the coacervating agents are released to the proximate bulk matrix of coa-nLyz microgels, locally elevated nLyz concentrations from nearby fast-releasing sol-nLyz microgels can trigger the coacervate re-association following Le Chatelier's principle. The re-associated coacervates behave in a manner similar to the suppressed near-linear release of coa-nLyz microgels, pointing to the existence of a lower threshold of NsL below which the overall release behaviors become indistinguishable from each other. Lastly, Kr exhibited an exponential growth trend while n showed a decreasing sigmoidal trend with increasing NsL. Therefore, exponential growth (EG) fitting and four-parameter logistic (4PL) sigmoidal fitting were applied to parameterize Kr and n as functions of NsL, respectively. The fitted analytical functions are provided in eqn (2A) and (2B).
| Kr(NsL) = Kr0·eq·NsL = 0.0003718·e6.996·NsL | (2A) |
 | (2B) |
Collectively, with both functions Kr(NsL) and n(NsL) obtained, Korsmeyer-Peppas release kinetic profiles of any arbitrary discrete combination of sol-nLyz and coa-nLyz unit ingredients can be estimated given the value of NsL. Conversely, a desired release profile can be projected onto a pair of Kr and n values, and the appropriate NsL can be obtained by optimizing both functions Kr(NsL) and n(NsL) to yield the desired release profile from proposed MHCs.
A demonstration of dual-protein release from MHCs via discrete combinations of nLyz- and pBSA-containing microgels
As a first step towards engineering multi-protein release from a single MHC system incorporating both nLyz and pBSA, we investigated the short-term release behavior of a pure sol-pBSA containing MHC system. As shown in Fig. 6, the pure sol-pBSA MHC indicated a diffusion-driven burst release behavior with relatively slower overall release rate compared to the sol-nLyz counterpart yet faster than the coa-nLyz counterpart over a 24 h period, indicating a ‘rate-reversed’ release behavior given that pBSA is a larger molecule than nLyz. This preliminary observation served as a motivation for the further experiments on dual-protein release using combinations of both nLyz- and pBSA-containing microgel unit ingredients to engineer the release profiles and timescale of the two proteins distinct in size. Moreover, the slower release behavior of pBSA may also result from the drag of pBSA experienced due to the PEG chain entanglement with the crosslinked PEG network along the path of diffusion while traversing through the bulk carrier matrix. | ||
| Fig. 6 Long-term cumulative release of MHCs packaged with three pure unit ingredients: sol-nLyz (blue solid curve with dots, retrieved from previous section results NsL = 1.0), coa-nLyz (red solid curve with black cross, retrieved from previous section results NsL = 0), and sol-pBSA (black solid curve with hollow hexagon) at batch condition with nsample = 3 for each group. Dashed curves indicate the fitted Korsmeyer-Peppas release kinetic models. | ||
Therefore, discrete combinations of sol-nLyz with sol-pBSA (hereafter abbreviated as SBSL) and coa-nLyz with sol-pBSA (abbreviated as SBCL) MHC systems, having microgel number ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, were then fabricated and tested. As shown in Fig. 7, the nLyz payload released from SBSL MHC showed rapid burst release behavior, with faster release rate compared to the pBSA payload as expected due to the size differences. In contrast, the nLyz released from SBCL MHC followed a near-linear release behavior while the pBSA followed a nearly similar release behavior to the case of the SBSL MHC. Most importantly, a clear rate-reversed release between nLyz and pBSA was observed in the case of SBCL MHC, as supported by the preliminary observation with a crossover time roughly at the 12 h time point.
:1, were then fabricated and tested. As shown in Fig. 7, the nLyz payload released from SBSL MHC showed rapid burst release behavior, with faster release rate compared to the pBSA payload as expected due to the size differences. In contrast, the nLyz released from SBCL MHC followed a near-linear release behavior while the pBSA followed a nearly similar release behavior to the case of the SBSL MHC. Most importantly, a clear rate-reversed release between nLyz and pBSA was observed in the case of SBCL MHC, as supported by the preliminary observation with a crossover time roughly at the 12 h time point.
