
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePharmaceutical salts of azole anti-fungal drugs: physicochemical behaviour and activity studies†
Hafsa
Qadri‡
 a,
Asif A.
Malik‡
b,
Aadil A.
Ahangar
b,
Manzoor Ahmad
Mir
*a,
Ajiaz A.
Dar
*b and
Abdul Haseeb
Shah
*a
a,
Asif A.
Malik‡
b,
Aadil A.
Ahangar
b,
Manzoor Ahmad
Mir
*a,
Ajiaz A.
Dar
*b and
Abdul Haseeb
Shah
*a
aDepartment of Bioresources, University of Kashmir, Hazratbal, Srinagar-190006, Jammu and Kashmir, India. E-mail: abdulhaseeb@kashmiruniversity.ac.in; drmanzoor@kashmiruniversity.ac.in
bCrystal Engineering Laboratory, Department of Chemistry, University of Kashmir, Hazratbal, Srinagar-190006, Jammu and Kashmir, India. E-mail: aijazku2015@gmail.com; daraijaz@uok.edu.in
First published on 8th July 2024
Abstract
Pharmaceutical cocrystal engineering is a potential and growing strategy for modulating the physicochemical and pharmacokinetic properties of drug molecules. This study aims to study the new solid forms of miconazole (MIC) and ketoconazole (KTC) prepared through the crystal engineering method of crystallization. Utilizing the understanding of the sulfonate-pyridinium synthon, molecular salts of MIC and KTC with naphthalene disulfonic acid (NDSA-2H) have been prepared and characterized through thermal, spectroscopic, microscopic, and diffraction methods. Both molecular salts, i.e., MIC-C and KTC-C, have been obtained as crystalline solids and their phase purity and formation have been established through diffraction studies. The new drug forms exhibit augmented thermal stability and aqueous solubility. Powder dissolution studies in an aqueous medium at pH 2 and pH 7 indicate a significant increase in thermal stability and aqueous solubility of the new drug forms compared to their drug precursors. Structural investigation of MIC-C validates the formation of the ionic sulfonate-pyridinium synthon involving proton transfer resulting in charge development, leading to enhancement in the physicochemical properties. In vitro studies show that KTC-C in addition to retaining most of the biological activities possesses antifungal potential comparable to that of the standard drug since it inhibited the growth of tested Candida strains without showing enhancement in host toxicity. Both the designed salts exhibit fluorescence properties inside Candida cells (in contrast to the standard drugs).
Introduction
We have been witnessing a worrying trend over the past few decades, wherein traditional antimicrobials fail to effectively treat infectious diseases.1,2 Urgency persists in developing novel therapeutics against human pathogens and averting resistance in the “post-antibiotic era”. Limited antifungal options for invasive infections and the spread of multidrug-resistant fungal strains heighten concerns, emphasizing the need for innovative solutions. The focus on fungal pathogens underscores the critical importance of addressing antimicrobial resistance.3 Invasive fungal infections, particularly those caused by Candida species, significantly contribute to global infectious disease mortality, impacting immunosuppressed individuals and those undergoing invasive medical procedures or prolonged ICU treatment.3 With a crude death rate of approximately forty percent despite appropriate antifungal treatments, Candida species, particularly Candida albicans, rank as the primary cause of all healthcare-associated bloodstream infections in the United States.3–6 In the oral cavity or gastrointestinal tract, C. albicans is typically present in healthy people as a harmless commensal; however, in people with severely compromised immunity, this pathogen can spread into the bloodstream and colonize internal organs, leading to potentially fatal systemic infections.6–8 Since the pathogen and humans are both eukaryotic, there are not many distinct biochemical targets that can be directed to build new antifungal drugs.3Due to the eukaryotic nature of fungal cells, difficulties with compound permeability across the fungal cell wall and membrane, and the lack of interest from the pharmaceutical sector, the creation of innovative antifungal drugs has often been delayed.9,10 In reality, since the middle of the 2000s, no new class of antifungals has been used in the clinical setting.11,12 There are now only a few primary classes of antifungal drugs that are licensed for the treatment of invasive mycoses, and the emergence of drug resistance in pathogen populations jeopardizes the effectiveness of these antifungal medications. Notably, the little arsenal of antifungals that are currently available to treat these severe infections is further threatened by the appearance of newly emerging drug-resistant species like Candida auris.3 Widespread antifungal use has spurred strong resistance in Candida species, showcasing adaptive strategies like up-regulated multidrug transporters, modified drug targets, and activated cellular stress responses in human fungal pathogens.3,13 Novel formulations aim to enhance traditional antifungal drugs, overcoming limitations such as toxicity and drug resistance. Promising new antifungals offer alternatives for treating superficial infections, potentially providing more effective options. These advancements provide hope for achieving improved pharmaceutical solutions against fungal infections.14
Crystal engineering (CE) is a promising strategy for developing materials with desired physicochemical properties.15 CE outweighs complex organic synthesis methods in terms of cost-effectiveness, environmental benignity, reduced time, and efficiency, making it a greener approach.16,17 The utilization of the principles and concepts of CE creates an immense opportunity in the direction of pharmaceutical chemistry, where drug co-crystals/salts with improved physicochemical and pharmacokinetic behavior can be designed and addressed.18–21 Co-crystal/salt formation is simple and involves the supramolecular complexation of a drug molecule of interest with a desirable and biologically safe coformer. Coformer selection, however, is key to modulating the behavior of new drug forms, and its selection is based on functional compatibility, which depends on hydrogen bonding tendency and the alignment of functionalities to form robust synthons.22 Supramolecular synthons commonly utilized for the design of salt formation include alcohol-pyridine, carboxylic acid-pyridine, acid-amide, etc. We have reported a very successful intermolecular interaction using the sulfonate-pyridinium synthon, which yields new solid forms with better optical, thermal, and solubility behaviour.23–29 The successful creation of the synthon requires the presence of pyridyl and sulfonate functionalities in the precursor molecules. Based on this understanding we have recently reported the development of organic salts of fluconazole with improved physicochemical properties and antifungal activities.30 Herein, we further utilize the sulfonate-pyridinium synthon for the development of organic salts of the commercial anti-fungal drugs MIC and KTC to explore the new solid forms in terms of their optical, physicochemical, and anti-fungal activities. The new drug forms MIC-C and KTC-C exhibit significantly improved thermal, optical, and solubility properties. Being class II drugs, solubility reports of salts of MIC and KTC have been reported by various groups earlier; however, to the best of our knowledge, detailed toxicity and antifungal activities of the new drugs have not been reported.31–36 Therefore, the detailed in vitro studies of these products have been reported. Both the salts show fluorescence inside cells. Moreover, KTC-C displayed potent antifungal efficacy and restricted the growth of various Candida strains at drug concentrations comparable to those of the parent drug without exhibiting any enhanced host cytotoxicity.
Results and discussion
Salts of MIC-C and KTC-C were obtained by slow evaporation of the methanolic solution of precursors at room temperature, as colorless blocks and pink flakes, respectively. Fourier-transform infrared (FT-IR) studies substantiate the formation of organic salts MIC-C and KTC-C. The asymmetric stretching of S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in MIC-C is at 1330 cm−1 and its symmetric stretching is at 1285 cm−1, while in KTC-C, the SO asymmetric and symmetric stretching are at 1365 and 1167 cm−1, respectively. The absorption bands at 1530–1620 cm−1 correspond to CC stretching and the peak at around 850–550 cm−1 corresponds to the presence of C–Cl stretching. New peaks in the product originate at around 1010–1030 cm−1, corresponding to the deprotonation of the sulfonic group, and subsequently, protonation of pyridyl nitrogen (N+–H) is observed as an absorption band in the range of 3040–3060 cm−1. The FT-IR spectra of the products MIC-C and KTC-C are provided in Fig. ES1.†1H NMR spectra of the products MIC-C and KTC-C are reported in DMSO-d6 and CD3OD, respectively. The formation of MIC-C and KTC-C in a 2
O in MIC-C is at 1330 cm−1 and its symmetric stretching is at 1285 cm−1, while in KTC-C, the SO asymmetric and symmetric stretching are at 1365 and 1167 cm−1, respectively. The absorption bands at 1530–1620 cm−1 correspond to CC stretching and the peak at around 850–550 cm−1 corresponds to the presence of C–Cl stretching. New peaks in the product originate at around 1010–1030 cm−1, corresponding to the deprotonation of the sulfonic group, and subsequently, protonation of pyridyl nitrogen (N+–H) is observed as an absorption band in the range of 3040–3060 cm−1. The FT-IR spectra of the products MIC-C and KTC-C are provided in Fig. ES1.†1H NMR spectra of the products MIC-C and KTC-C are reported in DMSO-d6 and CD3OD, respectively. The formation of MIC-C and KTC-C in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric ratio corresponding to the drug and NDSA-2H formers, respectively, for both salt forms, is confirmed by the comparison of the peak area ratios of protons in the crystal formers. Furthermore, both chemical shifts and relative peak intensity values are in agreement with their formation. The 1H-NMR spectra of MIC-C and KTC-C with peak assignments and intensity ratios are depicted in Fig. ES2 and ES3.† The formation and formulation of the products are sketched in Scheme 1.