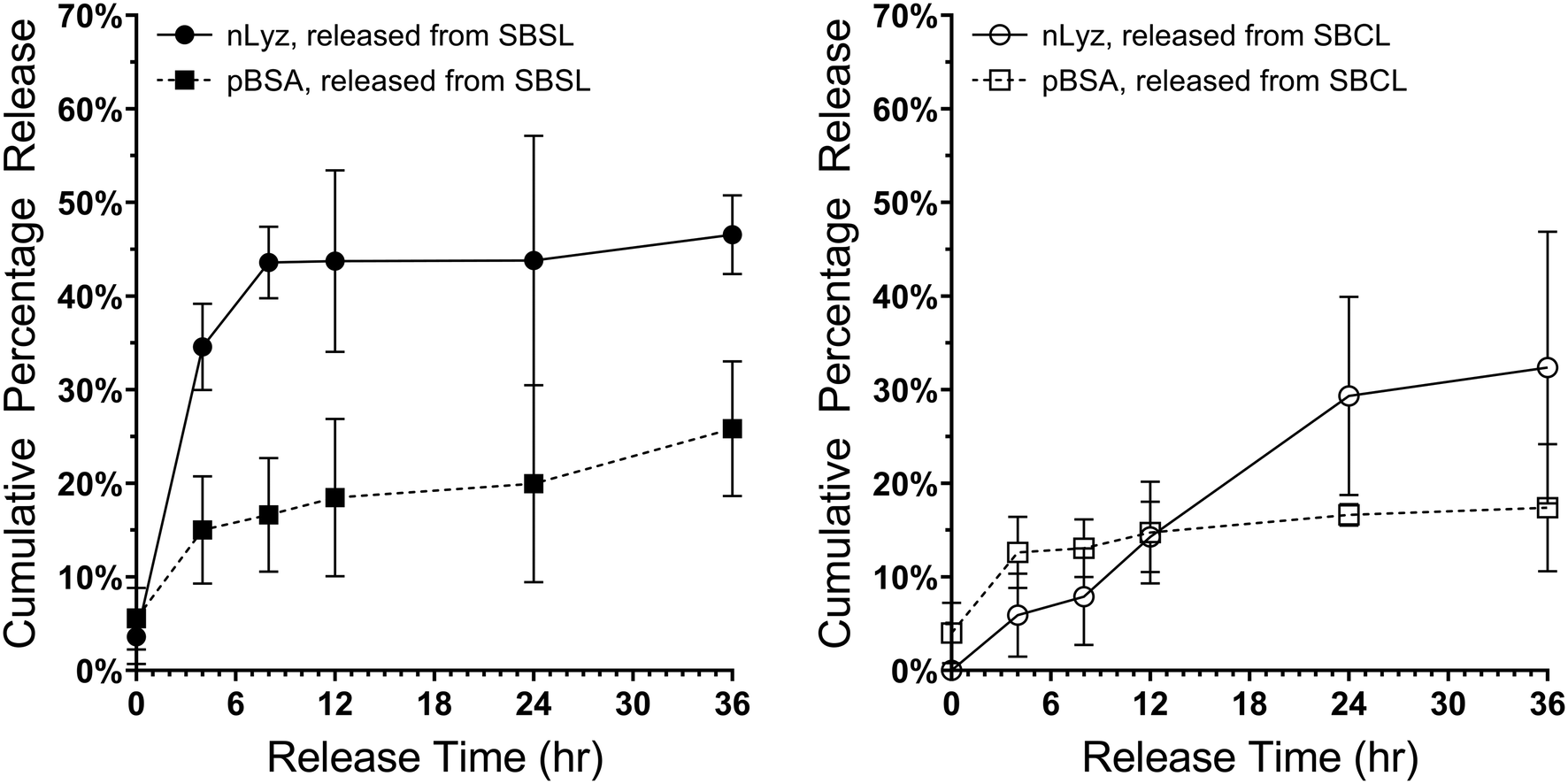 | ||
| Fig. 7 Long-term cumulative release of MHCs packaged with discrete combinations of nLyz- and pBSA-containing unit ingredients: sol-nLyz with sol-pBSA microgels in number ratio 1:1 (SBSL, left plot) and coa-nLyz with sol-pBSA microgels in number ratio 1:1 (SBCL, right plot). Both plots showed individual release profile of nLyz payloads (solid dots for SBSL and hollow circles for SBCL) and pBSA payloads (solid boxes for SBSL and hollow boxes for SBCL) at batch condition with nsample = 3 for each combination. The SBSL release plot indicated the intuitive size-exclusive diffusion-driven release rates that the smaller nLyz molecule released faster than the larger pBSA molecule, while the SBCL release plot indicated a rate-reversed release phenomenon with a crossover roughly at the 12-hr time point, due to the presence of a much slower releasing coa-nLyz ingredients comparing to sol-nLyz counterpart. | ||
Conclusion and future perspectives
In this report, we present fabricated microgels loaded with two different model proteins – nLyz and pBSA, with nLyz loaded in two distinct physical forms – as free dissolved solutes (sol-nLyz) having rapid burst release behavior, and as coacervates (coa-nLyz) having effectively slowed and sustained release behavior. The use of these microgels as ‘unit ingredients’ was demonstrated by packaging a variety of discrete combinations of microgels into a microgel/hydrogel composite (MHC) system as a proposed drug delivery system. These packaged MHC systems were then tested by in vitro long-term protein release, indicating both a tunable release of single nLyz payload with variable number ratio between sol-nLyz and coa-nLyz microgels, and a dual-protein release of nLyz and pBSA with a rate-reversed release behavior observed when slow-releasing coa-nLyz unit ingredients were used. Next, to quantify the tunability of nLyz release, Korsmeyer-Peppas kinetics model parameters, rate constant Kr and exponent n, were extracted as functions of the number fraction of sol-nLyz unit ingredients (NsL), where Kr was observed to increase exponentially with NsL while n was observed to decrease in a sigmoidal manner with increasing NsL. Our MHC DDS can accommodate various combinations of microgel unit ingredients, thus rendering simplicity to the formulation process, since no complicated and bespoke chemical modifications are required for the generation of new or intermediate release behavior, which can be achieved merely by varying the composition of each unit ingredient in the MHC.Given the relative simplicity and tunability of this concept, we envision the proposed MHC DDSs to be used as implantable drug depots (with degradable hydrogel matrices) or topically applicable wound dressings to deliver multiple therapeutic proteins (similar or distinct) in a sustained, controlled, and properly scheduled manner. Moreover, the proposed MHCs retain advantages of typical hydrogel materials, such as softness and high water-absorption capacity for enhanced biocompatibility, ability to protect proteins from in vivo enzymatic degradation, and the ability to localize/immobilize microgels that prevents them from migration after administration. Further, while we demonstrated coacervation as a means of incorporating proteins in a different