:1 stoichiometric ratio corresponding to the drug and NDSA-2H formers, respectively, for both salt forms, is confirmed by the comparison of the peak area ratios of protons in the crystal formers. Furthermore, both chemical shifts and relative peak intensity values are in agreement with their formation. The 1H-NMR spectra of MIC-C and KTC-C with peak assignments and intensity ratios are depicted in Fig. ES2 and ES3.† The formation and formulation of the products are sketched in Scheme 1.
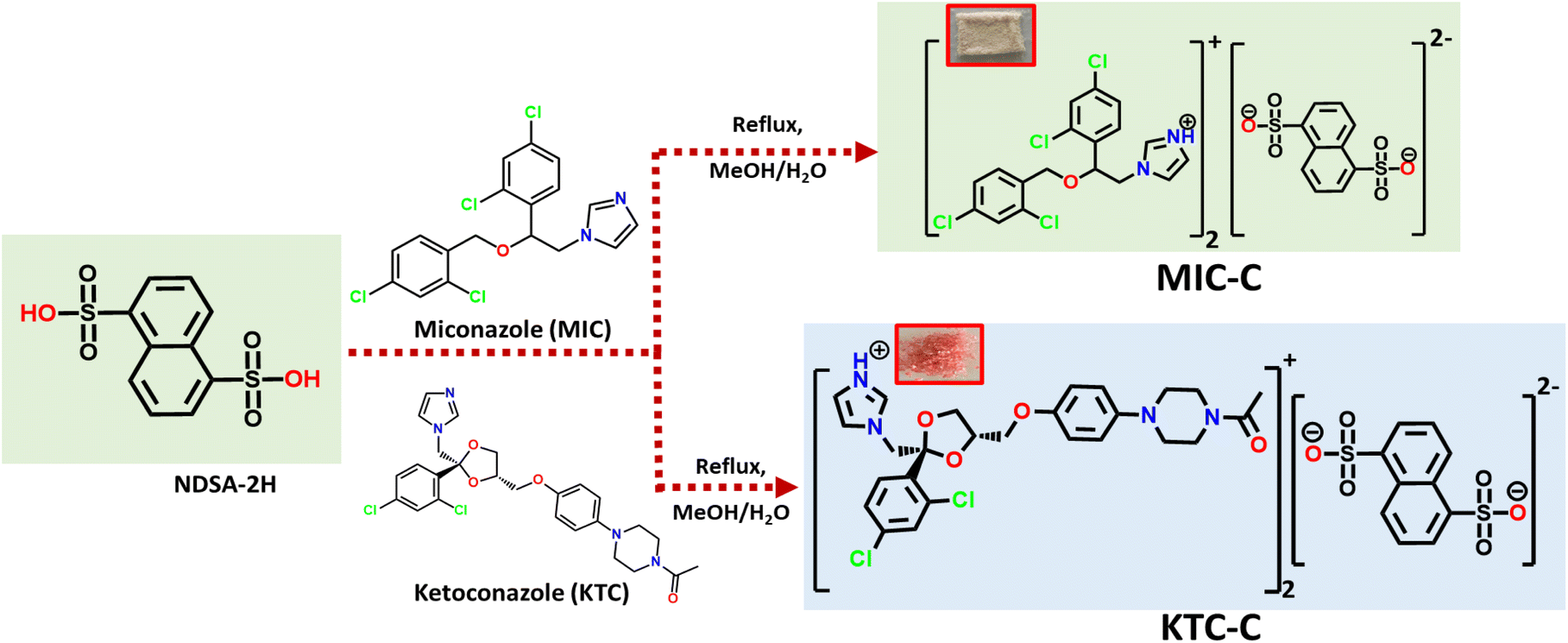 | ||
| Scheme 1 Synthetic procedure for pharmaceutical salts of MIC and KTC. | ||
MIC-C and KTC-C exhibit high thermal stability as indicated by their melting points (>220 °C) compared to the parent drugs, MIC (148–152 °C) and KTC (146 °C), which have been validated through DSC. Analysis of the TGA plots of MIC-C and KTC-C reveals no significant weight loss before the melting event, confirming their formation in non-solvated forms, Fig. ES4 and ES5.† No phase transitions were observed in the course of heating for MIC-C and KTC-C based on the DSC results and their melting occurs at 231 °C and 248 °C with onset temperatures of 228 °C and 236 °C, respectively, Fig. ES6.†
The organic salt forms have been characterized through diffraction methods using both single-crystal and powder forms. The powder X-ray diffraction (PXRD) of MIC-C and KTC-C substantiates their formation and phase purity. Experimental P-XRD data of MIC-C in comparison with its simulated data validated the phase retention upon size reduction, Fig. 1, and the P-XRD curve of KTC-C is provided in Fig. SE7.†
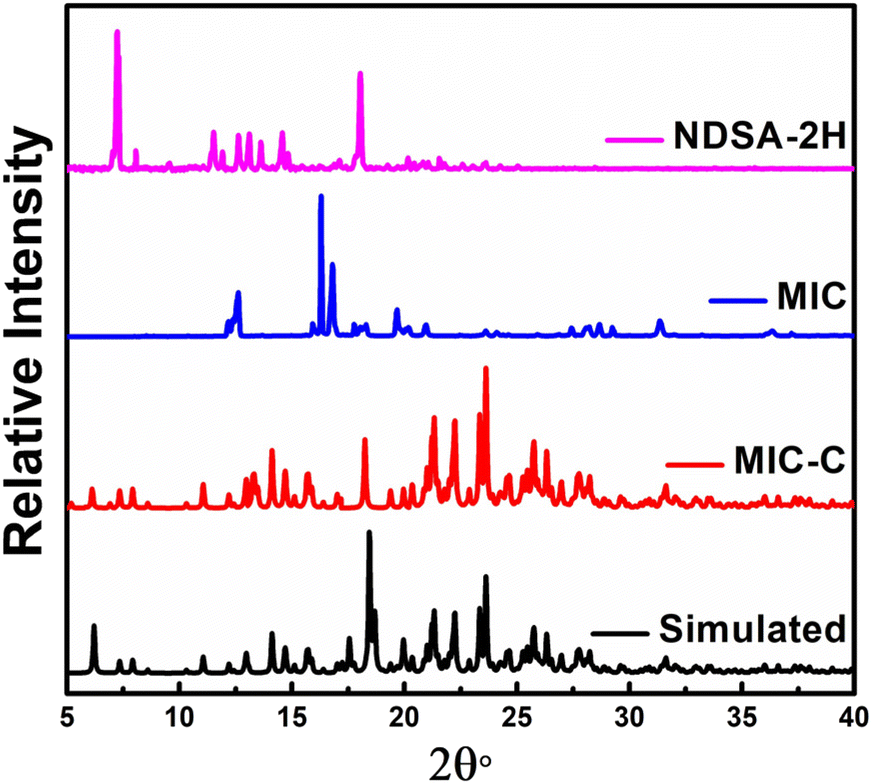 | ||
| Fig. 1 Experimental powder diffraction X-ray curves of MIC-C compared with its simulated pattern and precursors MIC and NDSA-2H. | ||
Solid-state structural analyses
Good-quality salts of KTC-C, suitable for single-crystal X-ray diffraction, could not be obtained despite multiple attempts, while MIC-C has been characterized crystallographically. The colorless blocks of MIC-C crystallize in a centrosymmetric triclinic space group, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , and the asymmetric unit is composed of two molecules of the MIC drug in the protonated form (MIC-H)+ and half a molecule of di-anionic organo-sulfonate (NDSA)2−. The existence of the crystal forms in the salt form is in agreement with the ΔpKa rule, as the difference between the crystal formers in both cases is more than 3. Miconazole has a pKa value of 6.7, ketoconazole is a dibasic compound with two pKa values of 2.91 and 6.5 and the pKa of 1,5-naphthalenedisulfonic acid is −0.51.37–39 Anticipated proton transfer between acid and base components occurs, and the new drug form exists as a salt with the formulation [(MIC)2+(NDSA)2−]. The augmented thermal stability of the organic salt form can be attributed to strong ionic interactions between the crystal components.
, and the asymmetric unit is composed of two molecules of the MIC drug in the protonated form (MIC-H)+ and half a molecule of di-anionic organo-sulfonate (NDSA)2−. The existence of the crystal forms in the salt form is in agreement with the ΔpKa rule, as the difference between the crystal formers in both cases is more than 3. Miconazole has a pKa value of 6.7, ketoconazole is a dibasic compound with two pKa values of 2.91 and 6.5 and the pKa of 1,5-naphthalenedisulfonic acid is −0.51.37–39 Anticipated proton transfer between acid and base components occurs, and the new drug form exists as a salt with the formulation [(MIC)2+(NDSA)2−]. The augmented thermal stability of the organic salt form can be attributed to strong ionic interactions between the crystal components.
The two sulfonate protons shift to the N3 and N6 atoms of the imidazole rings of two different molecules of MIC, and the proton transfer is validated by relaxed C–N–C bond angle values of 108.85 and 109.37°, respectively, around the protonated nitrogens. The N3 center of the protonated drug molecule forms a direct sulfonate-pyridinium bond with O6 of the sulfonate functionality N3–H3N⋯O6 [D⋯A: 2.694(1) Å; D–H⋯A: 163.19°] and further interacts with the sulfonate group of another neighbor by forming N3–H3⋯O2 [3.122(2) Å; 71.45(1)°] hydrogen bonds.
The protonated N6 center of another triazole ring of another molecule also forms a direct sulfonate-pyridinium interaction with the sulfonate oxygen O3 of the acid former: N6–HN6⋯O3 [1.810(3) Å; 152.70(2)°]. The molecular structure of MIC-C is provided in Fig. 2. Other than strong sulfonate-pyridinium interactions there are no significant intermolecular interactions, except for bifurcated halogen atom interactions around Cl11 and Cl15. Other halogen atoms Cl12, Cl13, Cl14, Cl16, Cl17, and Cl18 do not undergo significant interactions and are directed towards the voids in the reticular solid, while the protons on the dichlorobenzyl ring are sufficiently acidic to form hydrogen bonds with triazole nitrogens N1 and N4. π–π interactions are absent and the crystal lattice is stabilized by weak Cl⋯H and C⋯H dispersive interactions.
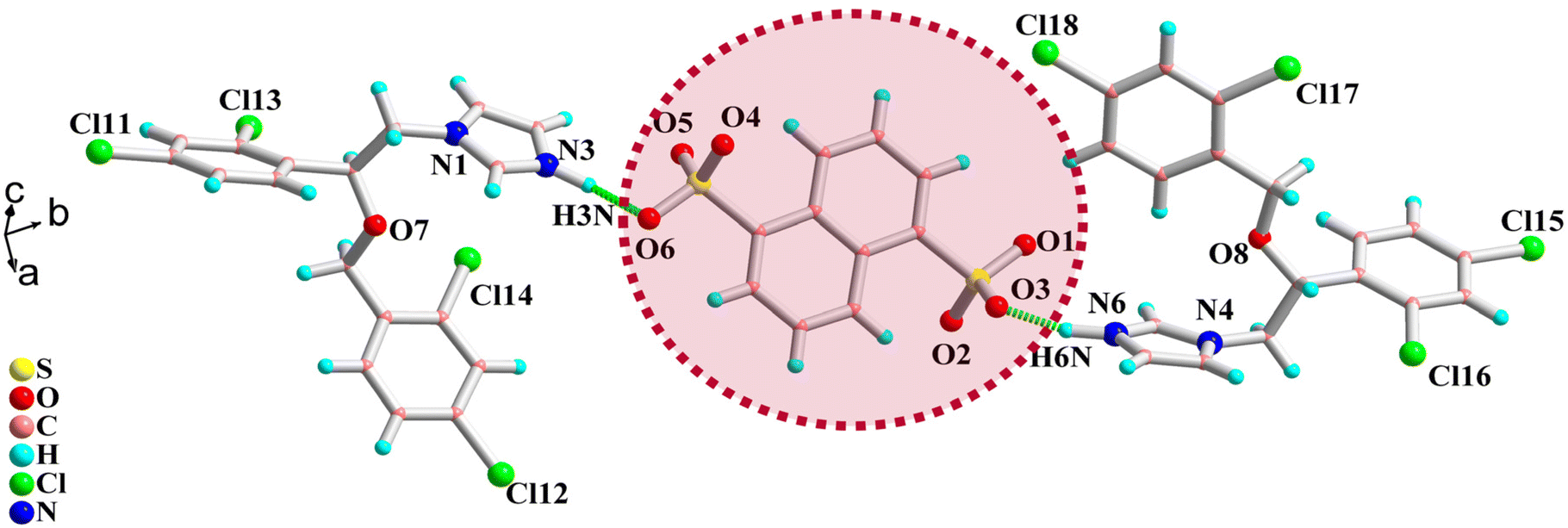 | ||
| Fig. 2 Molecular structure of MIC-C. | ||
Photo-physical studies
The photo-physical properties of the organic salts were recorded using a Shimadzu 2600 DRUV instrument. In the solid phase, the coformer NDSA-2H and the organic-salt of MIC-C moieties are colorless, while the organic salt of KTC-C appears light pink. In the solid-state absorption spectra, the MIC-C shows a broad peak at around 270 nm, which correlates to π–π* transition, while a redshift appears for KTC-C in comparison with the pristine KTC drug. The peak in KTC-C at around 400 nm may be attributed to charge transfer interactions, which are plausibly responsible for its pink color.The optical band gaps of the molecular salts have been obtained from Tauc plots after conversion through the Kubelka–Munk function indicating modulation of the energy difference between frontier orbitals. The KM absorption plot and band gap of molecular salts MIC-C and KTC-C compared with those of the parent drugs are given in Fig. 3A and B, respectively.
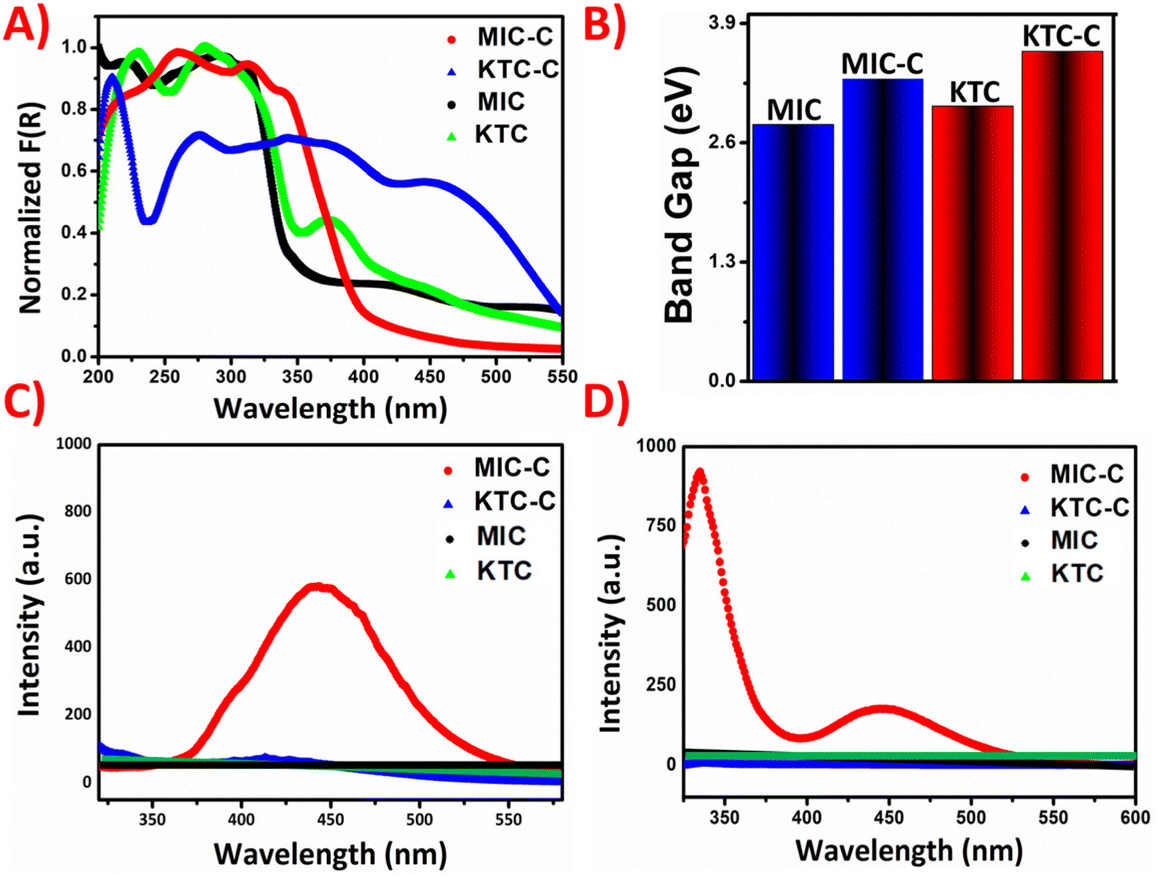 | ||
| Fig. 3 (A) Solid-state KM-absorbance of MIC-C and KTC-C compared with that of the parent drugs; (B) bar graph diagram representing the band gap of the molecular salts MIC-C and KTC-C compared with that of the parent drugs; (C) solid-state emission of the pharmaceutical organic salts MIC-C and KTC-C compared with that of the parent drugs and (D) solution-phase emission of MIC-C and KTC-C compared with that of the parent drugs (solvent used was water/MeOH (7:3 v/v)). | ||
Another interesting observation from structural and theoretical studies reveals that the MIC-C complex does not undergo significant π-stacking, a favorable situation for solid-state emission, which further prompted us to investigate the luminescence of the molecular salts. The luminescence behavior has been investigated in both the solid and solution phases. Emission turn-on has been observed in MIC-C in both solution and the solid state, compared to the non-emissive drug precursor, and KTC-C, respectively. The plausible reason responsible for the turn-on is aggregation-induced emission as shown in Fig. 3C and D. The optical images of the drug precursors and their salts under day and UV light are provided in Fig. 4.