physical state into the microgels, it is also possible to extend this idea to other physical states, such as the crystalline or amorphous states. In addition, bio-conjugated forms of proteins can also be incorporated readily to enable differential affinities within the hydrogel matrices. Hydrogel degradability, which can typically be built in through polymer modification, such as via the use of chemically modified dextran (by introducing ester moieties for hydrolysis, for example) can allow a further lever on protein release behavior. Moreover, alternative crosslinking chemistries such as bioorthogonal click chemistry (azide–alkyne cycloaddition, for instance) can be used to reduce the potential immobilization of proteins due to the VS–SH reaction. Meanwhile, moving beyond model proteins, and into the realm of therapeutic applications, we envision future work to study proteins with known therapeutic effects, such as immunoglobulin G (IgG), vascular endothelial growth factor (VEGF), interleukin-4 (IL-4) etc., to investigate the behaviours of proposed MHCs with proteins of distinct physicochemical properties and sizes. Thus, we envision that the idea of microgel ‘unit ingredients’ to be broadly generalizable and, crucially, expandable to a variety of complex and tunable release behaviours for customized clinical and biomedical applications. Finally, moving beyond this proof-of-concept work, and towards clinical applications, several additional aspects will be addressed in ongoing and future work, such as biocompatibility, cytotoxicity, long-term storage and release stability, and efficacy in in vivo animal models.
Experimental
Materials and methods
:1, and the mixture was vigorously mixed during the addition. The mixture was then extensively blended using a mechanical homogenizer with a stainless steel 25 mm quad-blade propeller at 2000 rpm for 20 min to a flowable and homogenous state without any obvious chunk of solids. The mixture was then collected into a bottle and vacuum degassed for 30 min. The degassed mixture was then stored and could be used directly in the manufacturing steps. To be used as the immobilizing bed, the YSF was poured onto plastic or glass Petri dishes for use without further modification.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) and a reactive thiol (–SH), which to some extent provided extra potential of chemical modifications with other functional groups coupled with reactive thiols. Herein, we adopted the vinylation using reagent DVS to synthesize Dex40k-VS from a reaction reported by Yu and Chau (Scheme 1),27a and next-step thiolation on Dex40k-VS using reagent DTT to synthesize Dex40k-SO2-TTSH from a reaction reported by Lau et al. (Scheme 2).27b For the description of synthesis and characterization details, please refer to ESI.†
C) and a reactive thiol (–SH), which to some extent provided extra potential of chemical modifications with other functional groups coupled with reactive thiols. Herein, we adopted the vinylation using reagent DVS to synthesize Dex40k-VS from a reaction reported by Yu and Chau (Scheme 1),27a and next-step thiolation on Dex40k-VS using reagent DTT to synthesize Dex40k-SO2-TTSH from a reaction reported by Lau et al. (Scheme 2).27b For the description of synthesis and characterization details, please refer to ESI.†