 | ||
| Fig. 4 Photographs of MIC-C and KTC-C under ambient light and UV light along with the crystal morphology. | ||
Solubility studies
Pharmaceutical solubility and bioavailability are critical factors in medication efficacy. Pharmaceuticals suffer from low water solubility due to their organic nature. We have discovered a remarkable increase in solubility of organic salts using the sulfonate-pyridinium supramolecular synthon. The expected crystallization of MIC-C and KTC-C motivated us to study their water solubility. The solubility of MIC-C and KTC-C vis-à-vis salt formers was explored to compare the apparent aqueous solubility at two physiological pH values of 2 and 7, and the results were replicated three times. The comparative apparent solubility values are depicted as histograms in Fig. 5.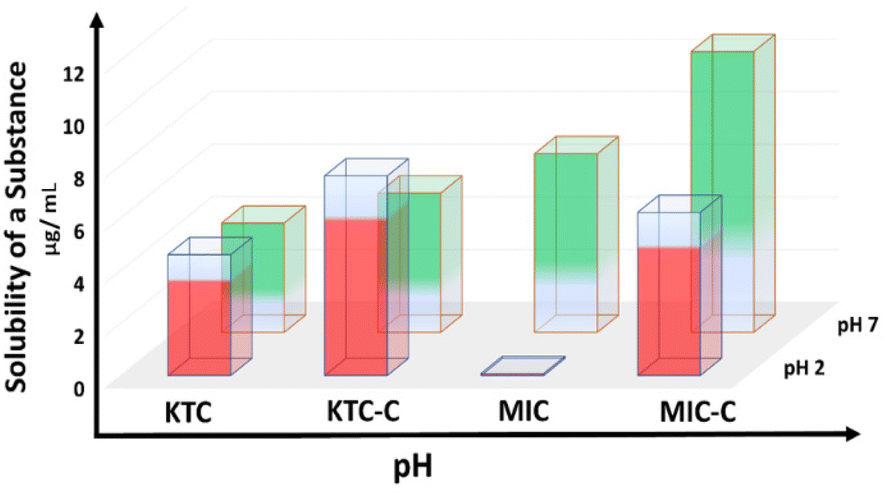 | ||
| Fig. 5 Bar graph showing the solubility enhancement in MIC-C and KTC-C over the parent drugs at pH values of 2 and 7. | ||
KTC-C exhibits better solubility at both pH values than the pure drug, as the absolute solubility values for KTC at pH 2 and 7 are 1300 and 1200 μg mL−1, respectively, while the solubility values for KTC-C are 2900 and 1700 μg mL−1 resulting in an increase of 123% and 42%, respectively. Meanwhile, the solubility values of MIC at pH 2 and 7 are 200 and 1800 μg mL−1 and those of MIC-C are 1500 and 2500 μg mL−1. This is a significant advantage that can help the biological efficiency of miconazole and ketoconazole drug activity, as better solubility of molecules can enhance their pharmacokinetic properties and bioavailability. The absorption spectra of miconazole and ketoconazole drugs at different physiological pH values of 2 and 7 are given in ES11 and 12.† The calculated approximate weight of the parent drug left after the solubility test is given in Table ES8.† The phases of the remaining solids of the parent drugs and salts MIC-C and KTC-C after solubility tests are given in Fig. ES13–16.†
Azole salt KTC-C shows antifungal activity comparable to that of the corresponding standard drug
The broth micro-dilution assay was used to investigate both pairs of drugs (KTC, KTC-C, MIC, and MIC-C) for their possible antifungal potential against various Candida strains. The MIC90 values (μg mL−1) for both drug pairs against several Candida strains are shown in Table 1. The findings of the broth micro-dilution assay reveal that KTC-C shows comparable activity to the standard drug KTC against most of the tested strains and that both pairs of drugs display efficacy against the majority of the tested strains, with MIC90 values ranging from 0.00195 to 0.25μg mL−1. Additional susceptibility tests, including the spot assay (Fig. ES8A†) and the disc diffusion assay (Fig. ES8B and Table ES1†), validate the results of the broth micro-dilution assay and show that each pair of drugs suppressed the growth of the majority of the tested Candida strains (Fig. ES8A and B†).| Drugs | C. albicans | C. parapsilosis | C. glabrata | C. auris | ADCdr1-GFP | ADCaMdr1-GFP | ADPdr5-GFP |
|---|---|---|---|---|---|---|---|
| KTC | 0.00195 | 0.0078 | 0.25 | 0.0625 | 0.03125 | 0.125 | 0.01562 |
| KTC-C | 0.00780 | 0.0078 | 0.25 | 0.0625 | 0.03125 | 0.250 | 0.03125 |
| MIC | 0.00195 | 0.0156 | 0.0156 | 0.0625 | 0.25 | .00390 | 0.125 |
| MIC-C | 0.00390 | 0.0625 | 0.0625 | 0.1250 | 0.25 | 0.0078 | 0.125 |
In the pristine state, KTC exhibits a 100% absolute concentration, while in the salt form, the effective concentration of KTC-C is less. Consequently, when comparing the MIC90 values of the two KTC forms, at equivalent MIC90 concentrations KTC-C provides a lower absolute concentration of KTC, suggesting enhanced drug efficacy. Conversely, when MIC90 values are doubled for KTC-C, its efficacy still matches that of KTC.
Assessing interactions between KTC, KTC-C, MIC, and MIC-C and other known antifungal agents
The potential interactions (synergism/antagonism) between KTC, KTC-C, MIC, and MIC-C and other known antifungals were determined by checkerboard assays. Based on FICI values the interactions between the drug combinations were investigated (Tables ES2A, B; ES3A, B; ES4A, B and ES5A, B†). Along with the WT Candida strains the study also employed drug transporter over-expressing yeast strains ADCDR1-GFP, ADPDR5-GFP, and ADCaMdr1-GFP. The azole drugs (KTC, KTC-C, MIC, and MIC-C) were tested for their interactions with caspofungin (an echinocandin) and amphotericin B (AmpB, a polyene) at sub-inhibitory concentrations against these fungal strains.At sub-inhibitory concentrations, KTC/KTC-C and caspofungin both exhibited synergistic effects against all the tested strains except for ADCDR1-GFP and ADPDR5-GFP (Table ES4A and B†). Meanwhile, the combinations of MIC/MIC-C and caspofungin showed synergistic effects against C. albicans, C. glabrata, and C. parapsilosiss only (Table ES5A and B†). Moreover, the combinations of MIC/MIC-C and AmpB showed no synergistic effect against any of the tested strains (Table ES3A and B†). However, KTC and AmpB combinations showed synergism only in the case of C. albicans and C. auris and the KTC-C and AmpB combinations showed synergism only in the case of C. albicans and C. glabrata (Table ES2A and B†). In general, both KTC-C and MIC-C displayed synergistic interactions comparable to those of the standard antifungals (KTC and MIC).
Analysis of Candida growth in the presence of KTC, KTC-C, MIC, and MIC-C
We next examined how effectively the drugs (KTC, KTC-C, MIC, and MIC-C) worked against the most common and emerging human fungal pathogens, C. albicans and C. auris, by following the Candida growth kinetics. To determine the effectiveness of the respective drugs, the OD600 value was evaluated at different time intervals after drug treatment. The outcomes showed that both the drug pairs (KTC, KTC-C and MIC, MIC-C) inhibited C. albicans and C. auris proliferation over a range of time. Additionally, the findings showed that both drug pairs had roughly comparable effectiveness against both the tested strains (Fig. 6A and B).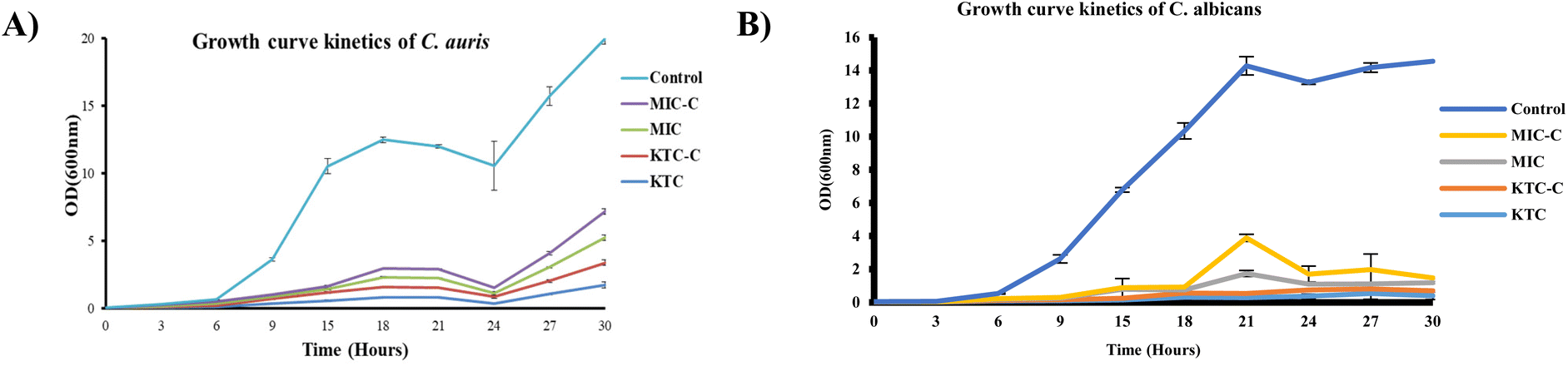 | ||
| Fig. 6 Growth analysis of C. auris (A) and C. albicans (B) in the presence of MIC, MIC-C, KTC and KTC-C. C. auris and C. albicans WT cells without any drug treatment were taken as respective controls. The data were collected in triplicate, and the results are presented as average ± SD. | ||
Azole salt KTC showed minimal host toxicity at effective concentrations
HEK293 and HepG2 cell lines were employed in the cell viability assay using the MTT reagent to determine the cytotoxicity of KTC, KTC-C, MIC, and MIC-C (Fig. 7A and B). Both the drug pairs (KTC, KTC-C, MIC, and MIC-C) were tested within the concentration range of 0.0625–8 μg mL−1. The findings showed that the drug pair KTC and KTC-C did not exhibit significant toxicity up to the maximum tested drug concentration of 8 μg mL−1 in the case of both HEK293 and HepG2 cell lines (Fig. 7A and B). Moreover, 64.7% of the cells were viable in the case of KTC-treated HEK293 cells and 59.8% in the case of KTC-C-treated HEK293 cells at 8 μg mL−1. Similarly, 61.84% of the cells were viable in the case of KTC-C-treated Hep G2 cells and 63.69% in the case of KTC-treated HEP G2 cells at 8 μg mL−1 drug concentration.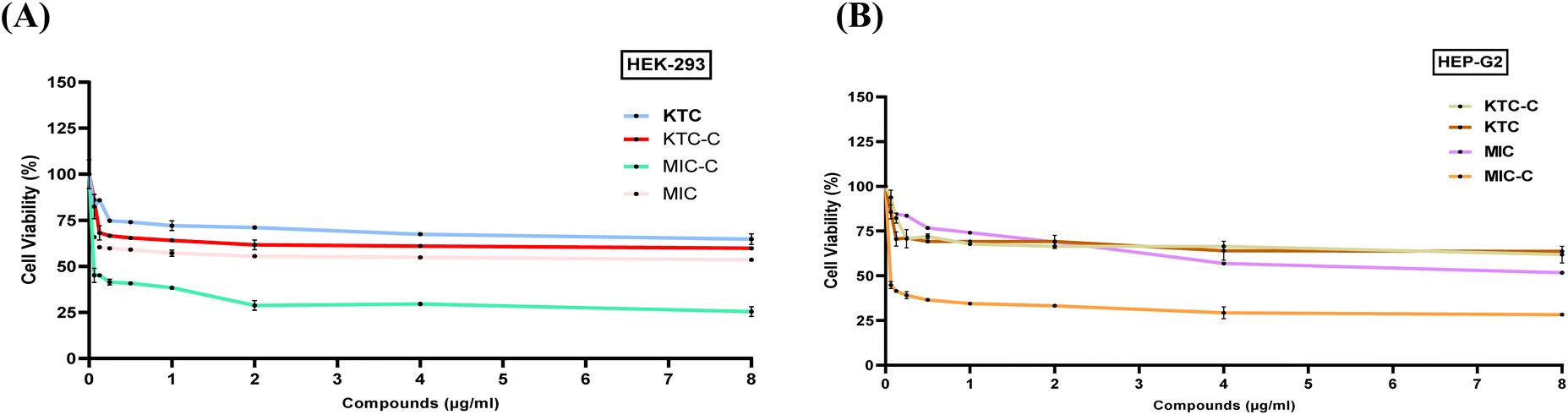 | ||
| Fig. 7 Cytotoxicity assay using (A) MIC, MIC-C, KTC and KTC-C in HEK-293 cells and (B) MIC, MIC-C, KTC and KTC-C in Hep-G2 cells. Both the drug pairs showed similar cytotoxicity patterns and the values were calculated, and the curves were generated using GraphPad PrismV8.4.3. The experiments were repeated thrice, and the findings are presented as average ± SD. | ||
In the case of the MIC/MIC-C drug pair, the findings revealed that MIC-C exhibited more cytotoxicity than MIC at the maximum tested drug concentration of 8 μg mL−1 in the case of both HEK293 and HepG2 cell lines. In the case of MIC-treated HEK293 cells, 53.63% of the cells were viable, while in the case of MIC-treated Hep G2 cells, 51.7% of the cells were viable at 8 μg mL−1. However, in the case of MIC-C-treated HEK293 cells, only 25.5% of the cells were viable, while in the case of MIC-C-treated Hep G2 cells, only 28.3% of the cells were viable at the same drug concentration. All the drugs (KTC, KTC-C, and MIC) except MIC-C (more cytotoxic) followed more or less similar mammalian cytotoxicity patterns and showed minimal cytotoxicity for both cell lines at their effective concentrations.
Assessing plasma membrane depolarisation and cell integrity in the presence of KTC, KTC-C, MIC, and MIC-C
Furthermore, we investigated whether the organic salts, under the influence of combined drug compositions, have any impact on the basic biology of Candida cells. To assess this, we checked plasma membrane depolarisation and cell integrity of Candida cells in the presence of drugs. To evaluate the impact of KTC, KTC-C, MIC, and MIC-C on the cell membrane polarization, we utilized DiBAC4 (3), a dye that penetrates depolarized cell membranes and binds to intracellular proteins that enhance its fluorescence.40 The staining of C. albicans cells with DiBAC4 (3) dye resulted in an increase in the relative fluorescence units in cell suspensions incubated for 1 h with the drugs at MIC/2 concentration (Fig. ES9A†). Although all the drug-treated C. albicans samples displayed only a slight increase in relative fluorescence at the tested concentration in comparison with the control (without any drug) there was no significant difference observed between the samples treated with the respective drug pairs (KTC, KTC-C, MIC, and MIC-C) indicating more or less an equal impact of the drugs on the cell membrane.To further evaluate the impact of drugs on the cell membrane integrity and intracellular enzyme activity of C. albicans cells, a fluorogenic substrate CFDA was used. The dye is absorbed by viable cells and, upon hydrolysis by intracellular esterases, produces carboxyfluorescein and its fluorescence signal can be measured. If a cell is injured, the hydrolysis activity is hampered.41 Our findings revealed that all the drug-treated C. albicans samples stained with CFDA displayed a decrease in relative fluorescence units (RFU) at MIC/2 concentration in comparison with the control sample (without any drug) but there was no significant difference observed between the cells treated with the respective drug pairs (KTC, KTC-C, MIC, and MIC-C) indicating the comparable impact of the drugs on the cell membrane integrity (Fig. ES9†). The decrease in fluorescence intensity was likely caused by a reduction in the intracellular retention of the dye. Moreover, in both the cases the lower impact of the drugs also points towards the fact that these azoles have different mechanisms of action (targets ergosterol biosynthesis) and normally do not target the cell membrane directly.