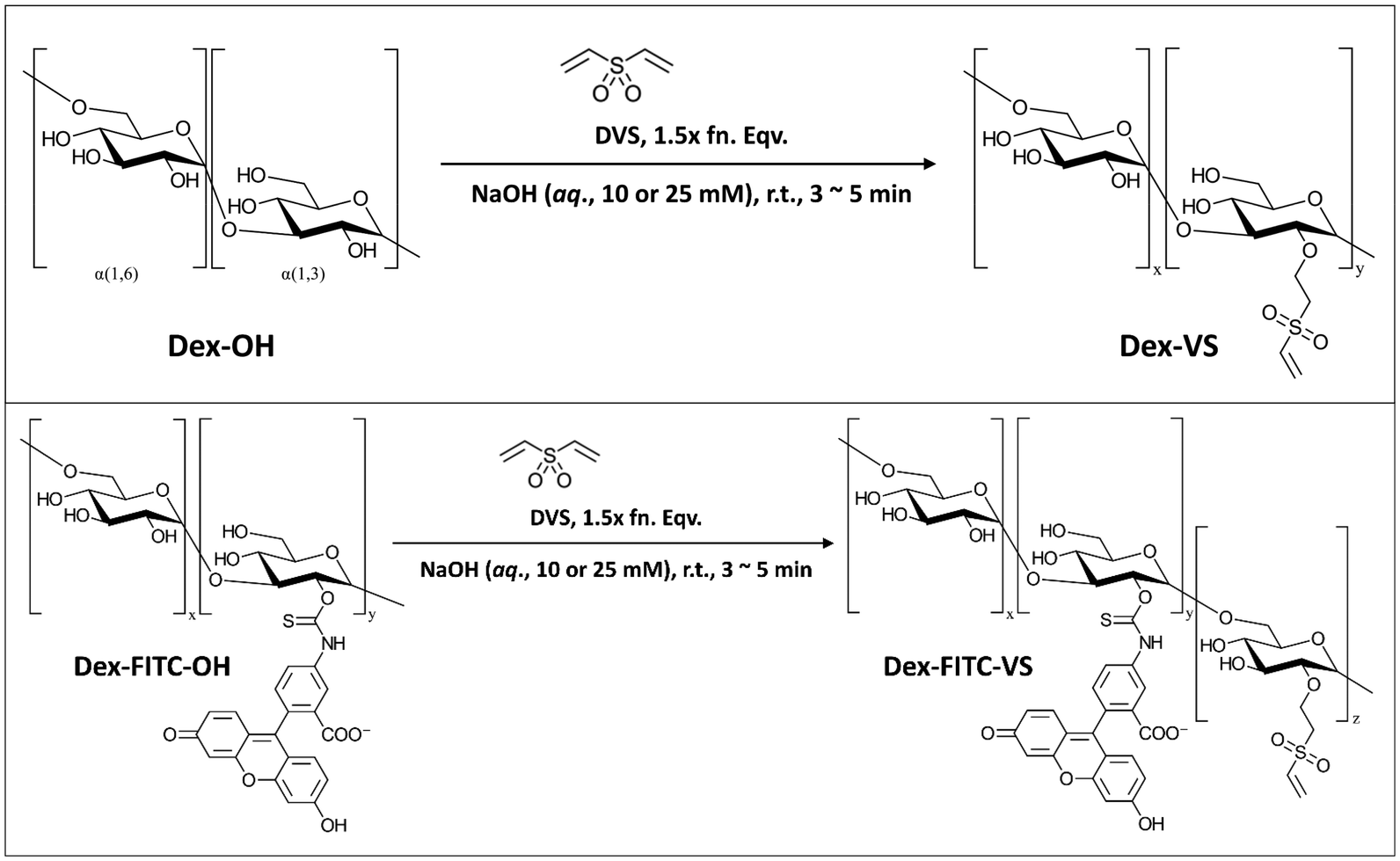 | ||
| Scheme 1 Synthesis of dextran vinyl sulfone (Dex40k-VS, top) and fluorescein conjugated dextran vinyl sulfone (Dex40k-FITC1%-VS, bottom) utilizing reagent divinyl sulfone (DVS) by Michael Addition of sulfonyl-activated vinyl (–CHCH2) onto hydroxyl (–OH) moieties of polysaccharide in ambient and basic condition.27a | ||
 | ||
| Scheme 2 Synthesis of dextran sulfonyl thiothreitol (Dex40k-SO2-TTSH) utilizing reagent DL-dithiothreitol (DTT) by Michael Addition of thiol (–SH) with sulfonyl-activated vinyl (–CHCH2) moieties of Dex40k-VS in ambient, slightly basic, and low-oxygen condition, with thiol in excess.27b | ||
 | ||
| Scheme 3 Synthesis of PEGylated BSA (mPEG2k-MALS-BSA, abbr. pBSA) utilizing reagent poly(ethylene glycol) monomethyl ether maleimide (mPEG2k-MAL) by Michael Addition of thiol (–SH) with carbonyl-activated vinyl (–CHCH–) moieties on the maleimide functional group in ambient and slightly basic condition, with maleimide in excess.28 | ||
:1 volumetric ratio); (iii) a droplet printing and mixing section, featuring a 2880 mm long outlet tube with a coiled segment composing “∞”-shape loops to enhance the internal mixing of droplets, from which droplets are printed into a Petri dish filled up with SiNP@(vPDMS:PMX-200) YSF as immobilizing bed to hold droplets for crosslinking and curing, prevent adjacent droplets merging, and maintain a spherical shape of droplets. All sections of tubing used in the system were PTFE tubing with 1.0 mm I.D. purchased from Diba (Omnifit®), and both cross- and T-junctions were PEEK plastic junctions purchased from IDEX Health & Science.
Experiment-wise, approximately 1.2 mL solution X, Y, and Z were freshly prepared, and the microfluidic system was assembled, and liquid streams were primed manually. Across all formulations, volumetric flow rates of the three aqueous solutions were kept identical, and the continuous phase volumetric flow rate was held with a continuous-to-discontinuous phase ratio kCD = 5.0. The CP was firstly pumped in (with QCP = 450 μL min−1) to fill up the droplet printing and mixing section and coat the tubing for roughly 10 min. After the CP coating, the buffer stream (Z) was turned on (with QZ, initial = 150 μL min−1) to fill up transferring tube and start to generate some blank buffer droplets to test the stability of the droplet generation section. With stable flow and droplet generation achieved, all three aqueous streams (X, Y, and Z) were pumped in (with QX = QY = QZ = 30 μL min−1) to start the microfluidic fabrication of microgels. The mixed droplets (pre-gelling Dex40k-VS:SO2-TTSH mixture) were manually discharged and printed into the YSF immobilizing bed for gelation and curing under room temperature overnight. Afterwards, the collection of gelled and cured microgels were done manually using a glass dropper to briefly screen the size and minimize residual YSF. Then, the collected microgels were thoroughly washed with anhydrous IPA for 6 to 8 times to remove the oily YSF residue while drying the microgels by antisolvent extraction, followed by vacuum evaporation in desiccator to evaporate residual IPA for roughly 30 min. After washing, the dried microgels were collected in a clean glass vial and stored in a dry box with silica gel desiccant under 4 °C for further usage.