Comparative fluorescence studies between drug pairs (KTC, KTC-C, MIC, and MIC-C)
The fluorescence characteristics of the drug pairs (KTC, KTC-C, MIC, and MIC-C) inside Candida cells were compared by treating the C. albicans cells with the respective drugs, and the fluorescence intensity between the drug-treated and control samples was examined using a fluorescence microscope. The findings (Fig. 8) demonstrated that C. albicans cells treated with the salt forms of the drugs (KTC-C and MIC-C) displayed enhanced fluorescence in comparison with the cells treated with the amorphous forms of the drugs (KTC and MIC) and control cells (without drug), indicating that the synthesized salts (KTC-C and MIC-C) have better fluorescence properties as observed within the cells. Notably, among the two salts, MIC-C displayed better fluorescence (Fig. 8). Additionally, this important feature of organic salts can be a very helpful characteristic for investigating the mechanism of drug efflux and/or retention by various fungal strains.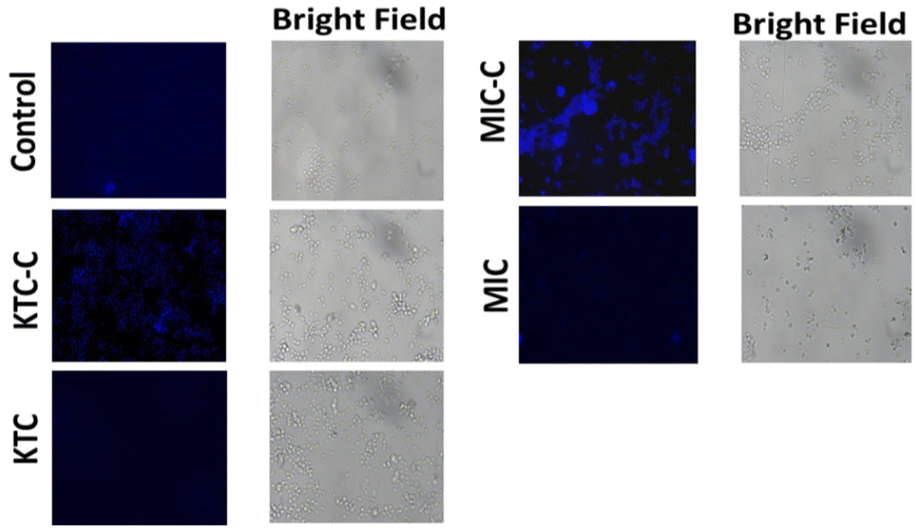 | ||
| Fig. 8 Fluorescence microscopy illustrations showing fluorescence inside C. albicans cells without the drug (control) treated with KTC and MIC along with their respective organic salt forms. | ||
Proteinase activity of Candida in response to KTC, KTC-C, MIC, and MIC-C
C. albicans and C. auris strains were investigated for proteinase activity (the ability of the Candida strains to secrete aspartyl proteinase on a solid medium). The strains exhibited proteinase activity but the activity varied significantly. When compared to the WT strains, the drug-treated Candida strains showed less proteinase activity. The difference in proteinase activity suggests the drug's ability to suppress the proteinase activity of Candida strains. The proteinase activity of WT C. albicans and C. auris treated with KTC, MIC, and their corresponding organic salts was comparatively less (Fig. ES10 and Table ES6A, B†). Moreover, compared to the amorphous forms, organic salts did not reveal significant differences in the proteinase activities indicating no significant loss of drug activities upon salt formation.Conclusion
The study describes two important pharmaceutical organic salts (KTC-C and MIC-C), their design and characterization, and a detailed investigation of their physicochemical properties. Better thermal stability, fluorescence, and water solubility are the key advantages of the modified crystalline drug forms compared to their precursors. A structure–property link for KTC-C and MIC-C has been established through structural and theoretical studies. Studies based on in vitro investigations showed that the synthesized organic salt KTC-C exhibits antifungal activity against a variety of human pathogenic Candida species, which is comparable to that of the parent drug. The current study shows that organic salts have significant antifungal potential and a mode of action similar to that of parent drugs. As a result, these salt forms (KTC-C in particular) can be used as alternatives to the parent drugs, to treat a variety of human pathogenic fungi because they show better thermo-stability and improved solubility and thus can be possibly used at low effective in vivo concentrations minimizing the host toxicity.Experimental
Methods and materials
1,5-Naphthalene-disulfonic acid (NDSA-2H) (>97%, Sigma-Aldrich), MIC (99% pure, TCI), and KTC (99% pure, TCI) were used as procured. Methanol was used for crystallization via the slow evaporation method. The melting point was determined using the MP70 melting point system capillary apparatus (Mettler Toledo) in one-end closed capillaries. Infrared spectroscopic data for molecular solids were obtained using BRUKER ALPHA FT-IR (4000–400 cm−1) in neat form.1H NMR spectra were recorded on a Bruker 500 MHz spectrometer in CD3OD (for KTC-C) and DMSO-d6 (for MIC-C) as the solvents, selected based on their solubilities. The chemical shifts (δ) are given in ppm and referenced to the internal standard tetramethylsilane (Si(CH3)4).
Solution and solid-state optical studies have been carried out on a Shimadzu 2600 spectrometer. For solid samples an integrating sphere attachment was used and data were recorded for samples coated on a BaSO4 medium and the spectra were acquired in the percent reflectance (%R) mode. Using the Kubelka–Munk function, the diffuse reflectance values were transformed into comparable absorption. Luminescence studies were recorded under ambient conditions using a Shimadzu RF-5301-PC spectrofluorometer with slit widths of 5/5 (excitation/emission), and the samples were prepared by coating the product powder on BaSO4 pellets. Thermal gravimetric analyses of these samples were performed on a Simultaneous Thermal Analyzer-STA (LINSEIS, USA 6807/8835/16) using an alumina crucible at a heating rate of 10 °C. DSC was carried out on a DSC 1 STARe system.
Synthesis of MIC-C
The NDSA-2H co-former (360 mg, 1 mmol) was taken in a beaker and dissolved in 10 mL of MeOH solvent. The miconazole drug (416 mg, 1 mmol) was also dissolved in 15 mL of MeOH, and its complete dissolution was ensured. The miconazole solution was then slowly added to the NDSA-2H solution with continuous stirring, and the resultant solution was concentrated upon heating for 20 min, filtered, and kept for crystallization. After a few days, white block-shaped salts were obtained. Yield. 65.7%, melting point >220 °C, FT-IR ν (cm−1) 1630, 1392, 1276, 1022, 1196 and 1146. UV-visible λmax = 272 nm. 1H NMR, δ, 400 MHz, DMSO-d6, ppm: δ 4.42 (d, 2H, –CH2), 4.54 (s, 2H, –CH2), 5.19 (t, 1H, –CH), 5.66 (s, 1H, N–H), 7.38 (d, 1H, Ar–H), 7.42 (d, 1H, Ar–H), 7.48 (s, 2H, Ar–H), 7.52 (d, 2H, Ar–H), 7.62 (d, 2H, Ar–H), 7.67 (s, 2H, Ar–H), 8.01 (d, 2H, Ar–H), 8.91 (d, 2H, Ar–H), 9.08 (s, 1H, –OH).Synthesis of KTC-C
The NDSA-2H co-former (360 mg, 1 mmol) was taken in a beaker and dissolved in 10 mL of MeOH solvent. The ketoconazole drug (531 mg, 1 mmol) was also dissolved in 15 mL of MeOH, and its complete dissolution was ensured. The ketoconazole solution was then slowly added to the NDSA-2H solution with continuous stirring, and the resultant solution was concentrated with heating for 20 min, filtered, and kept for crystallization. After 6–7 days, pink flake-shaped salts were obtained. Yield. 54.3%, melting point >220 °C, FT-IR, ν (cm−1) 1634, 1297, 1112, and 1156. UV-visible λmax = 470 nm. 1H NMR, δ, 400 MHz, CD3OD, ppm: δ 2.00 (s, 3H, –CH3), 2.83 (t, 2H, –CH2), 2.87 (t, 2H, –CH2), 3.19 (t, 2H, –CH2), 3.51 (t, 2H, –CH2), 3.71 (t, 2H, –CH2), 4.17 (q, 1H, C–H), 4.56 (d, 2H, –CH2), 4.62 (s, 2H, –CH2), 6.60 (d, 2H, Ar–H), 6.43 (d, 2H, Ar–H), 6.76 (d, 2H, Ar–H), 6.78 (d, 2H, Ar–H), 7.25 (d, 1H, Ar–H), 7.27 (d, 2H, Ar–H), 7.28 (s, 1H, Ar–H), 7.42 (d, 1H, Ar–H), 7.52 (d, 1H, Ar–H), 7.54 (s, 1H, Ar–H), 8.07 (d, 1H, Ar–H), 8.61 (s, 1H, N–H), 8.90 (d, 1H, Ar–H).SC-XRD studies
Single-crystal data of MIC-C were collected on a Rigaku Saturn 724+ CCD diffractometer using a graphite monochromator (Mo Kα, λ = 0.71075 Å). The selected crystals were mounted on the tip of a glass pin using mineral oil and placed in the cold flow produced by nitrogen gas. Complete hemispheres of data were obtained utilizing scans (0.3°, 16 s each frame). Rigaku Crystal Clear-SM Expert 2.1 software was used to calculate integrated intensities, which were then adjusted for absorption. SHELX software was used to solve and improve the structure. The structures were solved by direct methods and completed by iterative cycles of ΔF syntheses and full-matrix least-squares refinement against F. The crystal refinement parameters of MIC-C are provided in Table ES7.† Multiple attempts to diffract the molecular salt KTC-C were unsuccessful as the salts were too poor for evaluation.Powder X-ray diffraction
The P-XRD data were collected using a Rigaku D/Max-2500 X-ray diffractometer (Cu Kα radiation, λ 1.5406 Å) at 100 mA and 40 kV. The samples were scanned in reflection mode over the 2θ range of 5–50° with a step size of 0.02° at a scan rate of 8° min−1.Solubility studies
All absorbance values were calculated using a Shimadzu UV-2600 UV–visible spectrophotometer. To create the calibration standards, volumetric flasks were filled with precisely weighed amounts of the reference samples. The necessary amount of water was added to fill the volume, and the resulting solutions were sonicated to produce homogeneous solutions. A standard calibration curve was built using the stock solutions. These standard calibration curves were used to calculate the molar extinction coefficients (ε). Water was added to the volumetric flask along with the products until the saturation point was reached. For the absorbance measurements, the resulting solution was filtered and transferred to another volumetric flask. The concentrations of the products dissolved were calculated using the Beer–Lambert equation: A = εCl (A = absorbance measured using a UV–visible spectrophotometer, ε = molar extinction coefficient calculated from standard calibration curves, C = concentration of compounds dissolved, and l = path length).The apparent solubility was studied using a UV-spectrophotometer-2600 instrument. The precursors along with salts were taken at different pH values of 2 and 7 with concentrations ranging from 1 × 10−6 to 1 × 10−3 M to obtain the molar absorptivity values from their known concentrations using the Beer–Lambert law:
| A = εCl |
Thereafter, an unknown amount of substance is taken to obtain the absorbance and the value of the molar absorptivity constant (calculated above) is used to calculate the concentration of the substance.30
Reagents, drugs, cell lines, yeast strains, and growth conditions
The reagents of molecular biology or cell culture grade were utilized in this study. All the drugs (ketoconazole; KTC and miconazole; MIC) were purchased from Sigma-Aldrich. Dulbecco's modified Eagle's medium (DMEM), RPMI (Roswell Park Memorial Institute 1640 medium), and fetal bovine serum (FBS) were procured from Gibco and Thermo Fisher Scientific, United States, respectively. Dulbecco's modified Eagle's medium (DMEM, catalog # 11965-092, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin was used to culture all the mammalian cells (human embryonic kidney cell line HEK293 and the human lung carcinoma epithelial cells Hep-G2) employed in this study at 37 °C under 5% CO2 conditions. The cell lines were procured from the Cell Repository of National Centre for Cell Sciences (NCCS), Pune, India.Likewise, all the yeast strains [wild-type Candida (C. albicans ATCC-24433, C. glabrata ATCC-2001, C. parapsilosis ATCC 90018, and C. auris CBS10913) and drug transporter over-expressing yeast strains (ADCDR1-GFP, ADPDR5-GFP, and ADCaMDR1-GFP)] employed in the study were grown in yeast extract-peptone-dextrose (YEPD) media. Before each experiment, the strains were subcultured on YEPD solid medium (1% yeast extract, 2% peptone, 2% glucose, and 2% agar) at least twice at 37 °C to guarantee their viability and purity.
Drug susceptibility testing
Drug susceptibility was assessed by determining the minimal inhibitory concentration (MIC90) of the compounds and by performing spot and disc diffusion assays as explained below:Checkerboard assay
The checkerboard assay was carried out as previously described30 to check the possible interactions of KTC, KTC-C, MIC, and MIC-C in combination with conventional antifungal drugs at their sub-inhibitory concentrations. The method was performed to determine the fractional inhibitory concentration index (FICI) to study the possible interactions between the two compounds.Growth curve analysis
Briefly, following an overnight primary culture at 30 °C, a secondary culture was established [1 × 106 cells per ml (OD600 = 0.1)] in 50 ml YEPD liquid culture medium. After this, the respective drugs (at sub-MIC dosage) were added to this culture, and the cells were incubated at 30 °C. The drug-free culture served as growth control. Finally, the growth was observed by measuring OD600 at different time points (0, 3, 6, 9, 12…30 hours) by using a UV−Vis spectrophotometer.Drug cytotoxicity assay
An MTT (methyl thiazole tetrazolium)-based assay as previously described was performed to evaluate the cell viability of the cultured mammalian cell lines in the presence of drugs (KTC, KTC-C, MIC, and MIC-C).30 Briefly, the cells (HEK293 and Hep G2) were plated at a density of 3 × 103 cells per well in 96-well plates by using a hemocytometer, and upon reaching the desired confluence (90–95%), the cells were incubated with various drug concentrations of KTC, KTC-C, MIC, and MIC-C (ranging from 0.0625 to 8 μg mL−1) or vehicle alone (DMSO) for 24 hours in complete medium under standard culture conditions. After 24 hours of incubation period, the drug solutions were replaced with fresh media containing MTT 5 mg ml−1, and the growth inhibition was assessed based on the formula below:| % Inhibition = [1 − (OD of treated cells/OD of vehicle control cells)] × 100 |
Proteinase activity determination
The assay was performed as described previously.13,30 Briefly, solid YBD medium (yeast extract, bovine serum albumin, and glucose) plates in the presence (drug plates) or absence (control plates) of drugs were inoculated with C. albicans and C. auris strains (5 μL; OD600 = 0.1) cultured overnight in YEPD medium at 30 °C. The plates were then incubated at 37 °C for 4 days and the diameters of the colonies (a) and the clearance zone surrounding the colonies (b) were measured to assess the proteinase activity.Vitality and mortality assays
Fluorescent probes/dyes carboxyfluorescein diacetate (CFDA) (vitality-specific dye) and bis-(1,3-dibutyl barbituric acid) trimethine oxonol [DiBAC4(3)] (mortality-specific dye) were used to measure the physiological parameters like cytoplasmic and cell membrane integrity/intracellular enzyme activity and plasma membrane depolarisation.40,44,45Plasma membrane depolarisation/membrane potential alterations
To determine the alteration in the C. albicans cell membrane potential/polarisation state, the fluorescent dye DiBAC4(3) was used as described previously with some modifications.40,44,45 Briefly, aliquots of fungal cell suspensions (1 × 106 cells per ml (OD600 = 0.1) that had been pretreated for 1 hour with various concentrations of KTC, KTC-C, MIC, and MIC-C (at MIC/2) were mixed with the DiBAC4(3) dye (1.0 μM) and incubated for one hour in the dark and then washed with PBS. Finally, the intensity of the fluorescence was measured at an excitation wavelength of 488–492 nm and an emission wavelength of 510–518 nm using a fluorescence spectrophotometer.Cytoplasmic and cell membrane integrity/intracellular enzyme activity assay
The cytoplasmic and cell membrane integrity/intracellular enzyme activity was monitored with the fluorogenic substrate CFDA as described previously with some modifications.40,44,45 Briefly, aliquots of fungal cell suspensions (1 × 106 cells per ml; OD600 = 0.1) that had been pretreated for 1 hour with various concentrations (at MIC/2) of the compounds (KTC, KTC-C, MIC, and MIC-C) were incubated for one hour in the dark in the presence of CFDA dye (1.0 μM). The cells were then washed with PBS and finally, the intensity of the fluorescence was measured at an excitation wavelength of 490 nm and an emission wavelength of 520 nm using a fluorescence spectrophotometer.Author contributions
Conceptualization: AHS and AAD; formal analysis: HQ, AAM, AHS, and AAD; funding acquisition: AHS and AAD; investigation: HQ, AAM, and AAA; methodology: HQ, AAM, AAA, AHS, and AAD; supervision: AHS, AAD, and MAM; writing – original draft: HQ, AAM, and AAA; and writing – review & editing: AHS and AAD.Data availability
CIF files have been deposited with the Cambridge Crystallographic Data Centre (CCDC) viahttps://deposit.ccdc.cam.ac.uk/ under CCDC 2305701.† The remaining data supporting this article (and not included in the main file) have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by grants to AHS by the Department of Science and Technology (DST)-GoI under the INSPIRE Faculty Award (DST/INSPIRE/04/2015/001575) and the Science and Engineering Research Board (SERB)-GoI under Core Research grant (CRG/2023/002966). A. A. D. acknowledges the support of SERB-DST, New Delhi, in the form of a Core Research Grant (CRG/2022/003693). The funders have no role in “study design, data collection, and analysis, the decision to publish, or manuscript preparation.” HQ acknowledges the Senior Research Fellowship from the ICMR SFR (2021-9917) Project. A. A. M. and A. A. D. acknowledge the Department of Chemistry, University of Kashmir, for the facilities and support. The authors also acknowledge Dr Khalid Z. Masoodi, Transcriptomics Laboratory, SKUAST-K, for help with fluorescence microscopy. The authors would also like to acknowledge Prof. Raies Ahmad Qadri, Prof. Firdous A. Khanday, and Dr Altaf Bhat, University of Kashmir, for helping us with the work on human cell lines.References
- H. Qadri, A. H. Shah, S. M. Ahmad, B. Alshehri, A. Almilaibary and M. A. Mir, Saudi J. Biol. Sci., 2022, 29, 103376 CAS.