Characterizations
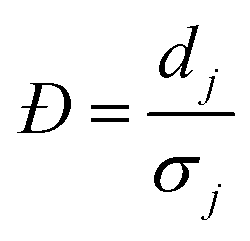 | (3A) |
For each type of microgel, a small fraction of collected microgels before washing were sampled for microscopic imaging. A dark-field image was taken by similar settings as the bright-field image, except for tuning white balance (R, G, B) = (1.31, 5.00, 1.77) to couple the fluorescence of fluorescein, and a UV lamp shining 5 cm above the sample at wavelength of 432 nm. In ImageJ, similar to the sizing procedure, for one type microgel (indexing microgel type as j), a visible and relatively spherical microgel (indexing each microgel sphere as i) was selected from the image, a diameter of the sphere was drawn manually across the visible symmetric axis of the green signal, and the RGB profile against pixels alongside this diameter was extracted using an ImageJ plugin graphical function. Next, using MATLAB, the G-profile against pixels was filtered through a moving mean with 3-pixel width window, and the overall average ( ) and standard deviation (
) and standard deviation (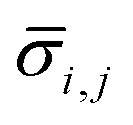 ) of filtered G-profile of this i-th sphere was computed. Then, a mixing index of this i-th sphere (ηi,j) was calculated by eqn (4A).
) of filtered G-profile of this i-th sphere was computed. Then, a mixing index of this i-th sphere (ηi,j) was calculated by eqn (4A).
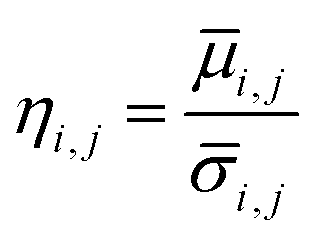 | (4A) |
Lastly, for each type of microgel, all visible spheres were measured following the identical procedures, and a box plot of all extracted ηi,j was generated to indicate the performance of in-droplet mixing of each formulation ( ) provided in ESI.†
) provided in ESI.†
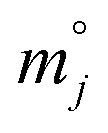 ) was calculated by eqn (4B).
) was calculated by eqn (4B).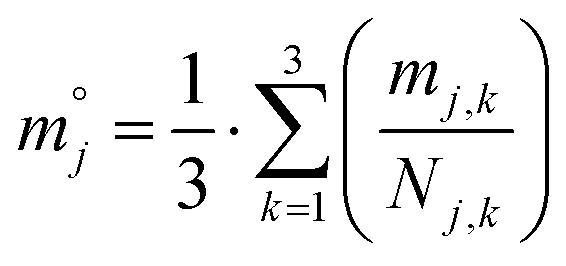 | (4B) |
:1:1 ratio in a centrifuge tube, and 600 μL of the gelling mixture was applied to the sampling plate of TA Instrument HR30 rheometer, with a stainless-steel Peltier cone plate (40.0 mm diameter, 1.99167° cone angle) at a sample gap of 50.0 μm and a controlled temperature of 21 °C. The flow peak hold measurement was kept at constant stress of 0.1 Pa and the viscosity was sampled every second in a span of 1500 s. The viscosity profile was plotted logarithmically with respect to time. A clear transition in the slope of viscosity increasing was spotted and an estimated interpolation of the transition point was performed for each profile as the estimated gelation time to report.30
 | (5A) |
Then, the percentage release (ωti) was calculated based on an estimation of total protein loading (m0) given by eqn (5B) with known masses of each type of microgels (Mj) weighed for the study.
 | (5B) |
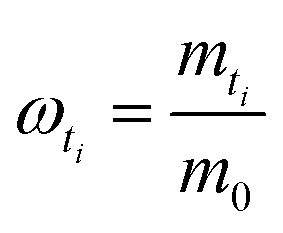 | (5C) |
 | (5D) |
:SH bulk carrier hydrogel matrix was fabricated following the exact protocol as described above for the macro-size hydrogel beads for swelling study (indexing each bead as j). Then, each bead was weighed M′j, 0 and placed in an Eppendorf 50.0 mL centrifuge tube. The tube was then filled up with 15.0 mL of RB, and a series of time point were sampled. At each timepoint, mass of each matrix was weighed and recorded as M′j, ti, which leads to a mass swelling ratio at timepoint ti calculated by eqn. (5D′). | (5D′) |
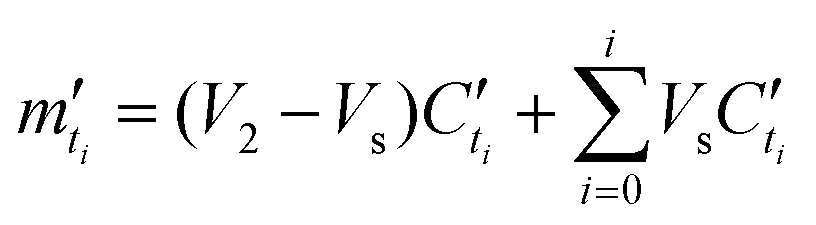 | (5A′) |
Then, the percentage release (ω′ti) was calculated based on an estimation of total protein loading (m′0) given by eqn. (5B′) with a known mass of each type of microgels (MsL, McL and MsB indicating sol-nLyz, coa-nLyz, and sol-pBSA microgels respectively) packaged in the designated MHC.