- H. Qadri, A. H. Shah and M. Mir, Curr. Drug Targets, 2021, 22, 1424–1436 CAS.
- Y. Lee, E. Puumala, N. Robbins and L. E. Cowen, Chem. Rev., 2020, 121, 3390–3411 CrossRef PubMed.
- H. Wisplinghoff, T. Bischoff, S. M. Tallent, H. Seifert, R. P. Wenzel and M. B. Edmond, Clin. Infect. Dis., 2004, 39, 309–317 CrossRef.
- M. A. Pfaller, D. J. Diekema, J. D. Turnidge, M. Castanheira and R. N. Jones, in Open forum infectious diseases, Oxford University Press, US, 2019, vol. 6, no. supplement_1, pp. S79–S94 Search PubMed.
- G. D. Brown, D. W. Denning, N. A. Gow, S. M. Levitz, M. G. Netea and T. C. White, Sci. Transl. Med., 2012, 4, 165rv113–165rv113 Search PubMed.
- G. D. Brown, D. W. Denning and S. M. Levitz, Science, 2012, 336, 647–647 CrossRef CAS.
- H. Qadri, M. F. Qureshi, M. A. Mir and A. H. Shah, Microbiol. Res., 2021, 247, 126725 CrossRef CAS.
- T. Roemer and D. J. Krysan, Cold Spring Harbor Perspect. Med., 2014, 4(5), a019703 CrossRef.
- D. S. Perlin, R. Rautemaa-Richardson and A. Alastruey-Izquierdo, Lancet Infect. Dis., 2017, 17, e383–e392 CrossRef.
- J. R. Perfect, Nat. Rev. Drug Discovery, 2017, 16, 603–616 CrossRef CAS PubMed.
- N. M. Revie, K. R. Iyer, N. Robbins and L. E. Cowen, Curr. Opin. Microbiol., 2018, 45, 70–76 CrossRef CAS.
- H. Qadri, A. H. Shah, M. A. Mir, M. F. Qureshi and R. Prasad, Fungal Genet. Biol., 2022, 161, 103713 CrossRef CAS.
- A. K. Gupta, M. Venkataraman and E. M. Quinlan, In Dermatophytes and Dermatophytoses, 2021, pp. 433–471 Search PubMed.
- E. Grothe, H. Meekes, E. Vlieg, J. Ter Horst and R. D. de Gelder, Cryst. Growth Des., 2016, 16, 3237–3243 CrossRef CAS.
- N. K. Duggirala, M. L. Perry, Ö. Almarsson and M. J. Zaworotko, Chem. Commun., 2016, 52, 640–655 RSC.
- A. A. Dar and S. Rashid, CrystEngComm, 2021, 23, 8007–8026 RSC.
- A. Mukherjee, Cryst. Growth Des., 2015, 15, 3076–3085 CrossRef CAS.
- G. Bolla and A. Nangia, Chem. Commun., 2016, 52, 8342–8360 RSC.
- G. Bolla, B. Sarma and A. K. Nangia, Chem. Rev., 2022, 122, 11514–11603 CrossRef CAS.
- M. Guo, X. Sun, J. Chen and T. Cai, Acta Pharm. Sin. B, 2021, 11, 2537–2564 CrossRef CAS.
- A. A. Ganie, P. Vishnoi and A. A. Dar, Cryst. Growth Des., 2019, 19, 2289–2297 CrossRef CAS.
- A. A. Ganie, A. A. Ahangar and A. A. Dar, Cryst. Growth Des., 2019, 19, 4650–4660 CrossRef CAS.
- A. A. Ganie and A. A. Dar, Cryst. Growth Des., 2021, 21, 3014–3023 CrossRef CAS.
- A. A. Dar and A. A. Ganie, Cryst. Growth Des., 2020, 20, 3888–3897 CrossRef CAS.
- A. A. Ahangar, R. Elancheran and A. A. Dar, J. Solid State Chem., 2022, 314, 123382 CrossRef CAS.
- A. A. Ganie, S. Rashid, A. A. Ahangar, T. M. Ismail, P. Sajith and A. A. Dar, Cryst. Growth Des., 2022, 22, 1972–1983 CrossRef CAS.
- A. A. Ganie, A. A. Ahangar, A. Dhir, A. K. Gupta and A. A. Dar, J. Phys. Chem. C, 2023, 127, 9257–9267 CrossRef CAS.
- A. A. Ahangar, A. A. Malik, I. Ahmad and A. A. Dar, Dyes Pigm., 2023, 111742 CrossRef CAS.
- A. A. Ahangar, H. Qadri, A. A. Malik, M. A. Mir, A. H. Shah and A. A. Dar, Mol. Pharm., 2023, 20, 3471–3483 CrossRef CAS.
- H. Yu, L. Zhang, M. Liu, D. Yang, G. He, B. Zhang, N. Gong, Y. Lu and G. Du, Pharmaceuticals, 2023, 16, 1349 CrossRef CAS.
- S. Hiendrawan, A. W. Hartanti, B. Veriansyah, E. Widjojokusumo and R. R. Tjandrawinata, Int. J. Pharm. Pharm. Sci., 2015, 7, 160–164 CAS.
- F. A. Martin, M. M. Pop, G. Borodi, X. Filip and I. Kacso, Cryst. Growth Des., 2013, 13, 4295–4304 CAS.
- K. Drozd, A. Manin, A. Voronin, D. Boycov, A. Churakov and G. Perlovich, Phys. Chem. Chem. Phys., 2021, 23, 12456–12470 RSC.
- S. Tsutsumi, M. Iida, N. Tada, T. Kojima, Y. Ikeda, T. Moriwaki, K. Higashi, K. Moribe and K. Yamamoto, Int. J. Pharm., 2011, 421, 230–236 CrossRef CAS.
- K. V. Drozd, A. N. Manin, D. E. Boycov and G. L. Perlovich, Pharmaceutics, 2022, 14, 1107 CrossRef CAS.
- S. Kumar, R. Nanda, P. Kuttepali and S. Sharma, Int. J. Pharm. Sci. Res., 2014, 5, 263 Search PubMed.
- A. Sharapova, S. Blokhina, M. Ol'khovich and G. Perlovich, J. Mol. Liq., 2022, 347, 118248 CrossRef CAS.
- X. Qiu, Y. Tian, W. Deng, F. Li, J. Hu, W. Deng, J. Chen, G. Zou, H. Hou and Y. Yang, Waste Manage., 2021, 136, 1–10 CrossRef CAS.
- N. Zorić, I. Kosalec, S. Tomić, I. Bobnjarić, M. Jug, T. Vlainić and J. Vlainić, BMC Complementary Altern. Med., 2017, 17, 1–10 CrossRef.
- J. Peter, D. Armstrong, C. A. Lyman and T. J. Walsh, J. Clin. Microbiol., 2005, 43, 3788–3792 CrossRef CAS PubMed.
- A. Ramzan, S. A. Padder, K. Z. Masoodi, S. Shafi, I. Tahir, R. U. Rehman, R. Prasad and A. H. Shah, Eur. J. Med. Chem., 2022, 240, 114609 CrossRef CAS PubMed.
- E. K. Dennis and S. Garneau-Tsodikova, Med. Mycol., 2019, 57, 874–884 CrossRef CAS.
- R. S. Liao, R. P. Rennie and J. A. Talbot, Antimicrob. Agents Chemother., 1999, 43, 1034–1041 CrossRef CAS.
- F. D. Gonelimali, J. Lin, W. Miao, J. Xuan, F. Charles, M. Chen and S. R. Hatab, Front. Microbiol., 2018, 9, 1639 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallographic data table of MIC-C, merged FT-IR spectra of MIC-C and KTC-C, 1H NMR spectra of MIC-C and KTC-C, TGA-DSC curves of MIC-C and KTC-C, and P-XRD plot of KTC-C. Combinatorial impact of salts compared with that of standard anti-fungals, disc diffusion and spot assays, and zone of inhibition and proteinase activity measurements. CCDC 2305701. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4pm00003j |
| ‡ Equal first authorship. |
| This journal is © The Royal Society of Chemistry 2024 |