 | (5B′) |
 | (5C′) |
Author contributions
Longfei Chen: conceptualization, data curation, formal analysis, investigation, methodology, visualization, and writing (original draft). Saif A. Khan: project administration, conceptualization, funding acquisition, supervision, resources, validation, and writing (review & editing).Data availability
Data for this article, including:(1) Visible-light microscopic sample images of microgels (sol-nLyz, coa-nLyz, sol-pBSA),
(2) Darkfield microscopic sample images of microgels (sol-nLyz, coa-nLyz, sol-pBSA),
(3) Proton nuclear magnetic resonance (1H NMR) spectra of synthesized polymers,
(4) Rheological flow peak-holding measurements,
(5) Scanning electron microscopy (SEM) images,
(6) Absorbance-based microplate measurement of Ellman's Assay, Bicinchoninic Acid (BCA) Assay, and Enzyme-Linked Immunosorbent Assay (ELISA), for both calibration curve and release studies, are available at Science Data Bank at https://doi.org/10.57760/sciencedb.11601.
MATLAB codes for this article, including an RGB profile plotter and analyzer (FluorPlotting.m) and an automatic batch analyzer for multiple RGB profiles (FluorMix.m) are available at GitHub at https://doi.org/10.5281/zenodo.13139858.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would sincerely express our appreciation to Dr Flore Mekki-Berrada for her assistance in troubleshooting the microfluidic system, assisting the microscopic imaging setup, and idealizing the assessing index of in-droplet mixing performance. We are grateful to Dr Arif Z. Nelson for his guidance in synthesizing hydrophobic yield stress fluids (YSF) for the droplet-embedded printing technique used for microgel generation. We thank Mr Yi Wei LEE for his kind assistance during the conduction of ELISA for both proteins, Ms Serene Chong for her kind assistance with the rheological measurements, and Mr Jun Ho MIN for his kind assistance with SEM sample preparation, operation, and imaging.References
- A. Vishwakarma, N. S. Bhise, M. B. Evangelista, J. Rouwkema, M. R. Dokmeci, A. M. Ghaemmaghami, N. E. Vrana and A. Khademhosseini, Engineering immunomodulatory biomaterials to tune the inflammatory response, Trends Biotechnol., 2016, 34(6), 470–482 CrossRef.
- (a) D. Wen, G. Chen, Q. Chen, P.Y Li, H. Cheng and Z. Gu, Engineering protein delivery depots for cancer immunotherapy, Bioconjugate Chem., 2019, 30(3), 515–524 CrossRef PubMed; (b) J. Honeychurch, E. J. Cheadle, S. J. Dovedi and T. M. Illidge, Immuno-regulatory antibodies for the treatment of cancer, Expert Opin. Biol. Ther., 2015, 15(6), 787–801 CrossRef; (c) D. M. Francis and S. N. Thomas, Progress and opportunities for enhancing the delivery and efficacy of checkpoint inhibitors for cancer immunotherapy, Adv. Drug Delivery Rev., 2017, 114, 33–42 CrossRef.
- H. J. Hoffmann, E. Valovirta, O. Pfaar, P. Moingeon, J. M. Schmid, S. H. Skaarup, L. O. Cardell, K. Simonsen, M. Larché, S. R. Durham and P. Sørensen, Novel approaches and perspectives in allergen immunotherapy, Allergy, 2017, 72(7), 1022–1034 CrossRef CAS.
- M. Feldmann and L. Steinman, Design of effective immunotherapy for human autoimmunity, Nature, 2005, 435(7042), 612–619 CrossRef CAS PubMed.
- K. K. Gupta, M. A. Khan and S. K. Singh, Constitutive inflammatory cytokine storm: a major threat to human health, J. Interferon Cytokine Res., 2020, 40(1), 19–23 CrossRef CAS.
- (a) L. Chung, D. R. Maestas Jr, F. Housseau and J. H. Elisseeff, Key players in the immune response to biomaterial scaffolds for regenerative medicine, Adv. Drug Delivery Rev., 2017, 114, 184–192 CrossRef CAS; (b) Y. Kuroda, T. Kawai, K. Goto and S. Matsuda, Clinical application of injectable growth factor for bone regeneration: a systematic review, Inflammation Regener., 2019, 39, 1–0 CrossRef CAS PubMed.
- (a) S. Barrientos, H. Brem, O. Stojadinovic and M. Tomic-Canic, Clinical application of growth factors and cytokines in wound healing, Wound Repair Regen., 2014, 22(5), 569–578 CrossRef; (b) J. Mao, L. Chen, Z. Cai, S. Qian, Z. Liu, B. Zhao, Y. Zhang, X. Sun and W. Cui, Advanced biomaterials for regulating polarization of macrophages in wound healing, Adv. Funct. Mater., 2022, 32(12), 2111003 CrossRef CAS; (c) A. J. Busti, J. S. Hooper, C. J. Amaya and S. Kazi, Effects of perioperative antiinflammatory and immunomodulating therapy on surgical wound healing, Pharmacotherapy, 2005, 25(11), 1566–1591 CrossRef CAS.
- (a) A. S. Cheung and D. J. Mooney, Engineered materials for cancer immunotherapy, Nano Today, 2015, 10(4), 511–531 CrossRef PubMed; (b) M. Strzelec, J. Detka, P. Mieszczak, M. K. Sobocińska and M. Majka, Immunomodulation—a general review of the current state-of-the-art and new therapeutic strategies for targeting the immune system, Front. Immunol., 2023, 14, 1127704 CrossRef.
- (a) J. Cheng, J. Liu, M. Li, Z. Liu, X. Wang, L. Zhang and Z. Wang, Hydrogel-based biomaterials engineered from natural-derived polysaccharides and proteins for hemostasis and wound healing, Front. Bioeng. Biotechnol., 2021, 9, 780187 CrossRef; (b) J. M. Zhang and J. An, Cytokines, inflammation, and pain, Int. Anesthesiol. Clin., 2007, 45(2), 27–37 CrossRef; (c) S. Lee and K. Margolin, Cytokines in cancer immunotherapy, Cancers, 2011, 3(4), 3856–3893 CrossRef PubMed; (d) A. D. Cook, A. D. Christensen, D. Tewari, S. B. McMahon and J. A. Hamilton, Immune cytokines and their receptors in inflammatory pain, Trends Immunol., 2018, 39(3), 240–255 CrossRef PubMed.
- A. Varanko, S. Saha and A. Chilkoti, Recent trends in protein and peptide-based biomaterials for advanced drug delivery, Adv. Drug Delivery Rev., 2020, 156, 133–187 CrossRef PubMed.
- (a) P. Davoodi, L. Y. Lee, Q. Xu, V. Sunil, Y. Sun, S. Soh and C. H. Wang, Drug delivery systems for programmed and on-demand release, Adv. Drug Delivery Rev., 2018, 132, 104–138 CrossRef PubMed; (b) A. M. Vargason, A. C. Anselmo and S. Mitragotri, The evolution of commercial drug delivery technologies, Nat. Biomed. Eng., 2021, 5(9), 951–967 CrossRef PubMed.
- J. Li and D. J. Mooney, Designing hydrogels for controlled drug delivery, Nat. Rev. Mater., 2016, 1(12), 1–7 Search PubMed.
- (a) S. Correa, A. K. Grosskopf, H. Lopez Hernandez, D. Chan, A. C. Yu, L. M. Stapleton and E. A. Appel, Translational applications of hydrogels, Chem. Rev., 2021, 121(18), 11385–11457 CrossRef CAS; (b) T. R. Hoare and D. S. Kohane, Hydrogels in drug delivery: Progress and challenges, Polymer, 2008, 49(8), 1993–2007 CrossRef CAS; (c) T. Vermonden, R. Censi and W. E. Hennink, Hydrogels for protein delivery, Chem. Rev., 2012, 112(5), 2853–2888 CrossRef CAS PubMed; (d) A. S. Hoffman, Hydrogels for biomedical applications, Adv. Drug Delivery Rev., 2012, 64, 18–23 CrossRef; (e) A. Mandal, J. R. Clegg, A. C. Anselmo and S. Mitragotri, Hydrogels in the clinic, Bioeng. Transl. Med., 2020, 5(2), e10158 CrossRef CAS; (f) X. Li, Y. Shou and A. Tay, Hydrogels for engineering the immune system, Adv. NanoBiomed Res., 2021, 1(3), 2000073 CrossRef CAS.
- (a) N. R. Richbourg and N. A. Peppas, The swollen polymer network hypothesis: Quantitative models of hydrogel swelling, stiffness, and solute transport, Prog. Polym. Sci., 2020, 105, 101243 CrossRef CAS; (b) N. R. Richbourg, M. Wancura, A. E. Gilchrist, S. Toubbeh and B. A. Harley, Cosgriff-Hernandez E, Peppas NA. Precise control of synthetic hydrogel network structure via linear, independent synthesis-swelling relationships, Sci. Adv., 2021, 7(7), eabe3245 CrossRef CAS; (c) C. C. Lin and A. T. Metters, Hydrogels in controlled release formulations: network design and mathematical modeling, Adv. Drug Delivery Rev., 2006, 58(12–13), 1379–1408 CrossRef CAS.
- (a) H. P. Erickson, Size and shape of protein molecules at the nanometer level determined by sedimentation, gel filtration, and electron microscopy, Biol. Proced. Online, 2009, 11, 32–51 CrossRef CAS; (b) J. K. Armstrong, R. B. Wenby, H. J. Meiselman and T. C. Fisher, The hydrodynamic radii of macromolecules and their effect on red blood cell aggregation, Biophys. J., 2004, 87(6), 4259–4270 CrossRef CAS PubMed.
- (a) C. A. Dreiss, Hydrogel design strategies for drug delivery, Curr. Opin. Colloid Interface Sci., 2020, 48, 1–7 CrossRef CAS; (b) L. Abune and Y. Wang, Affinity hydrogels for protein delivery, Trends Pharmacol. Sci., 2021, 42(4), 300–312 CrossRef CAS PubMed.
- Y. D. Limasale, P. Atallah, C. Werner, U. Freudenberg and R. Zimmermann, Tuning the local availability of VEGF within glycosaminoglycan–based hydrogels to modulate vascular endothelial cell morphogenesis, Adv. Funct. Mater., 2020, 30(44), 2000068 CrossRef.
- K. H. Bae, F. Lee, K. Xu, C. T. Keng, S. Y. Tan, Y. J. Tan, Q. Chen and M. Kurisawa, Microstructured dextran hydrogels for burst-free sustained release of PEGylated protein drugs, Biomaterials, 2015, 63, 146–157 CrossRef.
- (a) F. Artusio, A. Castellví, A. Sacristán, R. Pisano and J. A. Gavira, Agarose gel as a medium for growing and tailoring protein crystals, Cryst. Growth Des., 2020, 20(8), 5564–5571 CrossRef; (b) J. M. Schieferstein, P. Reichert, C. N. Narasimhan, X. Yang and P. S. Doyle, Hydrogel microsphere encapsulation enhances the flow properties of monoclonal antibody crystal formulations, Adv. Ther., 2021, 4(4), 2000216 CrossRef.
- S. Correa, A. K. Grosskopf, J. H. Klich, H. L. Hernandez and E. A. Appel, Injectable liposome-based supramolecular hydrogels for the programmable release of multiple protein drugs, Matter, 2022, 5(6), 1816–1838 CrossRef CAS PubMed.
- (a) D. Sivakumaran, D. Maitland and T. Hoare, Injectable microgel-hydrogel composites for prolonged small-molecule drug delivery, Biomacromolecules, 2011, 12(11), 4112–4120 CrossRef CAS PubMed; (b) D. Sivakumaran, D. Maitland, T. Oszustowicz and T. Hoare, Tuning drug release from smart microgel–hydrogel composites via cross-linking, J. Colloid Interface Sci., 2013, 392, 422–430 CrossRef CAS PubMed; (c) M. H. Chen, J. J. Chung, J. E. Mealy, S. Zaman, E. C. Li, M. F. Arisi, P. Atluri and J. A. Burdick, Injectable supramolecular hydrogel/microgel composites for therapeutic delivery, Macromol. Biosci., 2019, 19(1), 1800248 CrossRef PubMed.
- (a) A. S. Parmar and M. Muschol, Hydration and hydrodynamic interactions of lysozyme: effects of chaotropic versus kosmotropic ions, Biophys. J., 2009, 97(2), 590–598 CrossRef CAS; (b) A. Hawe, W. L. Hulse, W. Jiskoot and R. T. Forbes, Taylor dispersion analysis compared to dynamic light scattering for the size analysis of therapeutic peptides and proteins and their aggregates, Pharm. Res., 2011, 28, 2302–2310 CrossRef CAS PubMed.
- (a) Y. Yu and Y. Chau, Formulation of in situ chemically cross-linked hydrogel depots for protein release: from the blob model perspective, Biomacromolecules, 2015, 16(1), 56–65 CrossRef CAS; (b) J. Kim, Y. P. Kong, S. M. Niedzielski, R. K. Singh, A. J. Putnam and A. Shikanov, Characterization of the crosslinking kinetics of multi-arm poly (ethylene glycol) hydrogels formed via Michael-type addition, Soft Matter, 2016, 12(7), 2076–2085 RSC.
- (a) P. L. Ritger and N. A. Peppas, A simple equation for description of solute release I. Fickian and non-fickian release from non-swellable devices in the form of slabs, spheres, cylinders or discs, J. Controlled Release, 1987, 5(1), 23–36 CrossRef CAS; (b) P. L. Ritger and N. A. Peppas, A simple equation for description of solute release II. Fickian and anomalous release from swellable devices, J. Controlled Release, 1987, 5(1), 37–42 CrossRef CAS.
- (a) F. Comert, A. J. Malanowski, F. Azarikia and P. L. Dubin, Coacervation and precipitation in polysaccharide–protein systems, Soft Matter, 2016, 12(18), 4154–4161 RSC; (b) M. van de Weert, M. B. Andersen and S. Frokjaer, Complex coacervation of lysozyme and heparin: complex characterization and protein stability, Pharm. Res., 2004, 21, 2354–2359 CrossRef CAS PubMed.
- A. Z. Nelson, B. Kundukad, W. K. Wong, S. A. Khan and P. S. Doyle, Embedded droplet printing in yield-stress fluids, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(11), 5671–5679 CrossRef CAS.
- (a) Y. Yu and Y. Chau, One-step “click” method for generating vinyl sulfone groups on hydroxyl-containing water-soluble polymers, Biomacromolecules, 2012, 13(3), 937–942 CrossRef CAS PubMed; (b) C. M. L. Lau, G. Jahanmir, Y. Yu and Y. Chau, Controllable multi-phase protein release from in situ hydrolyzable hydrogel, J. Controlled Release, 2021, 335, 75–85 CrossRef CAS.
- F. M. Veronese and G. Pasut, PEGylation, successful approach to drug delivery, Drug Discovery Today, 2005, 10(21), 1451–1458 CrossRef CAS PubMed.
- G. Mathew, I. Mezić, S. Grivopoulos, U. Vaidya and L. Petzold, Optimal control of mixing in Stokes fluid flows, J. Fluid Mech., 2007, 580, 261–281 CrossRef CAS.
- R. Xu, E. J. Pope and J. D. Mackenzie, Structural evolution of sol-gel systems through viscosity measurement, J. Non-Cryst. Solids, 1988, 106(1–3), 242–245 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Chemical (polymer) syntheses and characterizations, sizing of microgels, quantified characterization of in-droplet mixing of microgels, rheological measurement, SEM images, swelling study with mesh size estimation, protein assays standard curves, and summary table of formulations and characterized data of microgels. See DOI: https://doi.org/10.1039/d4pm00080c |
| This journal is © The Royal Society of Chemistry 2024 |